

Abstract
Heregulin-mediated activation of HER4 initiates receptor cleavage (releasing an 80-kDa HER4 intracellular domain, s80HER4, containing nuclear localization sequences) and results in G2/M delay by unknown signaling mechanisms. We report herein that s80HER4 contains a functional cyclin B-like sequence known as a D-box, which targets proteins for degradation by APC/C, a multisubunit ubiquitin ligase. s80HER4 ubiquitination and ptoteosomal degradation occurred during mitosis but not during S-phase. Inhibition of an APC subunit (APC2) using siRNA knock-down impaired s80HER4 degradation. Mutation of the s80HER4 D-box sequence stabilized s80HER4 during mitosis, and s80HER4-dependent growth inhibition via G2/M delay was significantly greater with the D-box mutant. Polyomvirus middle-T antigen-transformed HC11 cells expressing s80HER4 resulted in smaller, less proliferative, more differentiated tumors in vivo than those expressing kinase-dead s80HER4 or the empty vector. Cells expressing s80HER4 with a disrupted D-box did not form tumors, instead forming differentiated ductal structures. These results suggest that cell cycle-dependent degradation of s80HER4 limits its growth inhibitory action, and stabilization of s80HER4 enhances tumor suppression, thus providing a link between HER4-mediated growth inhibition and cell cycle control.
Keywords: HER4, mitosis, D-box, cell growth, cancer
Introduction
Cell cycle progression involves programming signals that drive proliferation and checkpoint mechanisms that limit cell cycle progression. Progression through mitotic checkpoints requires the proteolytic degradation of certain key regulatory proteins, mediated by the anaphase promoting complex/cyclosome (APC/C), a multisubunit ubiquitin ligase that catalyzes the attachment of polyubiquitin chains onto substrates (1, 2), signaling their destruction by the 26S proteasome. Substrate specificity of APC/C is conferred by its association with coactivators, Cdc20 and Cdh1, each acting as a bridge between the substrate and APC/C. APC/C recognizes small motifs called D-boxes, A-boxes, or KEN boxes. The D-box bears the sequence RXXLXXXXN/D/E (with X being any amino acid), and is found in many proteins ubiquitinated by APC/C, e.g. cyclin B, polo-like kinase, and securin (3-6). To date, these motifs have not been reported in cell surface receptors. We report herein the presence of a functional D-box motif in the ErbB4/HER4 receptor tyrosine kinase (RTK).
The HER/ErbB family of RTKs consists of four members, HER1/ERBB1, HER2/ERBB2, HER3/ERBB3, and HER4/ERBB4. While HER1, -2, and -3 each contribute to aggressive tumor formation (7, 8), HER4 signaling may impair cellular proliferation of human breast cells and promote differentiation (9-13). In breast cancers, HER4 expression correlates with more differentiated tumor grade, longer survival, and positive prognostic indicators [estrogen receptor expression, decreased BrdU incorporation (14-20)]. HER4 is required for heregulin-mediated growth inhibition in human breast cancer cell lines (9), via delayed progression of cells through late G2 or early mitosis (21).
Each of the ErbB receptors has been reported to translocate to the nucleus. For example, HER4 has been detected in the nuclei of breast cancer cells and normal human and mouse mammary tissue (15, 18, 22-25). Unlike HER1, -2, and −3, which translocate to the nucleus as full-length receptors, only a proteolytically-derived intracellular domain of HER4, with a constitutively active kinase domain, becomes nuclear (26-29). Recent work shows that one HER4 isoform, JMa, undergoes a ligand-dependent, sequential HER4 proteolytic process. First HER4 (JMa) is cleaved by tumor necrosis alpha-converting enzyme, followed by a transmembrane γ-secretase cleavage, which releases the soluble 80-kDa cytoplasmic domain (s80HER4) into the cytosol (30-36). Once liberated, s80HER4 exhibits nuclear-cytoplasmic shuttling.
In this report, we examine nuclear s80HER4 regulation by demonstrating that proteolytic generation of s80HER4 from full-length HER4 was required for HER4-mediated growth inhibition, and that expression of s80HER4 induced growth inhibition via a delay in the G2/M phase of the cell cycle. We identified a D-box motif within s80HER4 that directs APC/C-mediated s80HER4 degradation during mitosis. Mutation of the HER4 D-box motif results in stabilization of s80HER4 and greater growth inhibition in response to ligand-activated HER4 or experimentally expressed s80HER4. Additionally, in vivo studies show that transformed HC11 cells expressing s80HER4 or a mutated D-box have decreased tumorigenicity as compared to parental cells.
Methods
Plasmids
The pLXSN vectors expressing human HER4, HER4KD, HER4V675A, and the pcDNA4/TO vectors expressing s80HER4 and s80KD are described elsewhere (21, 37). D-box sequence point mutations in pcDNA4/TO-s80 and pLXSN-HER4 were done with the primer 5′-TATTCAGGGTGATGATCcTATGAAGaTTCCCAGTCCAAATGAC [lowercase underlined nucleotides represent incorporated mutations; Quick-change Site-directed Mutagenesis Kit (Stratagene, La Jolla, CA)].
Cell culture and transfection
HeLa-TRex cells (InVitrogen), referred to as HeLa-rtTA cells, were cultured according to manufacturer's protocol, transfected with Fugene6 (Roche) and selected with Zeocin (5 μg/ml, InVitrogen) or G418 (400 μg/ml). Where indicated, cells were cultured in serum-free Dulbecco's Modified Eagle's Medium ± HRGβ1 (10 ng/ml; Genentech), TET (2 μg/ml), NOC (50 ng/ml), or HU (10 mM) (each from Sigma-Aldrich, St. Louis MO). Cells synchronized in NOC were collected by shake-off, pelleted by centrifugation, resuspended in media containing 10% serum ± CHX, TET, or HRG, where indicated, then re-plated. Where indicated, cells were transiently transfected with siRNA sequences directed against the human APC2 gene using siRNA transfection Reagent (Santa Cruz Biotechnologies). HC11, MCF-10A, and MDA-MB-453 cells expressing GFP and GFP-tagged s80HER4 have been described (Feng et al., submitted).
Western analysis and Immunoprecipitation
For membrane versus cytosolic separation, cells (108) were lysed in ice-cold buffer A [10 mm Tris-HCl, pH 7.4, 150 mm NaCl, 5 mm EDTA, and 250 mm sucrose, 1 mM sodium vanadate, and 1× protease inhibitor cocktail (Roche)] using a 19-gauge needle. The samples were centrifuged at 1500 × g, 4°C for 10 min to pellet nuclei, which were lysed in ice-cold buffer B [5 mm Tris-HCl, pH 7.4, 450 mm NaCl, 5 mm EDTA, and 250 mm sucrose, 1 mM sodium vanadate, and 1× protease inhibitor cocktail]. Membranes were pelleted from buffer A supernatants by centrifugation for 1 h at 100,000 × g in a Beckman ultracentrifuge. The resultant supernatant represented cytosolic fractions. The pellets, representing membrane fractions, were resuspended in 10 mm Tris-HCl, pH 7.4, 150 mm NaCl, 5 mm EDTA, 1% Triton X-100, 250 mM sucrose, 1 mM sodium vanadate and 1× protease inhibitor cocktail for 45 min at 4°C, then cleared by centrifugation. Whole cell extracts were prepared as described previously (21). HER4 was immunoprecipitated with α-HER4 Ab-132, or antibodies against cdc20, APC4 (Santa Cruz Biotechnologies), or cyclin B1 (Labvision Neomarkers, Fremont, CA), and protein A- or -A/G+ agarose beads (Santa Cruz Biotechnologies, Santa Cruz, CA). Immunoprecipitates were analyzed by immunoblotting using antibodies: GFP, APC4, APC2 (Santa Cruz Biotechnologies), Cyclin B or HER4 (Lab Vision Neomarkers) as previously described (21).
Fluorescence and GFP imaging
For GFP visualization, paraformaldehyde-fixed cells were examined with the Nikon Microphot FXA Upright fluorescence microscope. For immunofluorescence, cells were fixed in methanol, incubated 1h in HER4 antibody (Labvision Neomarkers), α-tubulin (Sigma-Aldrich), phospho-Ser10 histone H3 (Cell Signaling), or GFP (Santa Cruz Biotechnologies), then with rhodamine-conjugated goat anti-mouse and Alexa488-conjugated goat anti-rabbit antibody (Molecular Probes, Eugene, OR). Samples were mounted with Vectastain DAPI mounting media (Vector laboratories, Burlingame, VT).
