

Summary
Borrelia burgdorferi outer surface protein C (ospC) is required for the establishment of infection in mammals. However, its precise function remains controversial. The biologically active form of OspC appears to be a homodimer. Alpha helix 1 and 1′ of the apposing monomers form a solvent-accessible pocket at the dimeric interface that presents a putative ligand binding domain (LBD1). Here we employ site-directed and allelic-exchange mutagenesis to test the hypothesis that LBD1 is a determinant of OspC function in the mammalian environment. Substitution of residues E61, K60 and E63 which line LBD1 resulted in the loss of infectivity or influenced dissemination. Analyses of the corresponding recombinant proteins demonstrated that the loss of function was not due to structural perturbation, impaired dimer formation or the loss of plasminogen binding. This study is the first to assess the involvement of individual residues and domains of OspC in its in vivo function. The data support the hypothesis that OspC interacts with a mammalian derived ligand that is critical for survival during early infection. These results shed new light on the structure-functions relationships of OspC and challenge existing hypotheses regarding OspC function in mammals.
Introduction
Lyme borreliosis is caused by the spirochetal pathogens Borrelia burgdorferi, B. garinii and B. afzelii (Baranton et al., 1992; Benach et al., 1983; Burgdorfer et al., 1982; Marconi and Garon, 1992a, b; Postic et al., 1990; Postic et al., 1994; Welsh et al., 1992). Lyme disease spirochetes cycle between Ixodes ticks and mammals, with humans being incidental hosts (reviewed in (Barbour and Hayes, 1986; Clark and Hu, 2008; Tilly et al., 2008). Preventive strategies for Lyme disease are poorly developed and early diagnosis remains difficult (Earnhart and Marconi, 2008). If not promptly diagnosed and treated, serious sequelae can develop (Fish et al., 2008; Halperin, 2008; Puius and Kalish, 2008; Steere, 2001).
Differential gene expression by Lyme disease spirochetes is central to the adaptive responses required for completion of the enzootic cycle (Boardman et al., 2008; Caimano et al., 2007; Pal et al., 2008; Revel et al., 2002; Rogers et al., 2009). In feeding ticks, the spirochetes up-regulate numerous genes including outer surface protein C (OspC), a 21 kDa surface-exposed lipoprotein (Fuchs et al., 1992; Schwan et al., 1995) encoded by a stable and universal circular plasmid (cp26) (Jewett et al., 2007; Marconi et al., 1993a; Sadziene et al., 1993). OspC remains expressed through early stage infection in mammals and is then down-regulated (Fingerle et al., 1995; Masuzawa et al., 1994; Schwan and Piesman, 2000). Consistent with the expression of ospC at the tick-mammal interface, several studies indicate a strict correlation between OspC and an infectious phenotype (Fingerle et al., 2007; Gilmore and Piesman, 2000; Grimm et al., 2004; Pal et al., 2004; Tilly et al., 2006). However, the function of OspC remains the subject of considerable debate (Radolf and Caimano, 2008). It has been proposed that OspC plays an essential role in transmission by facilitating the translocation of spirochetes through the salivary glands of ticks (Pal et al., 2004). In contrast, others have suggested that OspC is not required in the tick but instead performs a critical function in mammals (Grimm et al., 2004; Stewart et al., 2006; Tilly et al., 2006; Tilly et al., 2007). Interestingly, OspC is not required for transmission of infection by tissue transplantation (Tilly et al., 2009). Based on these reports, it was postulated that OspC has an important function during initial host adaptation and upon its down-regulation, its function is assumed by other proteins that may include VlsE (Tilly et al., 2009). It has recently been proposed that OspC does not have a specific function but instead serves primarily to maintain membrane integrity in a non-specific fashion (Xu et al., 2008). Xu et al suggested that OspC can be functionally replaced by a variety of surrogate lipoproteins including DbpA, OspE and OspA (Xu et al., 2008). It is clear that conflicting interpretations exist regarding the role of OspC in Lyme disease spirochete pathogenesis.
Plasminogen and Salp15, mammalian and tick derived proteins, respectively, have been demonstrated to interact with OspC (Das et al., 2001; Hovius et al., 2007; Hovius et al., 2008b; Lagal et al., 2006; Pal et al., 2004). In other bacteria plasminogen binding has been demonstrated to facilitate tissue penetration and invasiveness (Lagal et al., 2006; Lahteenmaki et al., 1998; Lahteenmaki et al., 2001). The specific contribution of the plasminogen-OspC interaction in pathogenesis remains to be demonstrated as OspC is one of several plasminogen binding proteins produced by the Borrelia (Brissette et al., 2009; Coleman et al., 1995; Grosskinsky et al., 2009; Hovis et al., 2008; Lagal et al., 2006; Rossmann et al., 2007; Seling et al., 2009). Regarding Salp15, this tick derived protein has been demonstrated to have immunomodulatory activity (Anguita et al., 2002; Dai et al., 2009; Hovius et al., 2008a). Its interaction with OspC may serve to protect the Lyme disease spirochetes against antibody mediated killing (Hovius et al., 2008b; Ramamoorthi et al., 2005). Salp15 has also been demonstrated to bind to murine CD4 and DC-SIGN (Anguita et al., 2002; Hovius et al., 2008a). While the binding of Salp15 to OspC is well established, the ability of the Lyme disease spirochetes to infect mice by the needle inoculation route (a Salp15 independent approach) indicates that this interaction is not required to establish infection.
To unravel the function of OspC, a detailed understanding of OspC sequence-structure relationships will be required. OspC is phylogenetically diverse with >30 phyletic “types” having been defined (Brisson and Dykhuizen, 2004; Earnhart and Marconi, 2007; Seinost et al., 1999; Wang et al., 1999a). Within a phyletic type, OspC sequences differ by less than 5%. Variation at the inter-type level can be as high as ~45% (Earnhart et al., 2005; Eicken et al., 2001; Kumaran et al., 2001; Wang et al., 1999b). In spite of significant amino acid variability among OspC types, the proteins are structurally conserved (Eicken et al., 2001; Kumaran et al., 2001). OspC monomers consist of 5 alpha helices and 2 short β-strands. The monomers form a tight homodimer possessing a central four-helical bundle with an extensive buried interface (Eicken et al., 2001; Kumaran et al., 2001) (Fig.1.) While the dimer is thought to be the biologically active form of the protein, there is evidence suggesting that OspC also may form higher order oligomers that could be of biological relevance (Eicken et al., 2001; Zuckert et al., 2001). The OspC dimer possesses two solvent-accessible domains henceforth referred to as putative ligand binding domains 1 and 2 (LBD1 and LBD2; indicated in Fig.1C). LBD1, formed by the juxtaposition of alpha helix 1 and 1′ of the apposing monomers (Kumaran et al., 2001), is the primary focus of this study (Fig.1C and 1D). LBD1 is highly conserved with some residues being invariant (Earnhart and Marconi, 2007). LBD2, which occurs at the “crown” of the OspC dimer, varies in sequence among OspC types. The epitopes that define OspC type specific humoral responses are located within LBD2 (Fig.1C) (Buckles et al., 2006; Earnhart et al., 2005; Earnhart et al., 2007; Earnhart and Marconi, 2007).
Figure 1.

OspC structure, conservation and location of putative ligand binding domains. Apposing OspC monomers are presented in Panel A with conserved and variable residues indicated using a red to blue scale. Panel B presents a VAST alignment, visualized using CN3D 4.1 of type A (1GGQ; purple), type I (1F1M; yellow), and type E (1G5Z, gray) OspC proteins. Panel C depicts an OspC dimer with LBD1 and LBD2 indicated. Sequence conservation is indicated using the red to blue scale described in panel A. In panel D, the concavity algorithm (Capra et al., 2009) was employed to identify putative ligand binding pockets and to assess the probability that individual residues within LBD1 are involved in ligand binding. A blue-white-red scale of increasing predicted probability is presented. A combined ribbon diagram and surface projection of OspC (1GGQ) demonstrates the apposing helices that form LBD1 (left side of Panel E). The LBD1-containing region highlighted by the yellow box is magnified in the right half of panel E to display the spatial localization of residues investigated in this study (K60 in red, E61 in blue, E63 in green). Amino acids are labeled in one monomer only but indicated by color in both.
This study focused on the functions of OspC associated specifically with the mammalian environment. Here, we employed site-directed mutagenesis to substitute conserved residues within LBD1 and used allelic-exchange to replace the wild type copy of ospC with single and double amino acid substitution mutants in an isogenic background. The mutant strains were then employed to test the hypothesis that the in vivo functional role of OspC in the mammalian environment is mediated by specific residues within LBD1. A significant distinction between this study and earlier analyses that have employed gene deletion/interruption approaches (Grimm et al., 2004; Pal et al., 2004; Tilly et al., 2006) is that we studied the role of individual residues and domains of OspC without the potential complication of perturbation of membrane organization and integrity. The results presented here suggest that an essential function of OspC is to bind a small mammalian-derived ligand via LBD1. These analyses represent a significant methodological and conceptual step forward in dissecting the sequence-structure-function relationships of one of the most intensively studied virulence factors of the Lyme disease spirochetes.
