

Summary
Mutations in the btk gene encoding Bruton’s tyrosine kinase cause X-linked immune deficiency (XID), with impaired B-lymphocyte function as the major phenotype. Earlier we demonstrated that CBA/N-xid mice, unlike the wild type CBA mice, were not protected by BCG vaccination against tuberculosis (TB) infection. Since IFN-γ-producing T-cells and activated macrophages are key elements of anti-TB protection, it remained unclear how the mutation predominantly affecting B-cell functions interferes with responses along the T-cell – macrophage axis. Here we show that B cell deficiency leads to an abnormally rapid neutrophil migration towards the site of external stimulus. Using adoptive cell transfers and B cell genetic knock-out we demonstrate a previously unappreciated capacity of B cells to down-regulate neutrophil motility. In our system, an advanced capture of BCG by neutrophils instead of macrophages leads to a significant decrease in numbers of IFN-γ-producing T-cells and impairs BCG performance in XID mice. The defect is readily compensated for by the in vivo neutrophil depletion.
INTRODUCTION
Mutations in the gene encoding Bruton’s tyrosine kinase (Btk) cause severe X-linked agammaglobulinemia (XLA) in humans (1, 2), and a more mild X-linked immune deficiency (XID) in mice (3, 4). The XID phenotype in mice is due to a partial block of B-lymphocyte development which is caused by a missense mutation (R28C) in the N-terminal PH domain of Btk, leading to a total lack of B1 lymphocytes and a substantial decrease in numbers of conventional B2 lymphocytes (5, 6). Since the major phenotype of Btk deficiency is impaired B-lymphocyte development and function, interest in Btk immune functions was focused on B-cell responses (7–10). However, Btk is also expressed and functions in myeloid lineage cells (11–14), and B1a cells are essential not only for T-independent innate host responses but also for adaptive immunity (15). These observations have stimulated an investigation into a possible role for Btk in a broader range of immune responses.
In our early studies we demonstrated that CBA/LAcN-xid mice were much less efficiently protected by BCG vaccination against subsequent infection with virulent Mycobacterium tuberculosis compared to their CBA/Lac coisogenic counterparts. This was an intriguing observation, given the paucity of knowledge regarding genetic control of vaccine efficacy against any infection. The defect in CBA/N mice proved to be X-linked and was accompanied by a marked decrease in the T cell proliferative activity in response to mycobacterial antigens compared to the wild-type mice (16). Combining the generally accepted concept that effector and memory CD4+ T cells are the key elements of vaccine-induced protection against mycobacteria (17) with the apparent defect in B-cell functions associated with the xid mutation, we hypothesized that the antigen-presenting capacity of B cells was impaired in XID mice leading to insufficient T cell activation. After demonstrating equal capacities of CBA and CBA/N purified splenic B-cells to present mycobacterial and irrelevant antigens to T cell clones of appropriate specificities (16), we temporary put away the discovered phenomenon, lacking reasonable hypotheses and research tools to study it further. The renaissance of our interest was stimulated by the demonstration that btk gene mutations are expressed not only in B lymphocytes but in many other cells of immune system (11–14), although not in mature T cells (18). The latter suggests that alterations in the interactions between cell types other than T cells per se might account for the diminished BCG vaccine efficacy in XID mice.
Here we show that extremely rapid neutrophil migration towards the site of BCG injection in XID animals is a key feature of their response to BCG. Identical phenotype is expressed in CD19−/−, B-cell deficient mice. Using adoptive cell transfer, we demonstrate a previously unappreciated capacity of B cells to inhibit early neutrophil migration to the inflammatory site. In our system, this leads to alterations in BCG distribution among different types of phagocytes. As a consequence, after BCG vaccination the numbers of CD4+ T cells producing IFN-γ in the spleen are a significantly lower in XID compared to the wild type mice. In the lung, similar differences are observed exclusively following vaccination and subsequent infection, but not after vaccination alone. Using in vivo neutrophil depletion shortly before BCG injection, we demonstrate restoration of the vaccine performance in B cell-deficient animals.
MATERIALS AND METHODS
Mice, vaccination and infection
CBA/LacStoCit (hereafter – CBA), CBA/NCit (CBA/N), C57BL/6Jcit (B6) and B6.CD19−/− mice containing homozygous cre insertion in the cd19 coding sequence (19) were bred and maintained under conventional conditions with water and food provided ad libitum at the Animal Facilities of the Central Institute for Tuberculosis (Cit, Moscow, Russia), according to the guidelines of the Russian Ministry of Health, NIH Office of Laboratory Animal Welfare (OLAW) Assurance #A5502-11. Breeding pairs of CD19−/− mice were a kind gift of Dmitry Kuprash, Engelhardt Institute, Moscow. Mice of both sexes were used at 2 to 3 months of age; no sex-related phenotypic differences were noticed throughout the study.
Mice were vaccinated with 2 × 107 colony-forming units (CFU) of M. bovis BCG (Pasteur) in 0.3 ml of saline subcutaneously (s. c.) in the dorsum; a group of mice which received 2 × 105 BCG CFU was included in the experiment described in Fig. 1C. Mice were infected either intravenously (i. v.) with 5 × 105 CFU, or intratracheally (i. t.) with 104 CFU of mid-log-phase M. tuberculosis strain H37Rv (original stock was a kind gift of Dr. Gilles Marchal, Institute Pasteur, Paris), as described earlier (20, 21). Mortality of the mice was monitored daily starting day 14 following infection. To assess mycobacterial multiplication in spleens and lungs, 0.1 ml of serial 10-fold dilutions of sterile whole-organ 2-ml homogenates were plated onto Dubos agar, and colonies were counted after 18–20 days of incubation at 37°C. All experimental procedures were approved by the Institutional Animal Care and Use Committee.
Fig. 1. Defective phenotypes expressed in CBA/N mice and restoration of BCG performance using adoptive cell transfer approaches.

Compared to CBA mice, protection against TB challenge after BCG vaccination was significantly (P<0.01, Gohan’s criterion for survival curves) weaker in CBA/N-xid after injection of 5 × 105 M. tuberculosis CFU i. v. (A), or 104 CFU i. t. (B); N = 9 for each group, results presented as mean ± SEM. The defect did not depend upon vaccination dose (C: open bars – non-vaccinated control; black bars −2 × 105 CFU of BCG s. c.; hatched bars −2 × 107 CFU of BCG s. c., challenge − 5 × 105 CFU of M. tuberculosis i. v., N = 8 for each group, mean ± SEM). In CBA → CBA/N radiation bone marrow chimera (9.5 G irradiation, 2 × 107 donor BMC per recipient), protective effect of BCG was restored in terms of both mycobacterial lung CFU counts (D: N = 4 per group, 3 wk post-challenge, P<0.01, one of two similar experiments), and prolongation of survival time (E: N = 8 per group, 2 × 107 CFU of BCG, challenge − 5 × 106 CFU of M. tuberculosis i. v., P<0.05, Gohan’s criterion). Adoptive transfer of 2 × 107/mouse fetal liver (FL) cells from CBA donors to non-irradiated adult CBA/N recipients restored the numbers of B cells in their lymphoid organs to the levels characteristic for the w. t. mice (F) and was sufficient for restoration of BCG-provided protection (G).
