

Abstract
Unlike the proliferative action of other EGF receptor family members, HER4/ErbB4 is often associated with growth inhibitory and differentiation signaling. These actions may involve HER4 two-step proteolytic processing by intra-membraneous γ-secretase, releasing the soluble, intracellular 80kDa HER4 cytoplasmic domain, s80HER4. We demonstrate that pharmacologic inhibition of either γ-secretase activity or HER4 tyrosine kinase activity blocked heregulin-dependent growth inhibition of SUM44 breast cancer cells. We next generated breast cell lines stably expressing GFP-s80HER4 (GFP fused to the N-terminus of the HER4 cytoplasmic domain, residues 676–1308), GFP-CTHER4 (GFP fused to N-terminus of the HER4 C-terminus distal to the tyrosine kinase domain, residues 989–1308) or GFP alone. Both GFP-s80HER4 and GFP-CTHER4 were found in the nucleus, but GFP-s80HER4 accumulated to a greater extent and sustained its nuclear localization. s80HER4 was constitutively tyrosine phosphorylated and treatment of cells with a specific HER family tyrosine kinase inhibitor i) blocked tyrosine phosphorylation; ii) markedly diminished GFP-s80HER4 nuclear localization, and iii) reduced STAT5A tyrosine phosphorylation and nuclear localization as well as GFP-s80HER4:STAT5A interaction. Multiple normal mammary and breast cancer cell lines, stably expressing GFP-s80HER4 (SUM44, MDA-MB-453, MCF10A, SUM102, and HC11) were growth inhibited compared to the same cell line expressing GFP-CTHER4, or GFP alone. The s80HER4-induced cell number reduction was due to slower growth, as rates of apoptosis were equivalent in GFP, GFP-CTHER4, and GFP-s80HER4 expressing cells. Lastly, GFP-s80HER4 enhanced differentiation signaling as indicated by increased basal and prolactin-dependent β–casein expression. These results indicate that surface HER4 tyrosine phosphorylation and ligand-dependent release of s80HER4 are necessary, and s80HER4 signaling is sufficient for HER4-dependent growth inhibition.
Keywords: HER4, ErbB4, cytoplasmic domain, cell proliferation, mammary cell
INTRODUCTION
Drosophilia and C. elegans genomes encode a single EGF receptor-like molecule. Depending upon the cellular context and the expression of ligand or other accessory molecules, the single EGF receptor enhances cell proliferation or inhibits growth, and stimulates differentiation (1). Mammalian genomes contain four members of this receptor tyrosine kinase family: EGFR/HER1/ErbB1, HER2/ErbB2, HER3/ErbB3, and HER4/ErbB4. These four members, together with at least 10 ligands from two ligand families (the EGF and heregulin/neuregulin families), regulate numerous cellular functions, the most studied of which is proliferation but also include cell survival, motility, adhesion, differentiation and cell cycle inhibition (2–6). This complexity is, in part, due to the multiple ligands, which bind to receptors to produce receptor homodimers or heterodimers, with virtually all potential combinations of the four receptors. These activated receptor complexes stimulate well-known signaling cascades, including Ras-Raf MAP Kinase pathway and the PI3 kinase pathway (2–6). However, multiple other signaling pathways must be involved to achieve the diversity of biologic outcomes.
All four family members are expressed in breast epithelium and in many breast cancers. The EGF receptor, HER2 and HER3, appear, in general, to be involved in breast epithelial cell proliferation (3). In the mouse, the EGF receptor, HER2 and HER3, regulate mammary epithelial cell proliferation during puberty, while HER4 is activated during late pregnancy and lactation, and signals for differentiation (7–9). EGFR and HER2 have been studied extensively in experimental breast cancer models, as well as in human breast cancer samples. HER2 and EGFR overexpression or activation is associated with poor prognosis breast cancer, and molecular therapies targeting EGFR or HER2 have gained attention and, in some instances, success for the treatment of human breast cancer (3–5, 10).
HER4 was the last member of the family identified (11) and its relationship to breast cancer prognosis is still being defined (12). Most studies correlate HER4 expression with estrogen receptor positivity, lower tumor grade, and a better prognosis (13–16), but some studies report a poorer prognosis in subsets of HER4 positive breast cancers (17, 18). Newer findings regarding HER4 isoforms and their unique signaling and cellular processing may eventually explain these discrepancies in clinical correlation. HER4 RNA is alternatively spliced to yield four isoforms that may vary in signaling capability (19–21). Just proximal to the transmembrane region, an alternative splice creates the JM-a or JM-b isoform. JM-a, but not JM-b, is susceptible to proteolytic cleavage by tumor necrosis factor alpha converting enzyme (TACE) (22, 23). Several groups have shown that cleavage by TACE releases the extracellular domain and leads to a stochastic, second intramembrane cleavage event, performed by a γ-secretase-like molecule of the presenilin family (24, 25). This type of cleavage is characteristic of Notch, another transmembrane protein involved in growth and differentiation signaling (24). TACE leaves a membrane-associated m80kDaHER4, while the second, γ-secretase intramembraneous cleavage, releases the 80 kDa domain into the cytoplasm. Once released, three canonical nuclear localization sequences (NLS) and three nuclear export sequences (NES) can result in appearance of s80HER4 in the nucleus of tested cells (25).
Our group and others have shown that, in many HER4-expressing breast cells, heregulin treatment inhibits cell growth and can induce differentiation (26–31). Data from genetically engineered mice also suggests that HER4 is involved in mammary cell differentiation (9, 32), as mammary-specific HER4 gene deletion or inhibition by dominant negative HER4 expression retards mammary gland development and function. In contrast to studies demonstrating HER4-dependent growth inhibition, some reports show that HER4 activation can stimulate growth and cell survival (33–35), although several of these reports used other HER4 isoforms that may have distinct properties. How HER4 proteolytic processing impacts growth inhibition, differentiation, or proliferation signaling, and how it may relate to tumor suppression progression, or prognosis remains to be fully elucidated. Immunostaining, using C-terminus HER4 antibodies, reveals HER4 (presumably s80HER4) in the nucleus of normal human and mouse mammary cells, and nuclear HER4 has been detected by immunohistochemistry in breast cancer samples. In most, but not all, studies, nuclear HER4 staining correlates with a better prognosis (16, 36, 37).
The initial reports describing the sequential cleavage of HER4 by TACE and γ–secretase demonstrated its importance for growth inhibition of T47D breast cancer cells. But TACE and γ–secretase have also been reported to involve the cleavage of a number of other proteins including Notch (24), E-cadherin (38), CD44 (39), nectin-1α (40), and syndecan 3 (41), which may also influence mammary cells. Evidence of growth inhibition by the s80HER4 Cyt1 isoform across a range of normal and neoplastic breast cells is still lacking. In addition, several reports examining gene transactivation indicated that the HER4 C-terminus (beyond the tyrosine kinase domain) (25, 42) and the C-terminus of the EGF receptor (43) or HER2 (44), when fused to GAL4 DNA binding domain, are capable of stimulating GAL4 transactivation. This leaves open the possibility that the distal C-terminus may be the active moiety in nuclear HER4 signaling.
To further study the biologic signaling capabilities of the intracellular HER4 fragments, we have stably introduced GFP-tagged s80HER4 or the GFP-tagged HER4 C-terminus (the last 320 amino acids) into multiple normal mammary epithelial cells and breast cancer cells. We report that s80HER4 is constitutively tyrosine phosphorylated. In addition, the full cytoplasmic domain, s80HER4, which is kinase active, but not the HER4 C-terminus, inhibited cell proliferation, transactivated the promoter for the mammary specific gene, β-casein, and increased basal and prolactin-dependent β-casein mRNA expression. s80HER4-dependent growth inhibition was due to slower growth rather than apoptosis. s80HER4 accumulates in the nucleus to a greater extent than CTHER4, and inhibition of s80HER4 kinase activity diminished its nuclear localization and interaction with STAT5A, as well as tyrosine phosphorylation and nuclear localization of STAT5A.
RESULTS
γ-secretase inhibition abolishes heregulin-dependent SUM44 cell growth inhibition
We previously showed that both heregulin and HB-EGF inhibited the growth of SUM44 and other HER4-expressing breast cancer cells (31). Since SUM44 cells do not express EGFR and selective loss of HER2, HER3 tyrosine phosphorylation does not block the growth inhibitory action of ligand, HER4 is responsible for transmitting this signal (31,45). However, because a unique cellular processing scheme for certain HER4 isoforms has been described, the role of processing HER4 needs to be examined. This ligand-dependent, two-step proteolytic process releases the 80 kDa HER4 intracellular domain (s80HER4) into the cytoplasm. The cleavable isoform, JM-a, is expressed in SUM44 and could be subject to the extracellular domain cleavage performed by a TACE-like activity (22, 23, 46), followed by a second intramembranous cleavage, performed by a γ-secretase-like intramembraneous enzyme (25, 47). To determine if surface HER4 or intracellularly released s80HER4 is responsible for growth inhibition, we assessed whether HER4 cleavage and s80HER4 formation were required for this heregulin-dependent action. We first pre-incubated SUM44 with 100 nM HEDI, a commercially available γ-secretase inhibitor, for one hour prior to adding heregulin (10 ng/ml). Heregulin treatment diminished the increase of SUM44 cell number over a six-day culture period by approximately 50%; however, incubation with the γ-secretase inhibitor abolished this anti-proliferative effect (Fig. 1). Similar results were obtained by Carpenter and co-workers, using the γ-secretase inhibitor compound E in T47D breast cancer cells (25). Thus, γ-secretase activity is necessary for heregulin-dependent growth inhibition, but it remained formally possible that the γ-secretase activity blockade inhibited another signaling pathway (e.g. Notch, E-cadherin, or syndecan 3 cleavage) and not the HER4 cytoplasmic domain release.