Cell growth assays
Cells (0.1 × 106) were plated in 15-cm dishes at day 0 ± HRG1 or TET. Three plates per cell type per condition were collected by trypsinization and manually counted daily in duplicate.
Cell cycle analysis
Cells were collected by trypsinization, fixed in 75% MeOH, then stained with propidium iodide + RNase A (Becton-Dickenson). 10,000 stained nuclei per sample were analyzed in a FACSCalibur™ flow cytometer (Becton Dickinson). DNA histograms were modeled using Modfit-LT Software (Verity).
Tumor studies
HC11P cells were generated by transfection of pcDNA3-PyVmT (a gift from Dr. Bruce Cuevos, University of North Carolina Chapel Hill), followed by selection with hygromycin. Antibiotic-resistant clones were individually analyzed for PyVmT expression by northern analysis. A single clone, HC11P, was transfected with pcDNA4, pcDNA4-s80, pcDNA4-s80KD, and pcDNA4-s80db, selected with Zeocin, clonally expanded, and analyzed for expression. Pooled clones of cells (1× 106) were injected into the inguinal mammary glands of 6-week old BALB/c female mice, using sterile surgical procedures. Mice were examined weekly by palpation. Mice were sacrificed 15 weeks post-tumor injection, and tumor-bearing mammary glands were immediately fixed in formalin, paraffin-embedded, and sectioned for analysis.
Results
HRG-induced cleavage of HER4 to produce s80HER4 is required for HER4-mediated G2/M delay of HeLa cells
Ligand-mediated HER4 activation is known to result in HER4 cleavage and nuclear/cytoplasmic shuttling of the liberated 80 kDa intracellular domain. We demonstrated this in HeLa-rtTA cells (that do not express detectable HER4) transfected with a cDNA construct encoding wild-type human HER4 (Jma/Cyt1 splice isoform). Treatment of HeLa-HER4 cells with 10 ng/ml heregulin (HRG) for 2 hours (h) was followed by separation of the cells into membrane, cytoplasmic, and nuclear compartments. HER4 immunoprecipitates were analyzed for tyrosine phosphorylation and total HER4 expression by western analysis. HER4 immunoprecipitation from HRG-treated cells revealed ligand-dependent HER4 tyrosine-phosphorylation at 180 kDa in membranes from HeLa-HER4 cells, and the appearance of an 80-kD tyrosine-phopshorylated HER4 product in membrane, cytoplasmic, and nuclear extracts, but not in untreated, serum-starved HeLa-HER4 cells (Fig. 1A.1). We also examined the effects of HRG on cells expressing kinase-dead human HER4 (HER4KD) and on cells expressing a HER4 mutant (V675 to A). This V675 to A mutation that abolishes the HER4 intramembranous, γ-secretase cleavage site (referred to herein as HER4VA) (37). Tyrosine phosphorylation of membrane-bound, full-length HER4VA was detected in HRG-treated HeLa-HER4VA cells, as was a membrane-tethered 80 kDa HER4VA product, although to a lesser extent than what was observed for HRG-treated HeLa-HER4 cells. The 80 kDa HER4VA product was not detected in cytoplasmic or nuclear extracts from HRG-treated HeLa-HER4VA cells, suggesting that the initial HRG-dependent TACE-mediated cleavage of HER4VA occurred, but that the final intramembranous cleavage of HER4VA was not achieved, thus preventing the release of soluble s80HER4. As previously shown, HRG-treatment of cells expressing kinase-dead HER4KD did not result in tyrosine phosphorylation of HER4KD (9) The 80 kD HER4 product was not observed in HRG-treated HeLa-HER4KD cells, consistent with previous reports that kinase activity of HER4 is required to initiate TACE-mediated cleavage of HER4 in its extracellular stalk domain (Fig. 1A.1).
Figure 1. The intracellular domain of HER4, s80HER4, inhibits growth via G2/M delay in HaLa-rtTA cells A.
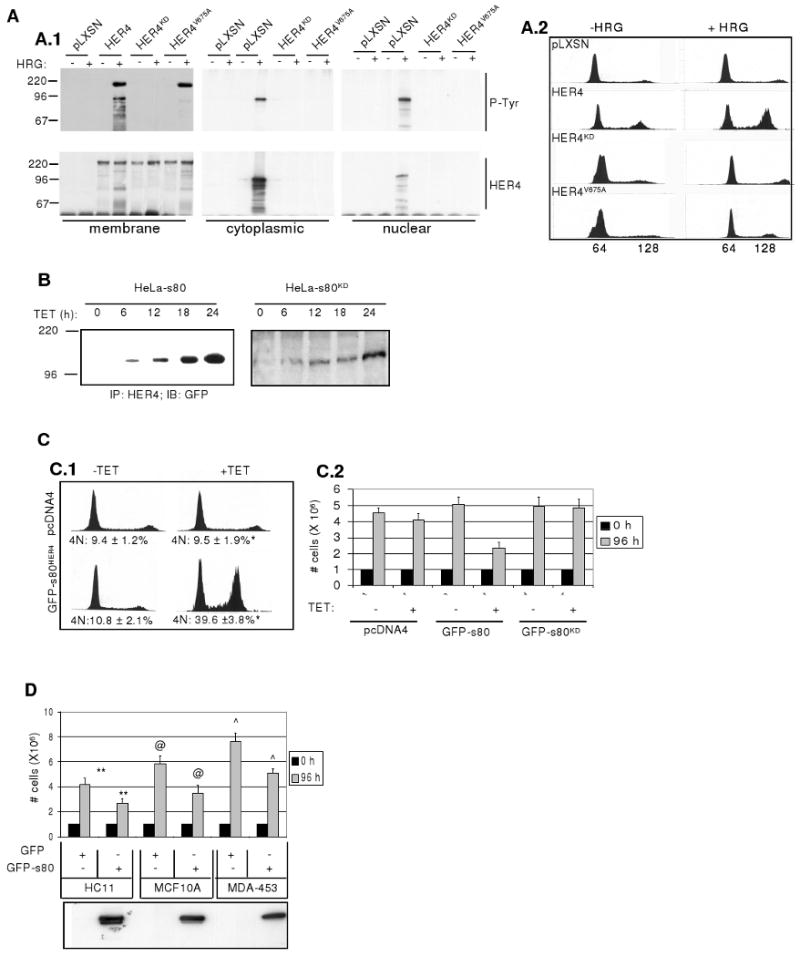
Mutation of the HER4 γ-secretase cleavage site inhibits formation of s80HER4 and interferes with HER4-mediated growth inhibition: A.1 Western analysis to detect phospho-tyrosine residues or total HER4 expression in HER4 immunoprecipitates (IPs) from membrane, cytoplasmic, or nuclear extracts from HeLa-rtTA cells expressing pLXSN, pLXSN-HER4, pLXSN-HER4KD, or pLXSN-HER4VA. Serum-starved cells were treated ± HRG (2h) before separation into cellular compartments using biochemical methods. Molecular weights are indicated at left. A.2: Cells grown in SF media ± HRG (48h) were stained with propidium iodide (PI) and analyzed by flow cytometry. Representative histograms are shown. B. Western analysis to detect GFP in HER4 IPs from HeLa-s80 and HeLa-s80KD cells cultured +TET for 0-24 h. C. Growth inhibition and G2/M delay in cells expressing GFP-s80HER4, but not in cells expressing GFP-s80KD. C.1: Representative histograms of HeLa-pcDNA4 and HeLa-s80 cells grown in SF media ±TET (48h), stained with PI and analyzed by flow cytometry. *P < 0.002, Student's unpaired T-test. C.2 Equal numbers of HeLa-pcDNA4, HeLa-s80, and HeLa-s80KD cells were plated, allowed to attach (24h), then treated for 0 or 96h ±TET in serum-free (SF) media. D. Equal numbers of HC11, MCF-10A, or MDA-453 cells expressing GFP-tagged s80 or GFP, were plated, allowed to attach (24h), then cultured for 0 or 96h in SF media. Each sample was counted in duplicate; each experiment was performed in triplicate. Values represent the average number of cells ± S.D. **P < 0.01; @P < 0.02; ˆP < 0.01; each calculated using Student's unpaired T-test Western analysis to detect s80HER4 in cell lysates is shown in lower panel.
HER4 is required for HRG-mediated growth inhibition in several breast cancer cells (9), resulting in an increased number of cells with 4N DNA (21). HeLa-pLXSN, -HER4, -HER4KD, and HER4VA cells were treated ± HRG for 48 h in serum-free media, to examine the effects of HRG on cell cycle distribution. While exogenous HER4 expression conferred HRG-mediated G2/M delay to HeLa cells, expression of HER4KD and HER4V675A did not allow for any observable HRG-mediated increases in cells with 4N DNA (Fig. 1A.2), suggesting that kinase activity and s80HER4 production are required for HRG-mediated cell cycle delay in HeLa-rtTA cells.