Results
Site-directed mutagenesis of LBD1 of OspC of B. burgdorferi B31-5A4
To test the hypothesis that residues within LBD1 are involved in the in vivo functional activities of OspC that are required for the establishment of infection in mammals, residues K60, E61 and E63, which are charged, highly conserved, and possess side chains that extend into the solvent accessible pocket of LBD1, were targeted for site-directed mutagenesis (Fig. 1E). These residues may serve as ligand contact points or be important in defining the physiochemical properties of LBD1 required for ligand binding. Residues E61 and E63, which are positively charged and hydrophilic, and the positively charged and hydrophilic residue, K60, were replaced with Q and Y residues (polar and hydrophilic), respectively. We reasoned that these substitutions would alter charge distribution, a key parameter in ligand binding, without significantly affecting secondary structure. A master allelic-exchange construct (designated as pCAEV1) was generated and ospC genes harboring amino acid substitutions were inserted. Selection was accomplished with a streptomycin resistance (strR) cassette downstream of the BbB21 open reading frame. In pCAEV1, the modified ospC genes are under the transcriptional control of a B. burgdorferi type A-ospC promoter (Alverson et al., 2003; Gilmore et al., 2001; Marconi et al., 1993a; Xu et al., 2007; Yang et al., 2005). The endogenous cp26-encoded wild type-type A ospC gene of B. burgdorferi B31-5A4 was replaced with the mutated genes or with a pCAEV1 construct harboring a wild type copy of ospC; the latter of which served as a control to verify that the general genetic manipulation procedures employed did not alter the properties of B. burgdorferi B31-5A4 (detailed below). As a negative control, the cp26-carried wild type copy of ospC was deleted from B. burgdorferi B31-5A4 and replaced with a resistance marker using the pΔospC strR vector (Fig. 2). After obtaining clonal populations of each mutant or control, plasmid content was determined through PCR using plasmid specific primer sets (data not shown) (Rogers et al., 2009). B31::ospC (wt), B31::ospC (K60Y), and B31::ospC (E61Q/E63Q) strains lost linear plasmid 21 (lp21) while the B31ΔospC, B31::ospC (E61Q) and B31::ospC (E63Q) strains retained the full complement of plasmids. As revealed by the infectivity data presented below, the loss of lp21 proved to be inconsequential. To verify that the allelic-exchange events did not result in unintended sequence changes, the introduced copy of ospC was amplified from the recombinant strains and the amplicons sequenced. No sequence alterations, other than those intended, were detected (data not shown).
Figure 2.

Schematic of the pCAEV1 and pΔospC strR vectors used for allelic-exchange replacement or gene deletion mutagenesis of ospC. A master construct, designated as pCAEV1, was generated to allow for the replacement of B. burgdorferi B31-5A4 type A ospC with ospC site-directed mutants. Panel A presents a schematic of pCAEV1 with a representative ospC gene inserted. Panel B presents a schematic of the pΔospC strR plasmid used to delete ospC from B. burgdorferi B31. The vectors were constructed as described in the text.
Analysis of the expression, surface presentation and growth rates of strains expressing site-directed mutants of OspC
To verify that the site-directed mutants of OspC were produced, exported to the cell surface and distributed in the cell in a manner similar to that of wild type OspC, immunoblot, proteinase K and immunofluorescence assays were performed. Immunoblot analyses demonstrated that OspC is produced at wild type levels by each modified strain (Fig. 3A). Proteinase K digestion assays verified the export to and presentation of OspC in the outer membrane (Fig. 3A). The periplasmic flagellar protein, FlaB, was not cleaved by proteinase K, demonstrating that membrane integrity was maintained during proteolysis (Fig.3A). This control served to further indicate that expression and presentation of the OspC site-directed mutants on the outer membrane of B31-5A4 did not, in and of itself, perturb membrane integrity and thus render cells more susceptible to proteolysis. IFA analyses demonstrated that the OspC proteins were uniformly distributed in the cell membrane. No aggregation or atypical clustering was observed (Fig. 3B). Lastly, growth curves revealed that the expression of the mutated OspC proteins does not modify growth rate (data not shown). Hence, all of the mutants displayed similar properties in vitro, indicating that there are no identifiable alterations that could potentially complicate comparative analyses of infectivity and dissemination of these strains in mice.
Figure 3.

Transgenic B. burgdorferi produce and present OspC at the cell surface. Control or transgenic strains of B. burgdorferi B31-5A4(indicated above each lane) were incubated with or without proteinase K, fractionated by SDS-PAGE, transferred to membranes and screened with anti-OspC or anti-FlaB antiserum (as indicated in Panel A). Panel B presents the results of immunofluorescent assays in which B. burgdorferi wild type or transgenic strains (indicated above each panel) were screened with anti-OspC antiserum. Methods were as described in the text.
Analysis of the infectivity of isogenic strains expressing site-directed substitution mutants of OspC
To determine if the production of mutated OspC proteins affects the ability of each strain to infect and disseminate, recombinant strains were administered to mice using needle inoculation. Ear punch biopsies and urinary bladders were obtained 4 weeks after inoculation. Positive cultures were obtained from all mice infected with B31-5A4(untransformed control), B31::ospC (wt), B31::ospC (E63Q) and B31::ospC (K60Y) (Table 2). In contrast, none of the mice injected with B31ΔospC, B31::ospC (E61Q/E63Q) or B31::ospC (E61Q) yielded positive cultures (Table 2). Since the sole genetic difference in strain B31::ospC (E61Q) relative to the wild type parental strain was a single amino acid substitution, it can be concluded that E61 is a critical determinant of OspC function in mammals.
Table 2.
Summary of culture results from mice inoculated with strains expressing wild-type and site-directed mutants of OspCa
| Week 2 | Week 4 | |||
|---|---|---|---|---|
| Ear | Bladder | Ear | Bladder | |
| B31-5A4(not transformed) | 5/5 | 5/5 | 5/5 | 5/5 |
| B31::ospC (wt) | 5/5 | 5/5 | 5/5 | 5/5 |
| B31ΔospC | 0/5 | 0/5 | 0/5 | 0/5 |
| B31::ospC (K60Y) | 4/5 | 5/5 | 4/5 | 5/5 |
| B31::ospC (E61Q/E63Q) | 0/5 | 0/5 | 0/5 | 0/5 |
| B31::ospC (E61Q) | 0/5 | 0/5 | 0/5 | 0/5 |
| B31::ospC (E63Q) | 5/5 | 5/5 | 5/5 | 5/5 |
the data are presented as the number of mice determined to be infected based on positive cultures of the organ indicated after 2 or 4 weeks of incubation in media.
Analysis of the immune response to strains expressing site-directed mutants of OspC
To determine if the inoculated mice developed a significant IgG response to B. burgdorferi (i.e., seroconversion), anti-B. burgdorferi IgG titers were determined by ELISA with immobilized B31-5A4whole cells serving as the test antigen. All strains that were infective, based on the cultivation results, displayed high level IgG titers whereas strains that yielded negative cultivation results had low titers (Fig. 4A). The low titer antibody response observed in these strains most likely reflects a response to the inoculum (1×104 cells). Immunoblot analyses yielded results consistent with the ELISAs (Fig. 4B). Sera from all infected mice reacted strongly with multiple B. burgdorferi proteins while no significant reactivity of the sera derived from cultivation negative mice was observed. The ELISA and immunoblot results are consistent with the infectivity data and serve to further establish that specific site-directed substitutions within LBD1 result in a non-infectious phenotype.
Figure 4.

Analysis of the antibody response to B. burgdorferi B31-5A4 wildtype or ospC transgenic strains in mice. ELISAs were conducted using serum harvested from mice 4 weeks after needle inoculation (infecting strain indicated along the X axis). The sera were serially diluted, screened against immobilized whole B. burgdorferi B31-5A4 wild type cells and titers calculated for each mouse at 1/3 ODmax (indicated by triangles). Geometric means are indicated by horizontal lines. In panel B, immunoblots of whole cell lysates of B. burgdorferi B31-5A4 wild type were screened with serum collected 4 weeks after inoculation with wild type or transgenic B. burgdorferi B31strains (as indicated above each lane).
Quantitative PCR (q-PCR) analysis of the dissemination of infectious strains expressing site-directed OspC substitution mutants
To assess the influence of site-directed substitutions within LBD1 on dissemination, spirochete burdens in tissues and organs of all infected mice were determined by q-PCR. The parental B31-5A4wild-type (untransformed), B31::ospC (wt) and B31::ospC (E63Q) strains disseminated to bladder, heart and joint (Fig. 5). The numbers of B31::ospC (E63Q) spirochetes in the bladders of infected mice were higher than those observed for all other strains; these differences were statistically significant. No significant differences in spirochete burdens were observed among the strains in tibiotarsal joints. Interestingly, very few B31::ospC (K60Y) spirochetes were detected in the heart. However, the difference was statistically significant only when compared with the wild type strain and hence some caution is in order in assessing this data. Nonetheless, this observation is noteworthy in light of recent findings suggesting that OspC is a determinant required for colonization of the heart (Antonara et al., 2007).
Figure 5.
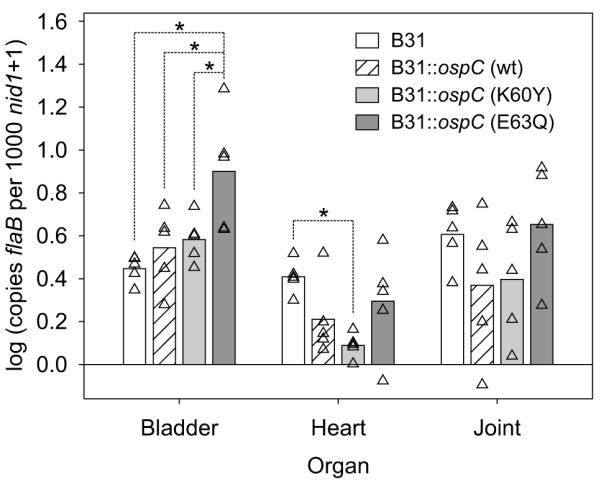
Quantitative PCR analysis of spirochete burdens in bladder, heart, and joint of infected mice. Mice were infected by needle inoculation and after 4 weeks organs were harvested. DNA was isolated from each organ and spirochete burden determined by amplification of the B. burgdorferi flaB gene (normalized to copies of the mouse nid1 gene). All analyses were repeated three times (in triplicate each time). Results from individual mice are shown as triangles, with the mean denoted by the bar. Statistical significance (p<0.05) between specific data sets (indicated by the lines above the bars) is denoted by *.