Fig. 1. Inhibition of γ-secretase blocks heregulin-induced growth inhibition.

SUM44 cells grown in fully complemented medium were treated with heregulin (10 ng/ml) or not in the presence or absence of HEDI at the concentrations shown for 6 days, then counted. Mean ± SD of triplicates are shown, representative of 3 independent experiments. Student’s t-test was used for statistical analysis. ***, P < 0.001 versus control.
Inhibition of HER4 tyrosine phosphorylation blocks the heregulin-dependent anti-proliferative effect
The process by which s80HER4 formation is regulated by ligand is not fully elucidated. Treatment of several cell lines with the phorbol ester, TPA, induces TACE-dependent HER4 extracellular domain cleavage, leaving the transmembrane anchor and a membrane-bound 80kDa HER4 (m80HER4) (22, 48). This species, at some rate, is released as s80HER4 by γ-secretase cleavage at a valine residue just inside the cytoplasmic membrane surface (25, 47). Presumably ligand-dependent (heregulin, HB-EGF, etc.), release of s80HER4 also requires TACE activation, followed by γ-secretase cleavage. The ligand-dependent cleavage process is either inefficient or subject to other control mechanisms, as ligand-dependent HER4 activation leaves the majority of HER4 at the cell surface as 180kDa holoenzyme for four or more hours (data not shown). Thus, inhibition of HER4 tyrosine kinase activation could block heregulin-dependent anti-proliferative effects, either by preventing ligand-dependent s80HER4 release or by inhibiting a necessary, sustained tyrosine kinase-dependent HER4 action at the cell surface, or both.
We investigated the necessity of HER4 tyrosine kinase activation in growth inhibition by incubating SUM44 cells for 60 minutes with the pan-EGF receptor family inhibitor, GW572016 (lapatinib), prior to addition of heregulin. In SUM44 cells (which do not express the EGF receptor), incubation with increasing doses of GW572016 resulted in inhibition of heregulin-dependent tyrosine phosphorylation of HER2, HER3 and HER4, all at similar doses, with near-maximal inhibition at 1.0 μM (Fig. 2A). This dose was used in SUM44 growth studies, and experiments demonstrated that 1.0 μ GW572016, incubated with cells for six days, nearly abolished heregulin-dependent growth inhibition (Fig. 2B). Taken with previous findings that HER4 homodimers mediate heregulin-induced growth inhibition, these results suggest that HER4 tyrosine phosphorylation is necessary for the growth inhibitory effects of HER4. Therefore, both tyrosine phosphorylation of HER4 and a γ-secretase-sensitive step appear necessary for HER4 growth inhibition.
Fig. 2. Inhibition of HER4 tyrosine phosphorylation blocks heregulin-induced growth inhibition.
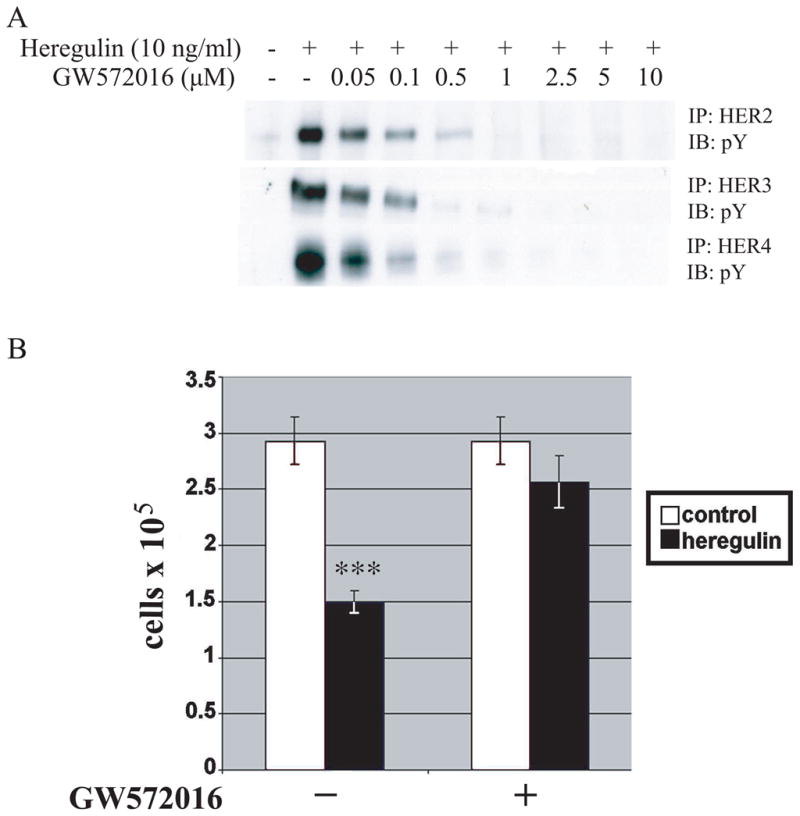
A, SUM44 cells were cultured for 6 days, treated with different doses of GW572016 for 1 h, and then with or without heregulin (10 ng/ml) for 15 minutes. The cells were lysed and the lysates were immunoprecipated with anti-HER2, anti-HER3, or anti-HER4 antibody and blotted with anti-phosphotyrosine antibody. B, SUM44 cells were grown with or without heregulin (10 ng/ml) in the presence or absence of GW572016 (1 μM) for 6 days, at which time the number of cells were counted. Mean ± SD of triplicates are shown, representative of 3 experiments. ***, P < 0.001 versus control.
Ectopic expression of GFP-s80HER4 and GFP-CTHER4
Because γ-secretase mediates release of s80HER4, and because inhibition of γ-secretase impaired heregulin-mediated growth inhibition, we studied whether s80HER4 could transduce the HER4 anti-proliferative effects in experiments that used the following constructs: 1) GFP-CTHER4, which consists of GFP fused at the N-terminus of the HER4 carboxy terminus (residues 989–1308); 2) GFP-s80HER4, GFP fused at the N-terminus of the entire HER4 cytoplasmic domain (residues 676–1308); and 3) and GFP alone (Fig. 3A schematic). We chose to place GFP at the N-terminus to avoid fusion to the HER4 C-terminal PDZ-binding domain, which may play a role in HER4 action. Note that GFP-CTHER4 lacks the kinase domain. These three constructs were cloned into pMSCV vector and packaged as retroviruses with VSVG coat proteins to enhance infectivity of epithelial cells. After infection and selection of pooled clones of stably transfected cells with puromycin, the level of expression was determined by immunoblotting SUM44 cell lysates with GFP antibody (Fig. 3B). GFP was expressed at high levels; GFP-CTHER4 and GFP-s80HER4 were expressed at lower but similar levels. Immunoprecipitation of HER4 with a polyclonal antibody directed at the HER4 C-Terminus, followed by immunoblotting with anti-phosphotyrosine antibody, demonstrated that GFP-s80HER4 was constitutively tyrosine phosphorylated, whereas GFP-CTHER4 was not (Fig. 3C). Carpenter has also recently reported that s80HER4 is constitutively tyrosine phosphorylated, and can homodimerize (49).
Fig. 3. Ectopic expression of GFP-s80HER4 and GFP-CTHER4.

A, Schematic representation of full-length HER4, GFP, GFP-CTHER4, and GFP-s80 HER4. B, Western analysis using anti-GFP antibody of lysates from SUM44 cells infected with retrovirus encoding GFP, GFP-CTHER4, or GFP-s80HER4. C, SUM44 cells stably expressing GFP, GFP-CTHER4, or GFP-s80HER4 were treated with or without heregulin (10 ng/ml) for 15 minutes, and lysed. The lysates were immunoprecipated with anti-HER2, anti-HER3, or anti-HER4 antibody and blotted with anti-HER2, anti-HER3, or anti-HER4 antibody respectively, or with anti-phosphotyrosine antibody. D, HC11 cells stably expressing GFP, GFP-CTHER4, or GFP-s80HER4 were treated with ± increasing concentrations of GW572016 for 1 h (top panel), or treated with different concentration of GW572016 for 1 h, then ± heregulin (10 ng/ml) for 15 minutes (bottom panel), and lysed. The lysates were immunoprecipated with anti-HER4 antibody and blotted with anti-phosphotyrosine antibody. E, COS-7 cells were transfected with full-length HER4 or GFP-s80HER4. Twenty-four h after transfection, the cells were treated with ± increasing concentrations of GW572016 for 1 h, lysed, and analysed for the level of tyrosine phosphorylation of HER4 or GFP-s80HER4 by using anti-phosphotyrosine antibody, and the level of HER4 or GFP-s80HER4 by using anti-HER4 antibody. Densitometry is shown in arbitrary units.
Next, we determined whether expression of the three constructs altered the ligand-independent or ligand-dependent phosphorylation of HER2, HER3, and HER4. In the absence of heregulin, there was little to no basal tyrosine phosphorylation of the three endogenous full-length receptors. Addition of heregulin induced tyrosine phosphorylation of full-length HER2, HER3, and HER4 in cells expressing GFP-s80HER4, GFP-CTHER4, or GFP (Fig. 3C). Thus, the phosphorylation level of the cell surface, transmembrane HER4 is not altered by expression of either the constitutively tyrosine phosphorylated, presumably dimeric, kinase active GFP-s80HER4 or GFP-CTHER4, which lacks the kinase domain. Figure 3C shows that membrane HER2, HER3, and HER4 are expressed and ligand-dependent tyrosine phosphorylated at roughly similar levels. The protein level of s80HER4 and GFP-CTHER4 are somewhat greater than endogenous HER4. CT is not phosphorylated, but s80HER4 is constitutively tyrosine phosphorylated at levels slightly less than that of ligand activated endogenous 180kDa HER4. This indicates that the s80HER4 kinase is less active or less efficiently dimerized in this cellular contest. More importantly, Figure 3 demonstrates that in these stably tranfected cell lines, s80HER4 is not functionally overexpressed and that the biologic effects described below do not result from substantial overexpression of exogenous s80HER4 kinase.
