

Abstract
The expression of the potent, constitutively activated EGFR variant, EGFRvIII, has been linked to breast cancer metastasis, but the mechanisms of EGFRvIII and CXCR4 crosstalk, which may facilitate breast cancer invasion, have never been explored. Here we report that CXCR4 expression is increased in breast cancer cells expressing EGFRvIII regardless of the ER/PgR status of the cells. Treatment of EGFRvIII-expressing breast cancer cells with the tyrosine kinase inhibitor, AG1478, reverses CXCR4 expression back to levels expressed in parental cells. In addition, expressing EGFRvIII enhances CXCL12/CXCR4-mediated invasion, which can be inhibited by CXCR4 inhibitors. Surprisingly, CXCR4 mRNA and its transcriptional regulator, HIF-1α, are up-regulated only in ER+/PgR+ estrogen-dependent EGFRvIII-expressing breast cancer cells, but not in ER-/PgR- or estrogen-independent cell lines, suggesting that HIF-1α and hormone receptor-mediated actions may have a role in the transcriptional regulation of CXCR4. We also demonstrate that p38 MAPK is one of the major down-stream signaling molecules responsible for EGFRvIII/CXCR4-mediated invasion as p38 MAPK activity was induced by CXCL12 stimulation under both normoxic and hypoxic conditions. More interestingly, inhibition of p38 MAPK activity significantly reduced CXCR4 expression and inhibited the invasive potential of EGFRvIII-expressing breast cancer cells, suggesting an essential role for p38 MAPK in EGFRvIII/CXCR4 induced invasion. Furthermore, CXCR4 is regulated post-translationally through decreased expression of AIP4 and β-arrestin 1/2, molecules involved in CXCR4 internalization, cellular trafficking, and degradation. These results provide a plausible mechanism for EGFRvIII-mediated invasion and establish a functional link between EGFRvIII and CXCR4 signaling pathways.
Keywords: EGFR, EGFRvIII, CXCR4, Invasion, Breast
Introduction
The biological and clinical significance of the Epidermal Growth Factor Receptor (EGFR) variant, EGFRvIII, in malignancies not associated with the central nervous system, continues to remain underdeveloped. Although recent studies have attempted to evaluate the clinical significance of EGFRvIII mRNA and protein expression (1;2), a conclusion on the clinical value of EGFRvIII expression in breast cancer is premature.
Our earlier studies have shown that the frequency of EGFRvIII expression increases with the pathogenesis of breast cancer, and no detectable levels of EGFRvIII were observed in normal tissues (3;4). We also demonstrated that high levels of EGFRvIII expression are capable of transforming 32D cells (non-tumorigenic hematopoeitic cells) and enhance tumorigenesis with ErbB2 (4;5). Constitutively activated EGFRvIII also mediates sustained signaling and activation of downstream signaling pathways, which is further enhanced with ErbB2 co-expression in these cells (4). Co-expression of EGFRvIII with ErbB2 has been detected in a subset of breast tumors, and furthermore, co-expression of EGFRvIII with ErbB2 enhances ErbB2 activation, and amplifies the down-stream signaling pathways observed in both EGFRvIII-expressing 32D cells and in breast cancer cells (4;5). Studies have also shown that EGFRvIII increases the motility of mouse fibroblasts (6), enhances the invasive potential of cancer cells (7–9), was detected in the peripheral blood of breast cancer patients (2), yet the molecular mechanisms by which EGFRvIII can enhance the invasive behavior of breast cancer cells remains unknown.
Chemokines exert their actions in responsive cells by activating their seven-transmembrane G-protein coupled receptors (GPCRs). CXCR4 was one of the chemokine receptors most frequently expressed in breast cancer cell lines, malignant breast tumors, and metastases (10). Stromal-derived factor-1 (CXCL12), the only ligand for CXCR4, is expressed in the lung, liver, bone marrow, lymph nodes, and to a lesser extent in the brain, and is believed to be responsible for breast cancer metastasis by directing CXCR4-expressing breast cancer cells to these organs (10). Upon stimulation with CXCL12, breast cancer cells show actin polymerization and pseudopodia formation, and migrate and invade in an CXCL12-directed manner (10).
Several studies have linked the expression of receptor tyrosine kinases (RTKs), such as ErbB-receptors, to CXCR4 expression in breast cancer. ErbB2 expression was shown to enhance the expression of CXCR4, which was required for ErbB2-mediated invasion in vitro and lung metastasis in vivo (11). CXCL12/CXCR4 signaling also transactivates EGFR and ErbB2 through Src activation in breast cancer cells (12). A high correlation between CXCR4 expression and the expression of EGFR and ErbB2 was observed in human breast tumor tissues and correlated with a poor overall survival rate in patients with breast cancer (11;13–16). In non-small cell lung cancer cells, activation of EGFR has been shown to up-regulate CXCR4 transcriptionally through increased expression and activity of HIF-1α (17). However, in breast cancer, mechanisms by which EGFR or EGFRvIII can regulate CXCR4 expression have yet to be established.
Here we report that EGFRvIII-expressing breast cancer cells have increased expression of CXCR4 and exhibit enhanced CXCL12/CXCR4-mediated invasion. CXCR4 in EGFRvIII-expressing breast cancer cells is regulated not only transcriptionally by its well-known transcriptional regulator, HIF-1α; it is also post-translationally regulated through multiple mechanisms. First, increased p38 activation was observed in EGFRvIII-expressing cells, and treatment with p38 inhibitors reduced CXCR4 expression and attenuated the invasive potential of EGFRvIII-expressing breast cancer cells. These results suggest that p38 MAPK plays a role in EGFRvIII/CXCR4 induced invasion. Finally, AIP4 and β-arrestin 1/2 are mediators of lysosomal degradation of CXCR4. We observed that EGFRvIII inhibits AIP4 and β-arrestin 1/2 expression to reduce the degradation of CXCR4, which eventually results in up-regulation of CXCR4 expression in EGFRvIII-expressing cells.
Material and Methods
Cell Culture and Reagents
The 32D mouse pro-B-lymphocyte cell line derivatives were grown in RPMI 1640 supplemented with 10% FBS and IL-3 supplied as 10% conditioned medium from the WEHI-3B mouse myelomonocytic leukemia cell line. MDA-MB-361, BT474, MCF-7, and SKBR3 breast carcinoma cell lines and their derivatives were maintained in IMEM supplemented with 10% FBS. Since endogenous EGFRvIII expression is lost in cancer cells under in vitro conditions (18), stable EGFRvIII-expressing cells were generated as previously described (5). Human recombinant CXCL12 and Epidermal Growth Factor (EGF) were purchased from R&D Systems (Minneapolis, MN). Tyrphostin AG1478, SB203850, Pertussis toxin (P.T.), AMD3100, and cycloheximide were purchased from Sigma-Aldrich (St. Louis, MO).
Immunoblot analysis
Breast cancer cells were plated in culture plates and grown to 50–80% confluence. Unless otherwise specified, cells were lysed after the removal of growth media. Some cultures were serum-starved overnight and then stimulated with CXCL12 or EGF for 5 minutes or treated with the tyrosine kinase inhibitor (TKI) AG1478 or cycloheximide for the specified times. Hypoxia experiments were performed in a computer monitored hypoxia chamber (94% nitrogen, 5% carbon dioxide, and 0.5 to 1% oxygen) for 24 hours. Cells were rinsed, lysed, and equal amounts of protein were then separated by SDS-PAGE and transferred to nitrocellulose membranes for western blot analysis. Antibodies against phospho-EGFR (Tyr1173), phospho-Akt (Ser473), phospho-p44/42 MAPK (Thr202/Tyr204), p44/42MAPK, phospho-p38 MAPK (Thr180/Tyr182), p38 MAPK, and Tubulin were purchased from Cell Signaling Technology (Danvers, MA); the antibody for HIF-1α was purchased from BD Biosciences; the antibodies for HIF-2α and HIF-1β were purchased from Novus Biologicals (Littleton, CO); antibodies against Akt (Akt1), AIP4, and β-Arrestin 1/2 were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA); the antibodies for EGFR and CXCR4 were purchased from NeoMarkers (Fremont, CA); and the antibody for GAPDH was purchased from Sigma-Aldrich. Densitometry measurements were performed using Scion Image software (Scion Corporation; Frederick, MD).
Fluorescence-Activated Cell Sorting (FACS) Analysis
Cells (0.5–1.0 × 106) were harvested and then stained for 1 hour with anti-CXCR4 (mab172 or mab173; R&D Systems) or anti-CXCR7 (R&D Systems) antibodies at 4°C. Stained cells were then washed with cold PBS. A secondary FITC-anti-mouse antibody (KPL; Gaithersburg, MD) was added for 30 minutes, and the CXCR4 or CXCR7 levels were quantified by flow cytometry.