Generation of recombinant OspC proteins harboring site-directed mutations: determination of alpha helical content, ability to dimerize and electrostatic surface charge maps
The possible effect of each substitution on OspC secondary structure was assessed using multiple predictive algorithms. Little or no alteration of secondary structure was predicted (data not shown). To directly assess the effect of each substitution on alpha helical content, recombinant proteins were generated and circular dichroism spectroscopic analyses were conducted (Fig. 6). The estimated alpha helical content of the wild type OspC protein (57.5% ± 4%) was found to be in excellent agreement with that determined by X-ray crystallography (~58%) (Eicken et al., 2001; Huang et al., 1999; Kumaran et al., 2001). Estimated percent alpha helical content values are indicated in Fig. 6. While the alpha helical content of E61Q decreased to some degree, the circular dichroism spectra of all other mutants were largely unaffected. To determine if the substitutions influenced the ability of OspC to dimerize, blue-native PAGE analyses were performed (Schagger et al., 1994) (Fig. 6, inset). All recombinant proteins efficiently dimerized and no residual monomer was detected. Collectively, these analyses indicate that the substitutions introduced into OspC did not significantly alter its structure or ability to dimerize.
Figure 6.
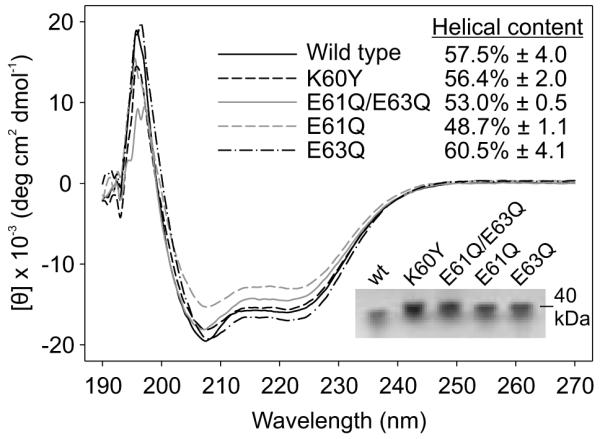
Circular dichroism and blue-native PAGE analyses of recombinant OspC site-directed substitution mutants. Recombinant protein was generated for wild type-type A OspC and each site-directed mutant as detailed in the text. The CD spectra are presented in the figure. The estimated alpha helical content of each protein is provided in the upper right inset. The results of blue native PAGE analyses, conducted to assess the ability of each recombinant protein to dimerize, are shown in the lower right inset. All methods were as described in the text.
To assess the possible impact of the amino acid substitutions on surface charge distribution, predictive surface charge maps were generated (Fig. 7). All substitution mutants displayed a surface charge map that differed in some domains from that observed for the wild type protein. The maps for the E61Q and E63Q mutants were nearly indistinguishable with both mutants developing an expansive positive surface charge zone in and around LBD1 relative to the wild type protein. The transition to a positively charged surface is even more evident in the E61Q/E63Q double mutant. This mutant also displayed a significant decrease in the distally located, negatively charged dome that is defined by LBD2. The K60Y protein also displayed significantly different surface charge distribution. Most of the positive charge clouds around the dimer were significantly reduced and transitioned to negative or neutral charge. The significance of the surface charge changes for each mutant are discussed below.
Figure 7.

Stereogram and electrostatic surface charge maps of wild type OspC and OspC site-directed mutants. A stereogram of a surface projection of the 1GGQ OspC crystal structure (Kumaran et al., 2001), in which the monomers are rotated 10 degrees, is presented in panel A. The residues selected for site-directed mutagenesis are indicated. In panel B the wild type-type A OspC dimer is shown in two rotations with the calculated electrostatic potential superimposed on the molecular surface (Swiss-PdbViewer 4.0.1; Poisson-Boltzman method; blue is positive, red negative). Electrostatic potential surface charge maps (panels C-F) are shown for OspC site-directed mutants (indicated below each panel). The charge maps for the mutants were superimposed onto the structure of wild type OspC.
Plasminogen binding by recombinant OspC proteins
Plasminogen has been demonstrated to bind to recombinant OspC with the affinity of the interaction significantly influenced by OspC sequence (Lagal et al., 2006). The determinants of OspC required for plasminogen binding have not yet been identified. To determine if substitutions introduced into LBD1 affect plasminogen binding, ELISA analyses were conducted. With the exception of E61Q/E63Q, the binding levels showed only minor differences for the mutated proteins; interestingly, E61Q/E63Q displayed 3 fold greater binding relative to wild type OspC (Fig. 8).
Figure 8.
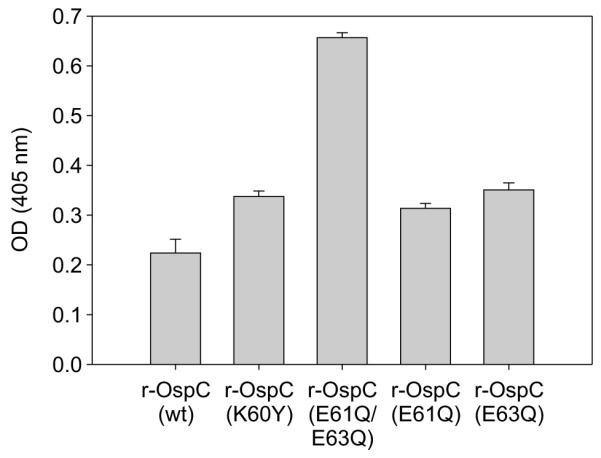
Plasminogen binding by recombinant OspC proteins. Human plasminogen, immobilized in ELISA plate wells, was screened with His-tagged wild type and mutant recombinant OspC proteins (indicated in the figure). Binding was detected with mouse-anti-His tag monoclonal antibody and peroxidase-conjugated goat-anti-mouse IgG. All methods are detailed in the text.
Discussion
Using gene deletion approaches it has been demonstrated that OspC is required for the Lyme disease spirochetes to establish infection in mice using tick or needle routes of inoculation (Grimm et al., 2004; Pal et al., 2004; Tilly et al., 2006). While these analyses have advanced our understanding of when OspC participates in the enzootic cycle its biological function remains unknown. In this study, we tested the hypothesis that LBD1 is a key determinant of OspC function in the mammalian environment.
Site-directed mutations were introduced into the type A ospC gene of B. burgdorferi and the mutated genes were transformed into an isogenic background. Residues K60, E61, and E63, which are invariant or highly conserved (Earnhart and Marconi, 2007) and which possess side chains that extend into the solvent accessible pocket of LBD1, were targeted for mutagenesis (see Fig. 1). Analysis of the resulting recombinant strains revealed that the expression, export, and presentation of the modified proteins on the cell surface were similar to the wild type strain. All strains maintained wild type growth rate and the full complement of plasmids (with the exception of the non-essential lp21 in some strains). Infectivity was assessed in mice through needle inoculation with cultivation and seroconversion serving as the read out. The parental wildtype B31-5A4strain (untransformed), B31::ospC (wt), B31::ospC (E63Q), and the B31::ospC (K60Y) strains were infectious while B31ΔospC, B31::ospC (E61Q/E63Q), and B31::ospC (E61Q) strains were non-infectious. It is striking that a single amino acid substitution at position 61 proved sufficient to render cells non-infectious, presumably by inactivating the critical functions associated with the OspC protein during the establishment of infection. For those strains that retained infectivity, we sought to determine if the amino acid substitutions introduced into LBD1 affect dissemination characteristics. The number of spirochetes present at different anatomical sites was determined by q-PCR. Higher numbers of B31::ospC (E63Q) in the urinary bladder and lower numbers of B31::ospC (K60Y) in the heart were observed. These results are consistent with the hypothesis that dissemination characteristics can be attributed, at least in part, to the physiochemical properties of OspC (Alghaferi et al., 2005; Brisson and Dykhuizen, 2004; Earnhart et al., 2005; Jones et al., 2006; Seinost et al., 1999; Wormser et al., 2008).
There are several possible explanations for the inability of the B31::ospC (E61Q) and B31::ospC (E61Q/E63Q) strains to establish infection. Since these strains are isogenic derivatives of the parental strain, the most direct explanation is that the loss of infectivity is a direct result of the modification of the OspC itself. The loss of infectivity would imply that OspC has a specific function during early stage infection that can not be fulfilled by any other Borrelia protein. As eluded to above, the biological properties of these and control strains suggest that they are not compromised in any way. At the molecular level, the amino acid substitutions could cause structural changes that ablate function. However, this possibility is not supported by secondary structure prediction analyses, circular dichroism and dimerization analyses that indicate that OspC structure and dimerization are largely unaffected. A second possibility is a direct role for residue E61 as a ligand contact point. Alternatively, E61 may indirectly influence the function of LBD1 through alteration of its local surface charge. While this possibility can not be ruled out, it is noteworthy that the CD spectra for the E63Q and E61Q mutants which are associated with infectivity and loss of infectivity, respectively, have nearly identical surface charge maps. In either case, it is clear that LBD1 and E61 are central in OspC’s in vivo function.
The calculated size of the solvent accessible pocket of LBD1 suggests its potential ligands would be relatively small (Eicken et al., 2001; Kumaran et al., 2001). It was postulated in an earlier study that the region of OspC, that we refer to in this study as LBD1, may be an aspartate binding domain (Eicken et al., 2001). The Salmonella typhimurium aspartate receptor, which is involved in chemotaxis and signaling (Yeh et al., 1996), possesses a four alpha helical bundle similar to that present in OspC. However, other than this limited structural similarity there is no discernable sequence homology between these proteins. While it seems unlikely that OspC binds aspartate, the general hypothesis that LBD1 is involved in the binding of small ligands is supported by its structural properties, the results presented herein and predictions obtained using the recently developed ConCavity algorithm (Capra et al., 2009). This algorithm, which combines information obtained from existing structural prediction algorithms, sequence conservation values, and the 3D shape analysis, has been reported to perform well in identifying ligand binding pockets and functionally relevant residues (Capra et al., 2009). While the ConCavity program was not available at the time that the rationale for the site-directed mutagenesis analyses described here were devised, its predictions are in excellent agreement with the experimental results present within. ConCavity identified LBD1 as the highest probability ligand binding pocket of OspC with residues K60, A64, E61, and E63 having the highest predicted scores for involvement in ligand binding (see Fig.1D). A64, which was not analyzed in this report, appears to be part of a stabilizing, hydrophobic shell that lines the outer edge of the solvent accessible pocket of LBD1. While the identity of the ligands that interact with LBD1 remain unknown, there is now strong and compelling evidence that LBD1 is a key functional determinant of OspC that plays an essential role in the establishment of infection in mammals.