ELISPOT assay
Single cell suspensions were obtained individually from lungs and spleens of mice exactly as described (20). Sterile filter Millipore plates were coated with rat antibody against murine IFN-γ (PharMingen, San Diego, CA), washed and blocked with RPMI-1640 containing 10% FCS (HiClone, Logan, UT). Cells from 3 individual animals per group were added to the wells with 4 doubling dilutions, starting 1 × 106 cells/well, and cultured for 48 h in medium alone or in the presence of 10μg/ml mycobacterium sonicate. Following staining with biotin-labeled rat antibody against murine IFN-γ (PharMingen), spots were counted using ELISPOT Bioreader 4000 Pro-X (BioSys, Karben, Germany), calculated and normalized for the bulk individual samples. The results are displayed as the mean ± SD per organ.
Adoptive transfers
To prepare radiation bone marrow chimera, CBA/N recipients were irradiated at 9.5Gr from a 60Co source and within 6h restored by the i. v. injection of bone marrow cells freshly isolated from femurs of CBA donors (one-donor-to-one-recipient transfer, 15–20 × 106 cells/mouse). Four control mice which did not receive protective cell transfer died at day 9–11 following irradiation with signs of acute bone marrow radiation disease. Recipients were rested for 6 wk, and used for vaccination/infection experiments. Fetal liver cells were obtained from 17–18-d CBA embryos, and 10 × 106/mouse were injected i. v. into adult CBA/N recipients 4 wk before starting vaccination procedures. Peritoneal cavity cells from CBA donors, either non-separated or depleted of plastic-adherent cells, were transferred into the peritoneal cavities of CBA/N recipients (5 × 106/mouse), and BCG was injected i. p. after a 15-h interval.
T-cell lines and proliferative response
2 × 106 immune cells isolated from popliteal lymph nodes of CBA and CBA/N mice, immunized in the foot pads with mycobacterial sonicate in incomplete Freund’s adjuvant 12d before, were cultured in 1 ml RPMI-1640 containing 10 % FCS, 10 mM HEPES, 4 mM L-glutamine, 5 × 10−5 M 2-ME, vitamins, piruvate, non-essential amino acids and antibiotics (all components – HiClone) in 24-well plates (Costar, Badhoevedorp, The Netherlands) for 14d in the presence of 10 μg/ml sonicate. Live immune cells (>93% viability by trypan blue exclusion) were isolated by centrifugation at 2500 g for 20 min at 23°C, on a 1.088 g/ml Lympholyte M gradient (Cedarlane Labs, Ontario, Canada), washed twice and counted. The next stimulation cycle was accomplished by co-culturing 2 × 105 isolated cells with irradiated (12 Gr) 1.5 × 106 splenic APC in the presence of sonicate for another 2–3 wk period. These cycles were repeated for 4–5 times, until the cells started to grow as a sonicate-specific T cell line (>99% of CD4+ T lymphocytes). Cell samples were kept at −150°C until used. To assess proliferative response, T-cells were thawed, washed, and 5 × 103 cells/well were cultured for a total of 48 h at 37°C, 5% CO2, in the presence of indicated numbers of APC obtained from peritoneal cavities (Fig. 2A). All cultures were performed in triplicate. For the last 18 h, cultures were pulsed with 0.5μCi/well methyl-[3H]-thymidine. Cultures were harvested onto glass microfibre filters using a cell harvester (Scatron, Oslo, Norway) for liquid scintillation counting. Results are expressed as counts per minute (cpm ± SD).
Fig. 2. Neutrophil migration towards the site of BCG injection in XID and wild type mice.

(A) CBA but not CBA/N peritoneal APC loaded with BCG in vivo for 2 h can present mycobacterial antigens to specific T cell lines (one of two similar experiments). (B, C) After i. p. BCG injection, in XID mice neutrophils migrate to the peritoneal cavity much more rapidly than in wild type mice (summary of 8 independent experiments, mean ± SD). The difference in migration speed is abrogated by the adoptive transfer of either whole (D) or non-adherent (E) population of CBA peritoneal exsudate cells (summary of 2 independent experiments, 3 mice in each group, N=6). (F) In the transwell system, CBA/N neutrophils migrate much faster, compared to CBA neutrophils, irrespective to migration stimuli (two independent experiments with mixtures of cells from 3 mice in each, P < 0.01, unpaired t-test).
Transmigration assay
Chemotaxis was evaluated in 24-well transwell plates with 3-μm-pore-size filters (Costar-Corning, Badhoevedorp, The Netherlands). Lower chambers were filled with either culture medium (supplemented RPMI-1640) alone, or medium containing 50 × 103 peritoneal plastic-adherent cells (>90% F4/80+, <2% Ly-6G+, Mph in Fig. 2E), or 50 × 103 non-adherent cells (4% F4/80+, 21% CD3+, 75% CD19+, <2% Ly-6G+, Lym in Fig. 2E), or a 1:1 mixture of these populations (Mph + Lym in Fig. 2E), with or without addition of 106 BCG CFU/well. A total of 5 × 105 cells isolated from peritoneal cavities 4 h after peptone stimulation (>96% Ly-6G+ PMN neutrophils) in 100 μl of medium were added to the upper chambers and allowed to migrate through the membrane for 1 h at 37°C. The cells were collected totally from the lower chambers, stained with anti-Ly-6G mAbs, and quantified by flow cytometry.
Chemokine levels in peritoneal exsudates
Peritoneal cavities of individual CBA and CBA/N mice (4 per group) were washed with 3 ml of heparin-containing saline at 30min and 90min after injection of either 1.0ml of saline containing 108 BCG CFU or saline alone (control). Peritoneal cells were removed by centrifugation, and the content of KC, MIP-2 and G-CSF in supernatants was assessed in ELISA format using commercially available kits (R & D Systems, Minneapolis, MN) according to the manufacturer’s instructions.
Gene expression evaluation
Total RNA from peritoneal cavity cells of individual mice was isolated using the commercial SV Total RNA Isolation System (Promega, Madison, WI). Reverse transcription of mRNA was carried out exactly as described (22). Quantitative real-time RT-PCR (qrt-PCR) with cDNA was performed using iCycler iQ Multicolor Real-Time PCR Detection System (BioRad, Hercules, CA). The following specific primers and TaqMan probes were purchased from DNA Synthesis, LLC (Moscow): csf3: F-GCTGCTGCTGTGGCAAAGT, R-AGCGCTGACAGTGACCAGG, probe: FAM-CACTATGGTCAGGACGAGAGGCCGTT-BHQ1; mip2: F-CACTCTCAAGGGCGGTCAAA, R-CAGGTCAGTTAGCCTTGCCTTT, probe: FAM-CCCTGGTTCAGAAAATCATCCAAAAGA-BHQ1; kc: F-TGTCAGTGCCTGCAGACCAT, R-GTGGCTATGACTTCGGTTTGG, probe: FAM-CATCCAGAGCTTGAAGGTGTTGCCCTC-BHQ1. The PCR reaction was performed in a 25μl final volume of water containing 2μl cDNA, 2.5 μl 10X TaqPol buffer (Promega), 1μl 5 mM dNTPs, 1μl 10μM forward and reverse primer mix, 0.5μl 10μM TaqMan probe, 0.5μl Taq DNA polymerase (5u/μl, Promega). PCR amplifications were performed in triplicate using an identical PCR program for all genes: 5 min at 94°C, followed by 50 cycles alternating 15 seconds at 94°C and 1 min at 60°C. Gene expression levels in the lung tissue of individual mice were normalized to those of β-actin (and confirmed against GAPDH). To quantify the results obtained by real-time PCR, the comparative threshold method was used exactly as described (23), with the expression of the results as mean fold increase ± SEM for groups of 3 mice in each of 3 independent experiments.