We made five distinct breast cell lines stably expressing GFP-CTHER4 or GFP-s80HER4 to test the ability of s80HER4 and the HER4 C-terminus (without kinase) to alter growth. Having demonstrated that GW572016 can inhibit HER4 in SUM44, we used the mouse mammary immortalized but non-neoplastic cell line, HC11, to determine if GW572016 could inhibit heregulin-dependent, full-length HER4 tyrosine phosphorylation and s80HER4 constitutive tyrosine phosphorylation. GW572016 inhibited both full-length HER4 and s80HER4 tyrosine phosphorylation in a similar dose dependent manner (Fig. 3D). To confirm this, we expressed both HER4 and s80HER4 at similar level by transient expression in COS-7 cells and examined the relative Ki inhibition by GW572016. The results showed that full length HER4 and s80HER4 were similarly inhibited by this pan-EGFR family inhibitor (Fig. 3E).
Subcellular localization of GFP, GFP-CTHER4 and GFP-s80HER4
The localization of GFP, GFP-CTHER4 and GFP-s80HER4 was examined using both live cell (GFP) and fixed cell analysis. Using live cell microscopy (Fig. 4A), it was observed that GFP and GFP-CTHER4 were distributed in cytoplasm and nuclei of HC11 cells, whereas GFP-s80HER4 was more concentrated in nuclei. Confocal microscopy (Fig. 4B) confirmed that GFP-s80HER4 was strongly localized to nuclei whereas GFP and GFP-CTHER4 were distributed between the cytoplasm and nuclei. Addition of leptomycin B, an antibiotic that slows nuclear export, to HC11 enhanced GFP-CTHER4 nuclear localization; GFP-s80HER4 was principally nuclear in the presence or absence of leptomycin B in HC11 cells.
Fig. 4. Localization of GFP, GFP-CTHER4, and GFP-s80HER4 in HC11 cells.

A, Live cell microscopy showing the localization of GFP fluorescence in HC11 cells stably expressing GFP, GFP-CTHER4, or GFP-s80HER4. Phase contrast is shown in lower panels. Bar, 100 μm, applies to each image. B, HC11 cells stably expressing GFP (a-c, j-l), GFP-CTHER4 (d-f, m-o) or GFP-s80HER4 (g-i, p-r), were treated without (a-i), or with (j-r) 20 ng/ml of leptomycin B for 24 h, then analyzed for GFP fluorescence by confocal microscopy. Subcellular distribution of GFP (a, j), GFP-CT (d, m), GFP-s80 (g, p), and DAPI (b, e, h, k, n, q) were visualized and captured. The merged pictures were also shown (c, f, i, l, o, r). Bar, 30 μm, applies to each image.
We performed similar analyses in GFP, GFP-CTHER4, and GFP-s80HER4 expressing SUM44 cells. Live cell microscopy of SUM44 cells expressing these constructs demonstrated that both GFP and GFP-CTHER4 appeared to be nearly equally distributed between the nucleus and cytoplasm. In contrast, GFP-s80HER4 accumulated to a greater degree in SUM44 cell nuclei (supplementary Fig. 1A). Live cells show the variability of expression levels in the cell populations, especially in the GFP-CTHER4 and GFP-s80HER4 cell lines, even though all cells are maintained in puromycin to continue selective pressure. Examination of fixed SUM44 cells, using confocal microscopy (supplementary Fig. 1B) showed again that GFP and GFP-CTHER4 are diffusely distributed in the cytoplasmic and nuclear compartments, whereas GFP-s80HER4 is more intensely localized in nuclei. With the addition of leptomycin B, GFP-CTHER4 is more concentrated in SUM44 nuclei, while GFP-s80HER4 is again maintained in nuclei (supplementary Fig. 1B).
The results in HC11 and SUM44 cells indicate that GFP-s80HER4 is more often found in the nucleus than GFP or GFP-CTHER4, either by increased nuclear import or decreased nuclear export. It is clear that GFP-s80HER4 is not always nuclear, indicating that there are other processes that regulate this transport. In this study, we observed that overexpression of s80HER4 caused a change in cell shape in monolayer cultures of both SUM44 and HC11 cells; the mechanism for this change is unknown. In a previous study, we demonstrated that in 3D matrigel culture, s80HER4 has profound effects on shape and lumen formation, presumably by sending differentiation signals (50).
Effects of s80HER4 kinase activity to nuclear localization and STAT5A activity
To determine if the s80HER4 tyrosine kinase activity affects nuclear-cytoplasmic shuttling of s80HER4, we incubated HC11 GFP-s80HER4 cells without or with 0.5 μM GW572016, a dose that does not abolish s80HER4 tyrosine phosphorylation, or with 5 μM GW572016, a dose sufficient to substantially reduce s80HER4 tyrosine phosphorylation (see Figs. 3D and 3E). As shown in Fig. 5A, 0.5 μM GW572016 did not affect the localization of GFP-s80HER4, but 5 μM GW572016 dramatically reduced the nuclear accumulation of GFP-s80HER4, resulting in cytoplasmic accumulation. To test the specificity of GW572016 inhibition of nuclear localization, COS-7 cells were transfected with GFP-s80HER4, p53-GFP, or GFP-histone, then treated without or with 10 μM GW572016 for 24 h. Again, as shown in Fig. 5B, 10 μM GW572016 greatly reduced the nuclear localization of GFP-s80HER4, but had no effect on the nuclear localization of p53-GFP or GFP-histone (0.5 μM GW572016 did not affect the nuclear localization of GFP-s80HER4 in COS-7 cells, data not shown). Whether GW572016 has other effects (other than HER4 tyrosine kinase inhibition) that regulate nuclear localization cannot be ruled out, but recent analyses showed that GW572016 is highly specific for the ErbB family (51) and the doses used do not alter the nuclear localization of p53 or histone.
Fig. 5. Effect of GW572016 on the localization of GFP-s80HER4 in HC11 cells.

A, HC11 cells stably expressing GFP-s80HER4 were treated without, or with 0.5 μM or 5 μM GW572016 for 24 h. Cells on the cover slip were fixed, stained with DAPI, then analyzed for GFP and DAPI fluorescence. The merged pictures were also shown. Bar, 30 μm, applies to each image. B, COS-7 cells were transfected with GFP-s80HER4, p53-GFP, or GFP-histone, and treated without or with 10 μM GW572016 for 24 h. Then, the cells were fixed, stained with DAPI, and analyzed for GFP and DAPI fluorescence. The merged pictures were also shown. Bar, 30 μm, applies to each image.
It has been reported that: i) HER4 and STAT5A are essential for breast development and lactation (9, 32), ii) HER4 and STAT5A co-immunoprecipitate (52), and iii) s80HER4 and STAT5A co-localize in nuclei (50, 53). We determined if GW572016 inhibition affected the co-immunoprecipitation of GFP-s80HER4 and STAT5A in co-transfected cells, and if GW572016 affected STAT5A tyrosine phosphorylation and nuclear localization. Fig. 6A shows that GFP-CTHER4 did not associate with STAT5A, while GFP-s80HER4 did. 5 μM and 10 μM GW572016 greatly decreased the tyrosine phosphorylation of GFP-s80HER4, and resulted in the decrease of the co-immunoprecipitation of GFP-s80HER4 and STAT5A. Furthermore, 5 μM and 10 μM GW572016 decreased s80HER4 and STAT5A tyrosine phosphorylation (Fig. 6B), while 0.5 μM GW572016 did not. Lastly, 5 μM GW572016 decreased nuclear localization of STAT5A in HC11 s80HER4 expressing cells (0.5 μM GW572016 did not alter nuclear STAT5A localization, data not shown). These results indicate that s80HER4 kinase activity can regulate STAT5A localization. As can be seen in Figure 3E, 5 μM GW572016 decreases GFP-s80HER4 expression minimally, but the decrement in tyrosine phosphorylation, and thus STAT5A nuclear localization, is much greater. When 10 μM GW572016 is used, s80HER4 expression is suppressed by ~25%, but tyrosine phosphorylation is almost totally abolished. Thus, GW572016 at high doses may have off-target effects, but the kinase inhibition and STAT5A changes are seen at doses below this.
Fig. 6. Effect of GW572016 on STAT5A and GFP-s80HER4 co-immunoprecipitation, STAT5A tyrosine phosphorylation and STAT5A nuclear localization.

A, COS-7 cells were co-transfected with STAT5A and GFP-CTHER4 or STAT5A and GFP-s80HER4, and treated without or with increasing concentrations of GW572016 for 40 hours. Cells were lysed with regular lysis buffer (137mM NaCl), and lysates immunoprecipitated with anti-HER4 or anti-STAT5 antibody. Immunoprecipitates were subject to gel electrophoresis and transferred and blotted with anti-HER4, anti-STAT5, or anti-phosphotyrosine antibody as indicated. B, COS-7 cells were co-transfected with GFP-s80HER4 and STAT5A, and treated without or with different concentrations of GW572016 for 40 hours. Cells were lysed in high salt (500 mM NaCl) lysis buffer, and the lysates were immunoprecipitated with anti-HER4 or anti-STAT5 antibody, electrophoresed, transferred, and blotted with anti-phosphotyrosine, anti-STAT5 or anti-HER4 antibody. C, HC11 cells stably expressing GFP-s80HER4 were treated without or with GW572016 (5 μM) for 24 h. Cells were fixed and visualized for GFP or rhodamine (using a rhodamine-labeled second antibody following the first antibody-antiSTAT5). The merged pictures are also shown. Bar, 30 μm, applies to each image.
