

Abstract
The catalytic enantioselective synthesis of densely functionalised organic molecules containing all-carbon quaternary stereocentres is a challenge to modern chemical methodology research. The catalytically controlled asymmetric α-alkylation of ketones represents another difficult task and has been of major interest to our and other research groups in the past. We now report a palladium-catalyzed enantioselective process that addresses both problems at once and allows the installation of vicinal all-carbon quaternary and tertiary stereocentres at the α-carbon of a ketone in a single step. This multiple bond forming process is carried out on readily available β-ketoester starting materials and proceeds via conjugate addition of an in situ-generated palladium enolate to activated Michael acceptors. In other words, the CO2-moiety of the substrate is displaced by a C-C fragment in an asymmetric cut-and-paste reaction. The products are obtained in high yield, diastereomeric ratio, and enantiomeric excess.
The enantioselective generation of carbon stereocentres having four other carbon substituents (also called quaternary or all-carbon quaternary stereocentres) is a task synthetic chemists are confronted with regularly1. Several methods have been developed to address this problem but catalytic approaches remain challenging2,3. Furthermore, when additional carbon stereocentres adjacent to the quaternary carbon are required, the available choices to install both within one reaction are extremely limited. While the combination of quaternary and secondary stereocentres can be introduced by aldol-, cycloaddition-, Michael- or even cross-coupling reactions, only a handful of catalytic methods exist that forge adjacent quaternary and tertiary carbon stereocentres. This motif was successfully introduced into organic molecules by α-functionalisation of carbonyl groups as demonstrated in recent reports. However, these approaches mostly rely on organic catalysts based on quinine or proline4,5,6,7 and only two enantioselective transition metal catalyzed examples have been published to date8,9. Interestingly, in both cases strongly coordinating substrates such as 1,3-diketones, β-ketoesters or α-cyanoesters are required to achieve good selectivity. Other examples, like the elegant work of Overman, demonstrated the feasibility of palladium catalysis to construct adjacent quaternary and tertiary or even vicinal quaternary stereocentres in a single double Heck-reaction10,11. These diastereoselective reactions employ an achiral catalyst and rely on substrate-directed stereocontrol. Palladium-catalyzed tandem reactions like this have found application in intramolecular diastereoselective coupling-reactions12. Intermolecular examples especially involving π-allyl-palladium species are less common, but some recent examples achieving high diastereoselectivities have been published13,14,15. Therefore, an approach to the formation of vicinal quaternary and tertiary stereocentres that employs a chiral palladium catalyst is appealing and one could imagine that an asymmetric reaction involving the formation of multiple bonds could achieve this goal.
The enantioselective α-functionalisation of ketones has been realised by means of palladium catalysis and over the past decade significant progress has been made in α-allylation16, vinylation17, and arylation18 reactions. A transition metal catalyzed α-alkylation of ketones has been reported by Jacobsen employing chromium-salen complexes as catalysts to yield quaternary stereocentres19,20. The requirement of tin-enolates as starting materials, which have to be synthesised in advance and are presumably toxic, is a strong limitation in this case.
Over the past few years, our group has been interested in novel methodologies for the generation of all-carbon quaternary stereocentres, which led to the development of an enantioselective palladium catalysed allylic α-alkylation of ketones. This process allows the construction of such quaternary carbon stereocentres starting from silylenolethers, enolcarbonates21 or readily available racemic β-ketoesters 1 (path a, Figure 1)22. We also demonstrated that the intermediate of this reaction, presumably a palladium enolate (A)23, can be intercepted by a strong electrophile like H+ to generate enantioenriched α-protonated products (path b)24,25.
Figure 1. Concept for the enantioselective functionalisation of palladium-enolates.

Three pathways for the palladium catalyzed generation α-functionalised ketones are shown. A palladium catalyst generated from [Pd2(dba)3] and ligand L undergoes oxidative addition to β-ketoester 1 following the loss of CO2 to form an enolate intermediate A. This palladium enolate can either react following path a to yield enantioenriched α-alkylated products or following path b in the presence of a proton source to give α-protonated products. The envisioned path c would form miscellaneous enantioenriched α-substituted ketones depending on other electrophilic additives "E+" that intercept enolate A.