It is important to note that OspC has been reported to bind at least two other ligands, the Salp15 protein of Ixodes ticks (Das et al., 2001; Hovius et al., 2007) and the mammalian-derived protein plasminogen (Lagal et al., 2006). The domains and residues of OspC that are involved in these interaction have not been identified. In this study, we did not specifically assess Salp15-OspC interactions since our focus was to examine the functional role of OspC in the mammalian environment. Based on the properties of LBD1 and the fact that none of the LBD1 site-directed mutants displayed attenuated plasminogen binding, it appears that plasminogen interacts with regions outside of LBD1. Interestingly, the E61Q/E63Q protein bound plasminogen at a higher level than wild type protein. Surface charge maps indicate that E61Q/E63Q has a significantly enhanced negative surface charge across the face of the dimer and a pronounced reduction in the negatively charged dome over LBD2. In that plasminogen binding to other proteins has been demonstrated to be mediated by electrostatic interactions (Rios-Steiner et al., 2001; Ye et al., 2001), it seems probable that the increased plasminogen binding by E61Q/E63Q reflects a more favorable surface charge on OspC. The results presented here highlight how sequence differences in natural OspC variants could effect plasminogen binding and possibly invasive potential. However, it is also evident that since the OspC mutant, E61Q, retains plasminogen binding ability but is non-infectious, it does not appear that plasminogen binding is the key function carried out by OspC. The extent to which the OspC-plasminogen interaction contributes to pathogenesis remains to be directly assessed. Such analyses are complicated by the fact that Borrelia produce several plasminogen binding proteins including OspE and CspZ (factor H binding proteins) (Brissette et al., 2009; Hovis et al., 2006).
Several opposing hypotheses have been offered regarding OspC function (reviewed in (Radolf and Caimano, 2008). It has been postulated that OspC does not have a specific or unique function and instead, by simple virtue of its abundance, serves to maintain membrane integrity (Xu et al., 2008). It was reported that the introduction of random lipoprotein genes (OspE, OspA, VlsE and DbpA) on an autonomously replicating plasmid restored infectivity to an ospC knock out strain in SCID mice (but less so in immunocompetent mice). From this it was concluded that structurally unrelated proteins, none of which harbor a ligand binding pocket analogous to LBD1, are functionally equivalent (i.e., the “surrogate hypothesis”). If OspC can, in fact, be replaced by randomly selected, unrelated Borrelia lipoproteins, it is not clear why OspC variants with single amino acid substitutions that are unaltered in structure and expressed at wild type levels, can not complement the putative membrane integrity maintenance function of OspC. In addition, if OspC could be replaced by other lipoproteins, it could be argued that ospC-deficient strains would have emerged in nature since there would be no selective advantage in maintaining such a gene. Extensive analyses of hundreds of Lyme disease isolates have clearly demonstrated that OspC is universal in all B. burgdorferi sensu lato complex species and homologs are carried by all relapsing fever spirochete species and isolates (Marconi et al., 1993b; Margolis et al., 1994). These facts, coupled with ospC phylogenetic analyses which indicate clear selective constraints on OspC sequence variation within specific domains, including LBD1 (Attie et al., 2007; Baranton et al., 2001; Earnhart and Marconi, 2007; Lagal et al., 2002; Lin et al., 2002; Qiu et al., 2008; Theisen et al., 1993), strongly argue against the “surrogate hypothesis”. Tilly et al. (2009) offered a slightly different interpretation on the “surrogate hypothesis” in which it was proposed that OspC does have a specific function and that once OspC is down-regulated in the mammalian environment, it is replaced by a surrogate protein that performs the same function. VlsE was suggested to be a possible OspC surrogate protein. However, as alluded to above, the divergent sequence and structure of VlsE and its lack of a LBD1-like domain, suggest that VlsE is an unlikely surrogate for OspC.
This report represents a significant step forward as it is the first to provide direct evidence that a specific domain of OspC is required for in vivo function. Specifically, we have demonstrated that residue E61 is required for infectivity and provide suggestive evidence that residues E63 and K60 influence dissemination. While we do not conclude that these are the only functionally important residues of OspC, the data firmly support the importance of LBD1 in OspC function. The results presented here challenge recent hypotheses regarding the role of OspC in Borrelia pathogenesis. Consistent with the phylogenetic, structural, and charge properties of OspC, we favor the hypothesis that OspC binds a small ligand that may be present in blood, extracellular fluid or possibly tissue that functions to facilitate the adaptive changes required for survival during the early stages of infection. Once the spirochetes adapt, OspC is no longer required and is down-regulated. Such a model is consistent with the requirement of OspC for establishing infection in mice by tick or needle inoculation routes but not by tissue transplantation. In the latter case, the spirochetes would already be “host-adapted” and would not require OspC. Although OspC lacks a transmembrane domain that might facilitate signaling, conformational changes in OspC, as a result of ligand binding, could trigger interactions with other cellular components that serve to transfer environmental signals and elicit adaptive responses. The approach employed here will allow for the further dissection of the essential determinants required for OspC function. With wild type and “loss of function” OspC mutant proteins in hand, it may be possible to devise high-throughput screening assays designed to identify OspC ligands that interact with LBD1 and thus unravel the function of OspC in Borrelia biology.
Experimental procedures
Site-directed mutagenesis of ospC
Site-directed mutations were introduced into the type A-ospC gene derived from B. burgdorferi B31-5A4 by overlap extension using mutagenic primers (Table 1) as previously described (Hovis et al., 2008). Four mutants were generated; K60Y, E61Q, E63Q and E61Q/E63Q (a double mutant). The wild type and mutant genes were annealed to the pET46 Ek/LIC vector and the plasmids were propagated as instructed by the supplier (Novagen). All constructs were validated by automated DNA sequencing (MWG Biotech).
Table 1.
Oligonucleotides used in this study
| Primer | Description | Sequence |
|---|---|---|
| pCAEV1 Upstream (+) | pCAEV1 vector construction | CAGAATGAGTTACTTCTGGATGG |
| pCAEV1 OspC 5′ (−) BspEI/MluI | PCAEV1 vector | GTACGCGTTTTTCCGGAATTATTACAAGATATAA |
| pCAEV1 OspC 3′ (+) BspEI | PCAEV1 vector | TCCGGATCAATATTATAAGATTAATTTGTTTTAAA |
| pCAEV1 BBB 21 3′ (−) AatII | PCAEV1 vector | GACGTCATCTCACATAAAACCAAAGAAACTAC |
| Spec/Strep (+) SalI | PCAEV1 vector | GTCGACTAATACCCGAGCTTCAAGGA |
| Spec/Strep (−) AatII | PCAEV1 vector | GACGTCATTATTTGCCGACTACCTTGG |
| pCAEV1 Downstream (+) SalI | PCAEV1 vector | GTCGACTTTAAAAAGTTGTTAAATAGACTTAACT |
| pCAEV1 Downstream (−) MluI | PCAEV1 vector | ACGCGTGGATATATGCAATCTTTAGTCCAG |
| Insert type A (+) BspEI | Amplification of insert | TCCGGAAAAGATGGGAATACATCTGCA |
| Insert type A (−) BspEI | Amplification of insert | TCCGGATTAAGGTTTTTTTGGACTTTCTG |
| Upstream confirm (+) | Transformation confirmation | CCTACGTTGTGATGAGACTTGATTT |
| Spec/strep 3′ (+) | Transformation confirmation | GGCGAGATCACCAAGGTAGTC |
| Insert sequencing (+) | OspC sequence | TAAAAAGGAGGCACAAATTAATG |
| Deletion upstream (+) | Deletion vector construction | GCAACAATCCAGTGTTTACAAAAACG |
| Deletion upstream (−) | Deletion vector construction | ACCGGTTCTGACGTCTAATTTGTGCCTCCTTTTT |
| Deletion downstream (+) | Deletion vector construction | GACGTCGTTGTGGCAGAAAGTCCAAAAAAACC |
| Deletion dow nstream (−) | Deletion vector construction | ACCGGTGCTGTTTAACGATTTATTTGATACTTTG |
| OspC (+) LIC | Amplification of OspC for | GACGACGACAAGATTAATAATTCAGGGAAAGAT |
| OspC (−) LIC | Amplification of OspC for | GAGGAGAAGCCCGGTTTAAGGTTTTTTTGGACT |
| OspC E61Q/E63Q (+) | Mutagenic primer | GTGAAACAGGTTCAAGCGTTG |
| OspC E61Q/E63Q (−) | Mutagenic primer | CAACGCTTGAACCTGTTTCAC |
| OspC K60Y (+) | Mutagenic primer | CAACGCTTCAACCTCATACAC |
| OspC K60Y (−) | Mutagenic primer | GTGTATGAGGTTGAAGCGTTG |
| OspC E61Q (+) | Mutagenic primer | GTGAAACAGGTTGAAGCGTTG |
| OspC E61Q (−) | Mutagenic primer | CAACGCTTCAACCTGTTTCAC |
| OspC E63Q (+) | Mutagenic primer | GTGAAAGAGGTTCAAGCGTTG |
| OspC E63Q (−) | Mutagenic primer | CAACGCTTGAACCTCTTTCAC |
| FlaB q-PCR primer (+) | q-PCR primer | CAGGTAACGGCACATATTCAGATGC |
| FlaB q-PCR primer (−) | q-PCR primer | CTTGGTTTGCTCCAACATGAACTC |
| Nid1 q-PCR primer (+) | q-PCR primer | CCAGCCACAGAATACCATCC |
| Nid1 q-PCR primer (−) | q-PCR primer | GGACATACTCTGCTGCCATC |
Allelic-exchange replacement of wild type ospC