Biologic action of GFP-s80HER4; growth inhibition of multiple breast cell lines
The majority of our studies have been performed in the SUM44 cell line, which exhibits a higher HER4 expression level than most breast cancer lines. In our previously published survey, SUM44 had the most robust heregulin-dependent growth inhibition of human breast cell lines tested (31). Having shown in Figures 1 and 2 that pharmacologic inhibition of γ-secretase and HER4 tyrosine kinase activity abrogated heregulin-dependent growth inhibition, we determined whether constitutive expression of s80HER4 would result in ligand-independent growth inhibition. First, a time course of cell growth using SUM44 cells stably expressing GFP, GFP-CTHER4 and GFP-s80HER4 showed slower growth of SUM44 s80HER4 cells at 7 days (Fig. 7A). Multiple replicates of this experiment demonstrated that constitutive GFP-s80HER4 expression resulted in a statistically significant 50%, decrease in growth rate, compared to cells expressing GFP and GFP-CTHER4 (Fig. 7B). The extent of growth inhibition was similar to that seen with the addition of heregulin to parental SUM44 cells (Fig. 1 and ref. (31)). To determine if this effect was limited to SUM44 cells, we stably expressed GFP, GFP-CTHER4 and GFP-s80HER4 in the following lines: MDA-MB-453 and SUM102 cells; both of which are human breast cancer cell lines with low-level or absent HER4 expression, respectively; a non-transformed human mammary cell line, MCF10A; and the non-transformed mouse mammary epithelial cell line, HC11. Figure 7C shows cell growth comparisons for each line, stably expressing GFP, GFP-CTHER4 or GFP-s80HER4. In each instance, growth rates in GFP and GFP-CTHER4 were similar; expression of GFP-s80HER4 reduced growth by approximately 50%. Thus, the tyrosine kinase domain-bearing s80HER4 mimics growth inhibition observed with heregulin treatment in HER4-expressing cells.
Fig. 7. Ectopic expression of GFP-s80HER4, but not GFP-CTHER4, decreases cell proliferation.

A, Time course of cell proliferation of SUM44 cells stably expressing GFP, GFP-CTHER4, or GFP-s80HER4. SUM44 cells (5000 cells per well) stably expressing GFP, GFP-CTHER4, or GFP-s80HER4 were plated in 96-well plates and cultured. At indicated time points, the relative cell numbers (OD values) were measured using MTS assay. Percentage increased OD value at each time point over the 20h time point are shown (Mean ± S.E.M. of triplicates). B, Cell proliferation of SUM44 cells stably expressing GFP, GFP-CTHER4, or GFP-s80HER4 after 7 days incubation. The percentages of increased OD value at the end of the incubation (7 days) to that at 20 hours are shown. The error bar represented S.E.M. of five independent experiments, each analyzed in triplicate. Student’s t-test was used for statistical analysis. ***, P < 0.001 versus GFP control. C, MTS assays showing cell proliferation of MDA-MB-453, MCF10, HC11 and SUM 102 cells stably expressing GFP, GFP-CTHER4 or GFP-s80HER4. OD values of the cells stably expressing GFP, GFP-CTHER4 or GFP-s80HER4 were determined at 20h and the end of the cell culture (MDA-MB-453, 5 days; MCF10A, 8 days; HC11, 3 days; SUM102, 4 days) using MTS assay. The percentages of increased OD value at the end of the incubation to that at 20 h are shown (the error bar represented S.E.M of triplicate samples). *, P < 0.05; **, P < 0.01 versus GFP control.
The anti-proliferative effects of GFP-s80HER4 were not caused by excess apoptosis
Data of others suggest that transient transfection of s80HER4 result in decreased cell number by inducing caspace activity and apoptosis (54). We measured apoptosis in several cell lines in which stable expression of s80HER4 reduced cell number during 3–7 day growth experiments. SUM44 cells stably expressing GFP, GFP-CTHER4, or GFP-s80HER4 were cultured for six days in serum-free growth factor-defined media, trypsinized, and equal cell numbers were used to detect histone-associated DNA fragment (apoptosis) using a sensitive ELISA assay. No statistically significant differences were detected in apoptosis rates between GFP, GFP-CTHER4, or GFP-s80HER4 cells. As a positive control, GFP-expressing cells were treated with 1.0 μM camptothecin for the final 24 hours of the culture. Camptothecin increased apoptosis by nine-fold (Fig. 8A). Similarly, HC11 cells, stably expressing GFP, GFP-CTHER4, or GFP-s80HER4, were plated and cultured in complete medium for one day, and then replaced with serum-free growth factor-defined medium and cultured for two days. Then the cells were trypsinized and equal cell numbers were used to detect apoptosis. No difference in apoptotic rates was detected between the three HC11 lines. Camptothecin treatment again dramatically increased apoptosis in GFP-HC11 cells (Fig. 8B). In both experiments, equal cell numbers were used, even though GFP-s80HER4 cells grew at half the rate, and the equal cell number represented a larger proportion of the culture. Still, rates of apoptosis were the same, suggesting that the expression of s80HER4 inhibits cell number increase by mechanisms other than stimulating apoptosis. Other experiments using cell lines stably or inducibly expressing s80HER4 have failed to show sub 2N DNA content on cell cycle analyses (45) or positive tunnel assays.
Fig. 8. Stable expression of GFP-s80HER4 does not induce apoptosis.

A, SUM44 cells stably expressing GFP, GFP-CTHER4 or GFP-S80HER4 were cultured in serum-free medium for 7 days. GFP expressing cells were also treated with 1 μM of camptothecin for 24 h before harvesting the cells as a positive control. Equal cell numbers were analyzed for apoptosis by ELIZA, which quantitatively measures cytoplasmic histone-associated DNA fragments. Results are expressed as mean±S.E.M. of three independent experiments; each sample being analyzed in duplicate. The Student’s t-test was used for statistical analysis. **, P = 0.006 versus GFP control. B, HC-11 cells stably expressing GFP, GFP-CTHER4 or GFP-S80HER4 were analyzed for apoptosis after 3 days as described above. A second plate of GFP expressing cells was treated with 1 μM of camptothecin for the final 24 h of the incubation. **, P = 0.04 versus GFP control.
GFP-s80HER4, but not GFP-CTHER4, stimulates β-casein promoter activity and protein expression
Previous reports demonstrate that CTHER4, when fused to the GAL4 DNA binding domain, transactivated a GAL4 reporter system (25) (42). Data from other groups have shown that a similar C-terminal construct from EGFR and HER2 also transactivated the GAL4 promoter (43, 44). Recently, work showed that HER4, transfected with STAT5A, stimulated β-casein promoter activity in MCF7B cells (53). In other work, we have demonstrated ligand- and HER4-dependent β-casein transcription is abrogated by preventing HER4 cleavage and s80HER4 production (50). To determine if promoter activation is stimulated by CTHER4 or s80HER4, we transiently co-transfected parental HC11 cells with plasmids containing GFP, GFP-CTHER4, or GFP-s80HER4, and pβ casein-lux, a reporter construct used to detect STAT5A-dependent gene expression (50), or pGL3 vector without β–casein promoter as control. While co-transfection with GFP or GFP-CTHER4 produced similar β-casein promoter activity, co-transfection with GFP-s80HER4 increased luciferase transcription from the β-casein promoter (Fig. 9A). This indicates (using a mammary-specific promoter system) that the entire HER4 cytoplasmic domain is more effective than the HER4 C-terminus at increasing transactivation. We next checked the β-casein mRNA levels in HC11 cells stably expressing GFP, GFP-CTHER4, or GFP-s80HER4 using quantitative reverse transcription-PCR (qRT-PCR). In the basal state, β-casein mRNA level in GFP-s80HER4 expressing cells was more than five-fold above that in GFP or GFP-CTHER4 expressing cells. Addition of 5 μg/ml of prolactin resulted in huge increases in all the HC11 cells, but GFP-s80HER4 expressing cells still produced approximately two-fold more β-casein mRNA above that seen in GFP or GFP-CTHER4 expressing cells (Fig. 9B). Lastly, we showed that prolactin-stimulated β–casein protein expression was greater in HC11 cells stably expressing GFP-s80HER4 (Fig. 9C). The effect of tyrosine kinase active s80HER4 on STAT5A dynamics and on β-casein expression strongly suggests a role for s80HER4 in the events regulation of mammary cell differentiation.
Fig. 9. GFP-s80HER4, but not GFP-CTHER4, activates β–casein promoter and increases β–casein mRNA and protein expression.

A, HC11 cells transfected with plasmids containing GFP, GFP-CTHER4 or GFP-S80HER4, and pβcasein-lux, a reporter construct in which a human β–casein promoter was fused to the upstream of luciferase reporter gene, or pGL3 vector without β–casein promoter, were lysed 48 h after transfection. The lysates were immunoprecipitated with anti-HER4 antibody and blotted with anti-phosphotyrosine antibody, and anti-HER4 antibody (top panel). The lysates from above were also used to determine luciferase activity by luciferase assay system (Promega). Results are expressed as mean±S.E.M. of three independent experiments with each sample being analyzed in duplicate (bottom panel). The Student’s t-test was used for statistical analysis. ***, P < 0.001 versus GFP control or GFP-CTHER4. B, β–casein mRNA levels as determined by quantitative reverse transcription-PCR (qRT-PCR). HC11 cells stably expressing GFP, GFP-CTHER4, or GFP-s80HER4 were cultured in normal culture medium for 2 days, changed to serum-free complemented medium containing 5 μg/ml insulin for 2 days, then treated with or without 5 μg/ml prolactin in the complemented medium for 2 days. Total RNA was extracted and qRT-PCR was performed using β–casein specific fluorescence-labeled oligonucleotide probes (the error bar represented S.E.M of triplicate samples). **, P ≤ 0.01 versus GFP control. C, HC11 cells stably expressing GFP, GFP-CTHER4, or GFP-s80HER4 were cultured as above, and treated with or without 5 μg/ml prolactin in the complemented medium for 2 days. The cells were lysed and the lysates were immuno-blotted with anti-β-casein or anti-α-tubulin antibody.