To expand this enolate-trapping approach toward the broader goal of establishing vicinal stereocentres and to provide a general approach for α-functionalisation of ketones, we contemplated the use of other prochiral electrophiles (path c). Therefore, an electrophilic additive had to be identified meeting the following crucial requirements: (1) no interference with the oxidative addition of the palladium(0) catalyst into the allylester functionality should occur; (2) the reaction of the palladium-enolate with the electrophilic additive must be preferred to the intramolecular allylic α-allylation; (3) a scavenger for the allyl fragment must be provided. In our first experiments carbon electrophiles like acyclic or cyclic enones were probed for their capability to intercept the intermediate enolate but in these cases only the regular allylic alkylation product was observed. Toward that end, we were drawn to earlier reports by Yamamoto of intercepted enolate alkylation by highly electrophilic conjugate acceptors such as benzylidenemalononitrile 2a (Table 1)15,26,27. Herein, we describe the development of an enantioselective palladium-catalysed decarboxylative alkylation of ketones that forges all-carbon quaternary stereocentres with an adjacent tertiary stereocentre in good yield and stereoselectivity. This reaction represents a rare example for an asymmetric palladium catalyzed tandem process.
Table 1.
Enantioselective decarboxylative enolate alkylation cascadea.
 | |||||||
|---|---|---|---|---|---|---|---|
| Entry | Substrate | Product | Time | Yieldb | dr(3:4)c | ee(3) | ee(4) |
| 1 | 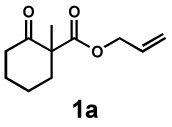 |
 |
24 h | 99% | 1 : 6.1 | 77% | 87% |
| 2 | 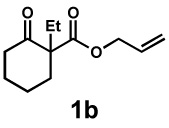 |
 |
48 h | 91% | 1 : 3.5 | 95% | 99% |
| 3d | 72 h | 88% | 1 : 3.4 | 88% | 97% | ||
| 4 | 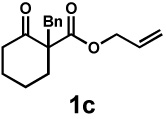 |
 |
40 h | 49% | 1 : 1.9 | 85% | 88% |
| 5e | 65 h | 65% | 1 : 1.9 | 93% | 94% | ||
| 6 | 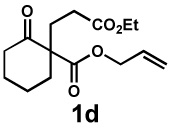 |
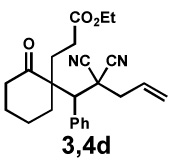 |
24 h | 56% | 1 : 3.3 | 82% | 89% |
| 7f | 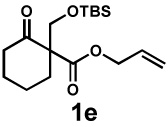 |
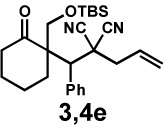 |
24 h | 56% | 1 : 1.3 | 69% | 70% |
| 8g | 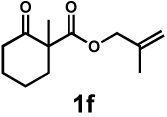 |
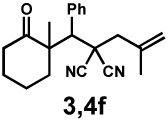 |
72 h | 57% | 1 : 2.4 | 75% | 81% |
| 9 | 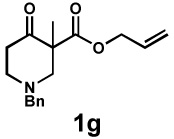 |
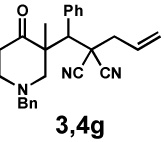 |
20 h | 97% | 1 : > 20 | - | 89% |
| 10 | 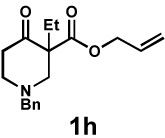 |
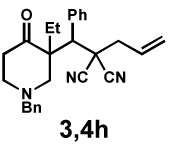 |
48 h | 99% | 1 : > 20 | 71% | 97% |
General reaction conditions: 1 (0.3–0.5 mmol), 2 (1.0 equiv), [Pd2(dba)3] (5 mol%), L2 (12.5 mol%), 1,4-dioxane (0.1 M), 40 °C.
Combined isolated yield.
Determined by 1H nmr.
Carried out on 1 mmol scale with 2.5 mol% [Pd2(dba)3] and 6.25 mol% L2.
Reaction performed at 23 °C.
Pd(pmdba)2 (10 mol%) was used as precatalyst.
Reaction performed at 60 °C.