Expression of s80HER4 decreases growth and G2/M transit of HeLa and breast cancer cells
Previous reports demonstrate that expression of s80HER4 (the Cyt1 splice variant) results in growth inhibition of multiple breast and breast cancer cells (21, 37) (Cook, et al, in preparation). We stably introduced into HeLa-rtTA cells a tetracycline (TET)-inducible pcDNA4 construct expressing the Cyt1 isoform of s80HER4 or kinase-dead s80KD, each tagged at the NH2-terminus with green fluorescent protein (GFP). Treatment of cells with TET induced target gene expression within 6-12 hours (h), reaching a maximum by 24 h (Fig. 1B). Induction of GFP-s80HER4 expression increased the proportion of HeLa cells with 4N DNA as compared to HeLa-pcDNA4 control cells treated with TET (Fig. 1C.1). Expression of GFP-s80HER4 for 96 h decreased the rate of total cell growth compared to TET-treated HeLa-pcDNA4 or HeLa-s80KD cells, expressing a GFP-tagged, kinase-dead variant of s80HER4 harboring a lysine 751-to-arginine mutation (Fig. 1C.2). To demonstrate the effects of s80HER4 in breast epithelial cells, we stably expressed GFP-tagged s80HER4 (or GFP alone) in HC11 cells (non-transformed mouse mammary cells), MCF10A cells (human non-transformed breast cells), and MDA-MB-453 cells (human breast cancer-derived cells). Cell numbers, measured 96 h after plating, revealed less cell growth in HC11, MCF10A, and MDA-MB-453 cells expressing GFP-s80HER4 than in these cells expressing GFP alone (Fig. 1D). We have demonstrated that the GFP-s80HER4-mediated decrease in overall cell growth was not due to increased apoptosis (Feng et al, in preparation). Taken together, these data suggest that s80HER4 production is both necessary and sufficient to decrease growth of HeLa and breast-derived cells.
Nuclear-cytoplasmic shuttling of GFP-s80HER4 is regulated with the cell cycle
After a 24 h TET treatment, we found GFP-s80HER4 simultaneously in both the nucleus and cytoplasm of 46.8% of all cells (950 cells counted), while a smaller percentage of cells displayed GFP-s80HER4 staining in only the nucleus (7.9%) or cytoplasm (30.0%); and some cells had no observable GFP-s80HER4 fluorescence (15.3%) (Fig. 2A). Although these experiments were performed in pooled clones of GFP-s80HER4-expressing cells, the heterogeneity of GFP-s80HER4 localization was also observed in clonally-derived populations examined individually (data not shown), indicating that the nuclear-cytoplasmic shuttling is a property of GFP-s80HER4 and not a property of different cell clones.
Figure 2. Cell-cycle-specific expression of s80HER4. A.
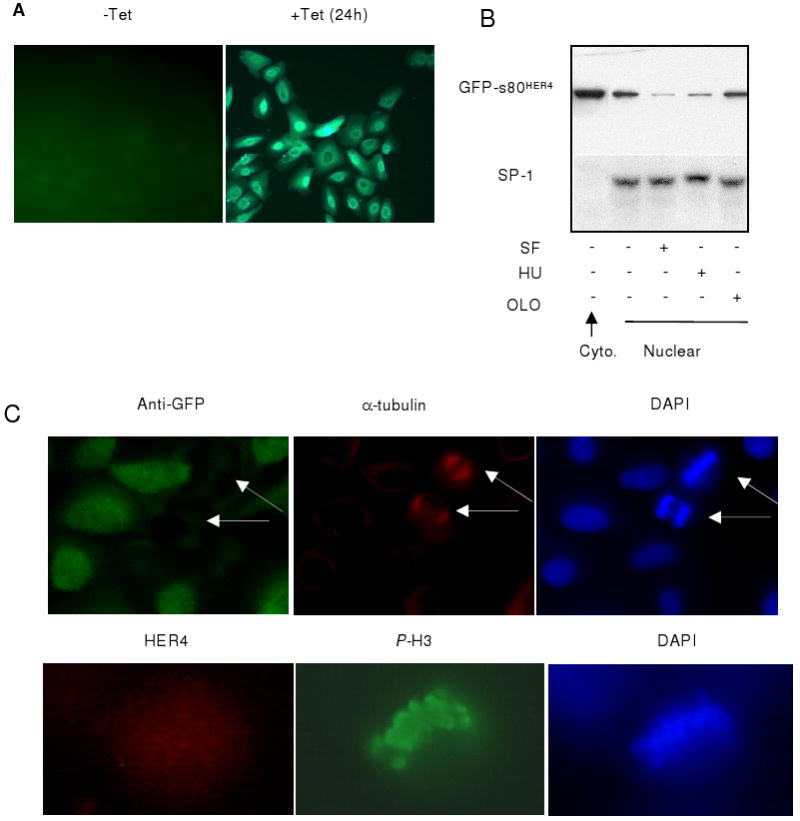
GFP imaging of HeLa-s80 cells cultured 24 h ± TET. B. HeLA-s80 cells were treated +TET for 24 h to induce expression of GFP-s80HER4, followed by 16 h treatment +TET in SF media, or +TET in 10% serum +HU, or +OLO. Cytoplasmic and nuclear extracts were examined by western analysis, using HER4 IP followed by immunoblotting for GFP (upper panels) or by analysis of extracts by immunoblotting for SP-1 (lower panel). C. Immunocytofluorescence of HeLa-s80 cells cultured +TET (48 h). Upper: Cells were dual-stained for GFP and α-tubulin. DAPI images shown at right. Lower: Immunocytofluoresence HER4 and phospho-Ser10 histone H3. DAPI staining shown at right.
Expression of GFP-s80HER4 was induced with TET for 24 h. We then analyzed expression of GFP-s80HER4 in nuclear extracts from cells that were randomly cycling for an additional 16 h, or that were synchronized for 16 h in G1 with serum-free media in the presence of TET, in S-phase with hydroxyurea (HU) + TET, or at the G2/M transition with olomoucine (OLO) + TET. In randomly cycling cells, cytoplasmic GFP-s80HER4 was observed at greater levels than nuclear GFP-s80HER4. We next examined whether relative levels of nuclear expression of GFP-s80HER4 were enriched in specific phases of the cell cycle. Lower levels of nuclear GFP-s80HER4 was observed in G1 and S-phase, as compared to the higher levels of nuclear GFP-s80HER4 found in cells enriched for the G2/M transition (Fig. 2B). To examine the expression of s80HER4 during mitosis (M), we used immunofluorescent staining of randomly cycling, TET-induced HeLa-s80 cells to detect the mitotic marker α-tubulin and GFP. Dual immunofluorescence revealed that of 47 cells harboring α-tubulin positive mitotic spindles, 0/47 cells simultaneously expressed GFP-s80HER4 (Fig. 2C). Phospho-Ser10-histone H3, another mitosis-specific marker, was examined as a marker of mitotic cells. 0 of 81 phospho-Ser-10 histone H3-positive cells were HER4-positive. These results suggest that GFP-s80HER4 expression decreases during mitosis.
Degradation of s80HER4 in mitosis
We synchronized TET-treated HeLa-s80 cells in mitosis (M) with nocodazole (NOC) for 16 h, then upon release of cells from mitosis, examined GFP-s80HER4 stability through the cell cycle by treating the cells with cycloheximide (CHX) to impair new protein synthesis at 0, 2, 4, 8, and 16 h after NOC release. Cells were harvested at 15-minute (min) intervals following CHX treatment and whole cell GFP-s80HER4 was analyzed, as breakdown of the nuclear membrane under these circumstances precludes analysis of nuclear GFP-s80HER4. While GFP-s80HER4 expression was detected at T=0, it was undetectable within 15 min of CHX treatment/nocodazole release (Fig. 3A, lanes 1-4), indicating that s80HER4 was rapidly degraded upon release from NOC. By 4 h and 8 h after NOC release, GFP-s80HER4 showed increasing stability (lanes 10 and 14 show GFP-s80HER4 15 min after release), and GFP-s80HER4 was stable through 45 min when examined 16 h after NOC release (lanes 17-20). While GFP-s80HER4 was rapidly degraded when cells were released in M, GFP-s80HER4 was highly stable in cells synchronized in S-phase with HU then released (Fig. 3B). This suggests that s80HER4 is more stable upon release from S-phase as compared to release from mitosis.
Figure 3. Degradation of s80HER4 during mitosis directed by APC/C. A.