DISCUSSION
The EGF receptor has been studied as the prototype for receptor tyrosine kinase proliferative signaling. The discovery that HER2/neu, the second member of the family, could be mutated to cause experimental carcinogenesis and was overexpressed in almost 20% of human breast cancers reinforced the idea that ErbB receptors regulate proliferation and, when activated, oncogenic progression. HER3/ErbB3 is often co-expressed with HER2 in human breast cancers and is implicated in proliferation and survival signaling.
HER4 is the one family member that has been associated with growth inhibition in many, but not all, cell culture models. We and others have used breast cancer cell lines under serum-free conditions to show that ligand-dependent activation of endogenous HER4 results in growth inhibition (32, (25) and, in responsive cells, a differentiation signal defined by the transcription of lactation genes (52). Our new results confirm the ability of HER4 to inhibit growth of breast cancer cells, and demonstrate that the intracellular domain of HER4, s80HER4, is sufficient to confer growth inhibition to a wide range of breast cells. However, there are examples in which ectopic expression or activation of HER4 results in a proliferative rather than a growth inhibitory response (37, 55); these have generally used non-breast cell models or an alternative, spliced cytoplasmic HER4 isoform (Cyt2).
Answers to the conundrum of differential HER4 responses in different cell types is beginning to emerge, with several discoveries. The first (by Elenius and co-workers) demonstrated multiple HER4 isoforms, resulting from alternative splicing in two regions, the extracellular juxtamembrane region (JM-a and JM-b) and a C-terminal insert region, CYT-1 (versus CYT-2), that changes signaling capabilities (19–21). The second discovery (by Carpenter’s and Kim’s groups) was the finding that the JM-a isoform, which contains a TACE cleavage site (22, 23), was susceptible to a second γ-secretase cleavage (1, 2). Our interest in HER4-dependent growth inhibition prompted us to look at the role of s80HER4 in that process.
Carpenter’s original report suggested that T47D growth could be inhibited by the heregulin-induced production of s80HER4, and that the nuclear localization and nuclear export sequences, found uniquely in HER4 among members of the EGF receptor family, could bring s80HER4 to the nucleus (25). Jones and co-workers showed that this translocation may result in co-translocation of STAT5A with s80HER4 into the nucleus, with potential consequences for STAT5A-inducible genes (53). Additionally, a HER4 C-terminus fusion protein, lacking the tyrosine kinase domain, was capable of transactivating a GAL4 promoter construct in co-transfection studies (25, 42). This is parallel to findings that a similar amino acid region in the C-terminus of both EGF receptor and HER2 can likewise transactivate GAL4 in co-transfection assays (43, 44). It has been speculated that s80HER4 might be cleaved in the nucleus to yield a transactivating product without the tyrosine kinase domain (56).
Our current work extends these recent results, first by using the SUM44 cell, our major model of heregulin and HB-EGF-dependent growth inhibition, to show that γ-secretase inhibition blocks the HER4 anti-proliferative effect. Likewise, we show that inhibition of the tyrosine kinase activity, by GW572016, also blocks the anti-proliferative effects. The blockade of heregulin-dependent HER4 tyrosine phosphorylation (e.g. in Figs. 2 and 3) could inhibit signaling, either by blocking surface HER4 signaling or by inhibiting the production of s80HER4 by TACE, and γ-secretase. While additional experiments are needed, our data suggest that s80HER4 can recapitulate ligand-dependent HER4 growth inhibition (Figs. 1, 2 and 7). However, at physiologic ligand and receptor levels, HER4-dependent growth inhibition may require both the production and action of s80HER4, as well as sustained HER4 cell surface tyrosine kinase activity.
Our data show that s80HER4 translocated and accumulated in the nucleus, in both live and fixed cells. We note that s80HER4 was not predominantly in the nucleus of every cell in which it was expressed, suggesting that there are complex mechanisms governing s80HER4 nuclear translocation. Tyrosine kinase activity appears to be at least one regulatory component, as GW572016 inhibition of s80HER4 constitutive tyrosine kinase activity dramatically impaired s80HER4’s nuclear accumulation. This constitutive ligand-independent, transmembrane HER4-independent, s80HER4 tyrosine phosphorylation indicates that the GFP-s80HER4 fusion protein has tyrosine kinase activity, a finding recently confirmed by others (49). We have confirmed the importance of s80HER4 tyrosine kinase activity in nuclear localization by showing the site-directed mutant s80HER4 lacking kinase activity also fails to localize in HC11 nuclei (50).
Lastly, our data indicate that s80HER4 inhibits growth and can enhance a STAT5-mediated transcription process to a greater extent than the HER4 C-terminal fragment. In five different breast/mammary cell lines, the expression of s80HER4, but not CTHER4, resulted in substantial growth inhibition. Growth inhibition under the conditions tested was not due to increased apoptosis. While published data suggests specific sequences within the cytoplasmic domain of HER4 can activate caspaces and apoptosis (54), our data indicate that chronic s80HER4 expression results in slower growth, without increasing apoptosis. Transactivation by the HER4 C-terminus, and other EGF receptor family C-termini, has been shown by several groups, using a GAL4 promoter transactivation assay (25, 42). Our data indicate that the mammary gland β-casein promoter is not effectively transactivated by CTHER4, whereas s80HER4, presumably in concert with STAT5A, can produce at least modest transactivation in this co-tranfection assay. Taken together, our results suggest that s80HER4, rather than CTHER4, is the biologically relevant entity in breast cells.
We do not know how growth inhibition or differentiated gene expression is regulated by s80HER4, but evidence is accumulating that HER4 is a uniquely endowed receptor tyrosine kinase, one in which precisely regulated proteolysis releases the cytoplasmic domain with an intact kinase activity. The encoded nuclear localization and export sequences are unique for the EGF receptor family and result in cytoplasmic nuclear shuttling with the nuclear localization appearing to depend upon an active kinase. The released cytoplasmic kinase domain is capable of continuous autophosphorylation, presumably until some physiologically relevant process turns it off or destroys s80HER4. The localization gives an indication that signals important to a cell’s fate, growth inhibition, and differentiation are played out in the cell nucleus. Future challenges include understanding the consequences of s80HER4 in human breast cancers where it is detected in a number of cases and appears to correlate with good prognosis tumors.
MATERIALS AND METHODS
Cell lines and cell culture
SUM44 and SUM102 cells were grown in serum-free growth factor-defined media as previously described (31). MDA-MB-453 cells and MCF10A cells were obtained from American Type Culture Collection (Manassas, VA). MDA-MB-453 and COS-7 cells were grown in Dulbecco modified Eagle Medium (Gibco BRL) supplemented with 10% fetal bovine serum (FBS). HC11 cells were grown in RPMI1640 with 10% FBS plus 5 μg/ml insulin (Gibco BRL), 10 ng/ml EGF (BD Biosciences, Bedford, MA) and antibiotics in a humidified incubator at 37°C with 5% CO2. All other cells were grown in a humidified incubator at 37°C with 10% CO2. Recombinant heregulin β1 was a gift from Genentech. Hydroxyethylene dihydropeptide isostere (HEDI) and prolactin were obtained from Sigma-Aldrich, St. Louis, MO.
Cell Growth Assay
Heregulin and inhibitor effects on SUM44 cells were evaluated by counting cells via hemacytometer as previously described (31). The cell growth assays in Fig. 7 were performed as following. 5,000 cells/well were plated in a 96 well plate, and cultured in serum-free growth factor-defined media at 37°C with 10% CO2. Cell number was analyzed using MTS [3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt] assay [CellTiter 96 Aqueous nonradioactive cell-proliferation assay kit (Promega)] according to manufacturer’s directions. Percentage increase = 100 × (ODend − OD20h)/OD20h.
Plasmid construction and transfection
The expression construct pLXSN-HER4 encoding full-length human HER4 (31) and pEGFP (Clontech) were used as templates for PCR amplification using Pfu DNA polymerase (Stratagene) to make constructs of GFP-CTHER4 (HER4 residues 989–1308) and GFP-s80HER4 (HER4 residues 676–1308) (Figure 3A). Both constructs were made by two overlapping PCRs. The primers generating GFP-CTHER4 were as following: 5′ GFP, cggggtaccatggtgagcaagggcgaggag; 3′ GFP, aagcttcatacgatcatcacccttgtacagctcgtccatgcc; 5′ CT, ggcatggacgagctgtacaagggtgatgatcgtatgaagc; 3′ CT, cggggtaccttacaccacagtattcc. The PCR products initially subcloned into pCDNA3 vector (Invitrogen), then subcloned into pMSCVpuro vector (Clontech). The primers generating GFP-s80HER4 were: 5′ GFP, ggaagatctgtcgccaccatggtgagcaagggc; 3′ GFP, ctttttgatgctcttccttctagtccggccggacttgtacag; 5′ s80HER4, ctgtacaagtccggccggactagaaggaagagcatcaaaaagaaaagagc; 3′ s80HER4, ttttccttttgcggccgcttacaccacagtattccggtg. The PCR product were subcloned into pMSCVpuro vector. All constructs were fully verified by DNA sequencing. The plasmid construct pMSCV-GFP, p53-GFP (57), and pQC-GFP-histone were kindly provided by Drs. Scott Hammond, Yanping Zhang, and James Bear (UNC Lineberger Comprehensive Cancer Center), respectively. Transfection was performed using FuGENE 6 (Roche Molecular Biochemicals) according to the manufacturer’s protocol.