Invasion assays
Invasion was measured using 24-well cell culture inserts with membranes with 8 µm pores and a matrigel-coating to mimic the basement membrane (BD Biosciences; San Jose, CA). Breast cancer cells were suspended in serum-free medium with 0.1% BSA and 2.0 × 105 cells were plated in the top part of the insert. The CXCR4 neutralizing antibody mab170 (1 or 10 µg/mL) (R&D Systems), AMD3100 (1 or 10 µg/mL), P.T. (0.25 µg/mL), SB203580 (10 µM) or the proper vehicle was added as pre-treatments and to the cell suspension. The inserts were placed in wells containing serum-free medium with 0.1% BSA, with or without CXCL12, or 10% FBS in IMEM. After incubation at 37°C for 48 hours, residual cells were wiped off the top of the membranes with cotton swabs, and invaded cells on the underside of the membranes were fixed and stained using the HEMA-3 kit (Fisher Diagnostics; Pittsburgh, PA). Cells were counted in 10 fields from three inserts per experimental condition. Experiments were performed in a minimum of two independent studies.
Quantitative real-time PCR
RNA was reverse transcribed from random hexamers using SuperScript® III Reverse transcriptase (Invitrogen; Carlsbad, CA). Real-time quantitative PCR was performed using the Real-time PCR system 7900 (Applied Biosystems; Foster City, CA). In brief, the PCR amplification reaction mixtures (25 µL) contained cDNA, RT2 PCR Primer Assay (SA Biosciences; Frederick, MD), and RT2 Real-Time SYBR Green Master Mix (SA Biosciences). The thermal cycle conditions included maintaining the reactions at 50°C for 2 minutes and at 95°C for 10 minutes, and then alternating for 40 cycles between 95°C for 15 seconds and 60°C for 1 minute. The relative gene expression for each sample was determined using the formula 2 (−δ Ct) = 2 (Ct (GAPDH)−Ct (target)), which reflected the target gene expression normalized to GAPDH levels.
Statistics
Statistical analysis was performed using ANOVA, followed by the Tukey test using SigmaStat software. Results were considered statistically significant at p<0.05.
Results
EGFRvIII increases CXCR4 expression in breast cancer cells
Expressing EGFRvIII is associated with increased proliferation, enhanced tumorigenesis and invasiveness of cancer cells (3–9). To determine whether ectopic expression of EGFRvIII alters CXCR4 expression to facilitate enhanced invasion of breast cancer cells, four EGFRvIII-expressing breast cancer cell lines were used for this study, including estrogen receptor (ER)-positive MDA-MB-361, BT474, MCF-7, and ER-negative SKBR3 cells. However, ectopically expressing EGFRvIII in MDA-MB-361 cells induces an estrogen-independent phenotype (19). EGFRvIII is constitutively active in breast cancer cells (Fig. 1a). Interestingly, regardless of the ErbB2 or ER status of these cells, all the cell lines expressing EGFRvIII had increased expression levels of CXCR4 in comparison to their parental cells based on FACS analysis. As shown in Fig. 1b, EGFRvIII-transfected MDA-MB-361 (MDA-MB-361/vIII) and BT474 (BT474/vIII) cells showed an increase in cell surface expression of CXCR4 in comparison to parental cells (MDA-MB-361/wt or BT474/wt). Similar observations were also observed in MCF-7 and SKBR3 transfected with EGFRvIII cDNA (see Supplemental Fig. 2). In order to determine whether ectopic expression of individual ErbB-receptors including EGFRvIII influences the expression of CXCR4, the hematopoietic 32D cell line was used since these cells do not endogenously express ErbB-receptors. Ectopically expressing EGFRvIII (32D/vIII) or ErbB2 (32D/ErbB2) enhances the expression of CXCR4, and the combination of EGFRvIII/ErbB2 expression (32D/vIII+ErbB2) also increases the CXCR4 protein levels in 32D cells (Fig. 1c). However, expressing full-length un-activated wild-type EGFR did not alter the expression levels of CXCR4 in 32D cells. We then further determined activated full-length EGFR had the ability to increase CXCR4 levels in 32D cells expressing EGFR (32D/EGFR). Stimulating 32D/EGFR cells with EGF for 24 hours also resulted in enhanced expression of CXCR4 in comparison with untreated 32D/EGFR cells (Fig. 1d). To confirm that the up-regulation of CXCR4 expression is due to the activity of EGFRvIII, EGFRvIII-expressing MDA-MB-361 and BT474 cells were treated with the TKI AG1478; Fig. 1e demonstrates that CXCR4 expression levels were remarkably reduced to the levels expressed in the parental breast cancer cells. An early study has shown that ErbB2 can enhance CXCR4 expression; CXCR4 is required for ErbB2-induced breast cancer cell invasion and metastasis (11). In order to further dissect whether the up-regulation of CXCR4 is due to constitutively activated EGFRvIII or ErbB2 in EGFRvIII-expressing cells, the anti-ErbB2 antibody Herceptin, and the ErbB2 tyrosine kinase inhibitor AG825, were used. Interestingly, inhibiting ErbB2 with Herceptin or AG825 had little to no effect on the increased CXCR4 expression in breast cancer cells expressing EGFRvIII (data not shown). These results suggest that up-regulation of CXCR4 is due to activated EGFRvIII.
Fig. 1.
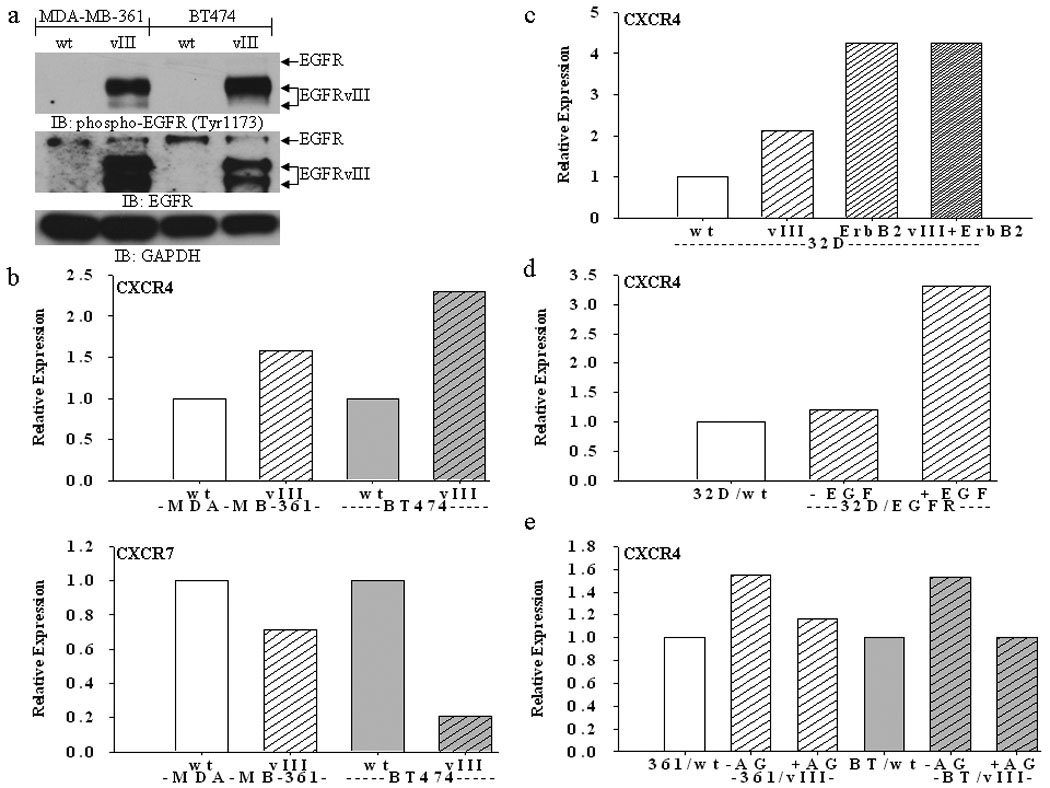
(a) Immunoblot analysis showing EGFRvIII is constitutively active in MDA-MB-361 and BT474 cells (parental cells are labeled “wt” and EGFRvIII-expressing cells are labeled “vIII”). (b) FACS analysis shows increased expression of CXCR4, but not CXCR7, in MDA-MB-361 and BT474 breast cancer cells stably expressing EGFRvIII. (c) FACS analysis shows increased expression of CXCR4 in 32D cells stably expressing EGFRvIII, ErbB2, and both EGFRvIII and ErbB2. (d) FACS analysis shows increased expression of CXCR4 in 32D cells stably expressing full-length EGFR (“32D/EGFR”) upon EGF (50 ng/mL) stimulation for 24 hours. (e) FACS analysis shows a reduction of CXCR4 in EGFRvIII-expressing MDA-MB-361 (“361”) and BT474 (“BT”) cells upon treatment with the TKI AG1478 (“AG”) (10 µM) for 24 hours. Bar graphs represent the relative expression based on the mean geometric fluorescence of cells in each group/control cells. Actual FACS plots are available in Supplemental Figure 1.