Neutrophil depletion in vivo and cell staining
CBA/N mice were injected i. p. with 100μg/mouse anti-Ly-6G, RB6-8C5 mAbs, kindly provided by S. Kaufmann (Max Plank Institute, Berlin). Control CBA mice were injected with either 100μg/mouse of irrelevant, isotype-matched, IgG2b mAbs against a potato virus, kindly provided by A. Avdienko (CIT, Moscow), or left untreated. Fifteen hours later the efficacy of neutrophil depletion (~99%) was assessed by flow cytometry of peripheral blood cells using FITC-conjugated anti-Ly-6G mAb 1A8 (PharMingen). Mice were vaccinated s. c. with 2 × 107 CFU of BCG, and challenged 5 wk later by the i. v. route with 5 × 105 CFU of M. tuberculosis H37Rv. Antibodies PE-anti-F4/80 (clone CI:A3-1, Caltag, Burlingame, CA), PE-anti-CD19 (clone 1D3, PharMingen), and PE-anti-CD3 (clone 145-2C11, PharMingen) were used to enumerate peritoneal macrophages, B-cells, and T cells, respectively.
RESULTS
Adoptive transfer of the wild type immunocytes restores BCG performance in XID mice
Impaired performance of BCG vaccine in CBA/N-xid mice was originally discovered in the acute TB model using high-dose intravenous challenge, which caused rapid mortality and can not be considered as adequately mimicking chronic TB infection (16). Before further studying the phenomenon, we tested BCG vaccine efficacy under more physiological conditions, i.e., in mice vaccinated with lower doses of BCG and infected with lower doses of M. tuberculosis delivered via i. v. and i. t. routes, as described earlier (20, 21). As shown in Fig. 1A–C, BCG vaccination provided a significantly stronger protection against subsequent TB challenge in the w. t. CBA, compared to CBA/N-xid mice, irrespective to the route and dose of infection, or the dose of vaccine. Under different experimental conditions, BCG vaccination efficacy in CBA/N mice varied between 0–35 per cent of that in CBA mice.
To find out whether it is possible to compensate for the defect of XID mice by the adoptive transfer of immunocytes from the w. t. coisogenic mice, we performed two types of experiments. First, we showed that the total replacement of immune system of CBA/N recipients with that of CBA donors in CBA → CBA/N radiation bone marrow chimeras restores the capacity of recipients to respond to BCG vaccination by decreasing the load of mycobacteria in their lungs (Fig. 1D) and increasing the survival time (Fig. 1E). Transfer of syngenic CBA/N cells to CBA/N recipients had no effect (data not shown), and a somewhat shorter survival time of all animals compared to other experiments was likely due to a general adverse effect of irradiation. Second, it was of interest to test whether a biased restoration of the B cell pool in XID mice is sufficient to compensate for the defect in response to BCG, given that the main difference between XID and w. t. mice is a marked B cell deficiency of the former. Thus, we transferred fetal liver cells from CBA donors into intact CBA/N recipients, being aware that the fetal liver is a good source of B cell precursors, especially of B-1a lymphocytes (5), and that the recipient’s T cells will continue normal functioning in non-irradiated recipients. Our results indicate that this type of transfer fully restored the B cell pool in lymphoid organs of CBA/N recipients to the w. t. level (Fig. 1F). Evaluation of BCG-induced protection in the recipient mice demonstrated a significant decrease in the lung CFU counts (data not shown) and an increase of survival time (Fig. 1G) compared to control animals. Thus, a biased restoration of B cell pool in XID mice was sufficient to normalize the protective effect of BCG vaccination.
In the absence of B cells neutrophils display increased migration towards the site of external stimulus
Since the capacities of purified CBA and CBA/N B-cells to present mycobacterial and irrelevant antigens to T cell clones were shown to be equal (16), we hypothesized that the defective BCG performance in XID animals could be due to an impaired processing of BCG and/or presentation of mycobacterial antigens to T cells by phagocytes. To address this issue, we compared the capacity of CBA and CBA/N peritoneal exudate cells loaded in vivo with live BCG to induce proliferative responses of mycobacteria-specific T cell lines. Mice were inoculated with BCG intraperitoneally, and after a 2-h interval peritoneal exudate cells were isolated, washed and co-cultured with T cells. Immediately after isolation, the viability of peritoneal cells did not differ between the two mouse strains, as assessed by the trypan blue exclusion. As shown in Fig. 2A, APC from CBA mice induced proliferation of both CBA and CBA/N T cells in a dose-dependent manner, whereas there was no response in the presence of CBA/N APC. This total lack of activity prompted us to re-evaluate the viability of the APC at the time when cells were put into co-culture with the T cells after washing and suspension procedures. It was found that the majority of peritoneal exsudate cells extracted from XID mice died within 20–40 min after isolation, so very small numbers of live APC of CBA/N origin were present in co-cultures.
Neutrophils are the cells which have very short life span and are extremely sensitive to physical manipulations. Therefore, we evaluated the neutrophil content among the cells which infiltrated the site of BCG injection in CBA and CBA/N mice. As shown in Fig. 2B, at 1.5–2.0 h post BCG injection neutrophils (myeloperoxidase-positive PMN cells) represented ~70 per cent of the total cell population in CBA/N mice, whereas in CBA animals their influx was substantially postponed, and the proportion of neutrophils remained lower than 20 per cent at 2 h post BCG injection, i. e., when APC were isolated in previous experiments. Moreover, 1.5 h after BCG injection the total number of infiltrating neutrophils was about 1 log higher in CBA/N compared to CBA mice (Fig. 2C), despite that the total cell counts were ~4-fold higher in CBA animals due to the presence of intact B cell population (not shown). The difference was not specific for BCG injection, since inoculation of zymosan particles resulted in similar inter-strain differences at the 1.5–2.0-h time point (data not shown). On the other hand, the difference totally depended upon an external stimulus, since there was no difference between naïve CBA and CBA/N mice regarding the ratio of PMN cells in bone marrow (40.8 ± 2.1% and 40.5 ± 1.8%, respectively), or in peripheral blood (20.3 ± 1.6% and 18.3 ± 1.9%, respectively).
In the next series of experiments we investigated whether adoptive transfers of cells from CBA to CBA/N mice, which were effective in restoration of protective response following BCG vaccination, changed the pattern of neutrophil influx to the site of BCG injection. We transferred into peritoneal cavities of CBA/N mice either a non-separated population (containing ~18% F4/80+ monocytes, ~17% CD3+ T cells, ~60% CD19+ B cells), or plastic adherent-depleted population (<4% F4/80+, ~21% CD3+, ~75% CD19+) of cells from the peritoneal cavities of CBA donors. Fifteen hours later, recipients and naïve CBA control mice received intraperitoneally 108 CFU of BCG, and the dynamics of neutrophil influx was compared between groups. As shown in Fig. 2C, D, both types of transfers substantially decreased the speed of neutrophil migration in CBA/N recipients, making them indistinguishable from CBA control mice. Since adherence to plastic removed about 90 per cent of the F4/80-positive monocytes from the transferred population leaving the effect of transfer unaltered, and CBA/N mice are not defective regarding T cell numbers and function, our results suggested that B cells play an important regulatory role in this system by decreasing the mobility of neutrophils triggered by an external stimulus. To confirm this function of B cells, we compared cell migration after i. p. BCG injection between B6.CD19−/− mice which display a severe deficiency of B cells (19) and the w. t. B6 mice. It was found that at 1.5 h post BCG injection the neutrophil content in peritoneal cavities was 54 ± 6 % and <2% (P < 10−6), respectively. This provides direct genetic evidence that B cells negatively regulate neutrophil migration.