Retrovirus Production, Infection and stable cell lines
A293T cells, seeded one day before at a density of 4–5 × 106 cells in 100-mm plates, were transfected with a packaging plasmid pCMV-VSVG and pUMVC3-gagpol (both vectors and A293T cells were kindly provided by Dr. Lishan Su, UNC Lineberger Comprehensive Cancer Center), and either pMSCV-GFP, pMSCV-GFP-CTHER4 or pMSCV- GFP-s80HER4 using FuGENE 6 (Roche Molecular Biochemicals) according to the manufacturer’s protocol. Viral supernatants were collected after 60 h of incubation, the last 48 h at 32°C with DMEM medium plus 2% FBS. Viral supernatants were filtered through a 0.45-μm-pore-size syringe filter, and were added with 8 μg of polybrene per ml to recipient cells which had been plated at 7 × 105 cells per 100-mm dish the day before. The cells were incubated at 37°C for 6 h in the filtered viral supernatant and then changed to normal growth medium. After 60 h of incubation, cells were selected in medium containing 2 μg/ml puromycin. Puromycin-resistant cells were pooled, and expression of the cDNA product was confirmed by western blotting. For the images of live cell microscopy in Fig. 4A and Fig. 5A, GFP positive cells were also selected by Modular Flow Cytometer (Cytomation Inc.).
Immunoprecipitation and immunoblot analysis
Cells were washed with cold phosphate-buffered saline and lysed in regular lysis buffer containing 20 mM Tris-HCl (pH 7.5), 50 mM sodium fluoride, 10% glycerol, 0.5% NP-40, 1 mM EDTA, 1mM EGTA, 20 mM β-glycerophosphate and 137 mM NaCl supplemented with sodium orthovanadate (1 mM) and Protease Inhibitors Cocktail (Roche), or high salt lysis buffer containing 20 mM HEPES, 50 mM NaF, 10% glycerol, 1% Triton, 5 mM EDTA, 500 mM NaCl, 1 mM sodium orthovanadate and protease inhibitors cocktail. Insoluble material was removed by centrifugation at 13,000 × g for 10 min at 4°C. Receptor proteins were precipitated for 3 h or overnight at 4°C with protein A/G or protein A agarose beads (Santa Cruz Biotechnologies, Santa Cruz, CA) and the following antibodies: HER2 [clone 9G6.10, mouse monoclonal antibody (Neomarkers, Inc.)]; HER3, polyclonal rabbit antisera raised by this lab against HER3 C-terminal; HER4, polyclonal rabbit antisera raised by this lab against recombinant gluthathione S-transferase fusion protein containing the C-terminal 80 amino acids of HER4; STAT5A (Zymed Laboratories). Immune complexes were washed three times with lysis buffer and denatured in sodium dodecyl sulfate (SDS) sample buffer. Protein samples were separated on a SDS-8% polyacrylamide gel and were electrophoretically transferred to a Sequi-blot polyvinylidene difluoride membrane (Bio-Rad). After blocking with 3% cold fish gelatin (Sigma), the membrane was probed overnight at 4°C or 1.5 h at room temperature with antiphosphotyrosine antibody (PY20; Santa Cruz Biotechnologies), HER4 antibody, HER2 antibody (Clone 2F12, Upstate, Lake Placid, NY), HER3 antibody (Ab-1, NeoMarkers, Fremont, CA), GFP antibody (Chemicon, Temecula, CA), STAT5A antibody, β-casein antibody (Santa Cruz Biotechnologies) or α–tubulin antibody (Santa Cruz Biotechnologies), washed three times with 0.1% Tween-20 in Tris-buffered saline, and detected with Enhanced ChemiLuminescence detection kit (Amersham Life Sciences).
Microscopy and Image acquisition
The images of live cell microscopy were captured using a Zeiss Axiovert 200 (LD A-Plan 20×/0.30 Ph1), and digitally acquired using a Zeiss AxioCam with Openlab 3.1.4 Zeiss imaging software. For fixed cell microscopy, the cells were grown on coverslips fixed in 3.7% formaldehyde, washed with PBS, and stained with DAPI (20 ng/ml), or STAT5A antibody and rhodamine red-conjugated donkey anti-mouse IgG (Jackson Immuno Research Laboratories). The coverslips were washed with PBS and mounted onto glass slides in fluorescent mounting media (DakoCytomation). Cells were visualized using either the Zeiss Axiovert 200 as above, or the Leica SP2 laser scanning confocal microscope with a 63x oil NA 1.40 Plan Apo lens (Nikon). For confocal microscopy, excitation of GFP and DAPI was performed using an argon ion laser at 488 nm and an UV laser at 364 nm respectively. Images were acquired and processed using Leica Confocal software. Minimal image processing was performed with Adobe Photoshop.
Apoptosis assay
Cell death was quantified by using the Cell Death Detection ELISA (Roche Molecular Biochemicals) which detects cytoplasmic histone-associated DNA fragments (mono- or oligonucleosomes). SUM44 cells stably expressing GFP, GFP-CTHER4 or GFP-s80HER4 were plated in 100 mm plates in serum-free medium changing the medium every other day. After seven days, the culture medium was saved to collect any floating cells. HC11 cells stably expressing GFP, GFP-CTHER4 or GFP-s80HER4 were plated in complete medium and cultured for 1 day, then the medium was replaced with serum-free growth factor-defined medium and cultured for 2 more days. At the end of the cell culture, the culture medium was saved to collect any floating cells. The attached cells were trypsinized off the plate and combined with the saved culture medium. Equal numbers of cells were obtained from each of the SUM44 or HC11 cells expressing GFP, GFP-CTHER4 or GFP-s80HER4, and then processed for apoptosis determination following the instructions of the supplier.
Quantitative reverse transcription-PCR (qRT-PCR)
qRT-PCR was performed as described previously (45). Briefly, total RNA was isolated from HC11 cells stably expressing GFP, GFP-CTHER4, or GFP-s80HER4 by using an RNeasy kit (QIAGEN) and was treated with RNase-free DNase (Ambion). β-casein primers and intervening fluorescent dye-labeled probes were designed using Primer Express software (ABI/Perkin-Elmer). Total RNA (10 ng) isolated from each cell line was assayed by real-time fluorescence qRT-PCR using an ABI PRISM 7900 instrument (PE Bio). Relative abundance of β-casein transcript was calculated by the formula: relative mRNA level = e(40-Ct).
Statistical analysis
Significant differences between treatment groups were assessed by Student’s t-test. P < 0.05 was considered statistically significant.
Supplementary Material
A, Live cell microscopy showing GFP fluorescence in SUM44 cells stably expressing GFP, GFP-CTHER4, or GFP-s80HER4. Phase contrast is shown in lower panels. Bar, 100 μm, applies to each image. B, SUM44 cells stably expressing GFP (a-c, j-l), GFP-CTHER4 (d-f, m-o) or GFP-s80HER4 (g-i, p-r) were treated without (a-i), or with (j-r) 20 ng/ml of leptomycin B for 24 h. The cells on the cover slip were analyzed by confocal microscopy for GFP fluorescence. Subcellular distribution of GFP (a, j), GFP-CTHER4 (d, m), GFP-s80HER4 (g, p), and DAPI (b, e, h, k, n, q) were visualized and captured. The merged pictures were also shown (c, f, i, l, o, r). Bar, 10 μm, applies to each image.
Acknowledgments
This work was supported by the Breast Cancer Research Foundation and NIH grant CA-112553.
We thank Drs. Young Whang and Zonghan Dai for helpful discussions. We thank Dr. Tona Gilmer of GlaxoSmithKline for her helpful comments and for providing lapatinib, GW572016. We also thank Drs. Scott Hammond for providing pMSCV-GFP, Yanping Zhang for providing p53-GFP, James Bear for providing GFP-histone, and Lishan Su for providing VSVG and gagpol plasmids. We appreciate the efforts of Carolyn Coffay in manuscript preparation. The confocal microscopy was done at Michael Hooker Microscopy Facility, University of North Carolina. The work was supported by the Breast Cancer Research Foundation and NIH grant CA-112553.
The abbreviations used are
- s80HER4
soluble 80 kDa HER4 cytoplasmic domain
- CTHER4
HER4 C-terminus beyond the tyrosine kinase domain, residues 989–1308
- NLS
nuclear localization sequence
- NES
nuclear exporting sequence
- STAT5A
signal transducer and activator of transcription 5A
- TACE
tumor necrosis factor-α-converting enzyme
- TPA
tetradecaonylphorbol-13-acetate
- HEDI
hydroxyethylene dihydropeptide isostere
- DAPI
4′,6-diamidino-2-phenylindole
- MAPK
mitogen-activated protein kinase
- PI3K
phosphatidylinositol 3-kinase
- VSVG
vesicular stomatitis virus glycoprotein
Footnotes
Publisher's Disclaimer: This is an un-copyedited author manuscript copyrighted by The Endocrine Society. This may not be duplicated or reproduced, other than for personal use or within the rule of “Fair Use of Copyrighted Materials” (section 107, Title 17, U.S. Code) without permission of the copyright owner, The Endocrine Society. From the time of acceptance following peer review, the full text of this manuscript is made freely available by The Endocrine Society at http://www.endojournals.org/. The final copy edited article can be found at http://www.endojournals.org/. The Endocrine Society disclaims any responsibility or liability for errors or omissions in this version of the manuscript or in any version derived from it by the National Institutes of Health or other parties. The citation of this article must include the following information: author(s), article title, journal title, year of publication and DOI.