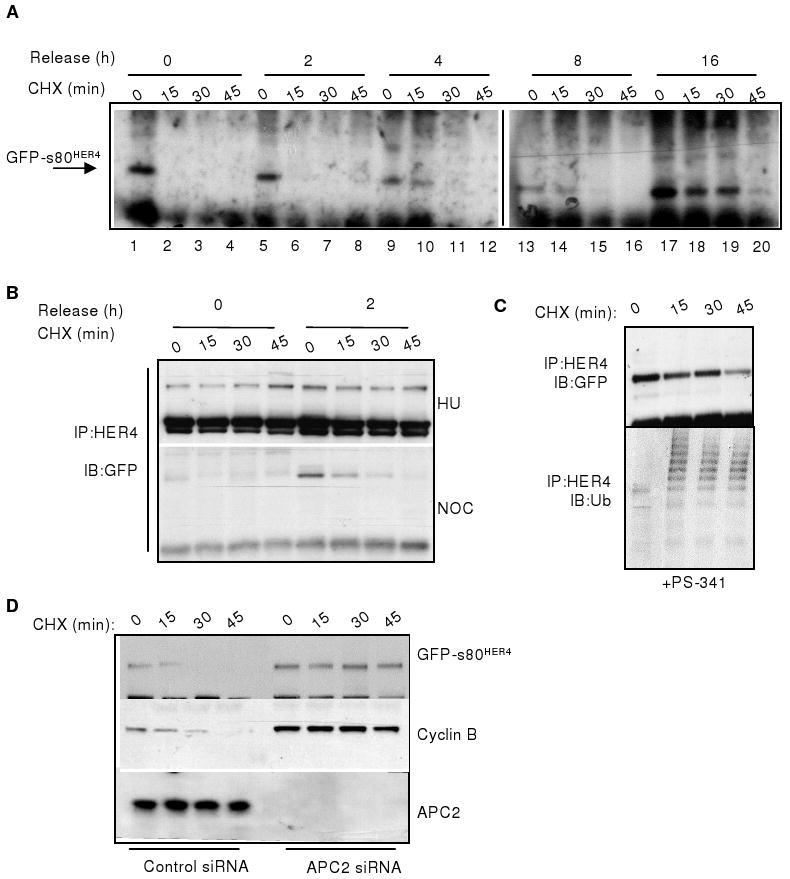
HeLa-s80 cells were cultured +NOC/+TET in 10% serum for 16 h. Cells were released into 10% serum +TET at time (T) =0, and treated +CHX at T=0, 2, 4, 8, and 16h after NOC release. Expression of GFP-s80HER4 was analyzed at 15 min intervals beginning at the time of CHX treatment, by HER4 IP followed by GFP immunoblot (IB). B. HeLa-s80 cells were synchronized +HU/+TET (upper panel) or +NOC/+TET (lower panel) in 10% serum for 16 h, then released into 10% serum +TET at T=0. Cells were treated +CHX at T=0 and 2h. Expression of GFP-s80HER4 was analyzed as described above. C. Cells were cultured +NOC/+TET in 10% serum for 16 h, with PS-341 added for the final 4 h. Cells were released into 10% serum +TET/+PS-341 at T=0. CHX was added at T=0. Expression of GFP-s80HER4 was analyzed as described above; blots were stripped then probed for ubiquitin (lower panel). D. HeLa-s80HER4 cells transfected with 10 nmol siRNA sequences targeting luciferase or APC2 were treated +NOC/+TET +10% serum for 16 h. Cells were released into 10% serum +TET/+CHX. Expression of GFP-s80HER4 and cyclin B were analyzed at 15 min intervals beginning at the time of CHX treatment, by IB of anti-HER4 or anti-cyclin B IP's using GFP or cyclin B antibodies, respectively. APC2 was analyzed by IB.
The APC/C directs proteasomal degradation of s80HER4 during mitosis
TET-treated HeLa-s80 cells were synchronized with NOC for 16 h, and treated with the proteasome inhibitor PS-341 for the final 4 h of synchronization. Cells were released from NOC in the presence of CHX at T=0, and stability of GFP-s80HER4 was measured as described above. Inhibition of the proteasome blocked degradation of s80HER4 through 45 minutes after release from NOC, and allowed for the accumulation of ubiquitinated GFP-s80HER4 (Fig. 3C), suggesting that s80HER4 is ubiquitinated during M and degraded by the proteasome.
The APC2 subunit, required for APC/C activity (38, 39), was knocked down using transfection of short interfering RNAs (siRNAs) into HeLa-s80HER4 cells (Fig. 3D). Transient transfection of 20 nmol APC2 siRNA (Santa Cruz Biotechnologies) impaired expression of APC2 protein in HeLa-s80HER4 cells. APC2 siRNA-transfected cells were grown in 10% serum in the presence of TET for 48 h, adding NOC to the cells for the final 16 h of culture to synchronize the cells in M. Cells were released from NOC and treated with CHX at T=0. Expression of GFP-s80HER4 was analyzed at 15-min intervals following NOC release/CHX treatment. GFP-s80HER4 was detected through 45 min following NOC release in cells lacking APC2 expression. In contrast, cells transfected with a control siRNA sequence showed decreased levels of GFP-s80HER4 expression 15 min after NOC release, and did not harbor detectable levels of GFP-s80HER4 at 30 min following NOC release. Similarly, expression of cyclin B, a known target of the APC/C, was stabilized in cells lacking APC2 expression, but not in cells transfected with a control siRNA sequence. This suggests that s80HER4, like cyclin B, is a target of the APC/C. This represents a novel regulatory mechanism of nuclear signaling for RTKs.
The D-box motif of s80HER4 is required for its APC/C-mediated degradation
Examination of the intracellular domain of human HER4 revealed a canonical D-box motif found in numerous proteins targeted by the APC/C for mitotic destruction, including cyclin B (Table 1). The D-box present in the HER4 intracellular domain is also seen in HER1/EGFR, but not in HER2 or HER3 (alignment per (40)). We introduced point mutations into the D-box motif of full-length HER4 and GFP-s80HER4 to eliminate the RXXLXXXXD consensus sequence, generating HER4db/s80db with sequences of AXXAXXXXD (Table 1). Expression constructs encoding GFP-tagged s80db were used to generate stable TET-inducible HeLa-rtTA cell lines.
Table 1.
Protein sequence of human cyclin B1 (NCBI accession number NM_031966) aligned with that of human HER4/s80HER4, human HER1, human HER2, and human HER3. Residues defining the D-box are bolded and underlined. Site-directed mutagenesis was performed to generate R-to-A and L-to-A mutations, shown in lowercase. * Alignment of ErbB sequences according to Plowman et al., 1993.
| Protein name | Amino acid #* | Sequence |
|---|---|---|
| Cyclin B1 | 40-60 | RPRTALGDIGNKVSEQLQAKM |
| HER4/s80HER4 | 990-1010 | |
| DDRMKLPSPNDSKFFQNLLDE | ||
| HER4db/s80db | 990-1010 | |
| DDaMKaPSPNDSKFFQNLLDE | ||
| HER1/EGFR | 960-980 | DERMHLPSPTDSNFYRALMDE |
| HER2/ErbB2 | 992-1012 | ED - LGP-ASPLDSTFYRSLLED |
| HER3/ErbB3 | 930-950 | SGPGIAPGPEPHGLTNKKLEE |
HeLa-s80HER4, HeLa-s80db, and HeLa-s80KD cells were treated with TET for 24 h to induce expression of GFP-s80HER4, GFP-s80KD, and GFP-s80db, respectively Constitutive tyrosine phosphorylation of GFP-s80HER4 and GFP-s80db was observed (Fig. 4A.1), consistent with a recent report that s80HER4 is an active tyrosine kinase (41). Tyrosine phosphorylation of GFP-s80KD was not observed (Fig. 4A.1), as expected. We synchronized TET-treated HeLa-s80db cells in M with NOC for 16 h, then released the cells in the presence of CHX at T=0. Expression of GFP-s80db was analyzed at 15 min intervals following NOC release/CHX treatment (Fig. 4A.2). Strong GFP-s80db expression was detected 45 min following CHX treatment after release into M, unlike GFP-s80HER4, which was undetectable within 15 min following CHX treatment and release into M (see Fig. 3A). Cyclin B was readily degraded upon NOC release in HeLa-s80db cells, suggesting that GFP-s80db did not impair the APC/C or proteasomal machinery (Fig. 4A.2).
Figure 4. Mutation of the s80HER4 D-box eliminates mitotic degradation of s80HER4 and enhances s80HER4-mediated growth inhibition. A.