Xid neutrophils have an increased motility
To further investigate the role of B cells in regulation of neutrophil mobility, cells were isolated from peritoneal cavities of CBA/N and CBA mice 3.5 h after peptone injection (>90% Ly-6G+ PMN) and put into the upper chambers of transwell plates. The lower chambers were supplied either with culture medium alone (control), or with different populations of peritoneal exudate cells from CBA mice, with or without BCG, as indicated in Fig. 2E. After 1 h of migration, the total number of Ly-6G+ cells that migrated into lower chambers was assessed by flow cytometry. As shown in Fig. 2E, neither combination of cells, or cells + BCG, in the lower chamber affected migration of neutrophils compared to the medium alone control. However, the migratory capacity of CBA/N neutrophils appeared to be 3–5-fold higher than that of their CBA counterparts, irrespective of the cell combinations in co-cultures. Analogous experiments performed with peritoneal cavity PMN obtained at 3.5 h after BCG injection provided identical results (not shown). Thus, we confirmed the results obtained in vivo regarding substantial differences in the mobility of neutrophils between the w. t. and XID mice, but, unlike the adoptive transfer experiments (Fig. 2C, D), we were unable to inhibit neutrophil migration in vitro. The most likely explanation for this discrepancy is that the inhibitory effect of B cells on neutrophil migration is not direct, but mediated via microenvironmental elements lacking in the in vitro setting (see Discussion).
Differences in neutrophil migration are not due to differences in the level of chemokine production immediately after BCG injection
It is well established that an orchestrated production of two CXC chemokines, KC and MIP-2, as well as the PMN growth factor G-CSF, is required to mobilize neutrophils into inflammatory sites (24, 25). Therefore, we assessed the levels of these mediators in peritoneal cavities of CBA and CBA/N mice shortly after BCG injection. Mice injected with PBS served as controls. The levels of all three factors in saline-injected control mice were below sensitivity of corresponding ELISA kits. However, as early as 30 min post BCG injection the contents of KC and MIP-2 grew dramatically in peritoneal cavities of CBA and CBA/N mice (KC: 1413 ± 304 and 847 ± 352pg/ml, respectively, P > 0.05; MIP-2: 865 ± 204 and 705 ± 169pg/ml, respectively, P > 0.05). By the 1.5-h time point, similarly assessed KC contents rapidly increased and reached very high (>2ng/ml) levels in mice of both strains (not shown). Such instant responses can not be based upon de novo protein syntheses and clearly point at the release of pre-synthesized factors from host cells. The lack of intrastrain differences is not surprising, since CXCL1 chemokines, like KC and MIP-2, are produced predominantly by epithelial and endothelial cells, which do not express Btk and are functionally identical in the wild type and XID mice. Thus, a delay of neutrophil influx in the wild type CBA mice in our system is likely due to its active inhibition rather than down-regulation of CXCL1 chemokine production by B cells.
Regarding G-CSF, the factor produced by bone marrow-derived cells, we were able to demonstrate interstrain differences which followed the pattern of neutrophil influx. Whereas the level of G-CSF in peritoneal cavities of intact or saline-injected mice of either strain was substantially lower than sensitivity of the test, after BCG injection significantly more G-CSF was present in CBA/N compared to CBA mice (485 ± 28 pg/ml and 279 ± 29 pg/ml, respectively, P = 0.023, unpaired t-test) at 1.5-h time point, i. e. when substantially more neutrophils arrived in peritoneal cavities of the former mice.
The expression of genes encoding neutrophil chemoattractants by peritoneal exsudate cells
Besides paracrine regulation of neutrophil trafficking by chemokines provided by several stromal sources, this response clearly displays an autocrine component (24). Therefore, we assessed the level of expression of genes encoding these molecules in the cells of peritoneal cavity before and after BCG injection. The results of quantitative RT-PCR were normalized at two sequential steps of experiment: total RNA was extracted from equal numbers of peritoneal cells (4 × 106 in each group), and equal quantities of RNA were used for the cDNA synthesis. As shown in Fig. 3, in the absence of BCG (time 0), cells washed out of the peritoneal cavity of the w. t. CBA mice expressed higher levels of mRNA for G-SCF (Fig. 3A) and KC (Fig. 3B) than their CBA/N counterparts. The levels of expression of MIP-2 (Fig. 3C), and IL-6 and IL-17 (not shown) mRNAs were not different between mice of the two strains. After BCG injection, rapid changes in gene expression occurred which paralleled the cellular phenotypes characteristic of the w. t. and XID mice. The level of gene expression for all three key chemokines, G-CSF (Fig. 3A), KC (Fig. 3B) and MIP-2 (Fig. 3C), increased dramatically within 45 min post-inoculation of BCG in XID mice, whereas in w. t. animals it was elevated only for MIP-2, but dropped for G-CSF, or remained unchanged for KC. At 1.5-2.0 h post BCG injection, the level of expression of all genes in w. t. and XID mice equalized (Fig. 3A-C), exactly as did the neutrophil content in their peritoneal cavities at a later time point (~3.5 h, Fig. 2B).
Fig. 3. Differences in chemokine gene expression between XID and w. t. mice.
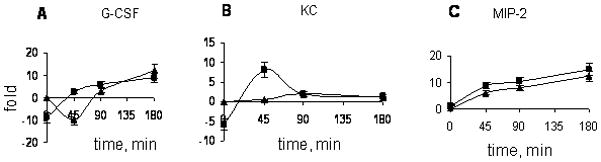
Early after BCG injection the expression of mRNA encoding key neutrophil-attracting factors G-CSF (A) and KC (B) increases in CBA/N but decreases in CBA mice (summary of three independent experiments, mixtures of RNA isolated from 3 mice in each, N=9). There was no difference in the dynamics of expression of mRNA for some other factors involved in neutrophil migration control, MIP-2 (C), IL-6 and IL-17 (not shown). Results are expressed as fold increase ± SEM compared to naïve CBA animals. Samples of RNA were analyzed by the quantitative real-time PCR assay, and gene expression levels in peritoneal cells were normalized to those of β-actin.
An impaired IFN-γ production by CD4 T in XID mice
Inter-strain differences in the dynamics of neutrophil arrival to the site of BCG injection resulted in a distinct distribution of cell-associated bacilli in CBA and CBA/N mice. Thus, after 2 h post intra-peritoneal injection, the vast majority of BCG was associated with mononuclear cells in the w. t. mice (Fig. 4A), but with PMN in the XID animals (Fig. 4B). This difference may influence the development of adaptive immunity against BCG, thus we examined how the capacity of CD4+ T cells to produce IFN-γ in response to mycobacterial antigens differed between CBA and CBA/N mice, given that this immune function is considered a critical component of anti-TB defense (17).
Fig. 4. BCG phagocytosis, T cell responses and restoration of BCG performance by the in vivo neutrophil depletion.