Disclosure Summary: The authors have nothing to declare.
References
- 1.Lacenere CJ, Sternberg PW. Regulation of EGF receptor signaling in the fruitfly D. melanogaster and the nematode C. elegans. Breast Dis. 2000;11:19–30. doi: 10.3233/bd-1999-11103. [DOI] [PubMed] [Google Scholar]
- 2.Yarden Y, Sliwkowski MX. Untangling the ErbB signalling network. Nat Rev Mol Cell Biol. 2001;2:127–137. doi: 10.1038/35052073. [DOI] [PubMed] [Google Scholar]
- 3.Earp HS, 3rd, Calvo BF, Sartor CI. The EGF receptor family--multiple roles in proliferation, differentiation, and neoplasia with an emphasis on HER4. Trans Am Clin Climatol Assoc. 2003;114:315–333. [PMC free article] [PubMed] [Google Scholar]
- 4.Roskoski R., Jr The ErbB/HER receptor protein-tyrosine kinases and cancer. Biochem Biophys Res Commun. 2004;319:1–11. doi: 10.1016/j.bbrc.2004.04.150. [DOI] [PubMed] [Google Scholar]
- 5.Holbro T, Hynes NE. ErbB receptors: directing key signaling networks throughout life. Annu Rev Pharmacol Toxicol. 2004;44:195–217. doi: 10.1146/annurev.pharmtox.44.101802.121440. [DOI] [PubMed] [Google Scholar]
- 6.Stern DF. ErbBs in mammary development. Exp Cell Res. 2003;284:89–98. doi: 10.1016/s0014-4827(02)00103-9. [DOI] [PubMed] [Google Scholar]
- 7.Schroeder JA, Lee DC. Dynamic expression and activation of ERBB receptors in the developing mouse mammary gland. Cell Growth Differ. 1998;9:451–464. [PubMed] [Google Scholar]
- 8.Troyer KL, Lee DC. Regulation of mouse mammary gland development and tumorigenesis by the ERBB signaling network. J Mammary Gland Biol Neoplasia. 2001;6:7–21. doi: 10.1023/a:1009560330359. [DOI] [PubMed] [Google Scholar]
- 9.Tidcombe H, Jackson-Fisher A, Mathers K, Stern DF, Gassmann M, Golding JP. Neural and mammary gland defects in ErbB4 knockout mice genetically rescued from embryonic lethality. Proc Natl Acad Sci U S A. 2003;100:8281–8286. doi: 10.1073/pnas.1436402100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hynes NE, Lane HA. ERBB receptors and cancer: the complexity of targeted inhibitors. Nat Rev Cancer. 2005;5:341–354. doi: 10.1038/nrc1609. [DOI] [PubMed] [Google Scholar]
- 11.Plowman GD, Culouscou JM, Whitney GS, Green JM, Carlton GW, Foy L, Neubauer MG, Shoyab M. Ligand-specific activation of HER4/p180erbB4, a fourth member of the epidermal growth factor receptor family. Proc Natl Acad Sci U S A. 1993;90:1746–1750. doi: 10.1073/pnas.90.5.1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gullick WJ. c-erbB-4/HER4: friend or foe? J Pathol. 2003;200:279–281. doi: 10.1002/path.1335. [DOI] [PubMed] [Google Scholar]
- 13.Pawlowski V, Revillion F, Hebbar M, Hornez L, Peyrat JP. Prognostic value of the type I growth factor receptors in a large series of human primary breast cancers quantified with a real-time reverse transcription-polymerase chain reaction assay. Clin Cancer Res. 2000;6:4217–4225. [PubMed] [Google Scholar]
- 14.Suo Z, Risberg B, Kalsson MG, Willman K, Tierens A, Skovlund E, Nesland JM. EGFR family expression in breast carcinomas. c-erbB-2 and c-erbB-4 receptors have different effects on survival. J Pathol. 2002;196:17–25. doi: 10.1002/path.1003. [DOI] [PubMed] [Google Scholar]
- 15.Witton CJ, Reeves JR, Going JJ, Cooke TG, Bartlett JM. Expression of the HER1–4 family of receptor tyrosine kinases in breast cancer. J Pathol. 2003;200:290–297. doi: 10.1002/path.1370. [DOI] [PubMed] [Google Scholar]
- 16.Barnes NL, Khavari S, Boland GP, Cramer A, Knox WF, Bundred NJ. Absence of HER4 expression predicts recurrence of ductal carcinoma in situ of the breast. Clin Cancer Res. 2005;11:2163–2168. doi: 10.1158/1078-0432.CCR-04-1633. [DOI] [PubMed] [Google Scholar]
- 17.Lodge AJ, Anderson JJ, Gullick WJ, Haugk B, Leonard RC, Angus B. Type 1 growth factor receptor expression in node positive breast cancer: adverse prognostic significance of c-erbB-4. J Clin Pathol. 2003;56:300–304. doi: 10.1136/jcp.56.4.300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bieche I, Onody P, Tozlu S, Driouch K, Vidaud M, Lidereau R. Prognostic value of ERBB family mRNA expression in breast carcinomas. Int J Cancer. 2003;106:758–765. doi: 10.1002/ijc.11273. [DOI] [PubMed] [Google Scholar]
- 19.Elenius K, Corfas G, Paul S, Choi CJ, Rio C, Plowman GD, Klagsbrun M. A novel juxtamembrane domain isoform of HER4/ErbB4. Isoform-specific tissue distribution and differential processing in response to phorbol ester. J Biol Chem. 1997;272:26761–26768. doi: 10.1074/jbc.272.42.26761. [DOI] [PubMed] [Google Scholar]
- 20.Elenius K, Choi CJ, Paul S, Santiestevan E, Nishi E, Klagsbrun M. Characterization of a naturally occurring ErbB4 isoform that does not bind or activate phosphatidyl inositol 3-kinase. Oncogene. 1999;18:2607–2615. doi: 10.1038/sj.onc.1202612. [DOI] [PubMed] [Google Scholar]
- 21.Junttila TT, Sundvall M, Maatta JA, Elenius K. Erbb4 and its isoforms: selective regulation of growth factor responses by naturally occurring receptor variants. Trends Cardiovasc Med. 2000;10:304–310. doi: 10.1016/s1050-1738(01)00065-2. [DOI] [PubMed] [Google Scholar]
- 22.Rio C, Buxbaum JD, Peschon JJ, Corfas G. Tumor necrosis factor-alpha-converting enzyme is required for cleavage of erbB4/HER4. J Biol Chem. 2000;275:10379–10387. doi: 10.1074/jbc.275.14.10379. [DOI] [PubMed] [Google Scholar]
- 23.Cheng QC, Tikhomirov O, Zhou W, Carpenter G. Ectodomain cleavage of ErbB-4: characterization of the cleavage site and m80 fragment. J Biol Chem. 2003;278:38421–38427. doi: 10.1074/jbc.M302111200. [DOI] [PubMed] [Google Scholar]
- 24.Ebinu JO, Yankner BA. A RIP tide in neuronal signal transduction. Neuron. 2002;34:499–502. doi: 10.1016/s0896-6273(02)00704-3. [DOI] [PubMed] [Google Scholar]
- 25.Ni CY, Murphy MP, Golde TE, Carpenter G. gamma -Secretase cleavage and nuclear localization of ErbB-4 receptor tyrosine kinase. Science. 2001;294:2179–2181. doi: 10.1126/science.1065412. [DOI] [PubMed] [Google Scholar]
- 26.Peles E, Bacus SS, Koski RA, Lu HS, Wen D, Ogden SG, Levy RB, Yarden Y. Isolation of the neu/HER-2 stimulatory ligand: a 44 kd glycoprotein that induces differentiation of mammary tumor cells. Cell. 1992;69:205–216. doi: 10.1016/0092-8674(92)90131-u. [DOI] [PubMed] [Google Scholar]
- 27.Bacus SS, Huberman E, Chin D, Kiguchi K, Simpson S, Lippman M, Lupu R. A ligand for the erbB-2 oncogene product (gp30) induces differentiation of human breast cancer cells. Cell Growth Differ. 1992;3:401–411. [PubMed] [Google Scholar]
- 28.Bacus SS, Gudkov AV, Zelnick CR, Chin D, Stern R, Stancovski I, Peles E, Ben-Baruch N, Farbstein H, Lupu R, et al. Neu differentiation factor (heregulin) induces expression of intercellular adhesion molecule 1: implications for mammary tumors. Cancer Res. 1993;53:5251–5261. [PubMed] [Google Scholar]
- 29.Culouscou JM, Plowman GD, Carlton GW, Green JM, Shoyab M. Characterization of a breast cancer cell differentiation factor that specifically activates the HER4/p180erbB4 receptor. J Biol Chem. 1993;268:18407–18410. [PubMed] [Google Scholar]
- 30.Daly JM, Jannot CB, Beerli RR, Graus-Porta D, Maurer FG, Hynes NE. Neu differentiation factor induces ErbB2 down-regulation and apoptosis of ErbB2-overexpressing breast tumor cells. Cancer Res. 1997;57:3804–3811. [PubMed] [Google Scholar]
- 31.Sartor CI, Zhou H, Kozlowska E, Guttridge K, Kawata E, Caskey L, Harrelson J, Hynes N, Ethier S, Calvo B, Earp HS., 3rd Her4 mediates ligand-dependent antiproliferative and differentiation responses in human breast cancer cells. Mol Cell Biol. 2001;21:4265–4275. doi: 10.1128/MCB.21.13.4265-4275.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Long W, Wagner KU, Lloyd KC, Binart N, Shillingford JM, Hennighausen L, Jones FE. Impaired differentiation and lactational failure of Erbb4-deficient mammary glands identify ERBB4 as an obligate mediator of STAT5. Development. 2003;130:5257–5268. doi: 10.1242/dev.00715. [DOI] [PubMed] [Google Scholar]
