

Abstract
Status epilepticus (SE), an unremitting seizure, is known to cause a variety of traumatic responses including delayed neuronal death and later cognitive decline. Although excitotoxicity has been implicated in this delayed process, the cellular mechanisms are unclear. Because our previous brain slice studies have shown that chemically induced epileptiform activity can lead to elevated astrocytic Ca2+ signaling and because these signals are able to induce the release of the excitotoxic transmitter glutamate from these glia, we asked whether astrocytes are activated during status epilepticus and whether they contribute to delayed neuronal death in vivo. Using two-photon microscopy in vivo, we show that status epilepticus enhances astrocytic Ca2+ signals for 3 d and that the period of elevated glial Ca2+ signaling is correlated with the period of delayed neuronal death. To ask whether astrocytes contribute to delayed neuronal death, we first administered antagonists which inhibit gliotransmission: MPEP [2-methyl-6-(phenylethynyl)pyridine], a metabotropic glutamate receptor 5 antagonist that blocks astrocytic Ca2+ signals in vivo, and ifenprodil, an NMDA receptor antagonist that reduces the actions of glial-derived glutamate. Administration of these antagonists after SE provided significant neuronal protection raising the potential for a glial contribution to neuronal death. To test this glial hypothesis directly, we loaded Ca2+ chelators selectively into astrocytes after status epilepticus. We demonstrate that the selective attenuation of glial Ca2+ signals leads to neuronal protection. These observations support neurotoxic roles for astrocytic gliotransmission in pathological conditions and identify this process as a novel therapeutic target.
Keywords: astrocyte, NMDA, metabotropic glutamate receptor, epilepsy, calcium, astrocytic glutamate release
Introduction
Traumatic head injury, stroke, or status epilepticus (SE) lead to the delayed death of neurons, which results in cognitive decline and later to epilepsy (Lemos and Cavalheiro, 1995). The cellular changes after the injury that lead to the delayed neuronal death are poorly defined. One of the precipitating injuries, SE, stimulates neuronal death lasting for days after the episode as well as the birth of new neurons, axon collateral sprouting, synaptogenesis, and reactive astrocytosis (Babb et al., 1984, 1991; de Lanerolle et al., 1989; Sutula et al., 1989; Houser, 1992, 1999; Houser et al., 1992; Houser and Esclapez, 1996; Fonseca et al., 2002; Garzillo and Mello, 2002). Because reactive astrocytes are associated with many disorders of the nervous system (Miller, 2005), we have become particularly interested in the potential role of astrocytes in delayed responses to injury.
Recent studies have demonstrated that astrocytes can release chemical transmitters, including glutamate and d-serine, which can regulate synaptic transmission and neuronal excitability through the activation of extrasynaptic NR2B-containing NMDA receptors (Araque et al., 2001; Haydon, 2001; Newman, 2003; Fellin et al., 2004). In addition to playing roles in physiology, this glial source of glutamate/d-serine has the potential to cause neuronal excitotoxicity because the activation of extrasynaptic NMDA receptors as well as NR2B-containing NMDA receptors has been shown to activate the neuronal cell death pathway (Hardingham et al., 2002; Zeron et al., 2002; Deridder et al., 2005; Gao et al., 2005).
Previous brain slice studies have shown that chemically induced epileptiform activity causes a sustained increase in Ca2+ signaling (Tian et al., 2005; Fellin et al., 2006) within astrocytes that outlasts the period of neuronal activity (Fellin et al., 2006). Because this prolonged elevation of glial Ca2+ signaling induces the release of glutamate, it has the potential to provide a sustained activation of NMDA receptors and possibly excitotoxicity. In this study, we focused our investigation in vivo where we asked whether SE induces astrocytic Ca2+ signals, and whether as a consequence the release of gliotransmitters could contribute to the delayed neuronal death that follows SE. We show that pilocarpine-induced SE stimulates a hyperactivation of the astrocyte by initiating Ca2+ signals that last for 3 d. Using pharmacological approaches that impact NMDA receptor-mediated gliotransmission, but not synaptic transmission, we demonstrate neuroprotection, allowing us to hypothesize that these glial Ca2+ signals initiate delayed neuronal death through activation of extrasynaptic NMDA receptors. To test this notion, we then attenuate Ca2+ signals in these glial cells in vivo using two independent Ca2+ buffers and demonstrate that astrocytic Ca2+ signaling, and thus Ca2+-dependent gliotransmission, contributes significantly to delayed neuronal death. This study provides new insights into pathological roles of astrocytes, implicating them as a critical signaling element that contributes in the induction of neuronal death.
Materials and Methods
All procedures were in strict accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the University of Pennsylvania Institutional Animal Care and Use Committee.
Slice preparation, patch-clamp recordings, and analysis.
Transverse brain slices (300–400 μm) were prepared from FVB/NJ mice (The Jackson Laboratory, Bar Harbor, ME) at postnatal days 12–45 as described previously (Pasti et al., 1997). After preparation, slices were incubated at 37°C for a recovery period of 1 h. The solution for slice cutting and incubation was as follows (in mm): 120 NaCl, 3.2 KCl, 1 NaH2PO4, 26 NaHCO3, 2 MgCl2, 1 CaCl2, 2.8 glucose, 2 Na-pyruvate, and 0.6 ascorbic acid at pH 7.4 with 95% O2 and 5% CO2. In the recording chamber, slices were continuously perfused with normal artificial CSF (ACSF) (in mm): 120 NaCl, 3.2 KCl, 1 NaH2PO4, 26 NaHCO3, 1 MgCl2, 2 CaCl2, 2.8 glucose, at pH 7.4 with 95% O2 and 5% CO2. Low Ca2+ solution was obtained by replacing CaCl2 with EGTA (0.25 mm). To elicit persistent epileptiform activity, in the experiments in Figure 4, J and K, pilocarpine was applied together with an additional epileptogenic stimulus as done previously (Rutecki and Yang, 1998). In this case, pilocarpine was perfused in 0 Mg2+-containing ACSF and in the presence of picrotoxin (100 μm). Pipette resistance was 3–4 MΩ. Intrapipette solution was as follows (in mm): 145 K-gluconate, 2 MgCl2, 5 EGTA, 2 Na2ATP, 0.2 NaGTP, 10 HEPES to pH 7.2 with KOH. Patch-clamp recordings were performed from layer 2/3 cortical neurons using standard procedures and MultiClamp-700B amplifiers (Molecular Devices, Union City, CA) or 2400 patch-clamp amplifiers (A-M Systems, Sequim, WA). Data were filtered at 1 kHz and sampled at 5 kHz with a Digidata 1320 interface and pClamp software (Molecular Devices). Experiments were performed at 30–35°C. Layer 2/3 pyramidal neurons were voltage-clamped at −60 mV. Evoked postsynaptic currents (EPSCs) were elicited by intracortical stimulation (0.1 Hz) with a bipolar tungsten electrode placed 100–200 μm beneath the recording pipette (approximately layer IV). The rise time of the NMDA component of the EPSCs and slow inward currents (SICs) was calculated with the 20–80% criterion and the decay time as the time constant of a single exponential fit. Extracellular field EPSPs were recorded with normal ACSF in the pipette and stimulating intracortical synapses as described previously for the patch-clamp recording. Data were filtered at 4 kHz and sampled at 20 kHz. Data analysis and fitting were performed with Clampfit 9.2 (Molecular Devices), Origin (Microcal Software, Northampton, MA), and SigmaPlot 8.0 (SPSS, Chicago, IL) software.
Figure 4.