Results
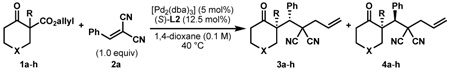
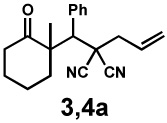
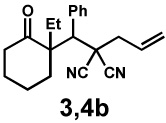
In our initial experiments, we found that exposure of β-ketoester (±)-1a28 to catalytic amounts of [Pd2(dba)3] (dba = dibenzylideneacetone) and (S)-t-BuPHOX (L1, PHOX = phosphinooxazoline)29 in the presence of electrophile 2a indeed followed the desired reaction pathway, efficiently producing diastereomers 3a and 4a (82% conversion), albeit with moderate diastereomeric ratio and enantiomeric excess (1:3.1 dr, 42% and 63% ee; see supporting information for details). Following careful optimisation, the combination of an electronically modified PHOX ligand (L2, 12.5 mol%)30 and [Pd2(dba)3] (5 mol%) in 1,4-dioxane at 40 °C was found to produce diastereomers in high enantioselectivity. In the presence of only one equivalent of electrophile 2a, the diastereomeric products 3a and 4a were isolated in 99% yield, 1:6.1 dr, 77% ee and 87% ee, respectively (Table 1, entry 1).
We then applied these conditions to a series of substrates with different substitution at the α-carbon, the allyl functionality or the backbone of the cyclic ketone. The enantiomeric excess improved significantly when the α-substituent was changed to an ethyl group and the desired product was isolated with 99% ee (entry 2). Other α-substituents such as benzyl groups, esters or protected alcohols were tolerated as well (entries 4–7) but in some cases, allylic alkylation of the enolate was observed as a side reaction, leading to decreased yields (entries 4, 6, and 7). This side reaction was suppressed by lowering the reaction temperature to 23 °C (entry 5). Moreover, the reaction tolerates substitution on the allyl fragment as well (entry 8). In addition to cyclohexanones, 1,4-piperidinone derived substrates also furnished the corresponding products in high yield and good enantioselectivity with remarkably high diastereomeric ratio (entries 9 and 10).
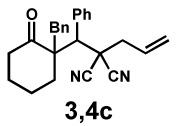
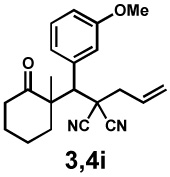
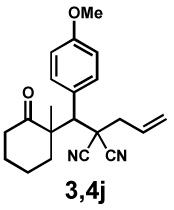
Next, the scope of the electrophile was explored. The reaction performed well with arylidenemalononitriles derived from m- and p-anisaldehyde, allowing the introduction of substituted aryl groups at the stereocentre β to the ketone (Table 2, entries 1 and 2). Methyl or dimethylamino substituents were introduced at the 4-position, as well (entry 3 and 4). Although the diastereo-and enantioselectivity were impressive in the latter case, the yield was again low due to significant allylic enolate alkylation. Here, a change of the palladium precursor to Pd(dmdba)2 (dmdba = 3,3',5,5'-dimethoxybenzylideneacetone) slightly increased the reaction outcome toward the conjugate addition product (entry 5). Importantly, the versatile 2-furanyl substituent was installed with high yield and enantioselectivity as well (entries 6 and 7). Finally, a benzodioxole and a styrene group were successfully introduced at the tertiary centre in good yield and selectivity (entries 8 and 9) with exclusive regioselectivity in the last case.
Table 2.
Asymmetric enolate alkylation of substrates 1a or 1b with different electrophilesa.