The CXCL12/CXCR4 axis contributes to EGFRvIII-mediated breast cancer cell invasion
Since both CXCR4 and EGFRvIII expression is associated with increased breast cancer metastasis (4;10), we investigated whether EGFRvIII enhanced CXCR4 expression resulted in increased CXCL12/CXCR4-mediated invasion. MDA-MB-361/vIII and BT474/vIII cells had enhanced CXCL12/CXCR4-mediated invasion in comparison to parental cells, which showed little to no invasive potential (Fig. 2a). Although, variations in MDA-MB-361 and BT474 cell lines and/or assay conditions may account for a lack of CXCL12-mediated invasion in parental cells, CXCL12-mediated migration was observed in parental cells (see Supplemental Fig. 3). Both a neutralizing anti-CXCR4 antibody and the small molecule CXCR4 inhibitor, AMD3100, completely abrogated the CXCL12-enhanced invasion observed in the BT474 breast cancer cells expressing EGFRvIII (Fig. 2b). MDA-MB-361/vIII cells display higher levels of invasion without a chemoattractant in the invasion assays, therefore, we determined whether inhibiting the CXCL12/CXCR4 axis would decrease this basal invasive potential. Using AMD3100 and the CXCR4 neutralizing antibody, we observed a significant decrease in basal invasive potential only in MDA-MB-361/vIII cells treated with AMD3100, while the parental MDA-MB-361 cells were unaffected by both inhibitors (Fig. 2c). This suggests that either an autocrine loop involving CXCL12 is inhibited by AMD3100 or that this small molecule inhibitor may have off-target or unknown effects on EGFRvIII-expressing breast cancer cells. Therefore, we examined whether Pertussis Toxin (P.T.), which inhibits the re-coupling of G-proteins to the receptors, inhibits serum-induced invasion. Fig. 2d demonstrates that the invasion of MDA-MB-361/vIII cells is significantly inhibited by a non-toxic concentration of P.T., confirming that the activity of GPCRs is essential to the overall invasive potential of EGFRvIII-expressing breast cancer cells.
Fig. 2.
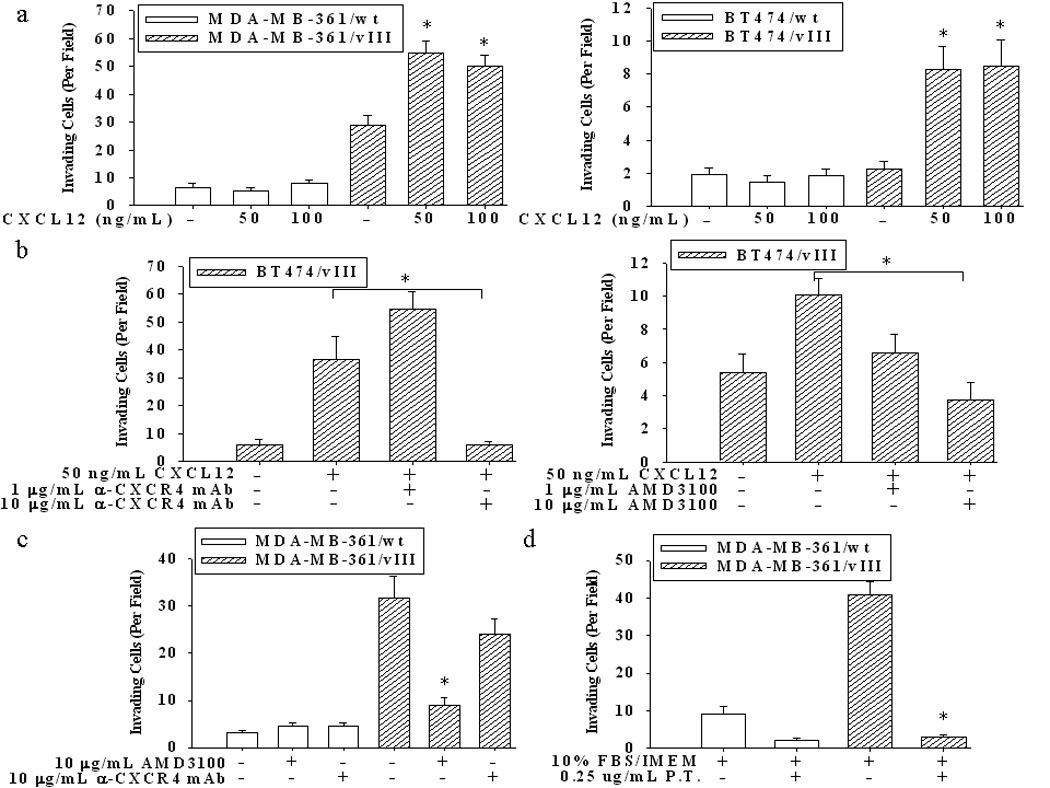
(a) Invasion of MDA-MB-361/vIII and BT474/vIII cells was significantly stimulated by CXCL12 (50–100 ng/mL) (* = p<0.001; ANOVA). Cells were plated in 0.1% BSA/IMEM in the upper chamber of transwells with membranes with 8 µm pores, and with CXCL12 in the lower chamber. After 48 hours of incubation, the membranes were fixed and stained, and cells counted on the underside of the membranes. (b) The addition of a CXCR4 neutralizing antibody (10 µg/mL) and the small molecule inhibitor AMD3100 (10 µg/mL) significantly inhibited the invasion of cells towards CXCL12 (* = p<0.001, ANOVA). (c) The addition of only AMD3100 (10 µg/mL) significantly inhibited the basal invasive potential of MDA-MB-361/vIII cells (* = p<0.001, ANOVA). (d) The addition of Pertussis Toxin (P.T.) (0.25 µg/mL) significantly inhibited the overall invasion of cells toward serum (* = p<0.001, ANOVA). The results shown are the mean and SE from 10 fields counted on triplicate membranes, and is representative of repeated experiments.
EGFRvIII up-regulates HIF-1α and CXCR4 under normoxic and hypoxic conditions
An early study indicated that CXCR4 expression is modulated under low oxygen tension, through the transcriptional activity of HIF-1α (17). Therefore, we next determined whether CXCR4 expression can be up-regulated in EGFRvIII-expressing breast cells under hypoxic conditions. BT474/vIII cells had an increased expression of CXCR4 under both normoxic and hypoxic conditions, as expected (Fig. 3a). In addition, HIF-1α protein levels were markedly increased in BT474 cells expressing EGFRvIII under both normoxic and hypoxic conditions (Fig. 3a; inset). Furthermore, up-regulation of HIF-1α was completely abolished with AG1478 treatment (Fig. 3b), indicating that the activity of EGFRvIII accounts for the up-regulation of HIF-1α. However, induction of HIF-1α protein was only observed in EGFRvIII-transfected ER+/progesterone receptor-positive (PgR+) estrogen-dependent cells (BT474/vIII and MCF-7/vIII), but not in the estrogen-independent cells (MDA-MB-361/vIII and SKBR3/vIII) (Fig. 3c) (19), suggesting they account for estrogen-mediated actions. These results lead us to hypothesize that although up-regulation of CXCR4 protein levels was observed in all the EGFRvIII-expressing cell lines regardless of their ER expression status, CXCR4 mRNA may be only increased in ER+/PgR+ estrogen-dependent cells, but not in the estrogen-independent cells. Indeed, investigation of CXCR4 transcripts has revealed that CXCR4 mRNA levels were up-regulated in only EGFRvIII-expressing ER+/PgR+ (BT474/vIII and MCF-7/vIII) breast cancer cells, but not in MDA-MB-361/vIII or SKBR3/vIII cells under normoxic conditions (Fig. 3d). Again, these observations are closely related to the maintenance of an estrogen-dependent phenotype in EGFRvIII-expressing breast cancer cells, as the estrogen-dependent MDA-MB-361 cells become estrogen-independent and mimic estrogen-receptor negative cells when forced to express EGFRvIII (19). SKBR3/vIII cells have decreased CXCR4 transcripts (Fig. 3d), yet have increased expression of CXCR4 at the cell surface (see Supplemental Fig. 2), suggesting that CXCR4 expression may also be regulated at the protein level. Moreover, up-regulation of CXCR4 mRNA levels was correlated with increased expression of HIF-1α, suggesting that a role of HIF-1α in the regulation of CXCR4 mRNA expression in ER+/PgR+ EGFRvIII-expressing breast cancer cell lines. Neither the expression of HIF-1β nor HIF-2α was altered in any of the cells (see Supplemental Fig. 4).
Fig. 3.