Gliotransmission but not intracortical synaptic transmission activates the NR2B-containing NMDA receptors of layer 2–3 pyramidal neurons. A, Whole-cell recording from a pyramidal neuron showing SICs induced by bath application of CHPG (0.5 mm). In these as well as in B–F, experiments are performed in the continuous presence of TTX (1 μm). B, Left, Percentage of neurons showing increased SIC frequency after application of DHPG (10–20 μm; 4 of 11 cells), CHPG (0.5–1 mm; 7 of 20), and CHPG and MPEP (0.5–1 mm and 50 μm, respectively; 1 of 17). In the latter experiments, the slice was preincubated with MPEP for 5 min before the application of CHPG and MPEP. The ability of MPEP to block actions of CHPG was tested using Fisher's exact test (p < 0.05). Right, Average frequency of SICs before and after DHPG (n = 4) and CHPG (n = 7) in the responsive neurons shown in the left panel. In this as well as in the other panels of this figure, *p < 0.05 and **p < 0.01. Error bars indicate SEM. C, Whole-cell recording from a pyramidal neuron showing SICs induced by photolysis of caged Ca2+ in single cortical astrocytes. D, Average frequency of SICs under control conditions, in the presence of d-AP5 (50 μm), and after d-AP5 washout in six cortical neurons showing SIC activity. SICs were stimulated by application of CHPG (0.5–1 mm) or low Ca2+ containing ACSF. E, F, Mean amplitude of SICs under control conditions, in the presence of ifenprodil (E; 3 μm) or NVP-AAM077 (F; 0.4 μm), and after drug washout. Data are normalized to the amplitude of SICs recorded under control conditions. See inset for representative SICs from the same cell under the different experimental conditions. Number of averaged SICs is 37, 48, and 70 from 9 cells for E, and 27, 25, and 13 from 7 cells for F, respectively. G, H, Representative experiments showing the time course of the NMDA EPSC-amplitude at basal condition (0 Mg2+-containing saline in the presence of 10 μm NBQX), during ifenprodil (G; 3 μm) and NVP-AAM077 (H; 0.4 μm) application, and after drug washout. Subsequent application of d-AP5 (50 μm) completely blocked the EPSC (data not shown). Inset, Average of 10 EPSCs in the different experimental conditions. EPSCs were evoked by positioning the stimulating electrode intracortically, 100–200 μm from the recording pipette. I, Ifenprodil (left; 3 μm) does not decrease the average amplitude of the NMDA EPSC (n = 6 cells), whereas NVP-AAM077 (right; 0.4 μm) drastically reduces its amplitude (n = 4 cells). Data are normalized to the average NMDA EPSC amplitude under control conditions. J, Epileptiform activity induced by acute application of pilocarpine (10 μm) triggers an increase in SIC frequency. Representative experiment showing membrane current under basal conditions (left), in the presence of pilocarpine (middle), and in the presence of pilocarpine and TTX (1 μm; right). Addition of TTX blocks pilocarpine-induced epileptiform activity and reveals the presence of SICs. Pilocarpine was always applied in the presence of 0 Mg2+ and 100 μm picrotoxin (see Materials and Methods). During epileptiform activity, unclamped action potentials were recorded and have been truncated for presentation purposes. K, Average SIC frequency under basal conditions in normal ACSF before perfusion of pilocarpine (left; open bar). After epileptiform activity was triggered by application of pilocarpine, 1 μm TTX was added to the saline to block neuronal activity. Thereafter, SIC frequency was measured again in 6 of 10 layer 2/3 neurons displaying SICs (left; filled bar). When TTX was added together with pilocarpine, no epileptiform activity was initiated in the slice and no significant increase in SIC frequency was observed in four of seven neurons displaying SICs (right).
Photolysis.
Transverse brain slices were obtained from 10- to 16-d-old C57BL/6 (The Jackson Laboratory). After preparation, slices were incubated at 33–35°C for a recovery period of 30–40 min. Slices were bulk loaded for 1.5 h at room temperature in the cutting solution containing fluo-4 AM (12.5 μg/ml), NP-EGTA-AM (12.5 μg/ml), and pluronic acid (1 μl/ml of 20% DMSO solution) saturated with 95% O2 and 5% CO2. The internal solution contained the following (in mm): 135 K-gluconate, 2.7 MgCl2, 1.7 NaCl, 1.1 EGTA, 0.1 CaCl2, 3.5 MgATP, 0.3 NaGTP, and 10 HEPES to pH 7.25 with KOH. The fluorescent dye Alexa 568 (0.1 mm) was added to internal solution to visualize the dendrites of the recorded neuron. Neuronal currents were monitored for 6 min before and after photolysis of one or two astrocytes that were close to neuronal dendrites, and the average SIC frequency was calculated in these time periods. Photorelease of Ca2+ was performed by a 3-μm-diameter UV pulse (351 and 364 nm) generated by an argon ion laser (Coherent Enterprise II; 100 ms duration for three times; power, 30 mW) connected by an optical fiber to an Uncager system (Prairie Technologies, Middleton, WI). Images were acquired using a Q-Imaging (Burnaby, British Columbia, Canada) cooled CCD camera and ImagePro software.
Pilocarpine-induced SE.
Male FVB/NJ mice (6–8 weeks old) from The Jackson Laboratory were used in these experiments. Behavioral seizures were assessed according to the scale of Racine (1972). Thirty minutes before the injection of pilocarpine, methylscopolamine (a muscarinic antagonist) was administered subcutaneously (1 mg/kg) to reduce adverse, peripheral effects. SE was induced through subcutaneous injection of 350 mg/kg pilocarpine-hydrochloride, a muscarinic agonist. Animal behavior and seizure activity was documented throughout the procedure. One hour after the onset of SE, diazepam (5 mg/ml) was administered subcutaneously to reduce seizure activity. Two groups of control mice were used, either those injected with saline or with a one-tenth dose of pilocarpine. For experiments in which we pharmacologically manipulated gliotransmission after SE, 2-methyl-6-(phenylethynyl)pyridine (MPEP) and ifenprodil (20 mg/kg), or [(R)-[(S)-1-(4-bromo-phenyl) ethylamino]-(2,3-dioxo-1,2,3,4-tetrahydro-quinoxalin-5-yl)methyl] phosphonic acid (NVP-AAM077) (2 mg/kg) were administered intraperitoneally to mice together with diazepam. (+)-5-Methyl-10,11-dihydro-SH-dibenzo[a,d]cyclohepten-5,10-imine maleate (MK-801) (1 mg/kg) was delivered 2 h after diazepam. Antagonists were subsequently administered intraperitoneally once daily for up to 3 d before killing. For experiments in which we selectively attenuate astrocytic calcium signal after SE, animals were anesthetized 4–5 h after the onset of SE using xylazine/ketamine. A craniotomy was made on one side of cortex and either BAPTA-AM (200 μm) or fluo-4 AM (500 μm) was locally applied to the cortical surface through the craniotomy. The cortex was then covered with 2% agarose containing either BAPTA-AM or fluo-4 AM, the incision was closed, and animals were allowed to recover, returned to the animal facility, and then killed 3 d later for Fluoro-Jade B (FJB) staining.
In vivo two-photon Ca2+ imaging.
Six- to 8-week-old FVB/NJ mice were anesthetized with urethane (1.5–2.0 mg/g body weight), held in a custom-made immobilization device, and a circular cranial window (2.0 mm diameter) was drilled in the skull overlying the barrel cortex. A metal frame, to attach the skull to the microscope platform, was attached to the skull using cyanoacrylate, and the dura was carefully removed. Two microliters of 50 μg of fluo-4 AM mixed with 5 μl of pluronic (20% pluronic plus 80% DMSO) was mixed with 30 μl of ACSF [containing the following (in mm): 120 NaCl, 10 HEPES, 3 KCl, 2 CaCl2, 1 MgCl2, 10 glucose; with pH 7.4] and was applied through the cranial window to the surface of the cortex. After 45–60 min, excess dye was removed by irrigation with ACSF. As described (Hirase et al., 2004; Nimmerjahn et al., 2004), this protocol leads to the selective labeling of astrocytes with the Ca2+ indicator, fluo-4. In some experiments, selectivity of labeling was confirmed using sulforhodamine 101 (SR101) (supplemental Fig. 4, available at www.jneurosci.org as supplemental material). One hundred microliters of 100 μm SR101 dissolved in ACSF was applied on the surface of the cranial window for 1–5 min before being washed away with ACSF. After 45–60 min, astrocytes were labeled selectively with SR101. Using two wide-field detectors, we confirmed colabeling of astrocytes with fluo-4 and SR101. To reduce movement artifacts, a glass coverslip was glued over the cranial window on the metal frame, and the gap between glass and cranial window was filled with 2% agarose premelted in ACSF solution.
After dye loading, mice were transferred to the microscope stage and were placed on a heating pad to keep the mice warmed at 37°C. In vivo two-photon imaging was performed using a Prairie Technologies (Middleton, WI) Ultima two-photon microscope attached to an Olympus (Tokyo, Japan) BX51 microscope equipped with a 60× water-immersion objective. A Chameleon Titanium:Sapphire laser (Coherent, Santa Clara, CA) was used for two-photon excitation. Excitation was provided at 820 nm and emission was detected by external photomultiplier tubes (525/70; DLCP 575; 607/45 nm). To reduce movement artifacts in each image, we sampled from a region of a full image and acquired each frame at 3.2 μs/pixel.
In some experiments, we coinjected MPEP together with the fluorescent dye rhodamine-dextran through the tail vein. A mixture of 200 μl of ACSF containing 10 mg/ml rhodamine-dextran and plus MPEP to yield a final concentration of MPEP of 1 μg/g was injected into the tail vein. Successful injection was confirmed by the rapid appearance of rhodamine in the vasculature within the cortex. We waited at least 30 min before recommencing Ca2+ imaging to ensure that the MPEP had crossed the blood–brain barrier. To study acute the effects of ifenprodil, 3,5-dihydroxyphenylglycine (DHPG), or 2-chloro-5-hydroxyphenylglycine (CHPG) on the Ca2+ signals, these drugs were applied to the cortical surface for 30 min before recommencing imaging. The cranial window was then refilled with 2% agarose containing the same concentration of drug.
The fluorescent signals were quantified by measuring the mean pixel intensities of the region of interest using MetaMorph software (Universal Imaging, West Chester, PA). Ca2+ changes were expressed as ΔF/F0 values, where F0 was the baseline fluorescence. To express the magnitude of Ca2+ signals without subjective selection of threshold values, we integrated the ΔF/F0 signal over a 300 s imaging period using Origin software. The resulting value is expressed as ΔF/F0 · s in all graphs.