A master allelic-exchange vector designated as pCAEV1 was generated using a pCR2.1 backbone and wild type or mutated ospC genes were inserted (Fig. 2). To create pCAEV1 the following regions of B. burgdorferi B31-5A4 cp26 were amplified: (1) region 5′ of ospC (includes BbB18 and extends through the signal sequence and first four amino acids of OspC; the amplicon harbors 3′ tandem BspEI and MluI sites), (2) region 3′ of ospC (includes BbB20 and BbB21 and flanking sequences; amplicon harbors 5′ BspEI and 3′ AatII sites) and (3) region 3′ of BbB21 (includes a portion of BbB22; amplicon harbors 5′ SalI and 3′ MluI sites). To allow for streptomycin resistance selection, aadA was amplified from pKFSS1 using primers with 5′ SalI and 3′ AatII sites (Frank et al., 2003) (Table 1). All amplicons were individually annealed into the pCR2.1 TOPO plasmid and the plasmids were propagated in E. coli. The plasmids were digested with the appropriate restriction enzymes to release the inserts. The inserts were ligated into the plasmid carrying the 5′ ospC-containing fragment in the pCR2.1 TOPO vector and transformed into E. coli (C2925; New England Biolabs) (Fig. 1A). To create the ospC allelic-exchange vectors, wild-type or ospC genes with site-directed mutations (generated as detailed above) were amplified with primers possessing 5′ and 3′ BspEI sites. The amplicons were cut with the appropriate restriction enzyme and ligated into pCAEV1 (Fig. 2A). The plasmids were propagated in Novablue E. coli cells (Novagen) and purified (HiSpeed Plasmid Midi; Qiagen). The resulting plasmids were introduced into B. burgdorferi B31-5A4 (kindly provided by Dr. Jon Skare) by electroporation essentially as previously described with some modifications (Samuels and Garon, 1993; Samuels et al., 1994). Cultures were grown at 33°C to mid-log phase in BSK-H medium with 6% rabbit serum (Sigma), collected by centrifugation, washed with cold Dulbecco’s PBS and washed twice with cold EPS buffer (93 g L−1 sucrose, 15% glycerol). The cells (5 × 107) were collected and suspended in 150 μL of EPS buffer. Fifty μL of the cell suspension was mixed with 20 μg of plasmid linearized with MscI and ScaI-HF (New England Biolabs). After 5 min on ice, the cells were electroporated (0.2 cm cuvette, 2.5 kV, 25 μF, 200 Ω), transferred to 10 mL BSK medium with 6% rabbit serum, and incubated overnight at 33 °C. The culture volumes were increased to 50 mL with fresh BSK media, streptomycin (50 μg mL−1) was added, and the cultures were maintained at 33°C for 2 to 4 weeks. Clonal populations were obtained by subsurface plating. The plasmid contents of the transformants were determined by PCR using plasmid-specific primer sets (McDowell et al., 2001; Rogers et al., 2009). To determine the sequence of the DNA that crossed over into cp26, the desired region was PCR amplified and sequenced (MWG Biotech).
Generation of a B. burgdorferi B31ΔospC strain
A pΔospC strR knockout vector was created by amplification of the upstream (1000 bp) and downstream (1025 bp) regions of ospC with primers harboring appropriate restriction sites. The upstream amplicon contains tandem AatII and AgeI sites at its 3′ end while the downstream amplicon has 5′ AatII and 3′ AgeI sites) (Table 1). The amplicons were cloned into pCR2.1 and the plasmids were propagated in Novablue E. coli. The plasmids were digested with AatII and AgeI, and the downstream fragment was ligated to the upstream-containing plasmid. Finally, the aadA gene (streptomycin/spectinomycin resistance) was excised from the pKFSS1-AatII plasmid (Frank et al., 2003) and ligated between the upstream and downstream sequences. Transformation, subsurface plating and plasmid screening were conducted as described above.
Assessment of growth curves
Fifteen mL of BSK-H complete media was seeded with approximately 5 × 105 cells mL−1 actively growing cells. The culture was maintained at 33°C (5% CO2) and cell counts (in triplicate) were conducted each day using dark-field microscopy.
Production of recombinant OspC proteins and anti-OspC antiserum
For recombinant protein production, BL21 (DE3) cells were transformed with the pET46 Ek/LIC plasmids carrying wild type or mutated ospC genes. Protein expression was induced with IPTG, the proteins purified by nickel chromatography, dialyzed against three changes of PBS, and quantified by the BCA assay (Pierce). To generate antiserum, C3H/HeJ mice were immunized with 10 μg of wild type recombinant OspC protein adsorbed to Imject alum (Pierce), with boosts at weeks 4 and 6. Serum was collected by tail bleed.
Proteinase K proteolysis and immunofluorescence assays
To assess the presentation of OspC at the B. burgdorferi cell surface, strains were cultivated at 27°C and then transferred to 37°C for 3 days to up-regulate ospC expression. The cells were harvested, washed twice with dPBS, and then the equivalent of 1 mL of culture at 0.3 OD600 was suspended in 1 mL of PBS with or without proteinase K (20 mg mL−1). The cell suspensions were incubated at 22°C (1 hr), phenylmethylsulfonyl fluoride was added (0.5 mg mL−1 methanol), the cells were harvested and suspended in 120 μL of SDS-PAGE sample buffer. The cell lysates (2 μL) were subjected to SDS-PAGE, transferred to membranes and screened with mouse-anti-OspC (1:20000) or with mouse-anti-FlaB (1:400000). Antibody binding was detected using peroxidase-conjugated goat-anti-mouse IgG and chemiluminescence (Supersignal West; Pierce).
The distribution of OspC on Borrelia cells was assessed by immunofluorescent microscopy. Cells were immobilized on slides (Superfrost Plus; Fisher), blocked with 3% BSA in PBS-T, and probed with mouse-anti-OspC antiserum (1:2000). Alexafluor 488-conjugated rabbit-anti-mouse IgG (1:200) served as the secondary antibody. The slides were mounted with Prolong Gold (Invitrogen) and the cells visualized using an Olympus BX51 fluorescence microscope.
Infectivity and dissemination analyses
Five C3H/HeJ mice per group were inoculated with 1×104 spirochetes of each strain by subcutaneous injection. After four weeks, the mice were euthanized and the heart, tibiotarsal joint, ear, bladder and blood were harvested. Portions of the ear and bladder biopsies were placed in BSK medium (supplemented with rifampicin, fosfomycin, and amphotericin B) and incubated at 37°C under 5% CO2. Each sample was visually assessed for spirochetes by dark-field microscopy after 2 and 4 weeks of incubation. To isolate DNA, approximately 0.5 g of each tissue or organ was digested with collagenase (2 mg mL−1; 4 h; 37°C) and then with proteinase K (0.1 mg mL−1; 16h; 55°C). The samples were extracted twice with phenol/chloroform/isoamyl alcohol, the DNA was precipitated using absolute ethanol, washed with 70% ethanol, suspended in water, and quantified by UV spectrophotometery. For q-PCR, the Borrelia flaB gene and the mouse nidogen 1 (nid1) gene were amplified with Sybr Green PCR master mix (Applied Biosystems) using an Opticon 2 DNA engine (MJ Research). PCR cycling conditions were: 94°C for 10:00, 40 cycles of 94°C for 15 secs, 56°C for 30 secs, 72°C for 30 secs, with fluorescence reads following the extension cycle. Relative numbers of gene copies were determined using a standard curve of Borrelia genomic DNA. To determine the extent of non-specific, background amplification, qPCR was also conducted using DNA extracted from the organs and tissues of uninfected mice. All samples were run in three replicates of triplicates, and the organ-specific background amplification was subtracted from the mean ratios. Statistical significance was assessed by ANOVA analysis of log-transformed data, with post-hoc testing by the Holm-Sidak method (SigmaPlot).
ELISA and Western blotting
The humoral response to inoculation was assessed by whole cell ELISA and Western blot analyses. For the ELISA analyses, B. burgdorferi B31-5A4 cells were collected, washed, suspended at 106 cells mL−1 (carbonate coating buffer) and immobilized onto ELISA plates (100 μL well−1; 4°C; overnight). The plates were blocked with 1% BSA in PBS-T. Serial dilutions of sera were applied (1 hr) and antibody binding detected using peroxidase-conjugated goat-anti-mouse IgG and ABTS chromogen. Titers were calculated as the inverse dilution corresponding to one third of the plateau optical density. For Western blot analyses, B. burgdorferi B31-5A4 whole cell lysates were separated by SDS-PAGE (Criterion, Bio-Rad) and blotted to PVDF membranes. The membranes were blocked with 5% nonfat dry milk in PBS-T and screened with mouse serum (1:1000 dilution). Antibody binding was detected using peroxidase-conjugated goat-anti-mouse IgG and chemiluminescence (Supersignal West; Pierce).
Circular dichroism analysis of OspC
Far-UV circular dichroism spectra of wild type and mutated recombinant OspC proteins were measured at 20°C in a Jasco J-715 spectropolarimeter (Jasco, Easton, MD, USA) (Chen et al., 1974). The protein samples (10 μM) were scanned in a 1 mm path-length cuvette using a 1 nm bandwidth, 8 s response time, and a scan rate of 20 nm/min. Three independent scans were made of each protein. Circular dichroism spectra were subtracted from background (buffer alone) and converted from circular dichroism intensity to molar ellipiticity (deg cm2 dmol−1).
Analysis of OspC dimerization using blue-native PAGE
To determine if the recombinant wild type and OspC site-directed mutant proteins undergo dimerization, one μg of each protein was equilibrated in blue-native PAGE loading buffer containing 0.02% Coomassie brilliant blue G-250 (CBB-G250), and electrophoresed in a Novex Bis/Tris NativePAGE gels 4-16% (Schagger et al., 1994). The cathode buffer (50mM tricine, 15mM bis/tris, pH 7.0) contained 0.002% CBB-G250 for the first half of the electrophoresis run. The anode buffer was 50mM bis/tris pH 7.0. The molecular mass of the recombinant OspC proteins was interpolated from a standard curve generated with NativeMark (Invitrogen) marker mix.