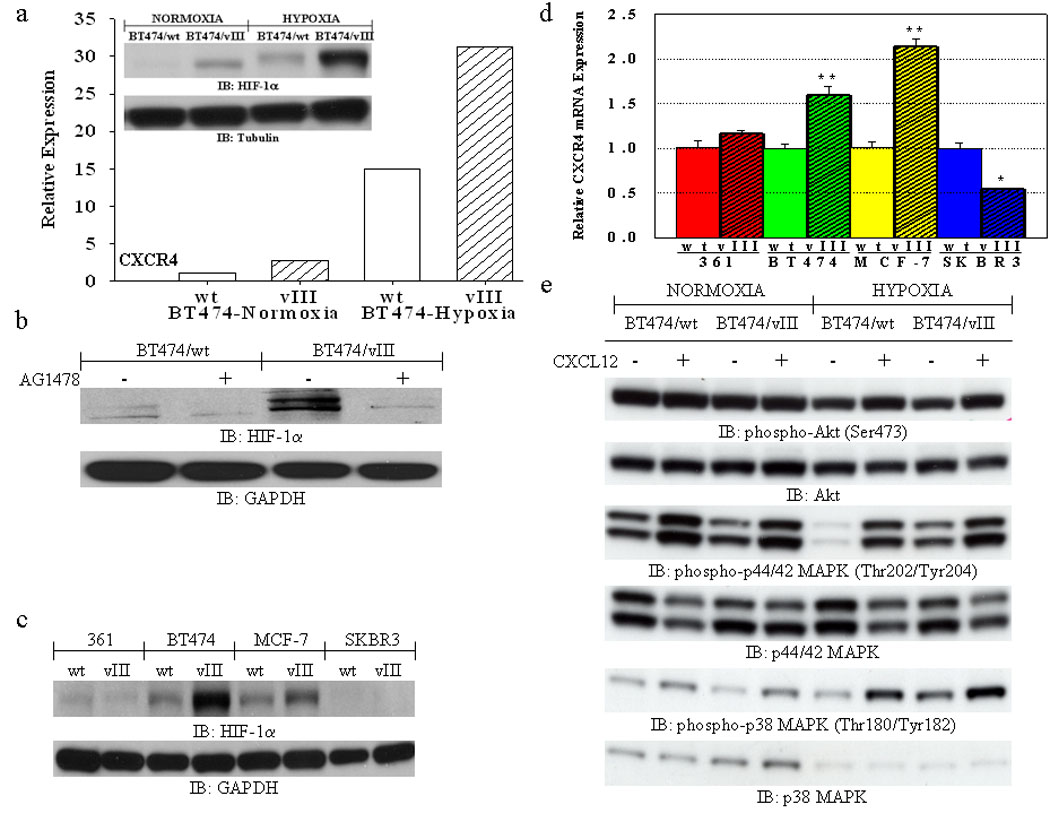
(a) FACS analysis shows increased expression of CXCR4 in BT474/vIII cells under both normoxic and hypoxic (24 hours) conditions in comparison to BT474/wt cells. Bar graphs represent the relative expression based on the mean geometric fluorescence of cells in each group/control cells. Actual FACS plots are available in Supplemental Figure 4. Inset, immunoblot analysis of lysates from BT474/wt and BT474/vIII cells under normoxic and hypoxic (24 hours) conditions showing increased expression of HIF-1α in BT474/vIII cells under both normoxic and hypoxic conditions. (b) Immunoblot analysis of lysates from BT474wt and BT474/vIII cells treated with the TKI AG1478 (10 µM) for 24 hours showing that inhibition of EGFRvIII activity can abolish increased HIF-1α expression in BT474/vIII cells under normoxic conditions. (c) Immunoblot analysis of lysates from MDA-MB-361 (“361”), BT474, MCF-7, and SKBR3 cells with and without EGFRvIII expression showing that HIF-1α is up-regulated in multiple breast cancer cells expressing EGFRvIII under normoxic conditions. (d) Quantitative real-time PCR analysis revealed significantly increased CXCR4 transcript levels only in BT474/vIII and MCF-7/vIII cells in comparison to parental cells (** = p<0.001; * = p<0.05, ANOVA). (e) Immunoblot analysis of lysates from BT474/wt and BT474/vIII cells showing that hypoxic conditions enhanced CXCL12-induced phosphorylation of Akt, p44/42 MAPK, and p38 MAPK. BT474/vIII cells also have higher basal levels of p44/42 MAPK and p38 MAPK activation under hypoxic conditions.
Next, we evaluated the CXCL12/CXCR4 signaling cascades under both normoxic and hypoxic conditions in EGFRvIII-expressing cells. Under normoxic conditions, the activity of p44/42 and p38 MAPK were enhanced by CXCL12 stimulation (Fig. 3e). Under hypoxic conditions, BT474/vIII cells exhibit higher basal levels of p44/42 and p38 MAPK activation, and activation of CXCL12/CXCR4 signaling cascades further enhances the activation of these down-stream signaling molecules as well as Akt (Fig. 3e).
Suppression of p38 MAPK activity decreases CXCR4 expression and reduces invasion in EGFRvIII-expressing breast cancer cells
The potentially enhanced p38 MAPK activity observed in EGFRvIII cells lead us to investigate the role of p38 MAPK in EGFRvIII-expressing cells. The activity of p38 MAPK is involved in the trafficking of GPCRs and ErbB-receptors and has been shown to be essential for breast cancer cell invasion (20–23). We observed that breast cancer cells expressing EGFRvIII have an increased basal activation of p38 MAPK and upon stimulation with EGF causes an even more enhanced activation of p38 MAPK (Fig. 4a). Hypoxic conditions also enhance the phosphorylation of p38 MAPK in EGFRvIII-expressing breast cancer cells (Fig. 3e). To further test the functional role of p38 MAPK in EGFRvIII-induced CXCR4 expression and invasion, a p38 MAPK inhibitor was used. Inhibition of p38 MAPK with SB203580 only slightly reduced the CXCR4 expression in MDA-MB-361 parental cells, whereas CXCR4 expression was down-regulated even more in MDA-MB-361/vIII cells (Fig. 4b). However, a down-regulation of EGFR, EGFRvIII, or ErbB2 at the cell surface was not observed under the same conditions (data not shown). Most importantly, treatment with SB203580 significantly attenuated the invasive potential of MDA-MB-361/vIII cells (Fig. 4c), indicating that p38 MAPK has an important role in the regulation of cell surface expression of CXCR4 and the enhanced invasive phenotype associated with EGFRvIII expression. Moreover, this effect on CXCR4 expression was not observed when MDA-MB-361/wt and MDA-MB-361/vIII cells were treated with the MAPK inhibitor PD98059 (data not shown).
Fig. 4.
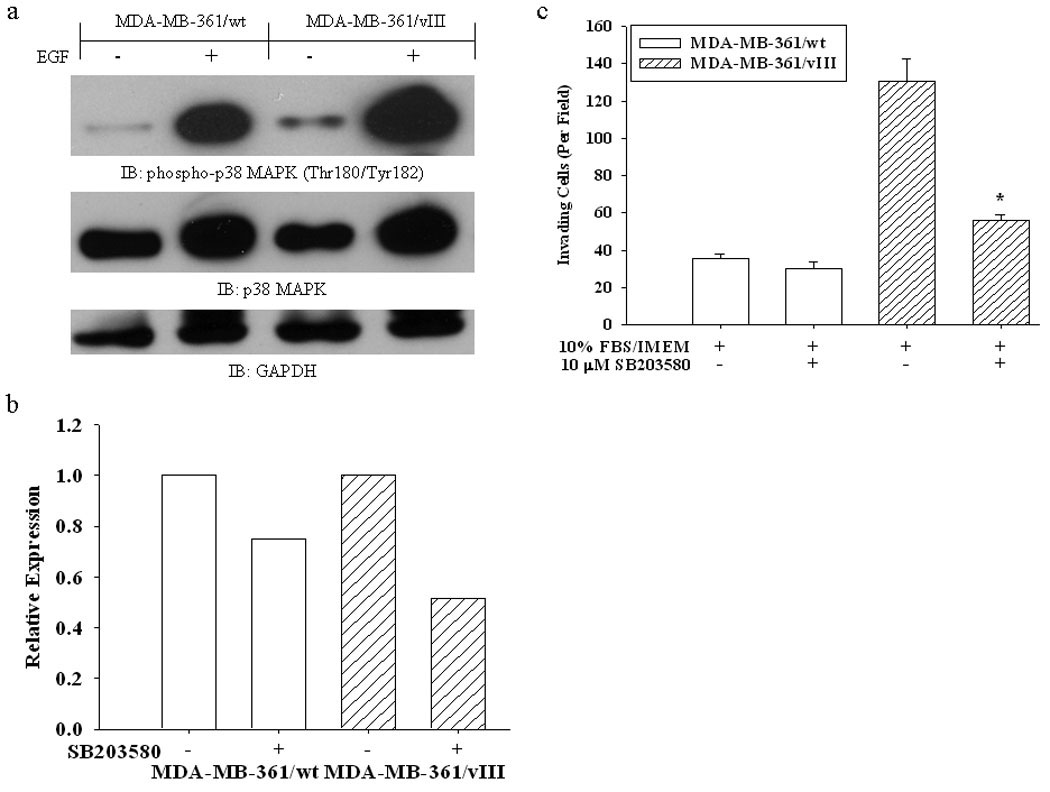
(a) Immunoblot analysis of lysates from serum-starved MDA-MB-361/wt and MDA-MB-361/vIII cells stimulated with EGF (50 ng/mL) for 5 minutes showing that MDA-MB-361/vIII cells have increased activation of p38 MAPK. (b) FACS analysis shows a pronounced decrease in the expression of CXCR4 in MDA-MB-361/vIII cells after 24 hours of treatment with the p38 inhibitor SB203580 (10 µM). Bar graphs represent the relative expression based on the mean geometric fluorescence of cells in each group/control cells. Actual FACS plots are available in Supplemental Figure 5. (c) The addition of SB203580 (10 µM) significantly inhibited the overall invasion of cells towards serum (* = p<0.001, ANOVA). The results shown are the mean and SE from 10 fields counted on triplicate membranes, and is representative of repeated experiments.