We recorded whisker-evoked potentials (EPs) using a glass electrode filled with ACSF and connected to a Swam IIC amplifier (Celica, Ljubljana, Slovenia). We stimulated mouse whiskers with a 100 ms air puff through a glass tube while the EP was being recorded. Data were acquired with a Digidata 1320 interface and pClamp software (Molecular Devices).
Transcardial perfusion and immunocytochemistry staining.
Mice were anesthetized with halothane and transcardially perfused first with ice-cold PBS and then 4% paraformadehyde in PBS (pH 7.4). After perfusion, the brain was postfixed in 4% paraformadehyde/PBS at 4°C for 30 min. It then was transferred to 30% sucrose overnight to prevent ice crystal formation. Coronal sections of the brain (20 μm) were cut on a cryostat (CM 3000; Leica, Nussloch, Germany), and were collected serially on precleaned slides and stored at −80°C until use. For Fluoro-Jade B staining, sections were washed with PBS three times and then immersed into 0.0001% Fluoro-Jade in 0.1% acetic acid solution at 4°C for 1 h. For double staining, sections were stained with either terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling staining kit (Upstate, Charlottesville, VA) or biotinylated mouse anti-NeuN (1:100) followed by the FJB staining procedure as described above. All sections were counterstained with 4,6-diamidino-2-phenylindole (DAPI) (1:1000; in PBS) and mounted using anti-fade mounting medium. Stained sections were viewed with epifluorescence and double-stained sections were examined using an Olympus Fluoview 1000 confocal microscope and analyzed using MetaMorph (Universal Imaging). Four random sections were selected from every animal. The number of FJB-labeled cells was counted bilaterally by a blinded investigator. To count the number of neurons in area CA3 and cortex cells, images of DAPI and NeuN staining in area CA3 of the hippocampus and 70–150 μm beneath the surface of cortex were acquired with a digital camera. Automated software was used to identify and count neuronal cell bodies based on the colocalization of NeuN with DAPI. Manual inspection confirmed the accuracy of this automated approach.
For studies of metabotropic glutamate receptor 5 (mGluR5) localization, sections obtained from GFAP-green fluorescent protein (GFP) transgenic mice (Zhuo et al., 1997) were cut (40 μm) and kept at −20°C in cryoprotective solution (30% sucrose, 30% ethylenglycol, 1% polyvinylpyrrolidone in PBS) until processing. After washing with PBS, sections were incubated in blocking solution (2 h; 2% normal horse serum, 0.5% BSA, and 0.3% Triton X-100 in PBS) and subsequently in blocking solution containing the primary antibody (rabbit anti-mGluR5; Chemicon International, Temecula, CA) for 48 h at 4°C. After washing in PBS, sections were incubated in blocking solution containing biotinylated anti rabbit antibody (2 h; room temperature), and then rinsed in PBS and incubated for 15 min in PBS containing Texas Red Avidin DCS (Vector Laboratories, Burlingame, CA). After being washed in PBS, sections were mounted using Vectashield mounting medium (Vector Laboratories). To study mGluR5 colocalization with astrocytes, we made an image mask based on GFP fluorescence to exclude and set to an intensity value of 0 those pixels that did not colocalize (supplemental Fig. 1, available at www.jneurosci.org as supplemental material).
Statistics.
n values reported in in vivo studies represent the number of animals. In our in vivo imaging experiments, Ca2+ measurements were generally made from 6–10 astrocytes per animal, and the data were averaged to obtain a single value per animal. Similarly in neuron and FJB counting studies, measurements were made on at least four sections, which were used to obtain a single value representative of that animal. When multiple comparisons were performed (for example, as shown in Fig. 5 and supplemental Fig. 3, available at www.jneurosci.org as supplemental material), ANOVA tests were performed and differences between individual groups were determined using the Newman–Keuls post hoc tests. Electrophysiological studies were tested for significance using the Student's t test unless stated otherwise. Statistical significance of data was reached at p < 0.05.
Figure 5.
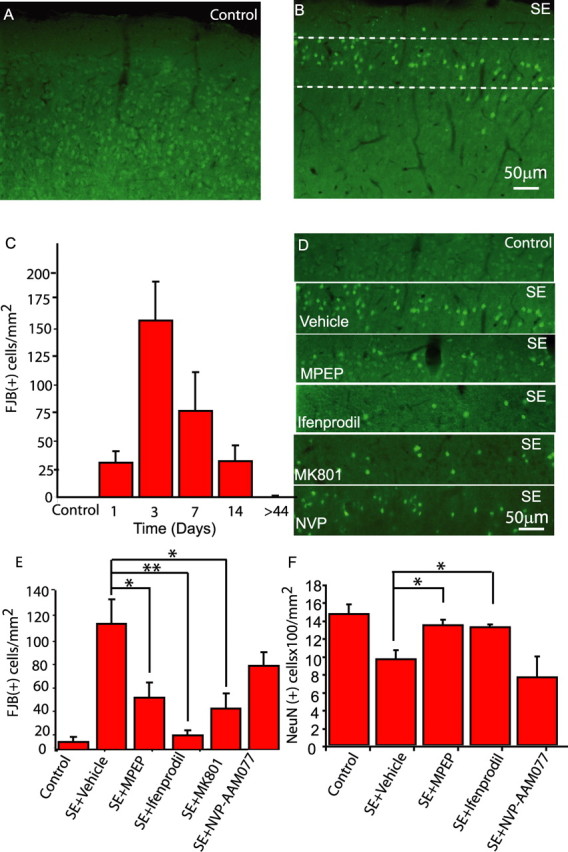
MPEP and ifenprodil protect neurons from SE-evoked death. FJB-stained sections of murine cortex from control (A) and mice 3 d after SE (B) show FJB labeling in layer 2/3, the region in which in vivo imaging and slice electrophysiology were performed. C, Time course of FJB labeling. D, The region identified by the dashed lines in B is shown from animals treated with glutamate receptor antagonists, as labeled. E, MPEP (20 mg/kg), ifenprodil (20 mg/kg), and MK-801 (1 mg/kg) significantly reduce the number of FJB-labeled cells 3 d after SE, whereas NVP-AAM077 (2 mg/kg) is not protective. F, In parallel experiments, the number of NeuN-labeled neurons in cortical layers 2/3 7 d after SE were counted using an automated system. Similar to results obtained with FJB, MPEP and ifenprodil, but not NVP-AAM077, protected neurons from delayed death that normally follows SE. *p < 0.05; **p < 0.01; n = 4–7 animals in each condition. Error bars indicate SEM.
Results
SE triggers delayed astrocytic Ca2+ signals in vivo during the latent period of epileptogenesis
Chemically induced epileptiform activity in brain slice preparations (Tian et al., 2005; Fellin et al., 2006) or in vivo (Hirase et al., 2004), in addition to evoking intense neuronal discharges, stimulates astrocytic Ca2+ signals. We recently demonstrated that chemically induced epileptiform activity induces a prolonged increase in astrocytic Ca2+ excitability that outlasts the period of epileptiform activity (Fellin et al., 2006), raising the potential for the excitability of the astrocyte to be altered in a long-term manner after seizure activity in vivo. We directly tested this hypothesis by using two-photon imaging of astrocytic Ca2+ (Hirase et al., 2004; Nimmerjahn et al., 2004) to ask whether seizures per se persistently modify the excitability of astrocytes. Layer 2/3 astrocytes in the barrel cortex were selectively loaded with the Ca2+ indicator fluo-4 either before or 2 or more days after pilocarpine-induced SE (Nimmerjahn et al., 2004). Intraperitoneal administration of pilocarpine into anesthetized mice induced astrocytic Ca2+ signals from a control value of 7.7 ± 1.6 (ΔF/F0 · s) to 43.2 ± 4.6 (n = 6) that are synchronized among networks of astrocytes (Fig. 1A,B) (n = 6 mice; p < 0.0001). To determine whether this acute change in Ca2+ signal might persist, we induced SE in mice and then imaged astrocytic Ca2+ 2 or more days later. Three days after SE, we observed a significant increase in astrocytic Ca2+ signals in mice that had entered SE compared with control mice that received either a saline injection or subthreshold injection of pilocarpine (Fig. 1C–E) (p < 0.001). This prolonged enhancement of Ca2+ excitability was detected for 3 d after SE, and then recovered to control levels thereafter (Fig. 1E).
Figure 1.