Assessment of plasminogen binding by wild type and site-directed mutant proteins
Plasminogen binding by recombinant OspC was assessed by ELISA (Lagal et al., 2006). ELISA plates (Costar 3590) were coated with 500 ng plasminogen (P7999, Sigma) in carbonate buffer (pH 9.6). The wells were blocked with 1% BSA in PBS-T. Recombinant OspC proteins (1 μg well−1 in blocking buffer) were incubated in triplicate wells for two hours at room temperature. Secondary detection of bound OspC was by mouse-anti-His-tag monoclonal antibody (Pierce), then by peroxidase-conjugated goat-anti-mouse IgG. Binding was quantified using ABTS chromogen.
Acknowledgments
We thank the members of the Marconi and Radolf labs for their helpful comments and suggestions. This work was supported in part by grants from the NIH, NIAID to RTM (AI-67830) and JDR (AI-29735).
Literature cited
- Alghaferi MY, Anderson JM, Park J, Auwaerter PG, Aucott JN, Norris DE, Dumler JS. Borrelia burgdorferi ospC heterogeneity among human and murine isolates from a defined region of northern Maryland and southern Pennsylvania: lack of correlation with invasive and noninvasive genotypes. J Clin Microbiol. 2005;43:1879–1884. doi: 10.1128/JCM.43.4.1879-1884.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alverson J, Bundle SF, Sohaskey CD, Lybecker MC, Samuels DS. Transcriptional regulation of the ospAB and ospC promoters from Borrelia burgdorferi. Mol Microbiol. 2003;48:1665–1677. doi: 10.1046/j.1365-2958.2003.03537.x. [DOI] [PubMed] [Google Scholar]
- Anguita J, Ramamoorthi N, Hovius JW, Das S, Thomas V, Persinski R, Conze D, Askenase PW, Rincon M, Kantor FS, Fikrig E. Salp15, an ixodes scapularis salivary protein, inhibits CD4(+) T cell activation. Immunity. 2002;16:849–859. doi: 10.1016/s1074-7613(02)00325-4. [DOI] [PubMed] [Google Scholar]
- Antonara S, Chafel RM, Lafrance M, Coburn J. Borrelia burgdorferi adhesins identified using in vivo phage display. Mol Microbiol. 2007;66:262–276. doi: 10.1111/j.1365-2958.2007.05924.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attie O, Bruno JF, Xu Y, Qiu D, Luft BJ, Qiu WG. Co-evolution of the outer surface protein C gene (ospC) and intraspecific lineages of Borrelia burgdorferi sensu stricto in the northeastern United States. Infect Genet Evol. 2007;7:1–12. doi: 10.1016/j.meegid.2006.02.008. [DOI] [PubMed] [Google Scholar]
- Baranton G, Postic D, Saint Girons I, Boerlin P, Piffaretti J-C, Assous M, Grimont PAD. Delineation of Borrelia burgdorferi sensu stricto, Borrelia garinii sp. nov., and group VS461 associated with Lyme borreliosis. Intern J Syst Bacteriol. 1992;42:378–383. doi: 10.1099/00207713-42-3-378. [DOI] [PubMed] [Google Scholar]
- Baranton G, Seinost G, Theodore G, Postic D, Dykhuizen D. Distinct levels of genetic diversity of Borrelia burgdorferi are associated with different aspects of pathogenicity. Res Microbiol. 2001;152:149–156. doi: 10.1016/s0923-2508(01)01186-x. [DOI] [PubMed] [Google Scholar]
- Barbour AG, Hayes SF. Biology of Borrelia species. Microbiol Rev. 1986;50:381–400. doi: 10.1128/mr.50.4.381-400.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benach JL, Bosler EM, Hanrahan JP, Coleman JL, Habicht GS, Bast TF, Cameron DJ, Ziegler JL, Barbour AG, Burgdorfer W, Edelman R, Kaslow RA. Spirochetes isolated from the blood of two patients with Lyme disease. N Engl J Med. 1983;308:740–742. doi: 10.1056/NEJM198303313081302. [DOI] [PubMed] [Google Scholar]
- Boardman BK, He M, Ouyang Z, Xu H, Pang X, Yang XF. Essential role of the response regulator Rrp2 in the infectious cycle of Borrelia burgdorferi. Infect Immun. 2008;76:3844–3853. doi: 10.1128/IAI.00467-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brissette CA, Haupt K, Barthel D, Cooley AE, Bowman A, Skerka C, Wallich R, Zipfel PF, Kraiczy P, Stevenson B. Borrelia burgdorferi infection-associated surface proteins ErpP, ErpA, and ErpC bind human plasminogen. Infect Immun. 2009;77:300–306. doi: 10.1128/IAI.01133-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brisson D, Dykhuizen DE. ospC diversity in Borrelia burgdorferi: Different hosts are different niches. Genetics. 2004;168:713–722. doi: 10.1534/genetics.104.028738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckles EL, Earnhart CG, Marconi RT. Analysis of antibody response in humans to the type A OspC loop 5 domain and assessment of the potential utility of the loop 5 epitope in Lyme disease vaccine development. Clin Vaccine Immunol. 2006;13:1162–1165. doi: 10.1128/CVI.00099-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgdorfer W, Barbour AG, Hayes SF, Benach JL, Grunwaldt E, Davis JP. Lyme disease--a tick-borne spirochetosis? Science. 1982;216:1317–1319. doi: 10.1126/science.7043737. [DOI] [PubMed] [Google Scholar]
- Caimano MJ, Iyer R, Eggers CH, Gonzalez C, Morton EA, Gilbert MA, Schwartz I, Radolf JD. Analysis of the RpoS regulon in Borrelia burgdorferi in response to mammalian host signals provides insight into RpoS function during the enzootic cycle. Mol Microbiol. 2007;65:1193–1217. doi: 10.1111/j.1365-2958.2007.05860.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capra JA, Laskowski RA, Thornton JM, Singh M, Funkhouser TA. Predicting protein ligand binding sites by combining evolutionary sequence conservation and 3D structure. PLoS Comput Biol. 2009;5:e1000585. doi: 10.1371/journal.pcbi.1000585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y-H, Yang JT, Chau KH. Determination of the helix and b form of proteins in aqueous solution by circular dichroism. Biochemistry. 1974;13:3350–3359. doi: 10.1021/bi00713a027. [DOI] [PubMed] [Google Scholar]
- Clark RP, Hu LT. Prevention of lyme disease and other tick-borne infections. Infect Dis Clin North Am. 2008;22:381–396. vii. doi: 10.1016/j.idc.2008.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman JL, Sellati TJ, Testa JE, Kew RR, Furie MB, Benach JL. Borrelia burgdorferi binds plasminogen, resulting in enhanced penetration of endothelial monolayers. Infect Immun. 1995;63:2478–2484. doi: 10.1128/iai.63.7.2478-2484.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai J, Wang P, Adusumilli S, Booth CJ, Narasimhan S, Anguita J, Fikrig E. Antibodies against a tick protein, Salp15, protect mice from the Lyme disease agent. Cell Host Microbe. 2009;6:482–492. doi: 10.1016/j.chom.2009.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das S, Banerjee G, DePonte K, Marcantonio N, Kantor FS, Fikrig E. Salp25D, an Ixodes scapularis antioxidant, is 1 of 14 immunodominant antigens in engorged tick salivary glands. J Infect Dis. 2001;184:1056–1064. doi: 10.1086/323351. [DOI] [PubMed] [Google Scholar]
- Earnhart C, Marconi RT. Lyme disease. In: Barrett AD, Stanberry LR, editors. Vaccines for Biodefense and Emerging and Neglected Diseases. Elsevier; 2008. [Google Scholar]
- Earnhart CG, Buckles EL, Dumler JS, Marconi RT. Demonstration of OspC type diversity in invasive human Lyme disease isolates and identification of previously uncharacterized epitopes that define the specificity of the OspC murine antibody response. Infect Immun. 2005;73:7869–7877. doi: 10.1128/IAI.73.12.7869-7877.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Earnhart CG, Buckles EL, Marconi RT. Development of an OspC-based tetravalent, recombinant, chimeric vaccinogen that elicits bactericidal antibody against diverse Lyme disease spirochete strains. Vaccine. 2007;25:466–480. doi: 10.1016/j.vaccine.2006.07.052. [DOI] [PubMed] [Google Scholar]
- Earnhart CG, Marconi RT. OspC phylogenetic analyses support the feasibility of a broadly protective polyvalent chimeric Lyme disease vaccine. Clin Vaccine Immunol. 2007;14:628–634. doi: 10.1128/CVI.00409-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eicken C, Sharma V, Klabunde T, Owens RT, Pikas DS, Hook M, Sacchettini JC. Crystal structure of Lyme disease antigen outer surface protein C from Borrelia burgdorferi. J Biol Chem. 2001;276:10010–10015. doi: 10.1074/jbc.M010062200. [DOI] [PubMed] [Google Scholar]
- Fingerle V, Hauser U, Liegl G, Petko B, Preac-Mursic V, Wilske B. Expression of outer surface proteins A and C of Borrelia burgdorferi in Ixodes ricinus. J Clin Microbiol. 1995;33:1867–1869. doi: 10.1128/jcm.33.7.1867-1869.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fingerle V, Goettner G, Gern L, Wilske B, Schulte-Spechtel U. Complementation of a Borrelia afzelii OspC mutant highlights the crucial role of OspC for dissemination of Borrelia afzelii in Ixodes ricinus. Int J Med Microbiol. 2007;297:97–107. doi: 10.1016/j.ijmm.2006.11.003. [DOI] [PubMed] [Google Scholar]
- Fish AE, Pride YB, Pinto DS. Lyme carditis. Infect Dis Clin North Am. 2008;22:275–288. vi. doi: 10.1016/j.idc.2007.12.008. [DOI] [PubMed] [Google Scholar]