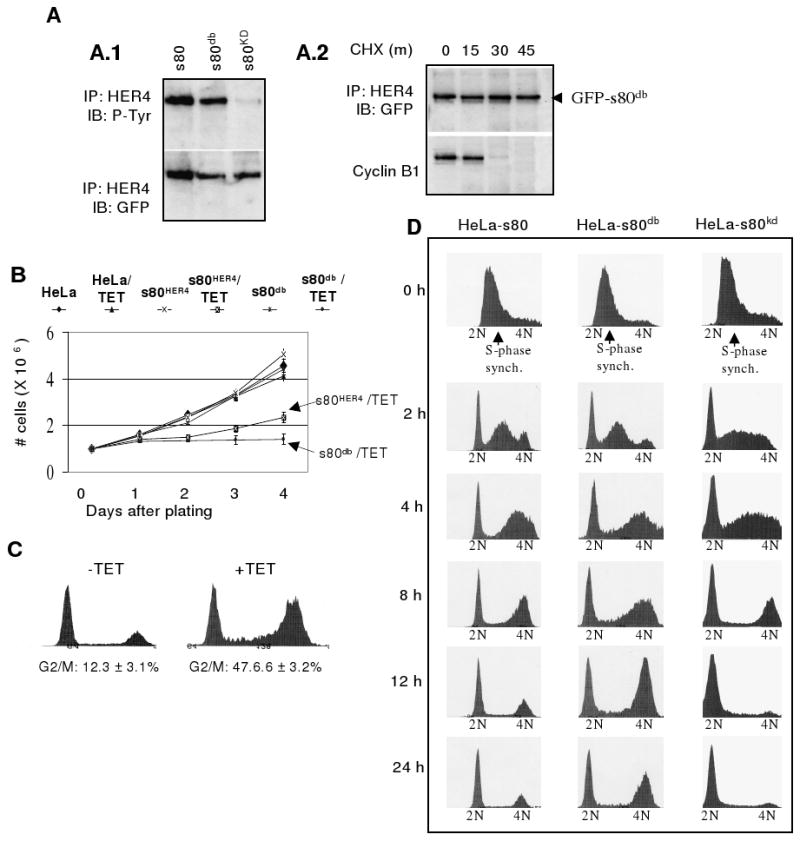
Mutation of the s80HER4 D-box stabilized s80HER4 during mitosis. A.1 Western analysis of HER4 IP's from HeLa-s80, -s80db, and -s80KD cells cultured 24 h +TET. IP's were analyzed for phospho-tyrosine or GFP. A.2: HeLa-s80db cells were cultured +NOC/+TET in 10% serum (16 h). Cells were released into 10% serum +TET/+CHX at T=0, and extracts collected at 15 min intervals. Expression of s80db and cyclin B was analyzed as described in Fig. 3. B. Equal numbers of cells were plated at day 0 ±TET. Cells were counted at 1-day intervals through 4 days. P= 0.011 comparing cell number of HeLa-s80HER4 +TET at day 4 to cell number of HeLa-s80db + TET at day 4, n=5, analyzed in triplicate. C. Cell cycle analysis of HeLa-s80db cells cultured ±TET (48h). > 10,000 nuclei were analyzed per condition; n=3; representative histograms shown. D. Cells were in 10% serum +HU/+TET for 16 h to synchronize in S-phase. Cells were released into 10% serum +TET at T=0 h. Cells were collected at 0, 2, 4, 8, 12, and 24 h following HU release. Cells were stained with propidium iodide and analyzed by flow cytometry as described above.
s80db impairs growth of HeLa cells
Equal numbers of HeLa-s80 and HeLa-s80db cells were plated ±TET and counted daily (Fig. 4B). Uninduced cells proliferated and cell number increased more than 4-fold in four days. TET-treated HeLa-s80 cells were less proliferative and only increased the cell number by 2.2-fold after 4 days in culture. TET-treated HeLa-s80db cells were markedly growth inhibited; cell number increased by only 1.4-fold (P= 0.011 as compared to TET-treated HeLa-s80 cells, n=5). Moreover, HeLa-s80db cells treated with TET for 48 h displayed a dramatic increase in the 4N-containing population compared to uninduced HeLa-s80db cells (Fig. 4C).
To examine the progression of HeLa-s80, -s80db, and -s80KD cells through the cell cycle over a 24 h period, TET-treated cells in 10% serum were synchronized in S-phase with HU, then released into media containing 10% serum +TET at T=0 h. Cells were collected at 0, 2, 4, 8, 12, and 24 h following HU release, and were analyzed at each time point by flow cytometry to determine the proportion of cells in each phase of the cell cycle. Thus, we were able to observe the progression out of S-phase, through G2/M, and into G1 as a function of time. At T=0 h, all cells harbored DNA content between 2N and 4N, suggesting that HU-mediated synchronization in S-phase was complete for each cell line (Fig. 4D). An increase in the amount of 2N and 4N-containing populations was evident for each cell line by 2 h. However, by 8 h following HU release, HeLa-s80db cells and HeLa-s80HER4 cells harbored a greater proportion of cells with 4N DNA as compared to HeLa-s80KD cells, suggesting that HeLa-s80KD cells progressed through G2/M more rapidly than did HeLa-s80 and HeLa-s80db cells. By 12 h there was substantial differences in the relative cell populations with 4N DNA (HeLa-s80db > HeLa-s80HER4 > HeLa-s80kd), a trend that was continued through 24 h. These results suggest that expression of s80db increases the time required for a cell to complete mitosis and return to G1.
Mutation of the D-box within full-length HER4 increases protein stability of proteolytically-derived s80HER4
To determine the consequences of the D-box mutation in full-length HER4, we produced this site-directed mutant (HER4db) and stably expressed HER4db in He-La cells. HRG stimulation of HeLa HER4db cells resulted in tyrosine phosphorylation of HER4db and production of s80db (Fig. 5A), similar to HeLa cells expressing wild-type HER4. This suggests that expression, kinase activity, and ligand-dependent proteolytic cleavage of HER4 are not impaired by mutation of the D-box. Immuno-localization of HER4 in HRG-treated HeLa-HER4db cells revealed cells with nuclear HER4db in the same microscopic field as other cells with strictly cytoplasmic HER4db (Fig. 5B). Similarly, wild-type HER4 localized to nuclear compartments and cytoplasmic compartments differentially in cells within the same microscopic field of HRG-treated HeLa-HER4 cells (Fig. 5B). These results are similar to the subcellular distribution of s80HER4 in populations of TET-induced HeLa-s80HER4 cells (see Fig. 2A). This suggests that proteolytically-derived s80HER4 displays complex regulation of nuclear-cytoplasmic shuttling, similar to that seen for exogenously expressed GFP-s80HER4. To determine if s80db derived from full-length HER4db displays greater stability than wild-type s80HER4 derived from full-length, wild-type HER4, HeLa-HER4 and HeLa-HER4db cells were synchronized in M using NOC in 10% serum for 16 h, then serum-starved in the presence of NOC for an additional 16 h. Cells were treated with HRG 1h prior to release. Finally, cells were released from NOC in the presence of HRG and CHX to examine the stability of proteolytically-derived s80HER4 and s80db at 15 minute intervals. While s80db was detected through 45 min following release from NOC, s80HER4 was not detected in cells released during M (Fig. 5C).
Figure 5. Mutation of the D-box within full length HER4 does not reduce s80HER4 production but does stabilize s80HER4. A.