| Entry | Electrophile | Product | Time | Yieldb | dr(3:4)c | ee(3) | ee(4) |
|---|---|---|---|---|---|---|---|
| 1 | 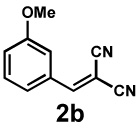 |
 |
68 h | 78% | 1 : 8.2 | 71% | 86% |
| 2 | 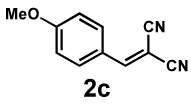 |
 |
36 h | 87% | 1 : 7.8 | 73% | 88% |
| 3 | 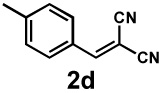 |
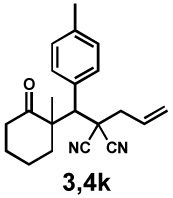 |
16 h | 76% | 1 : 6.2 | 75% | 87% |
| 4 | 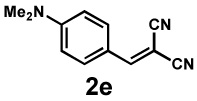 |
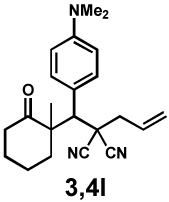 |
18 h | 22% | 1 : 8.9 | 78% | 99% |
| 5d | 18 h | 54% | 1 : >20 | nd | 99% | ||
| 6 | 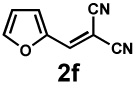 |
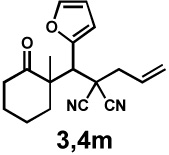 |
36 h | 87% | 1 : 3.5 | 65% | 81% |
| 7 | 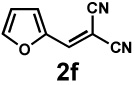 |
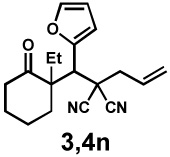 |
36 h | 92% | 1 : 2.3 | 89% | 96% |
| 8 | 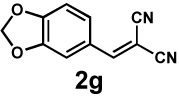 |
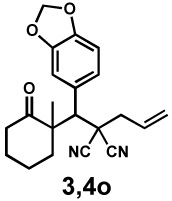 |
24 h | 99% | 1 : 14.0 | 58% | 95% |
| 9e | 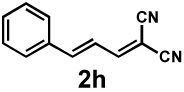 |
 |
24 h | 83% | 1 : 9.4 | 64% | 82% |
General reaction conditions: See Table 1.
Combined isolated yield.
Determined by 1H nmr.
Pd(dmdba)2 (10 mol%) was used as catalyst precursor and the reaction carried out on 0.1 mmol scale.
Only the 1,4-addition product was observed as determined by GHMBC-nmr.
As an initial probe to study the mechanism, we determined the absolute stereochemistry of diastereomeric products 3b and 4b to be (R,R) and (R,S) by single crystal X-ray analysis (Figure 2a, see supporting information for details). Interestingly, the absolute configuration at the α-quaternary centre is inverted, compared to the products from our standard asymmetric allylic alkylation (cf. 3b and 4b with 5)21, but matches the cyclohexanone products obtained from our protonation procedure (e.g., 6)24.
Figure 2. Absolute stereochemistry and limitations of the reported palladium catalysed enolate alkylation.

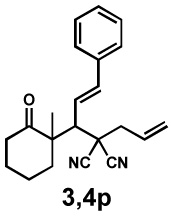
a, The absolute configuration of the major diastereomer (4) matches the prouct from the previously reprted palladium-catalyzed decarboxylative protonation (6) and is opposite to the outcome of the regular decarboxylative allylic alkylation (5). b, Electrophiles 2i and 2j also lead to product formation but with diminished yields and stereoselectivities. Products 3,4q however have three continuous quaternary and tertiary stereocentres. c, An alkyl substituted electrophile does not yield any product. Instead olefin isomerisation and allylic alkylation of 2k are observed. d, A mechanism in form of a catalytic cycle is proposed starting with an oxidative addition to the substrate involving decarboxylation followed by conjugate addition and reductive alkylation.
Strong electron-withdrawing groups on the Michael acceptor are necessary to achieve the desired conjugate addition. Although it was possible to employ reagents that differ from malononitriles like α-cyanoacetate and Meldrum's acid derivatives, the yields and stereoselectivities dropped significantly (Figure 2b). While products 3,4q were isolated only in 15% yield, it should be stated that in this case a series of two quaternary and one tertiary neighboring stereocentres were formed by a single palladium-catalyzed transformation, albeit with 64% ee. Notably, the conversion was significantly increased with the achiral, but more electronrich dppe ligand (96%) and the same two predominant diastereomers were observed as with ligand L2. Alternatively, inital attempts to employ alkyl-substituted acceptors were not successful, and instead, transfer of the allyl group to the alkylidenemalononitrile proceeded to yield adduct 7 (Figure 2c).
Discussion
In general, the diastereoselectivity is good for most products and some examples show remarkable results with only trace amounts of a second diastereomer formed. The amount of direct allylic alkylation, as observed in some cases, was lowered either by a temperature change or by employing a different palladium precursor and suggest a possible dependence on the coordination behaviour of the acceptor and the catalyst. Indeed, initial preliminary experimental results suggest a resting state that is different from the η1-allyl-palladium carboxylate observed for the enantioselective allylic alkylation of ketones and may involve coordination of the electrophile31. Detailed studies are currently underway and comment on the specific details of the process would be speculation at this stage. Although it is not possible to provide a detailed picture at this time, a plausible simplified catalytic cycle is outlined (Figure 2d). First, palladium(0) catalyst B reacts with substrate 1 under the loss of CO2 to form η1-allylpalladium enolate A. Next, conjugate addition to 2 occurs and results in stabilised intermediate C, consisting of a π-allyl-palladium cation and a deprotonated malononitrile. Reductive alkylation then yields the diastereomeric products 3,4 and regenerates catalyst B.