- Frank KL, Bundle SF, Kresge ME, Eggers CH, Samuels DS. aadA confers streptomycin resistance in Borrelia burgdorferi. J Bacteriol. 2003;185:6723–6727. doi: 10.1128/JB.185.22.6723-6727.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs R, Jauris S, Lottspeich F, Preac-Mursic V, Wilske B, Soutschek E. Molecular analysis and expression of a Borrelia burgdorferi gene encoding a 22 kDa protein (pC) in Escherichia coli. Mol Microbiol. 1992;6:503–509. doi: 10.1111/j.1365-2958.1992.tb01495.x. [DOI] [PubMed] [Google Scholar]
- Gilmore RD, Jr., Piesman J. Inhibition of Borrelia burgdorferi migration from the midgut to the salivary glands following feeding by ticks on OspC-immunized mice. Infect Immun. 2000;68:411–414. doi: 10.1128/iai.68.1.411-414.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilmore RD, Jr., Mbow ML, Stevenson B. Analysis of Borrelia burgdorferi gene expression during life cycle phases of the tick vector Ixodes scapularis. Microbes Infect. 2001;3:799–808. doi: 10.1016/s1286-4579(01)01435-6. [DOI] [PubMed] [Google Scholar]
- Grimm D, Tilly K, Byram R, Stewart PE, Krum JG, Bueschel DM, Schwan TG, Policastro PF, Elias AF, Rosa PA. Outer-surface protein C of the Lyme disease spirochete: a protein induced in ticks for infection of mammals. Proc Natl Acad Sci U S A. 2004;101:3142–3147. doi: 10.1073/pnas.0306845101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosskinsky S, Schott M, Brenner C, Cutler SJ, Kraiczy P, Zipfel PF, Simon MM, Wallich R. Borrelia recurrentis employs a novel multifunctional surface protein with anti-complement, anti-opsonic and invasive potential to escape innate immunity. PLoS ONE. 2009;4:e4858. doi: 10.1371/journal.pone.0004858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halperin JJ. Nervous system Lyme disease. Infect Dis Clin North Am. 2008;22:261–274. vi. doi: 10.1016/j.idc.2007.12.009. [DOI] [PubMed] [Google Scholar]
- Hovis KM, Tran E, Sundy CM, Buckles E, McDowell JV, Marconi RT. Selective binding of Borrelia burgdorferi OspE paralogs to factor H and serum proteins from diverse animals: possible expansion of the role of OspE in Lyme disease pathogenesis. Infect Immun. 2006;74:1967–1972. doi: 10.1128/IAI.74.3.1967-1972.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hovis KM, Freedman JC, Zhang H, Forbes JL, Marconi RT. Identification of an antiparallel coiled-coil/loop domain required for ligand binding by the Borrelia hermsii FhbA protein: additional evidence for the role of FhbA in the host-pathogen interaction. Infect Immun. 2008;76:2113–2122. doi: 10.1128/IAI.01266-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hovius JW, van Dam AP, Fikrig E. Tick-host-pathogen interactions in Lyme borreliosis. Trends Parasitol. 2007;23:434–438. doi: 10.1016/j.pt.2007.07.001. [DOI] [PubMed] [Google Scholar]
- Hovius JW, de Jong MA, den Dunnen J, Litjens M, Fikrig E, van der Poll T, Gringhuis SI, Geijtenbeek TB. Salp15 binding to DC-SIGN inhibits cytokine expression by impairing both nucleosome remodeling and mRNA stabilization. PLoS Pathog. 2008a;4:e31. doi: 10.1371/journal.ppat.0040031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hovius JW, Schuijt TJ, de Groot KA, Roelofs JJ, Oei GA, Marquart JA, de Beer R, van ’t Veer C, van der Poll T, Ramamoorthi N, Fikrig E, van Dam AP. Preferential protection of Borrelia burgdorferi sensu stricto by a Salp15 homologue in Ixodes ricinus saliva. J Infect Dis. 2008b;198:1189–1197. doi: 10.1086/591917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X, Link K, Koide A, Dunn JJ, Luft BJ, Koide S. 1H, 13C, and 15N NMR backbone assignments of 37 kDa surface antigen OspC from Borrelia burgdorferi. J Biomol NMR. 1999;14:283–284. doi: 10.1023/a:1008398527355. [DOI] [PubMed] [Google Scholar]
- Jewett MW, Byram R, Bestor A, Tilly K, Lawrence K, Burtnick MN, Gherardini F, Rosa PA. Genetic basis for retention of a critical virulence plasmid of Borrelia burgdorferi. Mol Microbiol. 2007 doi: 10.1111/j.1365-2958.2007.05969.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones KL, Glickstein LJ, Damle N, Sikand VK, McHugh G, Steere AC. Borrelia burgdorferi genetic markers and disseminated disease in patients with early Lyme disease. J Clin Microbiol. 2006;44:4407–4413. doi: 10.1128/JCM.01077-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumaran D, Eswaramoorthy S, Luft BJ, Koide S, Dunn JJ, Lawson CL, Swaminathan S. Crystal structure of outer surface protein C (OspC) from the Lyme disease spirochete, Borrelia burgdorferi. EMBO J. 2001;20:971–978. doi: 10.1093/emboj/20.5.971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagal V, Postic D, Baranton G. Molecular diversity of the ospC gene in Borrelia. Impact on phylogeny, epidemiology and pathology. Wien Klin Wochenschr. 2002;114:562–567. [PubMed] [Google Scholar]
- Lagal V, Portnoi D, Faure G, Postic D, Baranton G. Borrelia burgdorferi sensu stricto invasiveness is correlated with OspC-plasminogen affinity. Microbes Infect. 2006;8:645–652. doi: 10.1016/j.micinf.2005.08.017. [DOI] [PubMed] [Google Scholar]
- Lahteenmaki K, Virkola R, Saren A, Emody L, Korhonen TK. Expression of plasminogen activator pla of Yersinia pestis enhances bacterial attachment to the mammalian extracellular matrix. Infect Immun. 1998;66:5755–5762. doi: 10.1128/iai.66.12.5755-5762.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahteenmaki K, Kuusela P, Korhonen TK. Bacterial plasminogen activators and receptors. FEMS Microbiol Rev. 2001;25:531–552. doi: 10.1111/j.1574-6976.2001.tb00590.x. [DOI] [PubMed] [Google Scholar]
- Lin T, Oliver JH, Jr., Gao L. Genetic diversity of the outer surface protein C gene of southern Borrelia isolates and its possible epidemiological, clinical, and pathogenetic implications. J Clin Microbiol. 2002;40:2572–2583. doi: 10.1128/JCM.40.7.2572-2583.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marconi RT, Garon CF. Identification of a third genomic group of Borrelia burgdorferi through signature nucleotide analysis and 16S rRNA sequence determination. J Gen Microbiol. 1992a;138:533–536. doi: 10.1099/00221287-138-3-533. [DOI] [PubMed] [Google Scholar]
- Marconi RT, Garon CF. Phylogenetic analysis of the genus Borrelia: a comparison of North American and European isolates of Borrelia burgdorferi. J Bacteriol. 1992b;174:241–244. doi: 10.1128/jb.174.1.241-244.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marconi RT, Samuels DS, Garon CF. Transcriptional analyses and mapping of the ospC gene in Lyme disease spirochetes. J Bacteriol. 1993a;175:926–932. doi: 10.1128/jb.175.4.926-932.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marconi RT, Samuels DS, Schwan TG, Garon CF. Identification of a protein in several Borrelia species which is related to OspC of the Lyme disease spirochetes. J Clin Microbiol. 1993b;31:2577–2583. doi: 10.1128/jcm.31.10.2577-2583.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolis N, Hogan D, Cieplak W, Jr., Schwan TG, Rosa PA. Homology between Borrelia burgdorferi OspC and members of the family of Borrelia hermsii variable major proteins. Gene. 1994;143:105–110. doi: 10.1016/0378-1119(94)90613-0. [DOI] [PubMed] [Google Scholar]
- Masuzawa T, Kurita T, Kawabata H, Yanagihara Y. Relationship between infectivity and OspC expression in Lyme disease Borrelia. FEMS Microbiol Lett. 1994;123:319–324. doi: 10.1111/j.1574-6968.1994.tb07242.x. [DOI] [PubMed] [Google Scholar]
- McDowell JV, Sung SY, Labandeira-Rey M, Skare JT, Marconi RT. Analysis of mechanisms associated with loss of infectivity of clonal populations of Borrelia burgdorferi B31MI. Infect Immun. 2001;69:3670–3677. doi: 10.1128/IAI.69.6.3670-3677.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal U, Yang X, Chen M, Bockenstedt LK, Anderson JF, Flavell RA, Norgard MV, Fikrig E. OspC facilitates Borrelia burgdorferi invasion of Ixodes scapularis salivary glands. J Clin Invest. 2004;113:220–230. doi: 10.1172/JCI19894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal U, Dai J, Li X, Neelakanta G, Luo P, Kumar M, Wang P, Yang X, Anderson JF, Fikrig E. A differential role for BB0365 in the persistence of Borrelia burgdorferi in mice and ticks. J Infect Dis. 2008;197:148–155. doi: 10.1086/523764. [DOI] [PubMed] [Google Scholar]
- Postic D, Edlinger C, Richaud C, Grimont F, Dufresne Y, Perolat P, Baranton G, Grimont PAD. Two genomic species in Borrelia burgdorferi. Res Microbiol. 1990;141:465–475. doi: 10.1016/0923-2508(90)90072-x. [DOI] [PubMed] [Google Scholar]
- Postic D, Assous MV, Grimont PAD, Baranton G. Diversity of Borrelia burgdorferi sensu lato evidenced by restriction fragment length polymorphism of rrf (5S)-rrl (23S) intergenic spacer amplicons. Int J Syst Bacteriol. 1994;144:733–742. doi: 10.1099/00207713-44-4-743. [DOI] [PubMed] [Google Scholar]
- Puius YA, Kalish RA. Lyme arthritis: pathogenesis, clinical presentation, and management. Infect Dis Clin North Am. 2008;22:289–300. vi–vii. doi: 10.1016/j.idc.2007.12.014. [DOI] [PubMed] [Google Scholar]