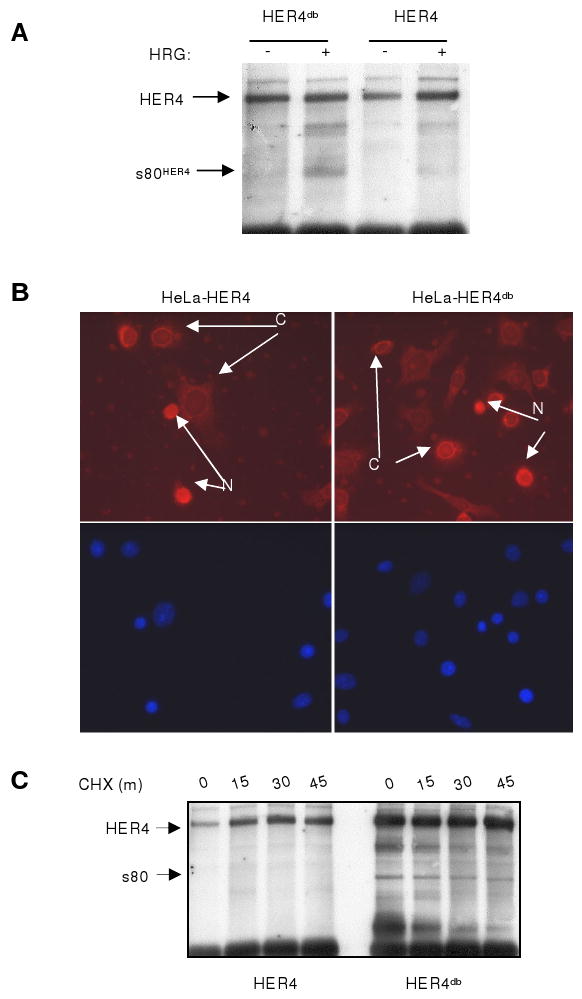
HeLa cells stably transfected with pLXSN-HER4 or -HER4db were serum-starved 16 h then treated +HRG (1h). HER4 IP's were analyzed for total HER4. B. Cells were serum-starved (16 h), treated +HRG (1h). Cells were analyzed for HER4 localization by immunocytofluorescence. DAPI staining is shown at bottom. N= nuclear localization of HER4. C= cytoplasmic localization of HER4. C. HeLa-HER4 and HeLa-HER4db cells were cultured in 10% serum +NOC (16 h), then in SF media +NOC (16 h), adding HRG for the final hour. Cells were released into SF media +HRG/+CHX at T=0 and cells were harvested at T=0, 15, 30, and 45 min. HER4 IPs were analyzed as described above.
Expression of s80db prevents tumor formation in vivo
Transformation of HC11 cells by overexpression of human ErbB2 confers tumorigenic potential to HC11 cells in vivo (42). We used a similar approach, transfecting HC11 cells with a cDNA construct encoding the Polyomavirus middle T antigen (PyVmT). A clonal population of HC11-PyVmT (HC11P) cells was then transfected with pcDNA4-s80HER4, -s80KD, -s80db, or empty pcDNA4. Antibiotic-selected, stably infected cell lines (1 × 106 cells) were injected into the inguinal mammary fatpads of 6-week old female BALB/c mice. Fifteen weeks following injection, the mammary gland was examined histologically. HC11P-pcDNA4 cells formed solid, poorly differentiated tumors (Fig. 6A). In contrast, tumors expressing s80HER4 were smaller, harboring numerous differentiated epithelial structures organizationally similar to lobulo-alveolar structures often seen in mouse mammary glands during early pregnancy. HC11P-s80KD cells formed highly proliferative solid tumors resembling tumors formed by HC11P-pcDNA4 cells. Immunohistochemical detection of proliferating cell nuclear antigen (PCNA) revealed that HC11P-s80KD and HC11P-pcDNA4 tumors harbored similar ratios of PCNA+ cells (P = 0.166, Student's unpaired T-test, n=5 tumors and 3 fields per tumor; Fig. 6B), whereas HC11P-s80 tumors exhibited three-fold fewer PCNA+ cells than vector controls (P < 0.001). Implanted HC11P-s80db cells formed large cystic structures of single epithelial layers surrounding large lumens filled with eosinophilic proteins, suggesting a degree of lactogenic differentiation (Fig. 6A). These did not appear to have malignant elements, and these structures exhibited the lowest percentage of PCNA+ cells (9%, Fig. 6B), with a statistically significant decrease in PCNA staining compared to HC11-s80 tumors (12.5%, P = 0.0371). No statistically significant differences were detected in the rate of cell death between the four groups, as measured by TUNEL analysis (Fig. 6B). These results suggest that expression of s80HER4 decreases growth of breast tumors in vivo, and mutation of the s80HER4 D-box enhances tumor suppression by s80HER4, perhaps by increasing the stability of s80HER4.
Figure 6. Decreased tumor formation in PyVmT-transformed HC11 cells expressing s80db. A.
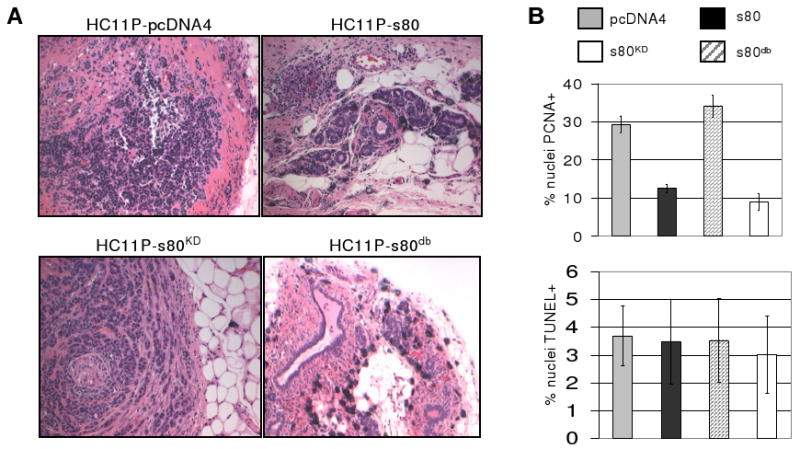
HC11P cells expressing s80HER4, s80KD, s80db, or the empty pcDNA4 vector were injected into the inguinal mammary fatpads of female BALB/c mice. Fifteen weeks post-implantation of cells, mice were euthanized and the mammary glands analyzed histologically; representative images are shown. B. Five tumor samples per group were analyzed by immunohistochemistry. PCNA positive (upper panel) or TUNEL positive nuclei (lower panel) were scored in 3 randomly chosen 400× fields per tumor section, for a total of 15 fields counted. HC11P-pcDNA4 and HC11P-s80KD had equivalent percentages of PCNA-positive nuclei (29.4±2.2 and 34.1±2.9, respectively). HC11P-s80 had a statistically significantly lower percentage of PCNA-positive cells (12.5±1.1, p<0.0001) as compared to vector alone. While HC11P-s80db percentage of PCNA-positive cells was the lowest of all groups (9.0±2.2), and was also significantly decreased when compared to HC11P-s80 (p<0.037).
Discussion
Recent evidence shows that ligand-mediated activation of the JMa HER4 isoform results in HER4 cleavage and liberation of the intracellular domain, s80HER4, which has the ability localize to the nucleus. Our studies demonstrate that nuclear-cytoplasmic shuttling of s80HER4 is regulated, in part, by a cell cycle-dependent process, culminating in its degradation during M. The destruction of s80HER4 during mitosis by the cell cycle machinery represents a novel regulatory mechanism for a receptor tyrosine kinase, and may relate directly to the ability of HER4 to slow cell growth by inhibiting progression through the G2/M transition or M.
Many proteins are degraded by the APC/C in a D-box-dependent manner. Of these, most have defined roles in regulating mitotic progression, including cyclin B, polo-like kinase, aurora A, and securin. Inhibition of APC/C-mediated degradation of these proteins results in delayed progression through M. For example, cyclin B1 in mammalian cells is degraded prior to anaphase (43) and expression of a nondegradable cyclin B1 harboring a mutation within the D-box delayed mitosis in mammalian cells and frog oocytes, by blocking the metaphase/anaphase transition (44, 45). Similarly, mutation of the s80HER4 D-box impaired its destruction and decreased the rate of G2/M progression, suggesting that destruction of the intracellular domain of HER4, like cyclin B, may be required for progression through the cell cycle in cells expressing nuclear HER4. The mechanism by which HER4 delays progression through G2/M is currently under investigation. However, our results are consistent with the idea that the intracellular domain of HER4-Cyt1 may indeed be involved in regulating the G2/M transition or progression through early M.
The discovery of ligand-dependent JMa HER4 proteolysis and release of a nuclear localizing soluble tyrosine kinase has resulted in multiple studies investigating the function of s80HER4. We and others have demonstrated s80HER4 growth inhibitory action (21, 33, 37); and have shown that HRG-mediated HER4 activity, as well as de novo expression of s80HER4, slows the progression of cells through G2/M. The data presented herein suggests a physiologic mechanism for the regulation of s80HER4 by APC-dependent ubiquitination and degradation during M-phase.