- 33.Tang CK, Concepcion XZ, Milan M, Gong X, Montgomery E, Lippman ME. Ribozyme-mediated down-regulation of ErbB-4 in estrogen receptor-positive breast cancer cells inhibits proliferation both in vitro and in vivo. Cancer Res. 1999;59:5315–5322. [PubMed] [Google Scholar]
- 34.Alaoui-Jamali MA, Song DJ, Benlimame N, Yen L, Deng X, Hernandez-Perez M, Wang T. Regulation of multiple tumor microenvironment markers by overexpression of single or paired combinations of ErbB receptors. Cancer Res. 2003;63:3764–3774. [PubMed] [Google Scholar]
- 35.Maatta JA, Sundvall M, Junttila TT, Peri L, Laine VJ, Isola J, Egeblad M, Elenius K. Proteolytic cleavage and phosphorylation of a tumor-associated ErbB4 isoform promote ligand-independent survival and cancer cell growth. Mol Biol Cell. 2006;17:67–79. doi: 10.1091/mbc.E05-05-0402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Srinivasan R, Gillett CE, Barnes DM, Gullick WJ. Nuclear expression of the c-erbB-4/HER-4 growth factor receptor in invasive breast cancers. Cancer Res. 2000;60:1483–1487. [PubMed] [Google Scholar]
- 37.Junttila TT, Sundvall M, Lundin M, Lundin J, Tanner M, Harkonen P, Joensuu H, Isola J, Elenius K. Cleavable ErbB4 isoform in estrogen receptor-regulated growth of breast cancer cells. Cancer Res. 2005;65:1384–1393. doi: 10.1158/0008-5472.CAN-04-3150. [DOI] [PubMed] [Google Scholar]
- 38.Marambaud P, Shioi J, Serban G, Georgakopoulos A, Sarner S, Nagy V, Baki L, Wen P, Efthimiopoulos S, Shao Z, Wisniewski T, Robakis NK. A presenilin-1/gamma-secretase cleavage releases the E-cadherin intracellular domain and regulates disassembly of adherens junctions. Embo J. 2002;21:1948–1956. doi: 10.1093/emboj/21.8.1948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Okamoto I, Kawano Y, Murakami D, Sasayama T, Araki N, Miki T, Wong AJ, Saya H. Proteolytic release of CD44 intracellular domain and its role in the CD44 signaling pathway. J Cell Biol. 2001;155:755–762. doi: 10.1083/jcb.200108159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kim DY, Ingano LA, Kovacs DM. Nectin-1alpha, an immunoglobulin-like receptor involved in the formation of synapses, is a substrate for presenilin/gamma-secretase-like cleavage. J Biol Chem. 2002;277:49976–49981. doi: 10.1074/jbc.M210179200. [DOI] [PubMed] [Google Scholar]
- 41.Schulz JG, Annaert W, Vandekerckhove J, Zimmermann P, De Strooper B, David G. Syndecan 3 intramembrane proteolysis is presenilin/gamma-secretase-dependent and modulates cytosolic signaling. J Biol Chem. 2003;278:48651–48657. doi: 10.1074/jbc.M308424200. [DOI] [PubMed] [Google Scholar]
- 42.Komuro A, Nagai M, Navin NE, Sudol M. WW domain-containing protein YAP associates with ErbB-4 and acts as a co-transcriptional activator for the carboxyl-terminal fragment of ErbB-4 that translocates to the nucleus. J Biol Chem. 2003;278:33334–33341. doi: 10.1074/jbc.M305597200. [DOI] [PubMed] [Google Scholar]
- 43.Lin SY, Makino K, Xia W, Matin A, Wen Y, Kwong KY, Bourguignon L, Hung MC. Nuclear localization of EGF receptor and its potential new role as a transcription factor. Nat Cell Biol. 2001;3:802–808. doi: 10.1038/ncb0901-802. [DOI] [PubMed] [Google Scholar]
- 44.Wang SC, Lien HC, Xia W, Chen IF, Lo HW, Wang Z, Ali-Seyed M, Lee DF, Bartholomeusz G, Ou-Yang F, Giri DK, Hung MC. Binding at and transactivation of the COX-2 promoter by nuclear tyrosine kinase receptor ErbB-2. Cancer Cell. 2004;6:251–261. doi: 10.1016/j.ccr.2004.07.012. [DOI] [PubMed] [Google Scholar]
- 45.Muraoka-Cook RS, Caskey LS, Sandahl MA, Hunter DM, Husted C, Strunk KE, Sartor CI, Rearick WA, Jr, McCall W, Sgagias MK, Cowan KH, Earp HS., 3rd Heregulin-dependent delay in mitotic progression requires HER4 and BRCA1. Mol Cell Biol. 2006;26:6412–6424. doi: 10.1128/MCB.01950-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhou W, Carpenter G. Heregulin-dependent trafficking and cleavage of ErbB-4. J Biol Chem. 2000;275:34737–34743. doi: 10.1074/jbc.M003756200. [DOI] [PubMed] [Google Scholar]
- 47.Lee HJ, Jung KM, Huang YZ, Bennett LB, Lee JS, Mei L, Kim TW. Presenilin-dependent gamma-secretase-like intramembrane cleavage of ErbB4. J Biol Chem. 2002;277:6318–6323. doi: 10.1074/jbc.M110371200. [DOI] [PubMed] [Google Scholar]
- 48.Vecchi M, Baulida J, Carpenter G. Selective cleavage of the heregulin receptor ErbB-4 by protein kinase C activation. J Biol Chem. 1996;271:18989–18995. doi: 10.1074/jbc.271.31.18989. [DOI] [PubMed] [Google Scholar]
- 49.Linggi B, Cheng QC, Rao AR, Carpenter G. The ErbB-4 s80 intracellular domain is a constitutively active tyrosine kinase. Oncogene. 2006;25:160–163. doi: 10.1038/sj.onc.1209003. [DOI] [PubMed] [Google Scholar]
- 50.Muraoka-Cook RS, Sandahl M, Husted C, Hunter D, Miraglia L, Feng SM, Elenius K, Earp HS., 3rd The Intracellular Domain of ErbB4 Induces Differentiation of Mammary Epithelial Cells. Mol Biol Cell. 2006;17:4118–4129. doi: 10.1091/mbc.E06-02-0101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fabian MA, Biggs WH, 3rd, Treiber DK, Atteridge CE, Azimioara MD, Benedetti MG, Carter TA, Ciceri P, Edeen PT, Floyd M, Ford JM, Galvin M, Gerlach JL, Grotzfeld RM, Herrgard S, Insko DE, Insko MA, Lai AG, Lelias JM, Mehta SA, Milanov ZV, Velasco AM, Wodicka LM, Patel HK, Zarrinkar PP, Lockhart DJ. A small molecule-kinase interaction map for clinical kinase inhibitors. Nat Biotechnol. 2005;23:329–336. doi: 10.1038/nbt1068. [DOI] [PubMed] [Google Scholar]
- 52.Jones FE, Welte T, Fu XY, Stern DF. ErbB4 signaling in the mammary gland is required for lobuloalveolar development and Stat5 activation during lactation. J Cell Biol. 1999;147:77–88. doi: 10.1083/jcb.147.1.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Williams CC, Allison JG, Vidal GA, Burow ME, Beckman BS, Marrero L, Jones FE. The ERBB4/HER4 receptor tyrosine kinase regulates gene expression by functioning as a STAT5A nuclear chaperone. J Cell Biol. 2004;167:469–478. doi: 10.1083/jcb.200403155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Vidal GA, Naresh A, Marrero L, Jones FE. Presenilin-dependent gamma -secretase processing regulates multiple ERBB4/HER4 activities. J Biol Chem. 2005;280:19777–19783. doi: 10.1074/jbc.M412457200. [DOI] [PubMed] [Google Scholar]
- 55.Cohen BD, Kiener PA, Green JM, Foy L, Fell HP, Zhang K. The relationship between human epidermal growth-like factor receptor expression and cellular transformation in NIH3T3 cells. J Biol Chem. 1996;271:30897–30903. doi: 10.1074/jbc.271.48.30897. [DOI] [PubMed] [Google Scholar]
- 56.Heldin CH, Ericsson J. Signal transduction. RIPping tyrosine kinase receptors apart. Science. 2001;294:2111–2113. doi: 10.1126/science.1067628. [DOI] [PubMed] [Google Scholar]
- 57.O’Keefe K, Li H, Zhang Y. Nucleocytoplasmic shuttling of p53 is essential for MDM2-mediated cytoplasmic degradation but not ubiquitination. Mol Cell Biol. 2003;23:6396–6405. doi: 10.1128/MCB.23.18.6396-6405.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A, Live cell microscopy showing GFP fluorescence in SUM44 cells stably expressing GFP, GFP-CTHER4, or GFP-s80HER4. Phase contrast is shown in lower panels. Bar, 100 μm, applies to each image. B, SUM44 cells stably expressing GFP (a-c, j-l), GFP-CTHER4 (d-f, m-o) or GFP-s80HER4 (g-i, p-r) were treated without (a-i), or with (j-r) 20 ng/ml of leptomycin B for 24 h. The cells on the cover slip were analyzed by confocal microscopy for GFP fluorescence. Subcellular distribution of GFP (a, j), GFP-CTHER4 (d, m), GFP-s80HER4 (g, p), and DAPI (b, e, h, k, n, q) were visualized and captured. The merged pictures were also shown (c, f, i, l, o, r). Bar, 10 μm, applies to each image.