EGFRvIII stabilizes CXCR4 protein levels by decreasing CXCR4 degradation
To elucidate whether de-regulation of CXCR4 degradation results in increased expression levels of CXCR4 in the EGFRvIII transfectants, we determined the rate of CXCR4 degradation. By inhibiting new protein synthesis using cycloheximide, we were able to show that CXCR4 protein levels were more stable in MDA-MB-361/vIII cells (Fig. 5a and b), The experiments were performed using both FACS (Fig. 5a) and immunoblotting (Fig. 5b) analysis as the detection of CXCR4 using immunoblotting techniques was found to be difficult for most of the breast cancer cells. These results suggest that enhanced CXCR4 protein stability may account for the up-regulation of CXCR4 in EGFRvIII expressing cells. Subsequently, we elucidated the mechanisms of enhanced CXCR4 protein stability through evaluation of the proteins that are involved in CXCR4 internalization, recycling, and degradation, such as the Nedd4-like E3 ubiquitin ligase for CXCR4, AIP4 (21;24;25), and β-Arrestin 1/2, which are involved in endosomal sorting of CXCR4 to the lysosome (21;26;27). Fig. 5c shows that AIP4 is highly expressed only in BT474/wt cells, and is down-regulated in BT474/vIII cells, while β-Arrestin 1/2 was shown to be universally down-regulated in all of the breast cancer cells expressing EGFRvIII (Fig. 5c). We found no changes in cortactin levels, a key regulator of CXCR4 internalization (28), nor any changes in ligand-induced internalization of CXCR4 (data not shown). Finally, we propose that EGFRvIII expression in breast cancer cells results in increased expression of CXCR4 and enhanced invasive potential by regulating CXCR4 at the transcriptional level through HIF-1α up-regulation and post-translationally by stabilizing CXCR4 expression through a reduction of molecules associated with CXCR4 internalization, cellular trafficking, lysosomal degradation and enhanced p38 MAPK activation (Fig. 6).
Fig. 5.
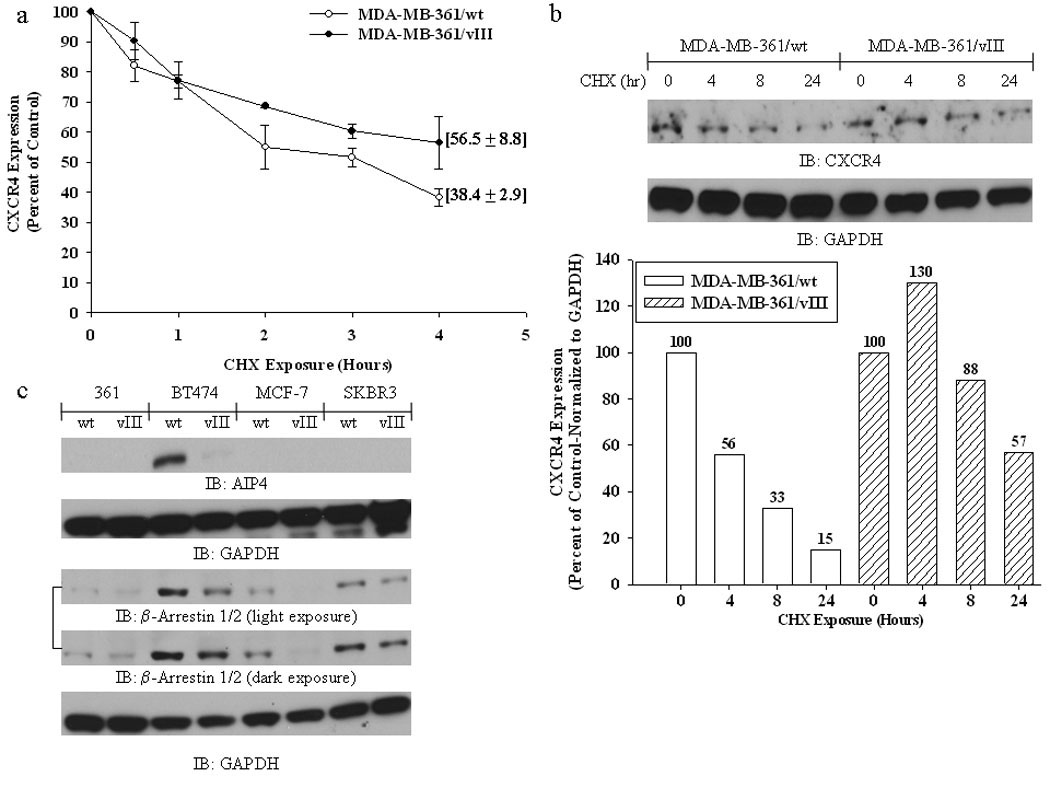
FACS (a) and immunoblot (b) analysis MDA-MB-361/wt and MDA-MB-361/vIII cells treated with the protein synthesis inhibitor cycloheximide (10 µg/mL) showing that EGFRvIII-expressing breast cancer cells have increased stability of CXCR4 protein levels. (b - bottom panel), densitometry measurements of CXCR4 immunoblot results. (c) Immunoblot analysis of lysates from MDA-MB-361 (“361”), BT474, MCF-7, and SKBR3 cells with and without EGFRvIII expression showing that AIP4 and β-arrestin 1/2 are down-regulated in multiple breast cancer cells expressing EGFRvIII. See Supplemental Fig. 6 for an additional β-arrestin 1/2 blot.
Fig. 6.
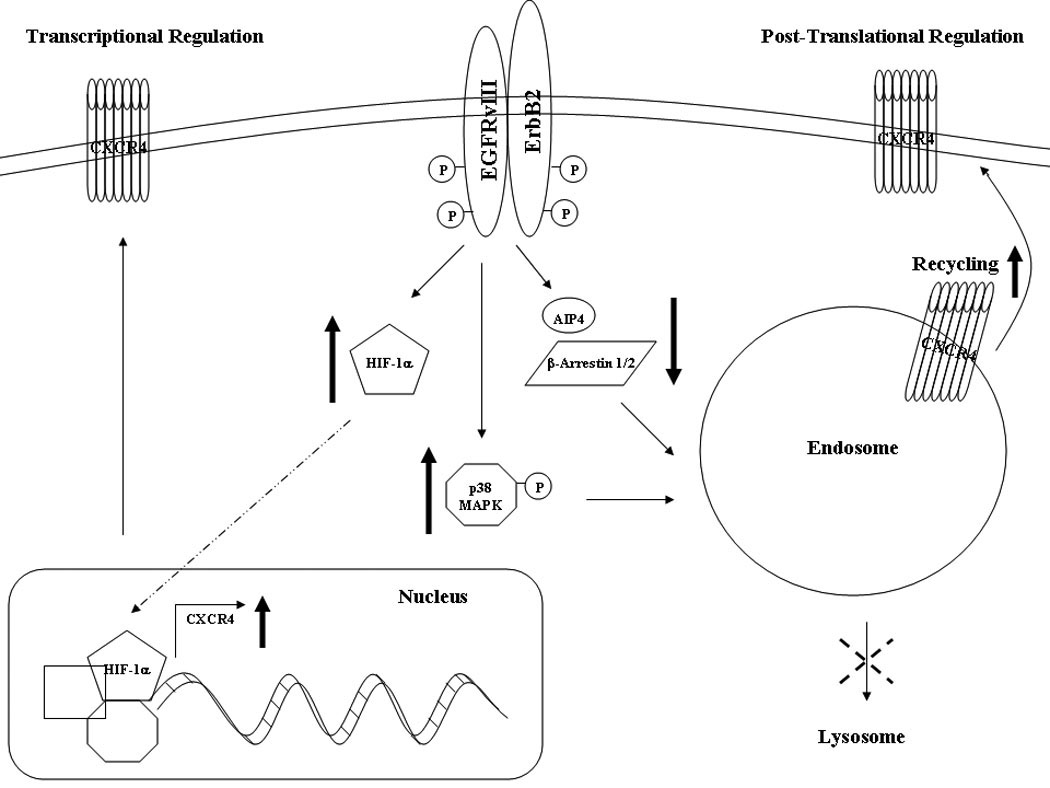
EGFRvIII enhances CXCR4 expression transcriptionally through the up-regulation of HIF-1α and post-translationally by increasing p38 MAPK activation and decreasing AIP4 and β-arrestin 1/2, leading to decreased CXCR4 internalization, cellular trafficking, and lysosomal degradation.
Discussion
In recent years, chemokines and their receptors have evolved into a major field in cancer research as these chemokine networks have been implicated in every aspect of cancer biology. CXCR4 plays a major role in ErbB-receptor mediated metastasis in several cancers, including ErbB2-mediated breast cancer (11). Here, we evaluated the relationship between CXCR4 and the naturally occurring EGFR variant, EGFRvIII, a potent oncogene that has been shown to be associated with breast cancer progression and metastasis (4). We have provided evidence that both an activated EGFR and ErbB2 have the ability to increase CXCR4 expression. We also demonstrated that up-regulation of CXCR4 is mediated by the activity of EGFRvIII, but not ErbB2 in all of the breast cancer model systems that we have examined. In addition to the role of CXCR4 in ErbB2-mediated breast cancer metastasis (11), our study has demonstrated that CXCR4 is likely to play an essential role in EGFRvIII-mediated invasion in breast cancer cells.