Other recent reports have suggested specific transcriptional roles for nuclear s80HER4 complexed with transcription factors or co-factors. These include a positive effect on estrogen receptor-mediated gene transcription in estrogen receptor positive breast cancer cells (46), on STAT5A-depdendent gene expression in differentiating mammary epithelial cells, and repression of astrocyte-specific gene transcription in astrocyte progenitor cells in the developing mouse brain (47). In these studies, s80HER4 colocalized in complexes on specific DNA elements, for example, on the progesterone receptor promoter with the estrogen receptor (ER) or the GFAP promoter with TAB2 and N-Cor (46, 47). Additionally, s80HER4 may enhance transcription factor activity through kinase-mediated phosphorylation. For example, s80HER4 phosphorylates the transcription factor, STAT5A, and enhances STAT5A-dependent transcription (12, 37). s80HER4 also facilitates movement of the transcriptional complex from cytoplasm to nucleus. Whether our postulated functional interaction with G2/M checkpoint cell cycle control mechanisms requires transcriptional control at specific promoters or tyrosine phosphorylation of other regulatory elements, remains to be determined. It will be of interest to determine whether altering s80HER4 mitotic destruction via the D-box mutation changes transcriptional modulators to address whether growth inhibition and transcriptional control by s80HER4 are distinct mechanistically.
The findings reported here may have clinical implications. Most clinical correlative studies of human breast cancer agree that nuclear HER4 correlates with positive prognostic indicators. We have shown the HER4 activation (9) or s80HER4 expression (21) (and see Fig. 1) inhibited breast cancer cell growth by decreasing the progression of cells through G2/M (21). Furthermore, we have demonstrated (Fig. 6) that s80HER4 slowed transformed breast cell proliferation when expressed in PyVmT-expressing HC11 cells. Ligand-mediated activation of HER4 results in s80HER4 release, nuclear-cytoplasmic shuttling, and growth inhibition; interference with proteolytic cleavage (HER4V675A, see Fig. 1) abrogates this action. Thus, s80HER4 production is an obligatory step for HER4-mediated growth inhibition. Moreover, s80HER4 expression is sufficient to confer growth inhibition to HeLa cells, or breast cancer cells, in the absence of full-length HER4 or ligand stimulation (21). The growth inhibitory signal of s80HER4 was strengthened by D-box mutation. Since the D-box is required for degradation during M, it is possible that s80HER4 impairs growth of cells by delaying the cell cycle in M, such that degradation of s80HER4 is required for mitotic progression. Although this hypothesis requires further investigation, these data demonstrate the potential link between nuclear HER4 signaling and regulation of mitotic progression. The suppression of PyVmT-induced tumor cell proliferation in vivo by s80HER4 and the increased effectiveness of the s80HER4 D-box mutant highlight the need for mechanistic studies, which in turn will help elucidate the therapeutic implications of s80HER4 expression in breast cancer.
Acknowledgments
This work was supported in part by the Breast Cancer Research Foundation, and grants from the National Institutes for Health, GM00678 and CA112553.
References
- 1.Harper JW, Burton JL, Solomon MJ. The anaphase-promoting complex: it's not just for mitosis any more. Genes Dev. 2002;16:2179–2206. doi: 10.1101/gad.1013102. [DOI] [PubMed] [Google Scholar]
- 2.Peters JM. The anaphase-promoting complex: proteolysis in mitosis and beyond. Mol Cell. 2002;9:931–943. doi: 10.1016/s1097-2765(02)00540-3. [DOI] [PubMed] [Google Scholar]
- 3.Glotzer M, Murray AW, Kirschner MW. Cyclin is degraded by the ubiquitin pathway. Nature. 1991;349:132–138. doi: 10.1038/349132a0. [DOI] [PubMed] [Google Scholar]
- 4.Visintin R, Prinz S, Amon A. CDC20 and CDH1: a family of substrate-specific activators of APC-dependent proteolysis. Science. 1997;278:460–463. doi: 10.1126/science.278.5337.460. [DOI] [PubMed] [Google Scholar]
- 5.Zachariae W, Nasmyth K. Whose end is destruction: cell division and the anaphase-promoting complex. Genes Dev. 1999;13:2039–2058. doi: 10.1101/gad.13.16.2039. [DOI] [PubMed] [Google Scholar]
- 6.Fang G, Yu H, Kirschner MW. The checkpoint protein MAD2 and the mitotic regulator CDC20 form a ternary complex with the anaphase-promoting complex to control anaphase initiation. Genes Dev. 1998;12:1871–1883. doi: 10.1101/gad.12.12.1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hynes NE, Lane HA. ERBB receptors and cancer: the complexity of targeted inhibitors. Nat Rev Cancer. 2005;5:341–354. doi: 10.1038/nrc1609. [DOI] [PubMed] [Google Scholar]
- 8.Stern DF. Tyrosine kinase signalling in breast cancer: ErbB family receptor tyrosine kinases. Breast Cancer Res. 2000;2:176–183. doi: 10.1186/bcr51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sartor CI, Zhou H, Kozlowska E, Guttridge K, Kawata E, Caskey L, Harrelson J, Hynes N, Ethier S, Calvo B, Earp HS., 3rd Her4 mediates ligand-dependent antiproliferative and differentiation responses in human breast cancer cells. Mol Cell Biol. 2001;21:4265–4275. doi: 10.1128/MCB.21.13.4265-4275.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Penington DJ, Bryant I, Riese DJ., 2nd Constitutively active ErbB4 and ErbB2 mutants exhibit distinct biological activities. Cell Growth Differ. 2002;13:247–256. [PubMed] [Google Scholar]
- 11.Tovey SM, Witton CJ, Bartlett JM, Stanton PD, Reeves JR, Cooke TG. Outcome and human epidermal growth factor receptor (HER) 1-4 status in invasive breast carcinomas with proliferation indices evaluated by bromodeoxyuridine labelling. Breast Cancer Res. 2004;6:R246–251. doi: 10.1186/bcr783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Williams CC, Allison JG, Vidal GA, Burow ME, Beckman BS, Marrero L, Jones FE. The ERBB4/HER4 receptor tyrosine kinase regulates gene expression by functioning as a STAT5A nuclear chaperone. J Cell Biol. 2004;167:469–478. doi: 10.1083/jcb.200403155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vidal GA, Naresh A, Marrero L, Jones FE. Presenilin-dependent gamma-secretase processing regulates multiple ERBB4/HER4 activities. J Biol Chem. 2005;280:19777–19783. doi: 10.1074/jbc.M412457200. [DOI] [PubMed] [Google Scholar]
- 14.Knowlden JM, Gee JM, Seery LT, Farrow L, Gullick WJ, Ellis IO, Blamey RW, Robertson JF, Nicholson RI. c-erbB3 and c-erbB4 expression is a feature of the endocrine responsive phenotype in clinical breast cancer. Oncogene. 1998;17:1949–1957. doi: 10.1038/sj.onc.1202107. [DOI] [PubMed] [Google Scholar]
- 15.Abd El-Rehim DM, Pinder SE, Paish CE, Bell JA, Rampaul RS, Blamey RW, Robertson JF, Nicholson RI, Ellis IO. Expression and co-expression of the members of the epidermal growth factor receptor (EGFR) family in invasive breast carcinoma. Br J Cancer. 2004;91:1532–1542. doi: 10.1038/sj.bjc.6602184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bacus SS, Chin D, Yarden Y, Zelnick CR, Stern DF. Type 1 receptor tyrosine kinases are differentially phosphorylated in mammary carcinoma and differentially associated with steroid receptors. Am J Pathol. 1996;148:549–558. [PMC free article] [PubMed] [Google Scholar]
- 17.Pawlowski V, Revillion F, Hebbar M, Hornez L, Peyrat JP. Prognostic value of the type I growth factor receptors in a large series of human primary breast cancers quantified with a real-time reverse transcription-polymerase chain reaction assay. Clin Cancer Res. 2000;6:4217–4225. [PubMed] [Google Scholar]