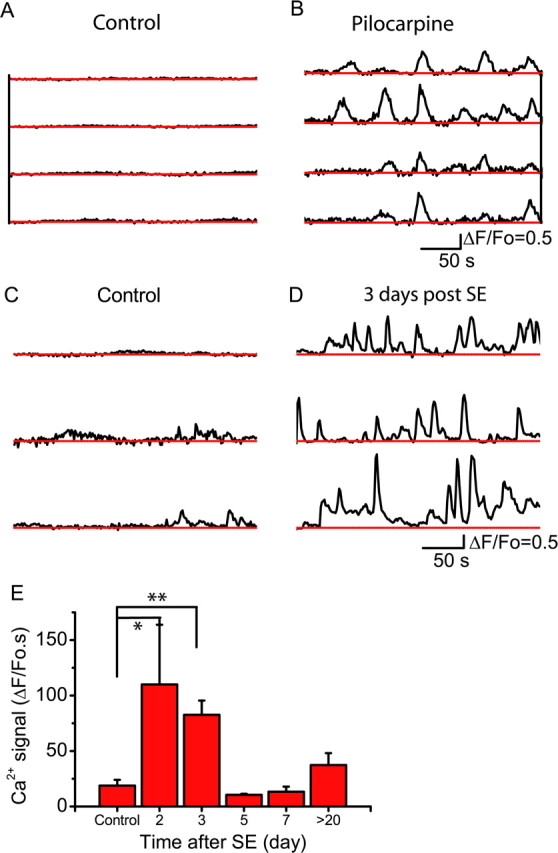
SE stimulates astrocytic Ca2+ signals. A, B, The time course of somatic Ca2+ signals recorded simultaneously in four astrocytes before (A) and after (B) acute injection of pilocarpine to the anesthetized mouse. Astrocytic Ca2+ signals (3 examples from 3 mice in each condition) recorded 3 d after a subthreshold injection of pilocarpine (Control) (C) or 3 d after SE (D). E, Time course of change in Ca2+ signals after SE reported as the integral of the ΔF/F0 signal (*p < 0.05; **p < 0.00005; n value is between 4 and 8 animals for each time point). Error bars indicate SEM.
mGluR5 mediates enhanced astrocytic calcium signals after SE
Because metabotropic glutamate receptors, and mGluR5 in particular, contribute to astrocytic Ca2+ signals in brain slices (Pasti et al., 1997; Parri et al., 2001; Bowser and Khakh, 2004; D'Ascenzo et al., 2007), we asked whether mGluR5 mediates the enhanced astrocytic Ca2+ signaling detected after SE. Anti-mGluR5 immunoreactivity showed colocalization with cortical astrocytes in GFAP-GFP transgenic mice raising the potential for direct mGluR5-mediated control of astrocytic Ca2+ signals (supplemental Fig. 1A–C, available at www.jneurosci.org as supplemental material). We therefore determined whether mGluR5 has the potential to stimulate astrocytic Ca2+ signals in vivo by local application of mGluR5 agonists to the cortex of control mice. Application of either DHPG (25 μm), a class I mGluR agonist, or CHPG (1 mm), a selective mGluR5 agonist, induced enhanced Ca2+ signaling within astrocytes (Fig. 2). Ca2+ signals that initiate within the processes of an astrocyte propagate through the cell and couple to adjacent astrocytes in the form of a Ca2+ wave (Fig. 2A) (n = 5 animals). CHPG-induced Ca2+ signals are attributable to the selective activation of mGluR5 because they are prevented by the mGluR5 antagonist MPEP (30 μm) (Fig. 2B,C). Because activation of mGluR5 does stimulate astrocytic Ca2+ signals, we determined whether mGluR5 is responsible for the enhanced astrocytic Ca2+ signals observed days after SE. Three days after SE, we measured astrocytic Ca2+ signals, and then injected the mGluR5 antagonist MPEP (1 mg/kg weight) through the tail vein, together with rhodamine-dextran as a positive label for successful injection (Fig. 3A). MPEP, which crosses the blood–brain barrier, significantly reduced the astrocytic Ca2+ signals (Fig. 3B,C) (n = 4 animals; p < 0.002). Because class I mGluR agonists/antagonists had inconsistent effects on intracortical synaptic transmission (supplemental Fig. 1D–G, available at www.jneurosci.org as supplemental material) yet reliably induce astrocytic Ca2+ signals (Fig. 2), these results demonstrate the importance of mGluR5 for the activation of Ca2+ signals in astrocytes after SE.
Figure 2.
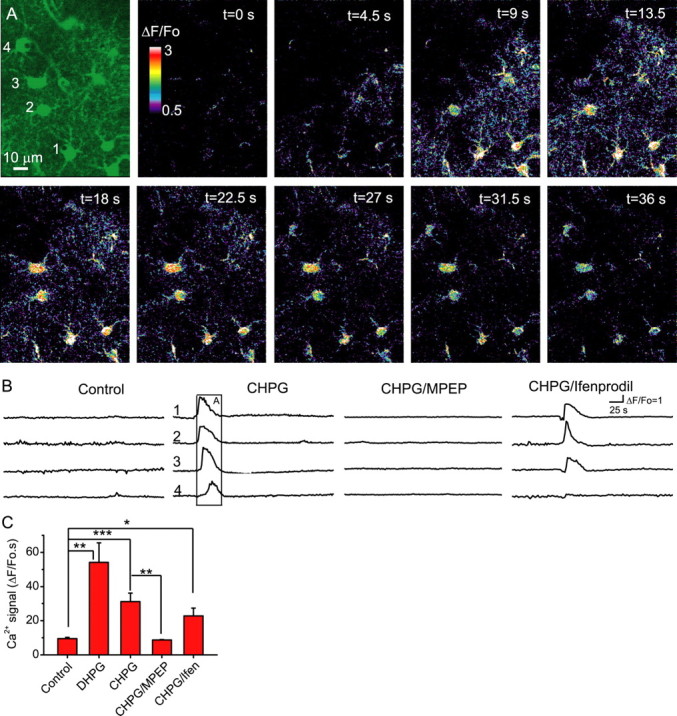
mGluR5 stimulates astrocytic Ca2+ signals in vivo. A, Sequential ΔF/F0 images showing that cortical astrocytes exhibit propagating Ca2+ waves in vivo when stimulated by the mGluR5 agonist CHPG (1 mm). B, Time courses of somatic Ca2+ fluorescence changes (ΔF/F0; examples of 4 cells in each case) in control animals in the absence and presence CHPG (1 mm) as well as with CHPG in the presence of MPEP (30 μm) and ifenprodil (10 μm). Note that the mGluR5 antagonist MPEP and not the NR2B NMDA receptor antagonist, ifenprodil, attenuate CHPG-induced Ca2+ signaling. The boxed region corresponds to the images in A. C, Astrocytic Ca2+ signals are stimulated by DHPG (25 μm) and CHPG (1 mm). The mGluR5 antagonist MPEP, but not the NR2B NMDA receptor antagonist ifenprodil, significantly attenuates CHPG-induced Ca2+ signals (n = 4–8 animals per group). *p < 0.05; **p < 0.02; ***p < 0.01. Error bars indicate SEM.
Figure 3.

Astrocytic Ca2+ signals in vivo in mice 3 d after SE are suppressed by the mGluR5 antagonist MPEP. A, Fluo-4-labeled astrocytes before (left) and after (right) the mouse was coinjected with MPEP and rhodamine-dextran through the tail vein. B, Calcium signals of a cell pair were inhibited after MPEP injection. C, Calcium signals before and after acute administration of MPEP (1 mg/kg tail vein injection), depicted as the ΔF/F0 integral. Also shown is the inhibition by four daily intraperitoneal administrations of MPEP (20 mg/kg). The NR2B NMDA receptor antagonist ifenprodil (20 μm) does not affect astrocytic Ca2+ signals. *p < 0.002 (n = 4 animals in each group except ifenprodil, n = 3). Error bars indicate SEM.
mGluR5-dependent activation of astrocytic Ca2+ signaling evokes NR2B-dependent NMDA receptor-mediated neuronal currents
Because of the potential for astrocytic Ca2+ signals to cause glutamate-mediated neuronal excitation, as has been described for thalamic (Parri et al., 2001), hippocampal pyramidal neurons (Angulo et al., 2004; Fellin et al., 2004; Kang et al., 2005; Perea and Araque, 2005), and medium spiny neurons of the nucleus accumbens (D'Ascenzo et al., 2007), we performed studies in acutely isolated brain slices to determine whether layer 2/3 cortical astrocytes excite cortical pyramidal neurons. Astrocytic Ca2+ signals evoked SICs mediated by NMDA receptors that can be identified based on four properties (Parri et al., 2001; Angulo et al., 2004; Fellin et al., 2004; Perea and Araque, 2005): their slow kinetics, their insensitivity to TTX and 2,3-dioxo-6-nitro-1,2,3,4-tetrahydrobenzoquinoxaline-7-sulfon-amide (NBQX), and their blockade by d-AP5. The class I mGluR agonist DHPG (10–20 μm) as well as the mGluR5 selective agonist CHPG (0.5–1 mm) induced TTX-insensitive SICs in layer 2/3 cortical neurons that were blocked by the mGluR5 antagonist MPEP (50 μm) (Fig. 4A,B) (n = 17; p < 0.05). These slow currents share kinetics typical of previously identified SICs (Fellin et al., 2004), which are one order of magnitude slower than NMDA-mediated synaptic-evoked currents (supplemental Table 1, available at www.jneurosci.org as supplemental material). In addition to exhibiting slow kinetics and an insensitivity to TTX, SICs fulfill the remaining pharmacological criteria to assign their origin to the astrocyte: they are mediated by the selective activation of NMDA receptors (NMDARs) because they are reversibly blocked by d-AP5 (50 μm) (Fig. 4D), whereas NBQX (10 μm) does not change the amplitude and kinetics of SICs (supplemental Table 1, available at www.jneurosci.org as supplemental material). To confirm the Ca2+ dependence of SICs, we photoreleased Ca2+ from the Ca2+ cage NP-EGTA that had been selectively loaded into astrocytes. Photolysis evoked a transient elevation of Ca2+ with similar properties to astrocytic Ca2+ signals induced by SE (Fig. 1B,D) that lasted for 7.5 ± 1.8 s (full-width half-maximum duration; mean ΔF/F0, 1.29 + 0.39) and evoked TTX-insensitive SICs in four of six cortical pyramidal neurons tested (Fig. 4C). These results are consistent with mGluR5 inducing astrocytic Ca2+ signals (Figs. 2, 3) that cause glial glutamate release, which in turn stimulates NMDA receptor-dependent SICs in cortical pyramidal neurons (Fig. 4).