CXCR4 expression is increased under hypoxic conditions by the transcriptional activity of the oxygen-dependent HIF-1α (17). However, non-small cell lung cancer cells were shown to up-regulate the expression of CXCR4 through EGFR-dependent HIF-1α protein stability and increased DNA binding and transcriptional activity at the CXCR4 promoter irrespective of oxygen levels (17). In this study, we report, for the first time, that EGFRvIII up-regulates HIF-1α protein levels and CXCR4 expression under both normoxic and hypoxic conditions in breast cancer cells. Increased CXCR4 transcripts were found to correlate with increased HIF-1α levels under normal oxygen tension in estrogen-dependent EGFRvIII-expressing breast cancer cells. Increased protein expression of other HIF-1α regulated genes, such as survivin (see Supplemental Fig. 7) (29), under normoxic conditions further supports the notion that increased HIF-1α activity is a novel signaling cascade in estrogen-dependent EGFRvIII-mediated oncogenesis. It is important to note that while EGF stimulation of parental breast cancer cells did increase HIF-1α levels at a short exposure time, EGF stimulation did not consistently increase CXCR4 levels in these cells at short or long exposure times (data not shown). In contrast, constitutively activated EGFRvIII mediates sustained downstream signaling (4), and therefore, may allow for HIF-1α protein accumulation under normal oxygen levels and consequently promote the transcription of CXCR4. Alternatively, increased HIF-1α protein and/or CXCR4 transcripts may be a distinct and unique hallmark of EGFRvIII signaling which is not obtained through the activation of full length EGFR in breast cancer. This is possible as it has been shown that full length EGFR and EGFRvIII do not share common signaling pathways in gliomas (30).
AIP4 overexpression suppresses CXCR4 expression in ErbB2-overexpressing breast cancer cells by decreasing the stability of CXCR4 protein levels (11). We have discovered that EGFRvIII decreases endogenous levels of AIP4 in BT474/EGFRvIII cells, but AIP4 was undetectable in other model systems. However, β-arrestin 1/2 expression level was reduced in all the EGFRvIII-expressing model systems. Since, β-arrestin 1/2 plays an essential role in the trafficking of several GPCRs, these results provide a possible mechanism in which the reduction of AIP4 and β-arrestin 1/2 expression promote an aberrant degradation of CXCR4, thereby leading to its up-regulation in EGFRvIII-expressing breast cancer cells. It should be mentioned that although we did not detect increases in CXCR7 or CXCR3 (data not shown) in EGFRvIII cells, other GPCRs involved in breast cancer metastasis may be up-regulated in EGFRvIII expressing breast cancer cells. The use of the GPCR inhibitor, Pertussis Toxin, was able to abolish the increased invasive potential of EGFRvIII-expressing breast cancers, suggesting that EGFRvIII-enhanced invasion may be the result of increased GPCR expression and/or signaling. However, knocking-down CXCR4 also decreases the invasive potential of EGFRvIII-expressing breast cancer cells (data not shown), implicating a significant role for CXCR4 in EGFRvIII-mediated invasion. These studies are currently underway.
Taken together, our results also show that induction of CXCR4 protein expression is regulated both transcriptionally and post-translationally. In ER negative and estrogen-independent breast cancer cells, EGFRvIII-induced CXCR4 expression is likely to be regulated at the post-translational level by increasing the synthesis rate of CXCR4 protein and enhancing the CXCR4 protein stability through decreasing the expression levels of those molecules involved in the cellular trafficking and degradation of CXCR4. In contrast, in estrogen-dependent ER+/PgR+ breast cancer cells, EGFRvIII-mediated up-regulation of CXCR4 expression may be regulated at the transcriptional level through up-regulation of HIF-1α, as well as at the post-translational level. Therefore, both transcriptional and post-translational mechanisms are likely responsible for EGFRvIII-mediated up-regulation of CXCR4 protein levels in EGFRvIII-expressing breast cancer cells.
Our results have also demonstrated that p38 MAPK is one of the major down-stream signaling molecules involved in EGFRvIII/CXCR4-mediated invasion. p38 MAPK activity was markedly induced by CXCL12 stimulation under both normoxic and hypoxic conditions. Inhibition of p38 MAPK activity significantly reduced CXCR4 levels and attenuated the invasive potential of EGFRvIII-expressing breast cancer cells, whereas MAPK inhibitors do not have this effect on CXCR4 expression. These results suggest that p38 MAPK plays a role in EGFRvIII/CXCR4 induced invasion.
Finally, glioma cell lines ectopically expressing EGFRvIII did not exhibit enhanced CXCR4 expression in comparison to parental cells (data not shown). This observation appears to be unique to our EGFRvIII breast cancer model system. It is likely that the glioma cells express higher basal levels of CXCR4 in comparison to breast cancer cells, which is also reflected in the highly invasive phenotype of glioma cells. EGFRvIII may only enhance CXCR4 expression in cancer cells that have low invasive potential, as is the case for the cancer cell lines used in our studies. These results suggest that CXCR4 has an essential role in enhanced invasion of EGFRvIII-expressing breast cancer cells. Hence, further investigations are required in order to fully elucidate the relationship between CXCR4 and EGFRvIII in breast cancer and whether targeting these two molecules with pre-existing or novel therapies will prevent breast cancer metastasis and potentially other oncogenic functions involving both CXCR4 and EGFRvIII.
Supplementary Material
Supplemental Fig. 1 (a) FACS analysis shows increased expression of CXCR4, but not CXCR7, in MDA-MB-361 and BT474 breast cancer cells stably expressing EGFRvIII [antibody 4-5H was used to detect EGFRvIII as previously described (4;31)]. Black lines, secondary antibody only; red lines, parental (“wt”) cells; blue lines, EGFRvIII (“vIII”) transfectants. (b) FACS analysis shows increased expression of CXCR4 in 32D cells stably expressing EGFRvIII, ErbB2, and both EGFRvIII and ErbB2. Black line, secondary antibody only; red line, parental cells; blue line, EGFRvIII transfectants; purple line, ErbB2 transfectants; green line; EGFRvIII and ErbB2 transfectants. (c) FACS analysis shows increased expression of CXCR4 in 32D cells expressing full-length EGFR upon EGF (50 ng/mL) stimulation for 24 hours. Black line, secondary antibody only; red line, parental cells; blue line, untreated EGFR transfectants; purple line, EGFR cells treated with EGF. (d) FACS analysis shows a reduction of CXCR4 in EGFRvIII-expressing MDA-MB-361 and BT474 cells upon treatment with the TKI AG1478 (10 µM) for 24 hours. Black lines, secondary antibody only; red lines, parental cells; blue lines, vehicle (DMSO) treated EGFRvIII transfectants; purple lines, EGFRvIII transfectants treated with AG1478. The x-axis represents fluorescence intensity and the y-axis represents cell counts.
Supplemental Fig. 2 (a) FACS analysis shows increased expression of CXCR4 in MCF-7 and SKBR3 breast cancer cells stably expressing EGFRvIII. Black lines, secondary antibody only; red lines, parental (“wt”) cells; blue lines, EGFRvIII (“vIII”) transfectants. The x-axis represents fluorescence intensity and the y-axis represents cell counts. (b) FACS results quantified. Bar graphs represent the relative expression based on the mean geometric fluorescence of cells in each group/control cells.
Supplemental Fig. 3 Migration of MDA-MB-361/wt and MDA-MB-361/vIII cells toward CXCL12 (50 ng/mL). Cells were plated in 0.1% BSA/IMEM in the upper chamber of transwells with membranes with 8 µm pores coated only with fibronectin (20 ng/mL), and with CXCL12 in the lower chamber. After 24 hours of incubation, the membranes were fixed and stained, and cells counted on the underside of the membranes. The results shown are the mean and SE from cells counted on triplicate membranes.
Supplemental Fig. 4 (a) FACS analysis shows increased expression of CXCR4 in BT474/vIII cells under both normoxic and hypoxic (24 hours) conditions in comparison to BT474/wt cells. Black line, secondary antibody only; red line, parental cells under normoxic conditions; purple line, EGFRvIII transfectants under normoxic conditions; blue line, parental cells under hypoxic conditions; green line, EGFRvIII transfectants under hypoxic conditions. The x-axis represents fluorescence intensity and the y-axis represents cell counts. (b) Immunoblot analysis of lysates from MDA-MB-361 (“361”), BT474, MCF-7, and SKBR3 cells with and without EGFRvIII expression showing that neither HIF-2α or HIF-1β is up-regulated in multiple breast cancer cells expressing EGFRvIII under normoxic conditions.
Supplemental Fig. 5 FACS analysis shows a pronounced decrease in the expression of CXCR4 in MDA-MB-361/vIII cells after 24 hours of treatment with the p38 inhibitor SB203580 (10 µM). Black lines, secondary antibody only; red lines, cells treated with vehicle control (DMSO); blue lines, cells treated with SB203580. The x-axis represents fluorescence intensity and the y-axis represents cell counts.
Supplemental Fig. 6 An additional immunoblot analysis of lysates from MDA-MB-361 (“361”), BT474, MCF-7, and SKBR3 cells with and without EGFRvIII expression showing that β-arrestin 1/2 is down-regulated in multiple breast cancer cells expressing EGFRvIII.
Supplemental Fig. 7 Immunoblot analysis of lysates from BT474/wt and BT474/vIII cells under normoxic and hypoxic (24 hours) conditions showing increased expression of HIF-1α and survivin in BT474/vIII cells under both normoxic and hypoxic conditions.