- 18.Srinivasan R, Gillett CE, Barnes DM, Gullick WJ. Nuclear expression of the c-erbB-4/HER-4 growth factor receptor in invasive breast cancers. Cancer Res. 2000;60:1483–1487. [PubMed] [Google Scholar]
- 19.Suo Z, Risberg B, Kalsson MG, Willman K, Tierens A, Skovlund E, Nesland JM. EGFR family expression in breast carcinomas. c-erbB-2 and c-erbB-4 receptors have different effects on survival. J Pathol. 2002;196:17–25. doi: 10.1002/path.1003. [DOI] [PubMed] [Google Scholar]
- 20.Witton CJ, Reeves JR, Going JJ, Cooke TG, Bartlett JM. Expression of the HER1-4 family of receptor tyrosine kinases in breast cancer. J Pathol. 2003;200:290–297. doi: 10.1002/path.1370. [DOI] [PubMed] [Google Scholar]
- 21.Muraoka-Cook RS, Caskey L, Sandahl M, Hunter D, Husted C, Strunk K, Sartor CI, Rearick W, Jr, McCall W, Sgagias M, Cowan K, Earp HS., 3rd Heregulin-dependent delay in mitotic progression requires HER4 and BRCA1. Mol Cell Biol. 2006 doi: 10.1128/MCB.01950-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Srinivasan R, Benton E, McCormick F, Thomas H, Gullick WJ. Expression of the c-erbB-3/HER-3 and c-erbB-4/HER-4 growth factor receptors and their ligands, neuregulin-1 alpha, neuregulin-1 beta, and betacellulin, in normal endometrium and endometrial cancer. Clin Cancer Res. 1999;5:2877–2883. [PubMed] [Google Scholar]
- 23.Long W, Wagner KU, Lloyd KC, Binart N, Shillingford JM, Hennighausen L, Jones FE. Impaired differentiation and lactational failure of Erbb4-deficient mammary glands identify ERBB4 as an obligate mediator of STAT5. Development. 2003;130:5257–5268. doi: 10.1242/dev.00715. [DOI] [PubMed] [Google Scholar]
- 24.Zhang M, Ding D, Salvi R. Expression of heregulin and ErbB/Her receptors in adult chinchilla cochlear and vestibular sensory epithelium. Hear Res. 2002;169:56–68. doi: 10.1016/s0378-5955(02)00339-8. [DOI] [PubMed] [Google Scholar]
- 25.Junttila TT, Sundvall M, Lundin M, Lundin J, Tanner M, Harkonen P, Joensuu H, Isola J, Elenius K. Cleavable ErbB4 isoform in estrogen receptor-regulated growth of breast cancer cells. Cancer Res. 2005;65:1384–1393. doi: 10.1158/0008-5472.CAN-04-3150. [DOI] [PubMed] [Google Scholar]
- 26.Lin SY, Makino K, Xia W, Matin A, Wen Y, Kwong KY, Bourguignon L, Hung MC. Nuclear localization of EGF receptor and its potential new role as a transcription factor. Nat Cell Biol. 2001;3:802–808. doi: 10.1038/ncb0901-802. [DOI] [PubMed] [Google Scholar]
- 27.Offterdinger M, Schofer C, Weipoltshammer K, Grunt TW. c-erbB-3: a nuclear protein in mammary epithelial cells. J Cell Biol. 2002;157:929–939. doi: 10.1083/jcb.200109033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang SC, Lien HC, Xia W, Chen IF, Lo HW, Wang Z, Ali-Seyed M, Lee DF, Bartholomeusz G, Ou-Yang F, Giri DK, Hung MC. Binding at and transactivation of the COX-2 promoter by nuclear tyrosine kinase receptor ErbB-2. Cancer Cell. 2004;6:251–261. doi: 10.1016/j.ccr.2004.07.012. [DOI] [PubMed] [Google Scholar]
- 29.Maatta JA, Sundvall M, Junttila TT, Peri L, Laine VJ, Isola J, Egeblad M, Elenius K. Proteolytic cleavage and phosphorylation of a tumor-associated ErbB4 isoform promote ligand-independent survival and cancer cell growth. Mol Biol Cell. 2006;17:67–79. doi: 10.1091/mbc.E05-05-0402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cheng QC, Tikhomirov O, Zhou W, Carpenter G. Ectodomain cleavage of ErbB-4: characterization of the cleavage site and m80 fragment. J Biol Chem. 2003;278:38421–38427. doi: 10.1074/jbc.M302111200. [DOI] [PubMed] [Google Scholar]
- 31.Vecchi M, Baulida J, Carpenter G. Selective cleavage of the heregulin receptor ErbB-4 by protein kinase C activation. J Biol Chem. 1996;271:18989–18995. doi: 10.1074/jbc.271.31.18989. [DOI] [PubMed] [Google Scholar]
- 32.Lee HJ, Jung KM, Huang YZ, Bennett LB, Lee JS, Mei L, Kim TW. Presenilin-dependent gamma-secretase-like intramembrane cleavage of ErbB4. J Biol Chem. 2002;277:6318–6323. doi: 10.1074/jbc.M110371200. [DOI] [PubMed] [Google Scholar]
- 33.Ni CY, Murphy MP, Golde TE, Carpenter G. gamma -Secretase cleavage and nuclear localization of ErbB-4 receptor tyrosine kinase. Science. 2001;294:2179–2181. doi: 10.1126/science.1065412. [DOI] [PubMed] [Google Scholar]
- 34.Ni CY, Yuan H, Carpenter G. Role of the ErbB-4 carboxyl terminus in gamma-secretase cleavage. J Biol Chem. 2003;278:4561–4565. doi: 10.1074/jbc.M210504200. [DOI] [PubMed] [Google Scholar]
- 35.Komuro A, Nagai M, Navin NE, Sudol M. WW domain-containing protein YAP associates with ErbB-4 and acts as a co-transcriptional activator for the carboxyl-terminal fragment of ErbB-4 that translocates to the nucleus. J Biol Chem. 2003;278:33334–33341. doi: 10.1074/jbc.M305597200. [DOI] [PubMed] [Google Scholar]
- 36.Carpenter G. Nuclear localization and possible functions of receptor tyrosine kinases. Curr Opin Cell Biol. 2003;15:143–148. doi: 10.1016/s0955-0674(03)00015-2. [DOI] [PubMed] [Google Scholar]
- 37.Muraoka-Cook RS, Sandahl M, Husted C, Hunter D, Miraglia L, Feng SM, Elenius K, Earp HS., 3rd The Intracellular Domain of ErbB4 Induces Differentiation of Mammary Epithelial Cells. Mol Biol Cell. 2006 doi: 10.1091/mbc.E06-02-0101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zachariae W, Shevchenko A, Andrews PD, Ciosk R, Galova M, Stark MJ, Mann M, Nasmyth K. Mass spectrometric analysis of the anaphase-promoting complex from yeast: identification of a subunit related to cullins. Science. 1998;279:1216–1219. doi: 10.1126/science.279.5354.1216. [DOI] [PubMed] [Google Scholar]
- 39.Kramer KM, Fesquet D, Johnson AL, Johnston LH. Budding yeast RSI1/APC2, a novel gene necessary for initiation of anaphase, encodes an APC subunit. Embo J. 1998;17:498–506. doi: 10.1093/emboj/17.2.498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Plowman GD, Culouscou JM, Whitney GS, Green JM, Carlton GW, Foy L, Neubauer MG, Shoyab M. Ligand-specific activation of HER4/p180erbB4, a fourth member of the epidermal growth factor receptor family. Proc Natl Acad Sci U S A. 1993;90:1746–1750. doi: 10.1073/pnas.90.5.1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Linggi B, Cheng QC, Rao AR, Carpenter G. The ErbB-4 s80 intracellular domain is a constitutively active tyrosine kinase. Oncogene. 2006;25:160–163. doi: 10.1038/sj.onc.1209003. [DOI] [PubMed] [Google Scholar]
- 42.Brandt R, Wong AM, Hynes NE. Mammary glands reconstituted with Neu/ErbB2 transformed HC11 cells provide a novel orthotopic tumor model for testing anti-cancer agents. Oncogene. 2001;20:5459–5465. doi: 10.1038/sj.onc.1204709. [DOI] [PubMed] [Google Scholar]
- 43.Clute P, Pines J. Temporal and spatial control of cyclin B1 destruction in metaphase. Nat Cell Biol. 1999;1:82–87. doi: 10.1038/10049. [DOI] [PubMed] [Google Scholar]
- 44.Wheatley SP, Hinchcliffe EH, Glotzer M, Hyman AA, Sluder G, Wang Y. CDK1 inactivation regulates anaphase spindle dynamics and cytokinesis in vivo. J Cell Biol. 1997;138:385–393. doi: 10.1083/jcb.138.2.385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chang DC, Xu N, Luo KQ. Degradation of cyclin B is required for the onset of anaphase in Mammalian cells. J Biol Chem. 2003;278:37865–37873. doi: 10.1074/jbc.M306376200. [DOI] [PubMed] [Google Scholar]
- 46.Zhu Y, Sullivan LL, Nair SS, Williams CC, Pandey AK, Marrero L, Vadlamudi RK, Jones FE. Coregulation of Estrogen Receptor by ERBB4/HER4 Establishes a Growth-Promoting Autocrine Signal in Breast Tumor Cells. Cancer Res. 2006;66:7991–7998. doi: 10.1158/0008-5472.CAN-05-4397. [DOI] [PubMed] [Google Scholar]
- 47.Sardi SP, Murtie J, Koirala S, Patten BA, Corfas G. Presenilin-Dependent ErbB4 Nuclear Signaling Regulates the Timing of Astrogenesis in the Developing Brain. Cell. 2006;127:185–197. doi: 10.1016/j.cell.2006.07.037. [DOI] [PubMed] [Google Scholar]