Because of the potential for astrocytic and synaptic glutamate to access distinct NMDA receptors (Fellin et al., 2004), we determined the relative sensitivity of SICs and intracortical evoked EPSCs to NVP-AAM077 (0.4 μm) and ifenprodil (3 μm), which preferentially inhibits NR2A/C/D- and NR2B-containing NMDA receptors, respectively (Williams, 1993; Auberson et al., 2002; Neyton and Paoletti, 2006). The amplitude of SICs was reversibly reduced by ifenprodil (p < 0.01), whereas they were insensitive to NVP-AAM077 (Fig. 4E,F). In contrast, intracortical NMDA receptor-mediated EPSCs were reversibly attenuated by NVP-AAM077 (Fig. 4H,I) (p < 0.01) but insensitive to ifenprodil (Fig. 4G,I). The selective attenuation of synaptic NMDA receptors by NVP-AMM077 and attenuation of SICs by ifenprodil show that gliotransmission, which is mediated by SICs, preferentially accesses extrasynaptic NR2B-containing NMDA receptors. Because NR2A- but not NR2C- or NR2D-containing NMDA receptors exhibit rapid kinetics (Vicini et al., 1998), it is likely that the NVP-AAM077-sensitive synaptic NMDA currents are mediated predominantly by NR2A-containing NMDA receptors. Thus, astrocytic, but not synaptic, glutamate preferentially accesses extrasynaptic NR2B subunit-containing NMDA receptors.
Delayed neuronal death is suppressed by antagonists of gliotransmission that do not affect synaptic transmission
Before evaluating the potential for astrocytes to contribute to delayed neuronal death, we first confirmed that pilocarpine is able to induce neuronally detected SICs as is predicted based on the ability of status epilepticus to induce astrocytic Ca2+ signals. Because it is not currently feasible to study SICs in vivo, we recorded from layer II/III cortical pyramidal neurons in brain slice preparations and included pilocarpine (10 μm) within reduced Mg2+ picrotoxin-containing ACSF to induce prolonged SE-like epileptiform events (Fig. 4J). After pilocarpine-induced epileptiform activity, we added TTX (1 μm) to block neuronal activity and reveal SICs. Pilocarpine-induced epileptiform activity significantly increased SIC frequency (Fig. 4K) compared with precontrol basal levels. This increase in SIC frequency required epileptiform activity because incubation in TTX along with initial pilocarpine-ACSF to prevent epileptiform discharges failed to significantly increase SIC frequency (Fig. 4K).
Given that SE in vivo evokes astrocytic Ca2+ signals, pilocarpine-induced epileptiform activity in situ stimulates SICs, and because glial glutamate activates NR2B subunit-containing NMDA receptors (Fig. 4), an NMDA receptor pathway implicated in mediating neuronal death (Hardingham et al., 2002), we investigated the potential for a linkage between gliotransmission and the delayed cell death that is known to follow SE (Borges et al., 2003). First, we confirmed the relative selectivity of pharmacological agents by monitoring astrocytic Ca2+ signals and whisker-deflection evoked potentials. In agreement with acute administration of MPEP (Fig. 3), daily intraperitoneal administration of MPEP (20 mg/kg), which commenced immediately after the termination of SE, reduced astrocytic Ca2+ signals (n = 4 animals; p < 0.0025) (Fig. 3C). In contrast, administration of MPEP had no impact on whisker-evoked potentials (supplemental Fig. 2A,D, available at www.jneurosci.org as supplemental material) (n = 4 animals) nor on intracortical synaptic transmission (supplemental Fig. 1E,G, available at www.jneurosci.org as supplemental material). Ca2+-dependent astrocytic glutamate acts on extrasynaptic NR2B-containing NMDA receptors (Fig. 4). Accordingly, acute cortical administration of ifenprodil (20 μm), an NR2B-selective antagonist, neither affected the whisker-evoked potentials (supplemental Fig. 2B,D, available at www.jneurosci.org as supplemental material) (n = 3 animals) nor astrocytic Ca2+ signals (Figs. 2C, 3C) (n = 3–4 animals), whereas as a positive control for drug access, TTX (1 μm) blocked evoked potentials (supplemental Fig. 2C,D, available at www.jneurosci.org as supplemental material) (n = 3 animals; p < 0.0002). Thus, MPEP and ifenprodil can be used selectively to prevent astrocytic Ca2+ signals and to attenuate astrocyte-evoked SICs without significant actions on either synaptic NMDAR currents or on whisker-evoked synaptic potentials and neuron-based integration.
We determined the time course of neuronal death that follows SE by labeling sections from animals with FJB (Schmued and Hopkins, 2000), a fluorescent label that selectively identifies dying cells. After SE, FJB-labeled neurons, which colabel with the neuronal marker NeuN, were observed in layer 2/3 of the cortex (Fabene et al., 2003) (Fig. 5A,B), the same cortical region in which in vivo imaging and slice electrophysiology was performed. The peak of FJB-labeled neurons correlated with the period of astrocytic Ca2+ signals with labeling declining thereafter (Fig. 5C). Because of this temporal correlation, we determined the relative roles for gliotransmission and synaptic transmission in mediating this neuronal death. One hour after the cessation of SE, we commenced four daily intraperitoneal injections of MPEP (20 mg/kg, i.p.), ifenprodil (20 mg/kg, i.p.), MK801 (1 mg/kg, i.p.), NVP-AAM077 (2 mg/kg, i.p.), or vehicle. Administration of ifenprodil (n = 4 animals) or MPEP (n = 4 animals), antagonists that attenuate gliotransmission but have no actions on NMDAR-mediated synaptic transmission or whisker-evoked field potentials, both caused significant neuronal protection (Fig. 5D,E) (ifenprodil, p < 0.01; MPEP, p < 0.05). In contrast, NVP-AAM077, which attenuates NMDAR-mediated synaptic transmission but not gliotransmission, did not provide neuronal protection (Fig. 5D,E). MK801, a use-dependent NMDAR antagonist that does not select between NR2B- or NR2A-containing NMDA receptor subunits offered intermediate protection compared with ifenprodil and NVP-AAM077. As an alternative approach to FJB labeling, we waited for 7 d after SE and identified and counted NeuN-positive neuronal cell bodies. Similar to the FJB data, ifenprodil (n = 4 animals; p < 0.05) and MPEP (n = 4 animals; p < 0.05), but not NVP-AAM077 (n = 3 animals), afforded significant protection against delayed neuronal death that normally follows SE (Fig. 5F). In agreement with these results in the cortex, we found a similar pattern of hippocampal pyramidal neuron protection by antagonists that attenuate gliotransmission (supplemental Fig. 3, available at www.jneurosci.org as supplemental material).
Neuronal protection afforded by attenuation of astrocytic Ca2+ signals
Although pharmacological evidence supports the hypothesis that gliotransmission contributes to delayed neuronal death, we sought alternative experimental strategies to selectively target the astrocyte. Because application of acetoxymethyl esters of Ca2+ indicators to the cortical surface selectively labels astrocytes (Nimmerjahn et al., 2004; Takano et al., 2006), we used this approach to attenuate astrocytic Ca2+ signaling and thus Ca2+-dependent gliotransmission. To evaluate the feasibility of the approach, we first loaded neurons and astrocytes with Ca2+ indicator using the bulk-loading technique (Stosiek et al., 2003) by ejecting fluo-4 AM from the tip of a pipette into layer 2/3 of the cortex. Subsequently, we applied BAPTA-AM together with the astrocyte selective dye SR101 to the cortical surface to load these compounds into astrocytes only. Figure 6A shows fluo-4-labeled neurons (green) and astrocytes (yellow) together with Ca2+ signals recorded in these cells (Fig. 6B). Surface application of BAPTA-AM had no impact on neuronal Ca2+ signals (Fig. 6B,C), as expected if surface application of this Ca2+ chelator led to selective loading into astrocytes. We then stimulated 1-(6-[(17β-methoxyestra-1,3,5[10]-trien-17-yl)amino]hexyl)-1H-pyrrole-2,5-dione (U73122) (10 μm)-sensitive phospholipase C-dependent astrocytic Ca2+ signals by application of ATP (0.5 mm) to the cortex. ATP-induced Ca2+ signals were significantly reduced by previous surface application of BAPTA-AM (p < 0.001). Because surface application of BAPTA-AM selectively attenuated astrocytic Ca2+ signals, we used this approach to determine whether astrocytic Ca2+ signals contribute to delayed neuronal death after SE.