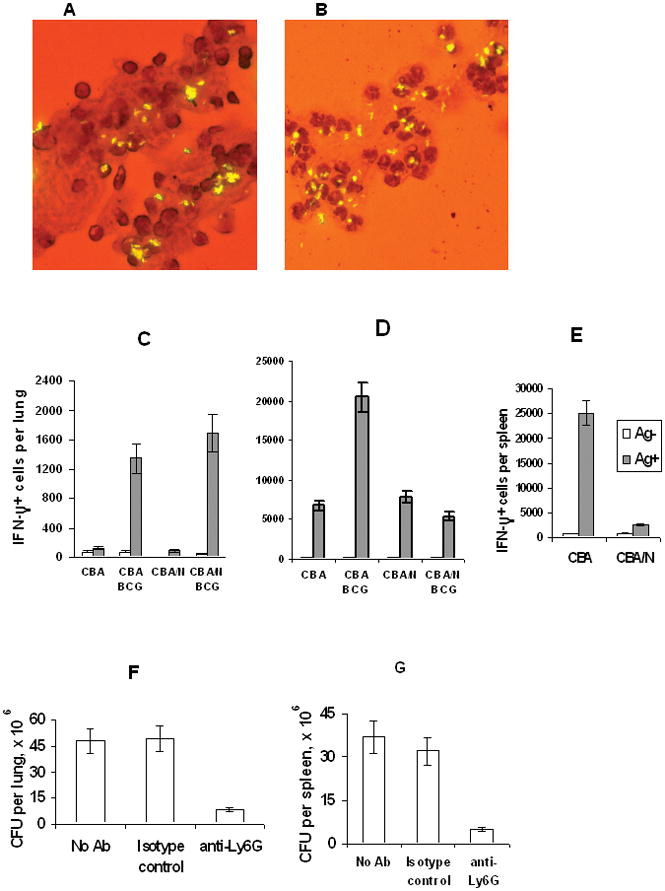
The vast majority of i. p. injected BCG is associated with mononuclear cells in CBA (A), but with PMN in CBA/N (B) mice, as demonstrated by auramine staining for mycobacteria with Gimsa counterstaining of cytospin preparations. Before challenge, BCG-vaccinated mice of the two strains did not differ by numbers of IFN-γ-producing CD4+ T cells in their lungs (C), but at week 3 post-infection the number of these cells in the lungs of the w. t. CBA mice was ~4 times (P < 0.01, Mann-Whitney U-test) higher (D). In the spleen, the numbers of IFN-γ-producing T cells differed ~10-fold (P < 0.001) after BCG vaccination in the absence of challenge (E). Magnetically sorted CD4+ lung T cells (C and D) or bulk spleen cells (E) from 3 individual mice in each group were analyzed using the ELISPOT assay; the results of one of two similar experiments are presented as mean ± SD. Neutrophil depletion prior to BCG vaccination resulted in significant (P < 0.01, Mann-Whitney U-test) ~8-fold reductions in CFU counts in lungs (F) and spleens (G) compared to control groups.
At week 5 after BCG vaccination, prior to TB challenge, mice of the two strains displayed low and equal numbers of IFN-γ-producing CD4+ T cells in their lungs (Fig. 4C). However, shortly after infection with virulent M. tuberculosis, a substantial 15-fold increase in numbers of IFN-γ positive cells was observed in CBA mice, whereas their CBA/N counterparts displayed only a 3-fold increase (Fig. 4D). Interestingly, while infection itself was a more powerful stimulus for IFN-γ production in the lungs than BCG vaccination (~5,000 and ~1,500 of IFN-γ-positive cells per lung, respectively), in the absence of BCG vaccination there was no difference between mice of the two strains. Thus, in the organ predominantly affected by infection (lung), the difference between the w. t. and XID mice became evident only after the secondary contact with mycobacteria. However, in the lymphoid organ responsible for the initiation of acquired immunity (spleen) the number of IFN-γ-positive cells grew 1-log higher in the w. t. compared to XID mice after BCG vaccination in the absence of challenge (Fig. 4E). Moreover, adoptive transfer of CBA fetal liver cells in CBA/N recipients resulted in a 3-fold increase in numbers of IFN-γ-positive cells in the lungs following vaccination and infection (data not shown). Thus, a biased restoration of B cell pool in XID mice was sufficient to normalize both the numbers of IFN-γ-producing cells and the protective effect of BCG vaccination (see above). Thus, a direct link between capacities to react normally to the vaccine by enhancing the numbers of IFN-γ producing T cells and to be protected against TB challenge was established in our model.
Neutrophil depletion in vivo restores BCG performance in XID mice
A major confounding variable between the sets of data presented above is the use of two different routes of BCG injection. Indeed, subcutaneous vaccination provides an adequate tool to demonstrate differences in BCG vaccination efficacy and to evaluate shifts in immune responses after adoptive transfers, but does not allow isolation of local cell populations before and after vaccination. On the other hand, intraperitoneal injection is perfect for the analyses at the cellular level, but poorly mimics a normal vaccination procedure. A straightforward approach to link the data on defective BCG performance in CBA/N-xid mice with an uncontrolled neutrophil influx is to remove neutrophils from XID animals at the very moment of vaccine injection, and to assess shifts in vaccination efficacy. To this end, we administered intraperitoneally anti-Ly-6G, RB6-8C5, and irrelevant, isotype-matched, antibodies to groups of CBA/N mice at day -1, vaccinated these as well as intact animals with 2 × 107 CFU of BCG s. c. at day 0, infected all animals i. v. with virulent M. tuberculosis 5 wk later, and compared CFU counts in lungs and spleens, as well as the content of IFN-γ-producing CD4+ T cells in the lungs, at week 3 post-challenge.
A single administration of RB6-8C5 antibodies appeared to be quite efficient for short-term neutrophil depletion, since 15 h after the administration the neutrophil counts in the peripheral blood of treated mice dropped ~99% compared to isotype controls, with total restoration of the neutrophil population 4 days afterwards (data no shown). As shown in Fig. 4F, G, neutrophil-depleted, BCG vaccinated XID mice showed highly significant (P = 0.012, Mann-Whitney U-test) ~8-fold reductions in CFU counts in lungs and spleens compared to control groups. Moreover, the number of IFN-γ-producing CD4+ T cells per lung was approximately 3-fold higher in neutrophil-depleted animals (data not shown). These results provides direct in vivo evidence that in mice defective in B cell-dependent neutrophil motility control, depletion of neutrophils substantially improves the efficacy of BCG vaccination.
DISCUSSION
The differences between XID and the wild type mice in the numbers of CD4+ T cells producing IFN-γ provide rational explanation of the difference in the vaccine performance but no mechanistic underpinning of the phenomenon, the search for which was the essence of this work.
While studying whether or not reduced T cell response in XID animals could be due to an impaired processing of BCG and/or presentation of mycobacterial antigens to T cells by phagocytes, we established that the two mouse strains differ profoundly regarding the speed of neutrophil influx at the site of BCG injection (Fig. 2). We demonstrated that adoptive transfers of peritoneal cells and, importantly, macrophage-depleted cells from CBA to CBA/N mice were sufficient to restore the control of neutrophil migration in XID mice (Fig. 2). Moreover, B-cell deficient CD19−/− mice showed alterations in neutrophil migration identical to those in XID mice, providing an independent genetic evidence of the inhibitory role of B-cells in neutrophil migration. In addition, results from the experiments with fetal liver cells and B-enriched peritoneal cells (Fig. 2) should be evaluated in the context of: (i) competitive disadvantage for other Btk-sufficient cell types transferred from CBA donors; (ii) unimpaired function of Btk-negative T cells in CBA/N mice (18). Taken together, these data strongly suggest that B cells play a previously underscored regulatory role in neutrophil locomotion.