In conclusion, a highly enantio- and diastereoselective palladium-catalyzed α-alkylation process has been developed that proceeds via the trapping of an intermediary palladium-enolate species by conjugate addition to a prochiral activated Michael acceptor. The reaction allows the asymmetric construction of densely functionalised molecules possessing an all-carbon quaternary centre neighboring a tertiary centre from simple racemic β-ketoester starting materials. Currently, expansion of this methodology to include other substrates and applications in multistep synthesis are in progress. Finally, mechanistic studies are also underway and will be reported in due course.
Methods
General Procedure
A flame-dried 50 mL Schlenk-tube was charged with [Pd2(dba)3] (13.7 mg, 0.015 mmol, 5 mol%) and L2 (22.2 mg, 0.0375 mmol, 12.5 mol%) under argon atmosphere and 3 mL of freshly distilled 1,4-dioxane were added. After stirring for 30 min at 23 °C, 1 (0.3 mmol, 1.0 equiv) and 2 (0.3 mmol, 1.0 equiv) were added simultaneously. The resulting yellow-green solution was stirred at the reported temperature for the reported amount of time (see Table 1 and 2). The consumption of starting material was monitored by tlc (KMnO4-stain) or by analysis of a small nmr sample. The solvent was removed under reduced pressure and the diastereomeric ratio determined by crude 1H nmr. Isolation and separation of products 3 and 4 was achieved by flash chromatography in hexanes/ethyl acetate mixtures in the given combined yields (Table 1 and 2). The enantiomeric excess was determined by either hplc or sfc analysis of the purified product (see supporting information).
Supplementary Material
Figure 3.


Acknowledgements
The authors wish to thank NIH-NIGMS (R01 GM 080269-01 and a postdoctoral fellowship to D.E.W.), the German Academic Exchange Service (DAAD, postdoctoral fellowship to J.S.), Abbott Laboratories, Amgen, Merck, Bristol-Myers Squibb, Boehringer Ingelheim, the Gordon and Betty Moore Foundation, and Caltech for financial support. We thank Sarah Reisman for helpful discussions. Lawrence Henling and Michael Day are gratefully acknowledged for X-ray crystallographic analysis. The Bruker KAPPA APEXII X-ray diffractometer was purchased via an NSF CRIF:MU award to the California Institute of Technology, CHE-0639094.
Footnotes
Author Contributions
J.S. planned and carried out the described experimental work and wrote the manuscript. D.E.W. and S.C.V. took part in the initial reaction development and screening experiments. B.M.S. initiated and directed the project. All authors commented on the manuscript.
Additional Information
Supplementary information and chemical compound information accompany this paper at www.nature.com/naturechemistry. Reprints and permission information are available online at http://npg.nature.com/reprintsandpermissions/.
Contributor Information
Jan Streuff, The Arnold and Mabel Beckman Laboratories of Chemical Synthesis, Division of Chemistry and Chemical Engineering, California Institute of Technology, MC 101-20, 1200 E. California Blvd., Pasadena, CA 91125 (USA), Fax: (1) 626-395-8436
David E. White, The Arnold and Mabel Beckman Laboratories of Chemical Synthesis, Division of Chemistry and Chemical Engineering, California Institute of Technology, MC 101-20, 1200 E. California Blvd., Pasadena, CA 91125 (USA), Fax: (1) 626-395-8436
Scott C. Virgil, Caltech Center for Catalysis and Chemical Synthesis, California Institute of Technology, 1200 E. California Blvd., Pasadena, CA 91125 (USA)
Brian M. Stoltz, The Arnold and Mabel Beckman Laboratories of Chemical Synthesis, Division of Chemistry and Chemical Engineering, California Institute of Technology, MC 101-20, 1200 E. California Blvd., Pasadena, CA 91125 (USA), Fax: (1) 626-395-8436.