Figure 6.

Attenuation of astrocytic Ca2+ signals by loading of Ca2+ chelators is neuroprotective. A, Two-photon overlay image (fluo-4, green; SR101, red; colocalization, yellow) of layer 2/3 neurons loaded with fluo-4 and astrocytes loaded with fluo-4 and SR101. Fluo-4 AM was injected into the cortex using the bulk-loading approach to label neurons and astrocytes, whereas SR101 was applied to the surface of the cortex to selectively label astrocytes. With neurons and astrocytes coloaded with fluo-4, BAPTA-AM was applied to the cortical surface to selectively load into astrocytes. Selectivity of loading was confirmed by monitoring neuronal and astrocytic Ca2+ signals. B, Spontaneous neuronal Ca2+ signals in control and BAPTA-loaded animals. C, Surface application of BAPTA-AM does not reduce neuronal Ca2+ signals. D–F, Application of ATP (0.5 mm; n = 4) to the cortex evokes robust Ca2+ signals in astrocytes (E), which are attenuated by previous application of BAPTA-AM to the cortical surface (n = 5; compare E, F). G, The phospholipase C inhibitor U73122 (10 μm; n = 4) attenuates ATP-induced Ca2+ signals, providing additional support for an astrocytic origin of these Ca2+ signals. H, Summary of ATP-induced Ca2+ signals. I, After status epilepticus, BAPTA-AM was applied to the cortical surface through a craniotomy to selectively attenuate astrocytic Ca2+ signals, and 3 d later, Fluoro-Jade B labeling of dying neurons was assessed both ipsilateral (BAPTA) and contralateral to the treatment. J, The selective loading of the Ca2+ chelator BAPTA into astrocytes resulted in fewer dying, FJB-labeled cells compared with contralateral untreated cortex (paired t test). Surface loading with the fluorescent Ca2+ indicator, fluo-4, caused similar neuroprotection, whereas application of vehicle did not. *p < 0.05; **p < 0.01; ***p < 0.001. Error bars indicate SEM.
To determine whether SE-induced astrocytic Ca2+ signals induce neuronal death, we injected mice with pilocarpine, and then 1 h later terminated SE with diazepam. Subsequently, we made a unilateral craniotomy through which we locally applied BAPTA-AM, or vehicle, to the cortical surface. Three days later, animals were cardiac perfused, sectioned, and labeled with FJB to count dying neurons. Selective loading of BAPTA into astrocytes reduced FJB labeling ipsilateral but not contralateral to the craniotomy (Fig. 6I,J). In parallel control mice, we applied the carrier to the cortex and found that FJB labeling was not significantly different from the contralateral unoperated cortex. Because Ca2+ indicators bind Ca2+, increase the Ca2+ buffering capacity of a cell, and thereby attenuate Ca2+ signals, we asked whether application of fluo-4 AM to the cortical surface, and the resulting selective labeling of astrocytes (supplemental Fig. 4, available at www.jneurosci.org as supplemental material), would be neuroprotective. Similar to experiments performed with BAPTA-AM, we found that the loading of fluo-4 selectively into astrocytes after SE, did afford significant unilateral protection as assessed by FJB labeling (Fig. 6J).
Discussion
We provide the first evidence supporting the concept that the release of the gliotransmitter glutamate from astrocytes can provide a stimulus that induces the delayed neuronal death. This conclusion is based on a sequence of experimental evidence: SE induces astrocytic Ca2+ signals that persist for 3 d (Fig. 1) and that are temporally correlated with the period of delayed neuronal death (Fig. 5C). Ca2+-stimulated glutamate-dependent gliotransmission acts selectively on extrasynaptic NR2B-containing NMDA receptors (Fig. 4), and ifenprodil, an NR2B selective antagonist, provides almost complete neuronal protection from delayed death (Fig. 5; supplemental Fig. 3, available at www.jneurosci.org as supplemental material). In contrast, NVP-AAM077, an antagonist of NR2A-containing NMDA receptors, which attenuates layer 2/3 synaptic NMDA currents, does not afford neuronal protection after SE. Although neuronal protection provided by ifenprodil is consistent with astrocytes stimulating neuronal death through the NR2B-containing NMDA receptor, it is also possible that enhanced synaptic activity resulting from SE leads to transmitter spillover allowing synaptic glutamate to access extrasynaptic receptors (Scimemi et al., 2004). In support of a gliotransmission origin of NR2B-mediated neuronal death is our observation of neuronal protection provided by the mGluR5 antagonist MPEP, which attenuates astrocytic Ca2+ signaling (Fig. 3B,C) and thus gliotransmission while not impacting synaptic transmission (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Undoubtedly, ifenprodil and MPEP could affect neuron-based signals and they could affect receptors in addition to NR2B and mGluR5. However, neither intracortical synaptic transmission (Fig. 4G,I; supplemental Fig. 1E, available at www.jneurosci.org as supplemental material) nor evoked potentials recorded in vivo were inhibited by these drugs (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). Regardless of such specificity issues, it is important to note that they reduce gliotransmission and are neuroprotective. This correlation provides a foundation to propose that astrocytes contribute to delayed neuronal death. To test this possibility, we used a cell type selective manipulation that attenuates astrocytic Ca2+ signals. Topical application of AM-conjugated BAPTA-based compounds to the cortical surface led to selective labeling of astrocytes (Hirase et al., 2004; Nimmerjahn et al., 2004; Gobel et al., 2007). Astrocytes located hundreds of micrometers beneath the cortical surface label with these compounds (supplemental Fig. 4, available at www.jneurosci.org as supplemental material). This labeling is thought to result from the loading of astrocytes adjacent to the cortical surface followed by dye transfer to adjacent astrocytes through gap junctions (Nimmerjahn et al., 2004). Imaging confirmed the selectivity of labeling because only astrocytic Ca2+ signals were attenuated by BAPTA. We went on to show that attenuation of astrocytic Ca2+ signals is neuroprotective. It should be noted that, in addition to BAPTA, the Ca2+ indicator fluo-4 was also neuroprotective. Although this might seem surprising, it must be remembered that fluo-4 is a fluorescent BAPTA derivative that binds Ca2+ with a similar affinity (345 nm) to BAPTA (160 nm). When taken together with the pharmacological evidence that MPEP and ifenprodil are neuroprotective, we suggest that SE evokes Ca2+ signals causing the release of gliotransmitters and the activation of NR2B-containing NMDA receptors that stimulate the cell death cascade and lead to the progressive loss of neurons. Because astrocytic Ca2+ signals induce the release of d-serine as well as glutamate, we cannot exclude the possibility that this coagonist of the NMDAR also contributes to neuronal excitotoxicity.
This and several previous studies have shown that neuronally detected SICs are evoked by astrocytes and in response to elevations in their internal Ca2+. In cultures, SICs require astrocytic Ca2+ elevations and are prevented by the loading of BAPTA into astrocytes (Araque et al., 1998). The photolytic release of Ca2+ in astrocytes evokes short latency (∼10 ms) NMDA receptor-dependent SICs in cocultured neurons (Parpura and Haydon, 2000). In brain slices, stimuli that elevate astrocytic Ca2+, such as ATP and mGluR5 agonists, cause a correlated increase in SIC frequency that is unaffected by acute application of tetanus neurotoxin holoprotein (Fellin et al., 2004) at a time when synaptic transmission, but not gliotransmission, is blocked, demonstrating their non-neuronal origin. In slice preparations, photolytic elevation of astrocytic Ca2+ causes NMDAR-dependent SICs (Fig. 4C) that are evoked when the Ca2+ signal propagating within the astrocytic processes arrives at the location of the neuronal dendrite (Fellin et al., 2004; D'Ascenzo et al., 2007). Additional support for an astrocytic origin is provided by the observation that intracellular dialysis of IP3 into single astrocytes evokes SICs in adjacent neurons (Kang et al., 2005). Because SICs have been observed in many brain regions including areas CA1 and CA3 of the hippocampus (Angulo et al., 2004; Fellin et al., 2004; Kang et al., 2005; Perea and Araque, 2005; Tian et al., 2005), the thalamus (Parri et al., 2001), the olfactory bulb (Kozlov et al., 2006), the nucleus accumbens (D'Ascenzo et al., 2007), and the cortex (this study), it is likely that they are a general property of the nervous system.