Neutrophils migrate in response to mycobacteria, engulf the bacilli and undergo apoptosis (28–31). Inter-strain differences in the dynamics of their arrival to the site of BCG injection resulted in a distinct pattern of distribution of bacilli: shortly after injection, the vast majority of BCG was associated with mononuclear cells in the w. t., but with polymorphonuclear cells in the XID animals (Fig. 4). This difference may influence the development of immune responses against BCG. While there is evidence that neutrophils effectively transport intradermally injected BCG into lymph nodes (32, 33), and that the interactions of BCG-infected neutrophils with dendritic cells and macrophages result in T cell cross-priming and pro-inflammatory reactions (34), immunological consequences of these events are not necessarily beneficial for the host. Mycobacteria-induced neutrophil activation leads to the acceleration of their apoptosis through mechanisms dependent on TLR2 and p38 MAP kinase (29). It was shown recently that, after ingestion of BCG-containing apoptotic neutrophils, macrophages intensively formed lipid bodies and produced large quantities of PGE2 and TGF-β which actively suppress T cell activation (35). Similar conclusions on inhibition of immune responses as a consequence of interaction between mycobacteria-activated apoptotic neutrophils and dendritic cells were drawn from another recent study (36). Reasoning along these lines, we studied in more detail the features of neutrophil migration in our system.
In vitro experiments demonstrated that the migratory capacity of CBA/N neutrophils is 3–5-fold higher than that of their CBA counterparts, irrespective of the cell combinations in co-cultures. Thus, we confirmed the results obtained in vivo regarding substantial differences in the mobility of neutrophils between the w. t. and XID mice, but, unlike the adoptive transfer experiments, we were unable to inhibit neutrophil migration in vitro (Fig. 2). The most likely explanation for this discrepancy is that the inhibitory effect of B cells on neutrophil migration is not direct, but mediated via microenvironmental elements lacking in the in vitro setting. Neutrophil migration from the bloodstream depends upon a complex network of chemokines, cytokines, integrins and selectins, some autocrine and some provided by endothelial and epithelial cells (37, 38). Thus, the conditions of neutrophil migration through the membrane in vitro differ profoundly from those during in vivo extravasation. However, a higher level of spontaneous migration of CBA/N neutrophils in our experiments is in good agreement with recent data demonstrating significantly enhanced PMN locomotion through membranes in the presence of the selective Btk inhibitor LFM-A13 (39). Taken together, these findings demonstrate that an enhanced motility of PMN is another phenotypic expression of xid mutation. It is not known why functionally competent Btk is needed for normal PMN locomotion, however, there is evidence that this kinase is involved in actin polymerization and cytoskeleton dynamics in B cells (40).
An orchestrated production of two CXC chemokines, KC and MIP-2, as well as the PMN growth factor G-CSF, are required to mobilize neutrophils into inflammatory sites (25). In response to BCG injection, high amounts of CXC chemokines were instantly released into peritoneal cavities of both XID and wild type mice, indicating that an inhibitory action of B cells onto neutrophil trafficking has features of active suppression rather than inhibition of chemokine production. An instant chemokine release in response to mycobacterial stimulus clearly indicates that there are rich depots of these molecules (one, very likely, is the liver – A. Gleiberman, personal communication) able to release pre-existing factors in response to different stimuli, including those provided by intracellular pathogens. This aspect of an early response to mycobacteria deserves further investigation.
Animal models have failed to demonstrate a clear role of neutrophils in response against mycobacteria, due primarily to conflicting data (41–43). However, more recently we and others using genetic approaches demonstrated deleterious rather than beneficial effects of these early inflammatory cells in the course of chronic mycobacterial infections (30, 44, 45). Similar results were obtained in the mouse model of leishmaniasis (46, 47). It is difficult to judge which feature of a strong neutrophil response is more deleterious for the host: a temporary shelter granted to a parasite (the “Trojan horse” concept, see 30, 46), or T cell response inhibition (35, 36), or both. Nevertheless, it is likely that an exaggerated neutrophil response is a kind of “biological mistake” when it occurs in the context of chronic infections caused by sophisticated intracellular pathogens. Data presented herein add the phenomenon of BCG capture by neutrophils to this line of evidence.
A few recent studies demonstrated that B cells have a suppressive effect on the inflammatory response (48, 49), in particular, CD5+ B1 cells specifically down-regulated T cell-mediated inflammation (49). Our results indicate that inhibition of neutrophil migration may be yet another regulatory B cell function, adding evidence to negative association between B cells and neutrophils in the host response to infection (50) – the phenomenon awaiting detailed mechanistic dissection.
Supplementary Material
Acknowledgments
We thank T. Radaeva and V. Sosunov for the expert help with qrt-PCR evaluations.
Footnotes
This work has been supported by the NIH grant AI078864 (to ASA), the Russian Foundation for Basic Research grant 07-04-00447 (to TKK), and the International Science and Technology grant 3626.
COMPETING INTERESTS STATEMENT
The authors declare that they have no competing financial interests.