Acknowledgements
This work was supported by the NIH Grant RO1 CA88871 (C.K. Tang). The authors thank the Flow Cytometry and Cell Sorting Core Facility Shared Resource of Lombardi Comprehensive Cancer Center, which is partially supported by National Institute of Health Grant 1P30-CA-51008 (Cancer Center Support Grant, to Lombardi Comprehensive Cancer Center). We would also like to thank Dr. Michael Johnson for usage of the hypoxia chamber.
Abbreviations
- EGFR
Epidermal Growth Factor Receptor
- GPCR
G-Protein Coupled Receptor
- RTK
Receptor Tyrosine Kinase
- EGF
Epidermal Growth Factor
- P.T.
Pertussis Toxin
- ER
Estrogen Receptor
- FACS
Fluorescence-Activated Cell Sorting
- TKI
Tyrosine Kinase Inhibitor
- PgR
Progesterone Receptor
Footnotes
Here we demonstrate that much like ErbB2, EGFRvIII increases the expression of the chemokine receptor CXCR4. However, EGFRvIII regulates CXCR4 transcriptionally and post-translationally, leading to increased CXCL12/CXCR4-mediated invasion in EGFRvIII-expressing breast cancer cells. This research may also provide more support for the production of new cancer therapeutic agents, which specifically target EGFRvIII and CXCR4 in tumors, and further support the initiation of pre-clinical and clinical trials studying the efficacy of combining novel, well-tolerated EGFR and CXCR4 targeting agents.
- EGFRvIII-expressing breast cancer cells have increased CXCR4 expression
- EGFRvIII-expressing breast cancer cells have enhanced CXCL12/CXCR4-mediated invasion
- Increased levels of HIF-1α, a transcriptional regulator of CXCR4, is correlated to increased CXCR4 transcripts in ER+/PgR+ EGFRvIII-expressing breast cancer cells
- Inhibition of p38 MAPK activity results in reduced CXCR4 expression and attenuated EGFRvIII-induced invasion
- CXCR4 is regulated post-translationally through decreased expression of AIP4 and β-arrestin 1/2, key molecules involved in CXCR4 internalization, cellular trafficking, and degradation
References
- 1.Nieto Y, Nawaz F, Jones RB, Shpall EJ, Nawaz S. Prognostic significance of overexpression and phosphorylation of epidermal growth factor receptor (EGFR) and the presence of truncated EGFRvIII in locoregionally advanced breast cancer. J Clin Oncol. 2007 Oct 1;25(28):4405–4413. doi: 10.1200/JCO.2006.09.8822. [DOI] [PubMed] [Google Scholar]
- 2.Silva HA, Abraul E, Raimundo D, Dias MF, Marques C, Guerra C, de Oliveira CF, Regateiro FJ. Molecular detection of EGFRvIII-positive cells in the peripheral blood of breast cancer patients. Eur J Cancer. 2006 Oct;42(15):2617–2622. doi: 10.1016/j.ejca.2006.03.033. [DOI] [PubMed] [Google Scholar]
- 3.Ge H, Gong X, Tang CK. Evidence of high incidence of EGFRvIII expression and coexpression with EGFR in human invasive breast cancer by laser capture microdissection and immunohistochemical analysis. Int J Cancer. 2002 Mar 20;98(3):357–361. doi: 10.1002/ijc.10224. [DOI] [PubMed] [Google Scholar]
- 4.Yu H, Gong X, Luo X, Han W, Hong G, Singh B, Tang CK. Co-expression of EGFRvIII with ErbB-2 enhances tumorigenesis: EGFRvIII mediated constitutively activated and sustained signaling pathways, whereas EGF-induced a transient effect on EGFR-mediated signaling pathways. Cancer Biol Ther. 2008 Nov 24;7(11) doi: 10.4161/cbt.7.11.6847. [DOI] [PubMed] [Google Scholar]
- 5.Tang CK, Gong XQ, Moscatello DK, Wong AJ, Lippman ME. Epidermal growth factor receptor vIII enhances tumorigenicity in human breast cancer. Cancer Res. 2000 Jun 1;60(11):3081–3087. [PubMed] [Google Scholar]
- 6.Pedersen MW, Tkach V, Pedersen N, Berezin V, Poulsen HS. Expression of a naturally occurring constitutively active variant of the epidermal growth factor receptor in mouse fibroblasts increases motility. Int J Cancer. 2004 Feb 20;108(5):643–653. doi: 10.1002/ijc.11566. [DOI] [PubMed] [Google Scholar]
- 7.Damstrup L, Wandahl PM, Bastholm L, Elling F, Skovgaard PH. Epidermal growth factor receptor mutation type III transfected into a small cell lung cancer cell line is predominantly localized at the cell surface and enhances the malignant phenotype. Int J Cancer. 2002 Jan 1;97(1):7–14. doi: 10.1002/ijc.1572. [DOI] [PubMed] [Google Scholar]
- 8.Lal A, Glazer CA, Martinson HM, Friedman HS, Archer GE, Sampson JH, Riggins GJ. Mutant epidermal growth factor receptor up-regulates molecular effectors of tumor invasion. Cancer Res. 2002 Jun 15;62(12):3335–3339. [PubMed] [Google Scholar]
- 9.Rodrigues S, Attoub S, Nguyen QD, Bruyneel E, Rodrigue CM, Westley BR, May FE, Thim L, Mareel M, Emami S, Gespach C. Selective abrogation of the proinvasive activity of the trefoil peptides pS2 and spasmolytic polypeptide by disruption of the EGF receptor signaling pathways in kidney and colonic cancer cells. Oncogene. 2003 Jul 17;22(29):4488–4497. doi: 10.1038/sj.onc.1206685. [DOI] [PubMed] [Google Scholar]
- 10.Muller A, Homey B, Soto H, Ge N, Catron D, Buchanan ME, McClanahan T, Murphy E, Yuan W, Wagner SN, Barrera JL, Mohar A, et al. Involvement of chemokine receptors in breast cancer metastasis. Nature. 2001 Mar 1;410(6824):50–56. doi: 10.1038/35065016. [DOI] [PubMed] [Google Scholar]
- 11.Li YM, Pan Y, Wei Y, Cheng X, Zhou BP, Tan M, Zhou X, Xia W, Hortobagyi GN, Yu D, Hung MC. Upregulation of CXCR4 is essential for HER2-mediated tumor metastasis. Cancer Cell. 2004 Nov;6(5):459–469. doi: 10.1016/j.ccr.2004.09.027. [DOI] [PubMed] [Google Scholar]
- 12.Cabioglu N, Summy J, Miller C, Parikh NU, Sahin AA, Tuzlali S, Pumiglia K, Gallick GE, Price JE. CXCL-12/stromal cell-derived factor-1alpha transactivates HER2-neu in breast cancer cells by a novel pathway involving Src kinase activation. Cancer Res. 2005 Aug 1;65(15):6493–6497. doi: 10.1158/0008-5472.CAN-04-1303. [DOI] [PubMed] [Google Scholar]
- 13.Cabioglu N, Sahin A, Doucet M, Yavuz E, Igci A, Yildirim O, Aktas E, Bilgic S, Kiran B, Deniz G, Price JE. Chemokine receptor CXCR4 expression in breast cancer as a potential predictive marker of isolated tumor cells in bone marrow. Clin Exp Metastasis. 2005;22(1):39–46. doi: 10.1007/s10585-005-3222-y. [DOI] [PubMed] [Google Scholar]
- 14.Cabioglu N, Yazici MS, Arun B, Broglio KR, Hortobagyi GN, Price JE, Sahin A. CCR7 and CXCR4 as novel biomarkers predicting axillary lymph node metastasis in T1 breast cancer. Clin Cancer Res. 2005 Aug 15;11(16):5686–5693. doi: 10.1158/1078-0432.CCR-05-0014. [DOI] [PubMed] [Google Scholar]
- 15.Cabioglu N, Gong Y, Islam R, Broglio KR, Sneige N, Sahin A, Gonzalez-Angulo AM, Morandi P, Bucana C, Hortobagyi GN, Cristofanilli M. Expression of growth factor and chemokine receptors: new insights in the biology of inflammatory breast cancer. Ann Oncol. 2007 Jun;18(6):1021–1029. doi: 10.1093/annonc/mdm060. [DOI] [PubMed] [Google Scholar]
- 16.Salvucci O, Bouchard A, Baccarelli A, Deschenes J, Sauter G, Simon R, Bianchi R, Basik M. The role of CXCR4 receptor expression in breast cancer: a large tissue microarray study. Breast Cancer Res Treat. 2006 Jun;97(3):275–283. doi: 10.1007/s10549-005-9121-8. [DOI] [PubMed] [Google Scholar]
- 17.Phillips RJ, Mestas J, Gharaee-Kermani M, Burdick MD, Sica A, Belperio JA, Keane MP, Strieter RM. Epidermal growth factor and hypoxia-induced expression of CXC chemokine receptor 4 on non-small cell lung cancer cells is regulated by the phosphatidylinositol 3-kinase/PTEN/AKT/mammalian target of rapamycin signaling pathway and activation of hypoxia inducible factor-1alpha. J Biol Chem. 2005 Jun 10;280(23):22473–22481. doi: 10.1074/jbc.M500963200. [DOI] [PubMed] [Google Scholar]
- 18.Lammering G, Valerie K, Lin PS, Hewit TH. Schmidt-Ullrich RK. Radiation-induced activation of a common variant of EGFR confers enhanced radioresistance. Radiother Oncol. 2004 Sep;72(3):267–273. doi: 10.1016/j.radonc.2004.07.004. [DOI] [PubMed] [Google Scholar]