In this study, we found distinct pharmacological sensitivity of NMDA receptors mediating synaptic transmission and gliotransmission. It is unlikely, however, that all synapses preferentially use NR2A- rather than NR2B-containing NMDA receptors. For example, in the hippocampus pharmacological studies have supported a role for NR2B-containing synaptic NMDA receptors (Kawakami et al., 2003; Scimemi et al., 2004). In a previous study in the rat cortex, NR2A-containing NMDA receptors were shown to mediate colossal–cortical synaptic transmission, whereas intracortical synapses made onto the same postsynaptic neurons were mediated by NR2B-containing NMDA receptors (Kumar and Huguenard, 2003). We found that intracortical synaptic NMDA receptors contained NR2A subunits. These differences may be because our studies were performed with older animals (up to postnatal day 48 compared with 21), in mice rather than in rats, and because we recorded from layer 2/3 rather than layer 5 pyramidal neurons (Kumar and Huguenard, 2003). Regardless of the subunit composition of synaptic NMDA receptors, of importance for this study is the observation that gliotransmission is suppressed by the NR2B antagonist ifenprodil, which is neuroprotective, providing one of several lines of evidence in support of a role for gliotransmission in neuronal excitotoxicity.
Because acute application of the mGluR5 agonist CHPG induces cell-wide Ca2+ signals and Ca2+ waves between cortical astrocytes of control mice (Fig. 2) with similar properties to those seen after SE (Fig. 1), we anticipate that after SE a rise in extracellular glutamate levels, perhaps resulting from immediate consequence of cellular damage during the seizure itself, induces the enhanced Ca2+ signaling of astrocytes. In turn, these signals lead to glutamate release and gliotransmission, which activates NR2B-containing NMDA receptors and initiates the cell death response. An alternative is the possibility of a role for neurons in a positive-feedback loop in which neuronal activity leads to mGluR5-dependent Ca2+ signals in astrocytes, which in turn leads to the release of the gliotransmitter glutamate that in return excites neurons through NMDA receptor function.
Our observation of enhanced astrocytic Ca2+ signaling in vivo after SE supports a previous cell culture study that investigated Ca2+ signaling in astrocytes isolated from a patient with Rasmussen's encephalitis, a rare form of intractable epilepsy. In that study, the tissue was isolated during surgery, and then plated into primary culture, and astrocytic Ca2+ signals were monitored thereafter. Initially, spontaneous Ca2+ signals were present, although their frequency declined during the following weeks (Manning and Sontheimer, 1997). Although control tissue was not available to make comparison measurements, taking these observations together with those presented in our study raises the potential of a general principle of glial Ca2+ signaling in which seizures enhance the Ca2+ excitability of astrocytes for prolonged periods that outlast the duration of the seizure itself.
Our demonstration that gliotransmission selectively activates NR2B-containing NMDA receptors, that these receptors are extrasynaptic and synaptic glutamate acts via NR2A-containing NMDA receptors suggests the potential for a general role for astrocytes in neuronal degeneration. Perhaps NR2B subunit-dependent neuronal degeneration in models of traumatic brain injury (Deridder et al., 2005) and Huntington's disease (Zeron et al., 2002) and during ischemic injury (Gao et al., 2005) is mediated by the release of glutamate from astrocytes. This role of NR2B subunit-containing NMDA receptors in mediating neuronal death together with the selective activation of these receptors by gliotransmission raises the potential of the astrocyte to contribute to neurodegeneration, especially because it is known that these glial cells become reactive during many neurological conditions including Alzheimer's and Parkinson's disease as well as during epilepsy (Miller, 2005).
Footnotes
This work was supported by the National Institute of Neurological Disorders and Stroke and National Institute of Mental Health (P.G.H., D.A.C.), the American Heart Association (S.D.), the Epilepsy Foundation (T.F.), and Italian University and Health Ministries Grant RBNE01RHZM_003 (G.C.).
P.G.H. has equity interest in Prairie Technologies, the company that manufactures the two-photon microscope used in some of the experiments reported in this study.
References
- Angulo MC, Kozlov AS, Charpak S, Audinat E. Glutamate released from glial cells synchronizes neuronal activity in the hippocampus. J Neurosci. 2004;24:6920–6927. doi: 10.1523/JNEUROSCI.0473-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araque A, Parpura V, Sanzgiri RP, Haydon PG. Glutamate-dependent astrocyte modulation of synaptic transmission between cultured hippocampal neurons. Eur J Neurosci. 1998;10:2129–2142. doi: 10.1046/j.1460-9568.1998.00221.x. [DOI] [PubMed] [Google Scholar]
- Araque A, Carmignoto G, Haydon PG. Dynamic signaling between astrocytes and neurons. Annu Rev Physiol. 2001;63:795–813. doi: 10.1146/annurev.physiol.63.1.795. [DOI] [PubMed] [Google Scholar]
- Auberson YP, Allgeier H, Bischoff S, Lingenhoehl K, Moretti R, Schmutz M. 5-Phosphonomethylquinoxalinediones as competitive NMDA receptor antagonists with a preference for the human 1A/2A, rather than 1A/2B receptor composition. Bioorg Med Chem Lett. 2002;12:1099–1102. doi: 10.1016/s0960-894x(02)00074-4. [DOI] [PubMed] [Google Scholar]
- Babb TL, Brown WJ, Pretorius J, Davenport C, Lieb JP, Crandall PH. Temporal lobe volumetric cell densities in temporal lobe epilepsy. Epilepsia. 1984;25:729–740. doi: 10.1111/j.1528-1157.1984.tb03484.x. [DOI] [PubMed] [Google Scholar]
- Babb TL, Kupfer WR, Pretorius JK, Crandall PH, Levesque MF. Synaptic reorganization by mossy fibers in human epileptic fascia dentata. Neuroscience. 1991;42:351–363. doi: 10.1016/0306-4522(91)90380-7. [DOI] [PubMed] [Google Scholar]
- Borges K, Gearing M, McDermott DL, Smith AB, Almonte AG, Wainer BH, Dingledine R. Neuronal and glial pathological changes during epileptogenesis in the mouse pilocarpine model. Exp Neurol. 2003;182:21–34. doi: 10.1016/s0014-4886(03)00086-4. [DOI] [PubMed] [Google Scholar]
- Bowser DN, Khakh BS. ATP excites interneurons and astrocytes to increase synaptic inhibition in neuronal networks. J Neurosci. 2004;24:8606–8620. doi: 10.1523/JNEUROSCI.2660-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Ascenzo M, Fellin T, Terunuma M, Revilla-Sanchez R, Meaney DF, Auberson YP, Moss SJ, Haydon PG. mGluR5 stimulates gliotransmission in the nucleus accumbens. Proc Natl Acad Sci USA. 2007;104:1995–2000. doi: 10.1073/pnas.0609408104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lanerolle NC, Kim JH, Robbins RJ, Spencer DD. Hippocampal interneuron loss and plasticity in human temporal lobe epilepsy. Brain Res. 1989;495:387–395. doi: 10.1016/0006-8993(89)90234-5. [DOI] [PubMed] [Google Scholar]
- Deridder MN, Simon MJ, Siman R, Auberson YP, Raghupathi R, Meaney DF. Traumatic mechanical injury to the hippocampus in vitro causes regional caspase-3 and calpain activation that is influenced by NMDA receptor subunit composition. Neurobiol Dis. 2005;22:165–176. doi: 10.1016/j.nbd.2005.10.011. [DOI] [PubMed] [Google Scholar]
- Fabene PF, Marzola P, Sbarbati A, Bentivoglio M. Magnetic resonance imaging of changes elicited by status epilepticus in the rat brain: diffusion-weighted and T2-weighted images, regional blood volume maps, and direct correlation with tissue and cell damage. NeuroImage. 2003;18:375–389. doi: 10.1016/s1053-8119(02)00025-3. [DOI] [PubMed] [Google Scholar]
- Fellin T, Pascual O, Gobbo S, Pozzan T, Haydon PG, Carmignoto G. Neuronal synchrony mediated by astrocytic glutamate through activation of extrasynaptic NMDA receptors. Neuron. 2004;43:729–743. doi: 10.1016/j.neuron.2004.08.011. [DOI] [PubMed] [Google Scholar]