- Qiu WG, Bruno JF, McCaig WD, Xu Y, Livey I, Schriefer ME, Luft BJ. Wide distribution of a high-virulence Borrelia burgdorferi clone in Europe and North America. Emerg Infect Dis. 2008;14:1097–1104. doi: 10.3201/eid1407.070880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radolf JD, Caimano MJ. The long strange trip of Borrelia burgdorferi outer-surface protein C. Mol Microbiol. 2008;69:1–4. doi: 10.1111/j.1365-2958.2008.06226.x. [DOI] [PubMed] [Google Scholar]
- Ramamoorthi N, Narasimhan S, Pal U, Bao F, Yang XF, Fish D, Anguita J, Norgard MV, Kantor FS, Anderson JF, Koski RA, Fikrig E. The Lyme disease agent exploits a tick protein to infect the mammalian host. Nature Letters. 2005;436:573–577. doi: 10.1038/nature03812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Revel AT, Talaat AM, Norgard MV. DNA microarray analysis of differential gene expression in Borrelia burgdorferi, the Lyme disease spirochete. Proc Natl Acad Sci. 2002;99:1562–1567. doi: 10.1073/pnas.032667699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rios-Steiner JL, Schenone M, Mochalkin I, Tulinsky A, Castellino FJ. Structure and binding determinants of the recombinant kringle-2 domain of human plasminogen to an internal peptide from a group A Streptococcal surface protein. J Mol Biol. 2001;308:705–719. doi: 10.1006/jmbi.2001.4646. [DOI] [PubMed] [Google Scholar]
- Rogers EA, Terekhova D, Zhang HM, Hovis KM, Schwartz I, Marconi RT. Rrp1, a cyclic-di-GMP-producing response regulator, is an important regulator of Borrelia burgdorferi core cellular functions. Mol Microbiol. 2009;71:1551–1573. doi: 10.1111/j.1365-2958.2009.06621.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossmann E, Kraiczy P, Herzberger P, Skerka C, Kirschfink M, Simon MM, Zipfel PF, Wallich R. Dual binding specificity of a Borrelia hermsii-associated complement regulator-acquiring surface protein for factor H and plasminogen discloses a putative virulence factor of relapsing fever spirochetes. J Immunol. 2007;178:7292–7301. doi: 10.4049/jimmunol.178.11.7292. [DOI] [PubMed] [Google Scholar]
- Sadziene A, Wilske B, Ferdows MS, Barbour AG. The cryptic ospC gene of Borrelia burgdorferi B31 is located on a circular plasmid. Infect Immun. 1993;61:2192–2195. doi: 10.1128/iai.61.5.2192-2195.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuels DS, Garon CF. Coumermycin A1 inhibits growth and induces relaxation of supercoiled plasmids in Borrelia burgdorferi, the Lyme disease agent. Antimicrob Agents Chemother. 1993;37:46–50. doi: 10.1128/aac.37.1.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuels DS, Mach K, Garon CF. Genetic transformation of the Lyme disease agent Borrelia burgdorferi with coumarin-resistant gyrB. J Bacteriol. 1994;176:6045–6049. doi: 10.1128/jb.176.19.6045-6049.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schagger H, Cramer WA, von Jagow G. Analysis of molecular masses and oligomeric states of protein complexes by blue native electrophoresis and isolation of membrane protein complexes by two-dimensional native electrophoresis. Anal Biochem. 1994;217:220–230. doi: 10.1006/abio.1994.1112. [DOI] [PubMed] [Google Scholar]
- Schwan TG, Piesman J, Golde WT, Dolan MC, Rosa PA. Induction of an outer surface protein on Borrelia burgdorferi during tick feeding. Proc Natl Acad Sci USA. 1995;92:2909–2913. doi: 10.1073/pnas.92.7.2909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwan TG, Piesman J. Temporal changes in outer surface proteins A and C of the lyme disease-associated spirochete, Borrelia burgdorferi, during the chain of infection in ticks and mice. J Clin Microbiol. 2000;38:382–388. doi: 10.1128/jcm.38.1.382-388.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seinost G, Dykhuizen DE, Dattwyler RJ, Golde WT, Dunn JJ, Wang IN, Wormser GP, Schriefer ME, Luft BJ. Four clones of Borrelia burgdorferi sensu stricto cause invasive infection in humans. Infect Immun. 1999;67:3518–3524. doi: 10.1128/iai.67.7.3518-3524.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seling A, Siegel C, Fingerle V, Jutras BL, Brissette CA, Skerka C, Wallich R, Zipfel PF, Stevenson B, Kraiczy P. Functional characterization of Borrelia spielmanii outer surface proteins that interact with distinct members of the human factor H protein family and with plasminogen. Infect Immun. 2009 doi: 10.1128/IAI.00691-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steere AC. Lyme disease. New England Journal of Medicine. 2001;345:115–125. doi: 10.1056/NEJM200107123450207. [DOI] [PubMed] [Google Scholar]
- Stewart PE, Wang X, Bueschel DM, Clifton DR, Grimm D, Tilly K, Carroll JA, Weis JJ, Rosa PA. Delineating the requirement for the Borrelia burgdorferi virulence factor OspC in the mammalian host. Infect Immun. 2006;74:3547–3553. doi: 10.1128/IAI.00158-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theisen M, Frederiksen B, Lebech AM, Vuust J, Hansen K. Polymorphism in ospC gene of Borrelia burgdorferi and immunoreactivity of OspC protein: implications for taxonomy and for use of OspC protein as a diagnostic antigen. J Clin Microbiol. 1993;31:2570–2576. doi: 10.1128/jcm.31.10.2570-2576.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilly K, Krum JG, Bestor A, Jewett MW, Grimm D, Bueschel D, Byram R, Dorward D, Vanraden MJ, Stewart P, Rosa P. Borrelia burgdorferi OspC protein required exclusively in a crucial early stage of mammalian infection. Infect Immun. 2006;74:3554–3564. doi: 10.1128/IAI.01950-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilly K, Bestor A, Jewett MW, Rosa P. Rapid Clearance of Lyme Disease Spirochetes Lacking OspC from Skin. Infect. Immun. 2007;75:1517–1519. doi: 10.1128/IAI.01725-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilly K, Rosa PA, Stewart PE. Biology of infection with Borrelia burgdorferi. Infect Dis Clin North Am. 2008;22:217–234. v. doi: 10.1016/j.idc.2007.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilly K, Bestor A, Dulebohn DP, Rosa PA. OspC-independent infection and dissemination by host-adapted Borrelia burgdorferi. Infect Immun. 2009;77:2672–2682. doi: 10.1128/IAI.01193-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G, van Dam AP, Dankert J. Evidence for frequent OspC gene transfer between Borrelia valaisiana sp. nov. and other Lyme disease spirochetes. FEMS Microbiol Lett. 1999a;177:289–296. doi: 10.1111/j.1574-6968.1999.tb13745.x. [DOI] [PubMed] [Google Scholar]
- Wang IN, Dykhuizen DE, Qiu W, Dunn JJ, Bosler EM, Luft BJ. Genetic diversity of ospC in a local population of Borrelia burgdorferi sensu stricto. Genetics. 1999b;151:15–30. doi: 10.1093/genetics/151.1.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsh J, Pretzman C, Postic D, Saint Girons I, Baranton G, McClelland M. Genomic fingerprinting by arbitrarily primed polymerase chain reaction resolves Borrelia burgdorferi into three distinct phyletic groups. Int J Syst Bacteriol. 1992;42:370–377. doi: 10.1099/00207713-42-3-370. [DOI] [PubMed] [Google Scholar]
- Wormser GP, Brisson D, Liveris D, Hanincova K, Sandigursky S, Nowakowski J, Nadelman RB, Ludin S, Schwartz I. Borrelia burgdorferi genotype predicts the capacity for hematogenous dissemination during early Lyme disease. J Infect Dis. 2008;198:1358–1364. doi: 10.1086/592279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Q, McShan K, Liang FT. Identification of an ospC operator critical for immune evasion of Borrelia burgdorferi. Mol Microbiol. 2007;64:220–231. doi: 10.1111/j.1365-2958.2007.05636.x. [DOI] [PubMed] [Google Scholar]
- Xu Q, McShan K, Liang FT. Essential protective role attributed to the surface lipoproteins of Borrelia burgdorferi against innate defences. Mol Microbiol. 2008;69:15–29. doi: 10.1111/j.1365-2958.2008.06264.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang XF, Lybecker MC, Pal U, Alani SM, Blevins J, Revel AT, Samuels DS, Norgard MV. Analysis of the ospC regulatory element controlled by the RpoN-RpoS regulatory pathway in Borrelia burgdorferi. J Bacteriol. 2005;187:4822–4829. doi: 10.1128/JB.187.14.4822-4829.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye Q, Rahman MN, Koschinsky ML, Jia Z. High-resolution crystal structure of apolipoprotein(a) kringle IV type 7: insights into ligand binding. Protein Sci. 2001;10:1124–1129. doi: 10.1110/ps.01701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh JI, Biemann HP, Prive GG, Pandit J, Koshland DE, Jr., Kim SH. High-resolution structures of the ligand binding domain of the wild-type bacterial aspartate receptor. J Mol Biol. 1996;262:186–201. doi: 10.1006/jmbi.1996.0507. [DOI] [PubMed] [Google Scholar]
- Zuckert WR, Kerentseva TA, Lawson CL, Barbour AG. Structural conservation of neurotropism-associated VspA within the variable Borrelia Vsp-OspC lipoprotein family. J Biol Chem. 2001;276:457–463. doi: 10.1074/jbc.M008449200. [DOI] [PubMed] [Google Scholar]