- 19.Zhang Y, Su H, Rahimi M, Tochihara R, Tang C. EGFRvIII-induced estrogen-independence, tamoxifen-resistance phenotype correlates with PgR expression and modulation of apoptotic molecules in breast cancer. Int J Cancer. 2009 Nov 1;125(9):2021–2028. doi: 10.1002/ijc.24540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim MS, Lee EJ, Kim HR, Moon A. p38 kinase is a key signaling molecule for H-Ras-induced cell motility and invasive phenotype in human breast epithelial cells. Cancer Res. 2003 Sep 1;63(17):5454–5461. [PubMed] [Google Scholar]
- 21.Moore CA, Milano SK, Benovic JL. Regulation of receptor trafficking by GRKs and arrestins. Annu Rev Physiol. 2007;69:451–482. doi: 10.1146/annurev.physiol.69.022405.154712. [DOI] [PubMed] [Google Scholar]
- 22.Qi X, Tang J, Loesch M, Pohl N, Alkan S, Chen G. p38gamma mitogen-activated protein kinase integrates signaling crosstalk between Ras and estrogen receptor to increase breast cancer invasion. Cancer Res. 2006 Aug 1;66(15):7540–7547. doi: 10.1158/0008-5472.CAN-05-4639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vergarajauregui S, San MA, Puertollano R. Activation of p38 mitogen-activated protein kinase promotes epidermal growth factor receptor internalization. Traffic. 2006 Jun;7(6):686–698. doi: 10.1111/j.1600-0854.2006.00420.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bhandari D, Robia SL, Marchese A. The E3 ubiquitin ligase atrophin interacting protein 4 binds directly to the chemokine receptor CXCR4 via a novel WW domain-mediated interaction. Mol Biol Cell. 2009 Mar;20(5):1324–1339. doi: 10.1091/mbc.E08-03-0308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Marchese A, Raiborg C, Santini F, Keen JH, Stenmark H, Benovic JL. The E3 ubiquitin ligase AIP4 mediates ubiquitination and sorting of the G protein-coupled receptor CXCR4. Dev Cell. 2003 Nov;5(5):709–722. doi: 10.1016/s1534-5807(03)00321-6. [DOI] [PubMed] [Google Scholar]
- 26.Cheng ZJ, Zhao J, Sun Y, Hu W, Wu YL, Cen B, Wu GX, Pei G. beta-arrestin differentially regulates the chemokine receptor CXCR4-mediated signaling and receptor internalization, and this implicates multiple interaction sites between beta-arrestin and CXCR4. J Biol Chem. 2000 Jan 28;275(4):2479–2485. doi: 10.1074/jbc.275.4.2479. [DOI] [PubMed] [Google Scholar]
- 27.Sun Y, Cheng Z, Ma L, Pei G. Beta-arrestin2 is critically involved in CXCR4-mediated chemotaxis, and this is mediated by its enhancement of p38 MAPK activation. J Biol Chem. 2002 Dec 20;277(51):49212–49219. doi: 10.1074/jbc.M207294200. [DOI] [PubMed] [Google Scholar]
- 28.Luo C, Pan H, Mines M, Watson K, Zhang J, Fan GH. CXCL12 induces tyrosine phosphorylation of cortactin, which plays a role in CXC chemokine receptor 4-mediated extracellular signal-regulated kinase activation and chemotaxis. J Biol Chem. 2006 Oct 6;281(40):30081–30093. doi: 10.1074/jbc.M605837200. [DOI] [PubMed] [Google Scholar]
- 29.Peng XH, Karna P, Cao Z, Jiang BH, Zhou M, Yang L. Cross-talk between epidermal growth factor receptor and hypoxia-inducible factor-1alpha signal pathways increases resistance to apoptosis by up-regulating survivin gene expression. J Biol Chem. 2006 Sep 8;281(36):25903–25914. doi: 10.1074/jbc.M603414200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhu H, Acquaviva J, Ramachandran P, Boskovitz A, Woolfenden S, Pfannl R, Bronson RT, Chen JW, Weissleder R, Housman DE, Charest A. Oncogenic EGFR signaling cooperates with loss of tumor suppressor gene functions in gliomagenesis. Proc Natl Acad Sci U S A. 2009 Feb 24;106(8):2712–2716. doi: 10.1073/pnas.0813314106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Han W, Zhang T, Yu H, Foulke JG, Tang CK. Hypophosphorylation of residue Y1045 leads to defective downregulation of EGFRvIII. Cancer Biol Ther. 2006 Oct;5(10):1361–1368. doi: 10.4161/cbt.5.10.3226. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Fig. 1 (a) FACS analysis shows increased expression of CXCR4, but not CXCR7, in MDA-MB-361 and BT474 breast cancer cells stably expressing EGFRvIII [antibody 4-5H was used to detect EGFRvIII as previously described (4;31)]. Black lines, secondary antibody only; red lines, parental (“wt”) cells; blue lines, EGFRvIII (“vIII”) transfectants. (b) FACS analysis shows increased expression of CXCR4 in 32D cells stably expressing EGFRvIII, ErbB2, and both EGFRvIII and ErbB2. Black line, secondary antibody only; red line, parental cells; blue line, EGFRvIII transfectants; purple line, ErbB2 transfectants; green line; EGFRvIII and ErbB2 transfectants. (c) FACS analysis shows increased expression of CXCR4 in 32D cells expressing full-length EGFR upon EGF (50 ng/mL) stimulation for 24 hours. Black line, secondary antibody only; red line, parental cells; blue line, untreated EGFR transfectants; purple line, EGFR cells treated with EGF. (d) FACS analysis shows a reduction of CXCR4 in EGFRvIII-expressing MDA-MB-361 and BT474 cells upon treatment with the TKI AG1478 (10 µM) for 24 hours. Black lines, secondary antibody only; red lines, parental cells; blue lines, vehicle (DMSO) treated EGFRvIII transfectants; purple lines, EGFRvIII transfectants treated with AG1478. The x-axis represents fluorescence intensity and the y-axis represents cell counts.
Supplemental Fig. 2 (a) FACS analysis shows increased expression of CXCR4 in MCF-7 and SKBR3 breast cancer cells stably expressing EGFRvIII. Black lines, secondary antibody only; red lines, parental (“wt”) cells; blue lines, EGFRvIII (“vIII”) transfectants. The x-axis represents fluorescence intensity and the y-axis represents cell counts. (b) FACS results quantified. Bar graphs represent the relative expression based on the mean geometric fluorescence of cells in each group/control cells.
Supplemental Fig. 3 Migration of MDA-MB-361/wt and MDA-MB-361/vIII cells toward CXCL12 (50 ng/mL). Cells were plated in 0.1% BSA/IMEM in the upper chamber of transwells with membranes with 8 µm pores coated only with fibronectin (20 ng/mL), and with CXCL12 in the lower chamber. After 24 hours of incubation, the membranes were fixed and stained, and cells counted on the underside of the membranes. The results shown are the mean and SE from cells counted on triplicate membranes.
Supplemental Fig. 4 (a) FACS analysis shows increased expression of CXCR4 in BT474/vIII cells under both normoxic and hypoxic (24 hours) conditions in comparison to BT474/wt cells. Black line, secondary antibody only; red line, parental cells under normoxic conditions; purple line, EGFRvIII transfectants under normoxic conditions; blue line, parental cells under hypoxic conditions; green line, EGFRvIII transfectants under hypoxic conditions. The x-axis represents fluorescence intensity and the y-axis represents cell counts. (b) Immunoblot analysis of lysates from MDA-MB-361 (“361”), BT474, MCF-7, and SKBR3 cells with and without EGFRvIII expression showing that neither HIF-2α or HIF-1β is up-regulated in multiple breast cancer cells expressing EGFRvIII under normoxic conditions.
Supplemental Fig. 5 FACS analysis shows a pronounced decrease in the expression of CXCR4 in MDA-MB-361/vIII cells after 24 hours of treatment with the p38 inhibitor SB203580 (10 µM). Black lines, secondary antibody only; red lines, cells treated with vehicle control (DMSO); blue lines, cells treated with SB203580. The x-axis represents fluorescence intensity and the y-axis represents cell counts.
Supplemental Fig. 6 An additional immunoblot analysis of lysates from MDA-MB-361 (“361”), BT474, MCF-7, and SKBR3 cells with and without EGFRvIII expression showing that β-arrestin 1/2 is down-regulated in multiple breast cancer cells expressing EGFRvIII.
Supplemental Fig. 7 Immunoblot analysis of lysates from BT474/wt and BT474/vIII cells under normoxic and hypoxic (24 hours) conditions showing increased expression of HIF-1α and survivin in BT474/vIII cells under both normoxic and hypoxic conditions.