- Fellin T, Gomez-Gonzalo M, Gobbo S, Carmignoto G, Haydon PG. Astrocytic glutamate is not necessary for the generation of epileptiform neuronal activity in hippocampal slices. J Neurosci. 2006;26:9312–9322. doi: 10.1523/JNEUROSCI.2836-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonseca CG, Green CR, Nicholson LF. Upregulation in astrocytic connexin 43 gap junction levels may exacerbate generalized seizures in mesial temporal lobe epilepsy. Brain Res. 2002;929:105–116. doi: 10.1016/s0006-8993(01)03289-9. [DOI] [PubMed] [Google Scholar]
- Gao J, Duan B, Wang DG, Deng XH, Zhang GY, Xu L, Xu TL. Coupling between NMDA receptor and acid-sensing ion channel contributes to ischemic neuronal death. Neuron. 2005;48:635–646. doi: 10.1016/j.neuron.2005.10.011. [DOI] [PubMed] [Google Scholar]
- Garzillo CL, Mello LE. Characterization of reactive astrocytes in the chronic phase of the pilocarpine model of epilepsy. Epilepsia 43. 2002;(Suppl 5):107–109. doi: 10.1046/j.1528-1157.43.s.5.40.x. [DOI] [PubMed] [Google Scholar]
- Gobel W, Kampa BM, Helmchen F. Imaging cellular network dynamics in three dimensions using fast 3D laser scanning. Nat Methods. 2007;4:73–79. doi: 10.1038/nmeth989. [DOI] [PubMed] [Google Scholar]
- Hardingham GE, Fukunaga Y, Bading H. Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat Neurosci. 2002;5:405–414. doi: 10.1038/nn835. [DOI] [PubMed] [Google Scholar]
- Haydon PG. GLIA: listening and talking to the synapse. Nat Rev Neurosci 2001. 2001;2:185–193. doi: 10.1038/35058528. [DOI] [PubMed] [Google Scholar]
- Hirase H, Qian L, Bartho P, Buzsaki G. Calcium dynamics of cortical astrocytic networks in vivo. PLoS Biol. 2004;2:E96. doi: 10.1371/journal.pbio.0020096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houser CR. Morphological changes in the dentate gyrus in human temporal lobe epilepsy. Epilepsy Res Suppl. 1992;7:223–234. [PubMed] [Google Scholar]
- Houser CR. Neuronal loss and synaptic reorganization in temporal lobe epilepsy. Adv Neurol. 1999;79:743–761. [PubMed] [Google Scholar]
- Houser CR, Esclapez M. Vulnerability and plasticity of the GABA system in the pilocarpine model of spontaneous recurrent seizures. Epilepsy Res. 1996;26:207–218. doi: 10.1016/s0920-1211(96)00054-x. [DOI] [PubMed] [Google Scholar]
- Houser CR, Swartz BE, Walsh GO, Delgado-Escueta AV. Granule cell disorganization in the dentate gyrus: possible alterations of neuronal migration in human temporal lobe epilepsy. Epilepsy Res Suppl. 1992;9:41–48. [PubMed] [Google Scholar]
- Kang N, Xu J, Xu Q, Nedergaard M, Kang J. Astrocytic glutamate release-induced transient depolarization and epileptiform discharges in hippocampal CA1 pyramidal neurons. J Neurophysiol. 2005;94:4121–4130. doi: 10.1152/jn.00448.2005. [DOI] [PubMed] [Google Scholar]
- Kawakami R, Shinohara Y, Kato Y, Sugiyama H, Shigemoto R, Ito I. Asymmetrical allocation of NMDA receptor epsilon2 subunits in hippocampal circuitry. Science. 2003;300:990–994. doi: 10.1126/science.1082609. [DOI] [PubMed] [Google Scholar]
- Kozlov AS, Angulo MC, Audinat E, Charpak S. Target cell-specific modulation of neuronal activity by astrocytes. Proc Natl Acad Sci USA. 2006;103:10058–10063. doi: 10.1073/pnas.0603741103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar SS, Huguenard JR. Pathway-specific differences in subunit composition of synaptic NMDA receptors on pyramidal neurons in neocortex. J Neurosci. 2003;23:10074–10083. doi: 10.1523/JNEUROSCI.23-31-10074.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemos T, Cavalheiro EA. Suppression of pilocarpine-induced status epilepticus and the late development of epilepsy in rats. Exp Brain Res. 1995;102:423–428. doi: 10.1007/BF00230647. [DOI] [PubMed] [Google Scholar]
- Manning TJ, Jr, Sontheimer H. Spontaneous intracellular calcium oscillations in cortical astrocytes from a patient with intractable childhood epilepsy (Rasmussen's encephalitis) Glia. 1997;21:332–337. [PubMed] [Google Scholar]
- Miller G. Neuroscience. The dark side of glia. Science. 2005;308:778–781. doi: 10.1126/science.308.5723.778. [DOI] [PubMed] [Google Scholar]
- Newman EA. New roles for astrocytes: regulation of synaptic transmission. Trends Neurosci. 2003;26:536–542. doi: 10.1016/S0166-2236(03)00237-6. [DOI] [PubMed] [Google Scholar]
- Neyton J, Paoletti P. Relating NMDA receptor function to receptor subunit composition: limitations of the pharmacological approach. J Neurosci. 2006;26:1331–1333. doi: 10.1523/JNEUROSCI.5242-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimmerjahn A, Kirchhoff F, Kerr JN, Helmchen F. Sulforhodamine 101 as a specific marker of astroglia in the neocortex in vivo. Nat Methods. 2004;1:31–37. doi: 10.1038/nmeth706. [DOI] [PubMed] [Google Scholar]
- Parpura V, Haydon PG. Physiological astrocytic calcium levels stimulate glutamate release to modulate adjacent neurons. Proc Natl Acad Sci USA. 2000;97:8629–8634. doi: 10.1073/pnas.97.15.8629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parri HR, Gould TM, Crunelli V. Spontaneous astrocytic Ca2+ oscillations in situ drive NMDAR-mediated neuronal excitation. Nat Neurosci. 2001;4:803–812. doi: 10.1038/90507. [DOI] [PubMed] [Google Scholar]
- Pasti L, Volterra A, Pozzan T, Carmignoto G. Intracellular calcium oscillations in astrocytes: a highly plastic, bidirectional form of communication between neurons and astrocytes in situ. J Neurosci. 1997;17:7817–7830. doi: 10.1523/JNEUROSCI.17-20-07817.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perea G, Araque A. Properties of synaptically evoked astrocyte calcium signal reveal synaptic information processing by astrocytes. J Neurosci. 2005;25:2192–2203. doi: 10.1523/JNEUROSCI.3965-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Racine RJ. Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr Clin Neurophysiol. 1972;32:281–294. doi: 10.1016/0013-4694(72)90177-0. [DOI] [PubMed] [Google Scholar]
- Rutecki PA, Yang Y. Ictal epileptiform activity in the CA3 region of hippocampal slices produced by pilocarpine. J Neurophysiol. 1998;79:3019–3029. doi: 10.1152/jn.1998.79.6.3019. [DOI] [PubMed] [Google Scholar]
- Schmued LC, Hopkins KJ. Fluoro-Jade B: a high affinity fluorescent marker for the localization of neuronal degeneration. Brain Res. 2000;874:123–130. doi: 10.1016/s0006-8993(00)02513-0. [DOI] [PubMed] [Google Scholar]
- Scimemi A, Fine A, Kullmann DM, Rusakov DA. NR2B-containing receptors mediate cross talk among hippocampal synapses. J Neurosci. 2004;24:4767–4777. doi: 10.1523/JNEUROSCI.0364-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stosiek C, Garaschuk O, Holthoff K, Konnerth A. In vivo two-photon calcium imaging of neuronal networks. Proc Natl Acad Sci USA. 2003;100:7319–7324. doi: 10.1073/pnas.1232232100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutula T, Cascino G, Cavazos J, Parada I, Ramirez L. Mossy fiber synaptic reorganization in the epileptic human temporal lobe. Ann Neurol. 1989;26:321–330. doi: 10.1002/ana.410260303. [DOI] [PubMed] [Google Scholar]
- Takano T, Tian GF, Peng W, Lou N, Libionka W, Han X, Nedergaard M. Astrocyte-mediated control of cerebral blood flow. Nat Neurosci. 2006;9:260–267. doi: 10.1038/nn1623. [DOI] [PubMed] [Google Scholar]
- Tian GF, Azmi H, Takano T, Xu Q, Peng W, Lin J, Oberheim N, Lou N, Wang X, Zielke HR, Kang J, Nedergaard M. An astrocytic basis of epilepsy. Nat Med. 2005;11:973–981. doi: 10.1038/nm1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicini S, Wang JF, Li JH, Zhu WJ, Wang YH, Luo JH, Wolfe BB, Grayson DR. Functional and pharmacological differences between recombinant N-methyl-d-aspartate receptors. J Neurophysiol. 1998;79:555–566. doi: 10.1152/jn.1998.79.2.555. [DOI] [PubMed] [Google Scholar]
- Williams K. Ifenprodil discriminates subtypes of the N-methyl-d-aspartate receptor: selectivity and mechanisms at recombinant heteromeric receptors. Mol Pharmacol. 1993;44:851–859. [PubMed] [Google Scholar]
- Zeron MM, Hansson O, Chen N, Wellington CL, Leavitt BR, Brundin P, Hayden MR, Raymond LA. Increased sensitivity to N-methyl-d-aspartate receptor-mediated excitotoxicity in a mouse model of Huntington's disease. Neuron. 2002;33:849–860. doi: 10.1016/s0896-6273(02)00615-3. [DOI] [PubMed] [Google Scholar]
- Zhuo L, Sun B, Zhang CL, Fine A, Chiu SY, Messing A. Live astrocytes visualized by green fluorescent protein in transgenic mice. Dev Biol. 1997;187:36–42. doi: 10.1006/dbio.1997.8601. [DOI] [PubMed] [Google Scholar]