References
- 1.Tsukada S, Saffran DC, Rawlings DJ, Parolini O, Allen RC, Klisak I, Sparkes RS, Kubagawa H, Mohandas T, Quan S, et al. Deficient expression of a B cell cytoplasmic tyrosine kinase in human X-linked agammaglobulinemia. Cell. 1993;72:279–290. doi: 10.1016/0092-8674(93)90667-f. [DOI] [PubMed] [Google Scholar]
- 2.Vetrie D, Vorechovský I, Sideras P, Holland J, Davies A, Flinter F, Hammarström L, Kinnon C, Levinsky R, Bobrow M, et al. The gene involved in X-linked agammaglobulinemia is a member of the scr family of protein-tyrosine kinases. Nature. 1993;361:226–233. doi: 10.1038/361226a0. [DOI] [PubMed] [Google Scholar]
- 3.Rawlings DJ, Saffran DC, Tsukada S, Largaespada DA, Grimaldi JC, Cohen L, Mohr RN, Bazan JF, Howard M, Copeland NG, et al. Mutation of unique region of Bruton’s tyrosine kinase in immunodeficient XID mice. Science. 1993;261:358–361. doi: 10.1126/science.8332901. [DOI] [PubMed] [Google Scholar]
- 4.Thomas JD, Sideras P, Smith CI, Vorechovsky I, Chapman V, Paul WE. Colocalization of X-linked agammaglobulinemia and X-linked immunodeficiency genes. Science. 1993;261:355–358. doi: 10.1126/science.8332900. [DOI] [PubMed] [Google Scholar]
- 5.Hardy RR, Hayakawa K. CD5+ B cells, a fetal B cell lineage. Adv Immunol. 1994;55:297–339. doi: 10.1016/s0065-2776(08)60512-x. [DOI] [PubMed] [Google Scholar]
- 6.Khan WN, Alt FW, Gerstein RM, Malynn BA, Larsson I, Rathbun G, Davidson L, Müller S, Kantor AB, Herzenberg LA, et al. Defective B cell development and function in Btk-deficient mice. Immunity. 1995;3:283–299. doi: 10.1016/1074-7613(95)90114-0. [DOI] [PubMed] [Google Scholar]
- 7.Kerner JD, Appleby MW, Mohr RN, Chien S, Rawlings DJ, Maliszewski CR, Witte ON, Perlmutter RM. Imaired expansion of mouse B cell progenitors lacking Btk. Immunity. 1995;3:311–312. doi: 10.1016/1074-7613(95)90115-9. [DOI] [PubMed] [Google Scholar]
- 8.Takata M, Kurosaki T. A role for Bruton’s tyrosine kinase in B cell antigen receptor-mediated activation of phospholipase C-γ2. J Exp Med. 1996;184:31–40. doi: 10.1084/jem.184.1.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Petro JB, Rahman SM, Khan WN. Bruton’s tyrosine kinase is required for activation of IκB kinase and nuclear factor κB in response to B cell receptor engagement. J Exp Med. 2000;191:1745–1754. doi: 10.1084/jem.191.10.1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bajpai UD, Zhang K, Teutsch M, Sen R, Wortis HH. Bruton’s tyrosine kinase links the B cell receptor to nuclear factor κB activation. J Exp Med. 2000;191:1735–1744. doi: 10.1084/jem.191.10.1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hata D, Kawakami Y, Inagaki N, Lantz CS, Kitamura T, Khan WN, Maeda-Yamamoto M, Miura T, Han W, Hartman SE, Yao L, Nagai H, Goldfeld AE, Alt FW, Galli SJ, Witte ON, Kawakami T. Involvement of Bruton’s tyrosine kinase in FcεR1-dependent mast cell degranulation and cytokine production. J Exp Med. 1998;187:1235–1247. doi: 10.1084/jem.187.8.1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Quek LS, Bolen J, Watson SP. A role for Bruton’s tyrosine kinase (Btk) in platelet activation by collagen. Curr Biol. 1998;8:1137–1140. doi: 10.1016/s0960-9822(98)70471-3. [DOI] [PubMed] [Google Scholar]
- 13.Mukhopadhyay S, Mohanty M, Mangla M, George A, Bal V, Rath S, Ravindran B. Macrophage effector functions controlled by Bruton’s tyrosine kinase are more crucial than the cytokine balance of T cell responses for microfilarial clearance. J Immunol. 2002;168:2914–2921. doi: 10.4049/jimmunol.168.6.2914. [DOI] [PubMed] [Google Scholar]
- 14.Mangla M, Khare A, Vineeth V, Panday NN, Mukhopadhyay A, Ravindran B, Bal V, George A, Rath S. Pleiotropic consequences of Bruton’s tyrosine kinase deficiency in myeloid lineages lead to poor inflammatory responses. Blood. 2004;104:1191–1197. doi: 10.1182/blood-2004-01-0207. [DOI] [PubMed] [Google Scholar]
- 15.Wardemann H, Boehm T, Dear N, Carsetti R. B-1a B cells that link the innate and adaptive immune responses are lacking in the absence of the spleen. J Exp Med. 2002;195:771–780. doi: 10.1084/jem.20011140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nikonenko BV, Apt AS, Mezhlumova MB, Avdienko VG, Yeremeev VV, Moroz AM. Influence of the mouse Bcg, Tbc-1 and xid genes on resistance and immune responses to tuberculosis infection and efficacy of bacille Calmette-Guerin (BCG) vaccination. Clin Exp Immunol. 1996;104:37–43. doi: 10.1046/j.1365-2249.1996.d01-643.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Flynn JL, Chan J. Immunology of tuberculosis. Ann Rev Immunol. 2001;19:93– 129. doi: 10.1146/annurev.immunol.19.1.93. [DOI] [PubMed] [Google Scholar]
- 18.Yang WC, Collette Y, Nunes JA, Olive D. Tec kineses: a family with multiple roles in immunity. Immunity. 2000;12:378–382. doi: 10.1016/s1074-7613(00)80189-2. [DOI] [PubMed] [Google Scholar]
- 19.Rickert RC, Rajewsky K, Roes J. Impairment of T-cell-dependent B-cell responses and B-1 cell development in CD19-deficient mice. Nature. 1995;376:352–355. doi: 10.1038/376352a0. [DOI] [PubMed] [Google Scholar]
- 20.Lyadova IV, Eruslanov EB, Khaidukov SV, Yeremeev VV, Majorov KB, Pichugin AV, Nikonenko BV, Kondratieva TK, Apt AS. Comparative analysis of T lymphocytes recovered from the lungs of mice genetically susceptible, resistant and hyperresistant to Mycobacterium tuberculosis-triggered disease. J Immunol. 2000;165:5921–5931. doi: 10.4049/jimmunol.165.10.5921. [DOI] [PubMed] [Google Scholar]
- 21.Eruslanov EB, Majorov KB, Orlova MO, Mischenko VV, Kondratieva TK, Apt AS, Lyadova IV. Lung cell responses to M. tuberculosis in genetically susceptible and resistant mice following intratracheal challenge. Clin Exp Immunol. 2004;135:19–28. doi: 10.1111/j.1365-2249.2004.02328.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Radaeva TV, Kondratieva EV, Sosunov VV, Majorov KB, Apt AS. A human-like TB in genetically susceptible mice followed by the true dormancy in a Cornell-like model. Tuberculosis (Edinb) 2008;88:576–585. doi: 10.1016/j.tube.2008.05.003. [DOI] [PubMed] [Google Scholar]
- 23.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 24.Furze RC, Rankin SM. Neutrophil mobilization and clearance in the bone marrow. Immunology. 2008;125:281–288. doi: 10.1111/j.1365-2567.2008.02950.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wengner AM, Pitchford SC, Furze RC, Rankin SM. The coordinated action of G-CSF and ELR + CXC chemokines in neutrophil mobilization during acute inflammation. Blood. 2008;111:42–49. doi: 10.1182/blood-2007-07-099648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huang H, Li F, Cairns CM, Gordon JR, Xiang J. Neutrophils and B cells express XCR1 receptor and chemotactically respond to lymphotactin. Biochem Biophys Res Commun. 2001;281:378–382. doi: 10.1006/bbrc.2001.4363. [DOI] [PubMed] [Google Scholar]
- 27.Wang CR, Liu MF, Huang YH, Chen HC. Up-regulation of XCR1 expression in rheumatoid joints. Rheumatology (Oxford) 2004;43:569–573. doi: 10.1093/rheumatology/keh147. [DOI] [PubMed] [Google Scholar]