References
- 1.Das JP, Chechik H, Marek I. A unique approach to aldol products for the creation of all-carbon quaternary stereocenters. Nature Chem. 2009;1:128–132. doi: 10.1038/nchem.131. [DOI] [PubMed] [Google Scholar]
- 2.Christoffers J, Baro A. Quaternary Stereocenters, Challenges and Solutions in Organic Synthesis. Wiley-VCH: Weinheim; 2005. [Google Scholar]
- 3.Trost BM, Jiang C. Catalytic enantioselective construction of all-carbon quaternary stereocenters. Synthesis. 2006:369–396. [Google Scholar]
- 4.Bencivenni G, et al. Targeting structural and stereochemical complexity by organocascade catalysis: Construction of spirocyclic oxindoles having multiple stereocenters. Angew. Chem. Int. Ed. 2009;48:7200–7203. doi: 10.1002/anie.200903192. [DOI] [PubMed] [Google Scholar]
- 5.Wu F, Li H, Hong R, Deng L. Construction of quaternary stereocenters by efficient and practical conjugate additions to α,β-unsaturated ketones with a chiral organic catalyst. Angew. Chem. Int. Ed. 2006;45:947–950. doi: 10.1002/anie.200502658. [DOI] [PubMed] [Google Scholar]
- 6.Li H, et al. Stereocontrolled creation of adjacent quaternary and tertiary stereocenters by a catalytic conjugate addition. Angew. Chem. Int. Ed. 2005;44:105–108. doi: 10.1002/anie.200461923. [DOI] [PubMed] [Google Scholar]
- 7.Mase N, Thayumanavan R, Tanaka F, Barbas CF., III Direct asymmetric organocatalytic Michael reactions of α, α-disubstituted aldehydes with β-nitrostyrenes for the synthesis of quaternary carbon-containing products. Org. Lett. 2004;6:2527–2530. doi: 10.1021/ol049196o. [DOI] [PubMed] [Google Scholar]
- 8.Hamashima Y, Hotta D, Sodeoka M. Direct generation of nucleophilic chiral palladium enolate from 1,3-dicarbonyl compounds: Catalytic enantioselective Michael reaction with enones. J. Am. Chem. Soc. 2002;124:11240–11241. doi: 10.1021/ja027075i. [DOI] [PubMed] [Google Scholar]
- 9.Taylor MS, Jacobsen EN. Enantioselective Michael additions to α,β-unsaturated imides catalyzed by a salen-Al complex. J. Am. Chem. Soc. 2003;125:11204–11205. doi: 10.1021/ja037177o. [DOI] [PubMed] [Google Scholar]
- 10.Abelman MM, Overman LE. Palladium-catalyzed polyene cyclizations of dienyl aryl iodides. J. Am. Chem. Soc. 1988;110:2328–2329. [Google Scholar]
- 11.Overman LE, Paone DV, Stearns BA. Direct stereo-and enantiocontrolled synthesis of vicinal stereogenic quaternary carbon centers. Total syntheses of meso- and (−)-chimonanthine and (+)-calycanthine. J. Am. Chem. Soc. 1999;121:7702–7703. [Google Scholar]
- 12.Poli G, Giambastiani G, Heumann A. Palladium in organic synthesis: Fundamental transformations and domino processes. Tetrahedron. 2000;56:5959–5989. [Google Scholar]
- 13.Balme G, Bossharth E, Monteiro N. Pd-Assisted multicomponent synthesis of heterocycles. Eur. J. Org. Chem. 2003:4101–4111. [Google Scholar]
- 14.Shu W, Jia G, Ma S. Palladium-catalyzed three-component cascade cyclization reaction of bisallenes with propargylic carbonates and organoboronic acids: Efficient construction of cis-fused bicyclo[4.3.0]nonenes. Angew. Chem. Int. Ed. 2009;48:2788–2791. doi: 10.1002/anie.200805422. [DOI] [PubMed] [Google Scholar]
- 15.Patil NT, Huo Z, Yamamoto Y. Palladium-catalyzed decarboxylative aza-Michael addition-allylation reactions between allyl carbamates and activated olefins. Generation of quaternary carbon adjacent to secondary amine carbon center. J. Org. Chem. 2006;71:6991–6995. doi: 10.1021/jo061110c. [DOI] [PubMed] [Google Scholar]