- 28.Aleman M, Garcia A, Saab MA, de la Barrera SS, Finiasz M, Abbate E, Sasiain MC. Mycobacterium tuberculosis-induced activation accelerates apoptosis in peripheral blood neutrophils from patients with active tuberculosis. Am J Respir Cell Mol Biol. 2002;27:583–592. doi: 10.1165/rcmb.2002-0038OC. [DOI] [PubMed] [Google Scholar]
- 29.Aleman M, Schierloh P, de la Barrera SS, Musella RM, Saab MA, Baldini M, Abbate E, Sasiain MC. Mycobacterium tuberculosis triggers apoptosis in peripheral neutrophils involving toll-like receptor 2 and p38 mitogen protein kinase in tuberculosis patients. Infect Immun. 2004;72:5150–5158. doi: 10.1128/IAI.72.9.5150-5158.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Eruslanov EB, I, Lyadova V, Kondratieva TK, Majorov KB, Scheglov IV, Orlova MO, Apt AS. Neutrophil responses to Mycobacterium tuberculosis infection in genetically susceptible and resistant mice. Infect Immun. 2005;73:1744–1753. doi: 10.1128/IAI.73.3.1744-1753.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Persson YA, Blomgran-Julinder R, Rahman S, Zheng L, Stendahl O. Mycobacterium tuberculosis-induced apoptotic neutrophils trigger a pro-inflammatory response in macrophages through release of heat scock protein 72, acting as synergy with the bacteria. Microbes Infect. 2008;10:233–240. doi: 10.1016/j.micinf.2007.11.007. [DOI] [PubMed] [Google Scholar]
- 32.Abadie V, Badell E, Douillard P, Ensergueix D, Leenen PJ, Tanguy M, Fiette L, Saeland S, Gicquel B, Winter N. Neutrophils rapidly migrate via lymphatics after Mycobacterium bovis BCG intradermal vaccination and shuttle live bacilli to the draining lymph nodes. Blood. 2005;106:1843–1850. doi: 10.1182/blood-2005-03-1281. [DOI] [PubMed] [Google Scholar]
- 33.Morel C, Badell E, Abadie V, Robledo M, Setterblad N, Gluckman JC, Gicquel B, Boudaly S, Winter N. Mycobacterium bovis BCG-infected neutrophils and dendritic cells cooperate to induce specific T cell responses in humans in mice. Eur J Immunol. 2008;38:437–447. doi: 10.1002/eji.200737905. [DOI] [PubMed] [Google Scholar]
- 34.Aleman M, de la Barrera SS, Schierloh PL, Alves L, Yokobori N, Baldini M, Abbate E, Sasiain MC. In tuberculous pleural effusions, activated neutrophils undergo apoptosis and acquire a dendritic cell-like phenotype. J Infect Dis. 2005;192:399–409. doi: 10.1086/431680. [DOI] [PubMed] [Google Scholar]
- 35.D’Avila H, Roque NR, Cardoso RM, Castro-Faria-Neto HC, Melo RCN, Bozza PT. Neutrophils recruited to the site of Mycobacterium bovis BCG infection undergo apoptosis and modulate lipid body biogenesis and prostaglandin E2 production by macrophages. Cell Microbiol. 2008;10:2589–604. doi: 10.1111/j.1462-5822.2008.01233.x. [DOI] [PubMed] [Google Scholar]
- 36.Aleman M, de la Barrera SS, Schierloh P, Yokobori N, Baldini M, Musella R, Abbate E, Sasiain M. Spontaneous or Mycobacterium tuberculosis-induced apoptotic neutrophils exert opposite effects on the dendritic cell-mediated immune response. Eur J Immunol. 2007;37:1524–1537. doi: 10.1002/eji.200636771. [DOI] [PubMed] [Google Scholar]
- 37.Galligan C, Yoshimura T. Phenotypic and functional changes of cytokine- activated neutrophils. Clin Immunol Allergy. 2003;83:24–44. doi: 10.1159/000071555. [DOI] [PubMed] [Google Scholar]
- 38.Luu NT, Rainger G, Buckley CD, Nash GB. CD31 regulates direction and rate of neutrophil migration over and under endothelial cells. J Vasc Res. 2003;40:467–479. doi: 10.1159/000074296. [DOI] [PubMed] [Google Scholar]
- 39.Zen K, Liu Y. Role of different protein kinases in fMLP-induced neutrophil transmigration. Immunobiol. 2008;213:13–23. doi: 10.1016/j.imbio.2007.07.001. [DOI] [PubMed] [Google Scholar]
- 40.Sharma S, Orlowski G, Song W. Btk regulates B cell receptor-mediated antigen processing and presentation by controlling actin cytoskeleton dynamics in B cells. J Immunol. 2009;182:329–339. doi: 10.4049/jimmunol.182.1.329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Appelberg R, Castro AG, Gomes S, Pedrosa J, Silva MT. Susceptibility of beige mice to Mycobacterium avium: role of neutrophils. Infect Immun. 1995;63:3381–3387. doi: 10.1128/iai.63.9.3381-3387.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pedrosa J, Saunders BM, Appelberg R, Orme IM, Silva MT, Cooper AM. Neutrophils play a protective nonphagocytic role in systemic Mycobacterium tuberculosis infection of mice. Infect Immun. 2000;68:577–583. doi: 10.1128/iai.68.2.577-583.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fulton SA, Reba SM, Martin TD, Boom WH. Neutrophil mediated mycobacteriocidal immunity in the lung during Mycobacterium bovis BCG infection in C57BL/6 mice. Infect Immun. 2002;70:5322–5327. doi: 10.1128/IAI.70.9.5322-5327.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Keller C, Hoffmann R, Lang R, Brandau S, Hermann C, Ehlers S. Genetically determined susceptibility to tuberculosis in mice causally involves accelerated and enhanced recruitment of granulocytes. Infect Immun. 2006;74:4295–4309. doi: 10.1128/IAI.00057-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Beisiegel M, Kursar M, Koch M, Loddenkemper C, Kuhlmann S, Zedler U, Stäber M, Hurwitz R, Kaufmann SHE. Combination of host susceptibility and virulence of Mycobacterium tuberculosis determines dual role of nitric oxide in the protection and control of inflammation. J Infect Dis. 2009;199:1222–1232. doi: 10.1086/597421. [DOI] [PubMed] [Google Scholar]
- 46.van Zandbergen G, Klinger M, Mueller A, Dannenberg S, Gebert A, Solbach W, Laskay T. Cutting edge: neutrophil granulocyte serves as a vector for Leishmania entry into macrophages. J Immunol. 2004;173:6521–6525. doi: 10.4049/jimmunol.173.11.6521. [DOI] [PubMed] [Google Scholar]
- 47.Peters NC, Egen JG, Secundino N, Debrabant A, Kimblin N, Kamhawi S, Lawyer P, Fay MP, Germain RN, Sacks D. In vivo imaging reveals an essential role for neutrophils in leishmaniasis transmitted by sand flies. Science. 2008;321:970–974. doi: 10.1126/science.1159194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Moritoki Y, Zhang W, Tsunayama K, Wakabayashi K, Yang GX, Bowlus C, Ridgway WM, Ueno Y, Ansari AA, Coppel PL, Mackay IB, Flavell RA, Gershwin ME, Lian ZX. B cells suppress the inflammatory response in a mouse model of primary biliary cirrhosis. Gastroenterology. 2009;136:1037–1047. doi: 10.1053/j.gastro.2008.11.035. [DOI] [PubMed] [Google Scholar]
- 49.Yanaba K, Bouaziz JD, Haas KM, Poe JC, Fujimoto M, Tedder TF. A regulatory B cell subset with a unique CD1dhiCD5+ phenotype controls T cell- dependent inflammatory responses. Immunity. 2008;28:639–650. doi: 10.1016/j.immuni.2008.03.017. [DOI] [PubMed] [Google Scholar]
- 50.Smelt SC, Cotterell EJ, Engwerda CR, Kaye PM. B cell-deficient mice are highly resistant to Leishmania donovani infection, but develop neutrophil-mediated tissue pathology. J Immunol. 2000;164:3681–3688. doi: 10.4049/jimmunol.164.7.3681. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.