- 16.Mohr JT, Stoltz BM. Enantioselective Tsuji allylations. Chem.-Asian J. 2007;2:1476–1491. doi: 10.1002/asia.200700183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chieffi A, Kamikawa K, Åhman J, Fox JM, Buchwald SL. Catalytic asymmetric vinylation of ketone enolates. Org. Lett. 2001;3:1897–1900. doi: 10.1021/ol0159470. [DOI] [PubMed] [Google Scholar]
- 18.Liao X, Weng Z, Hartwig JF. Enantioselective α-arylation of ketones with aryl triflates catalyzed by difluorphos complexes of palladium and nickel. J. Am. Chem. Soc. 2008;130:195–200. doi: 10.1021/ja074453g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Doyle AG, Jacobsen EN. Enantioselective alkylation of acyclic α,α-disubstituted tributyltin enolates catalyzed by a {Cr(salen)} complex. Angew. Chem., Int. Ed. 2007;46:3701–3705. doi: 10.1002/anie.200604901. [DOI] [PubMed] [Google Scholar]
- 20.Doyle AG, Jacobsen EN. Enantioselective alkylations of tributyltin enolates catalyzed by Cr(salen)Cl: Access to enantiomerically enriched all-carbon quaternary centers. J. Am. Chem. Soc. 2005;127:62–63. doi: 10.1021/ja043601p. [DOI] [PubMed] [Google Scholar]
- 21.Behenna DC, Stoltz BM. The enantioselective Tsuji allylation. J. Am. Chem. Soc. 2004;126:15044–15045. doi: 10.1021/ja044812x. [DOI] [PubMed] [Google Scholar]
- 22.Mohr JT, Behenna DC, Harned AM, Stoltz BM. Deracemization of quaternary stereocenters by Pd-catalyzed enantioconvergent decarboxylative allylation of racemic β-ketoesters. Angew. Chem. Int. Ed. 2005;44:6924–6927. doi: 10.1002/anie.200502018. [DOI] [PubMed] [Google Scholar]
- 23.Keith JA, et al. The inner-sphere process in the enantioselective Tsuji allylation reaction with (S)-t-Bu-phosphinooxazoline ligands. J. Am. Chem. Soc. 2007;129:11876–11877. doi: 10.1021/ja070516j. [DOI] [PubMed] [Google Scholar]
- 24.Marinescu SC, Nishimata T, Mohr JT, Stoltz BM. Homogeneous Pd-catalyzed enantioselective decarboxylative protonation. Org. Lett. 2008;10:1039–1042. doi: 10.1021/ol702821j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mohr JT, Nishimata T, Behenna DC, Stoltz BM. Catalytic enantioselective decarboxylative protonation. J. Am. Chem. Soc. 2006;128:11348–11349. doi: 10.1021/ja063335a. [DOI] [PubMed] [Google Scholar]
- 26.Patil NT, Yamamoto Y. Palladium-catalyzed cascade reactions of highly activated olefins. Synlett. 2007:1994–2005. [Google Scholar]
- 27.Shim J-G, Nakamura H, Yamamoto Y. Palladium catalyzed regioselective β-acetonation-α-allylation of activated olefins in one shot. J. Org. Chem. 1998;63:8470–8474. [Google Scholar]
- 28.Mohr JT, Krout MR, Stoltz BM. Preparation of (S)-2-allyl-2-methylcyclohexanone. Org. Synth. 2009;86:194–211. doi: 10.15227/orgsyn.086.0194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Helmchen G, Pfaltz A. Phosphinooxazolines-a new class of versatile, modular P,N-ligands for asymmetric catalysis. Acc. Chem. Res. 2000;33:336–345. doi: 10.1021/ar9900865. [DOI] [PubMed] [Google Scholar]
- 30.White DE, Stewart IC, Grubbs RH, Stoltz BM. The catalytic asymmetric total synthesis of elatol. J. Am. Chem. Soc. 130;2008:810–811. doi: 10.1021/ja710294k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sherden NH, Behenna DC, Virgil SC, Stoltz BM. Unusual allylpalladium carboxylate complexes: Identification of the resting state of catalytic enantioselective decarboxylative allylic alkylation reactions of ketones. Angew. Chem. Int. Ed. 2009;48:6840–6843. doi: 10.1002/anie.200902575. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.