

Abstract
Patients coinfected with human immunodeficiency virus (HIV) and hepatitis C virus (HCV) develop more rapid fibrosis than those infected with HCV only. In HIV/HCV-coinfected patients, fibrosis progression correlates with HIV RNA levels, suggesting a direct role of HIV in liver fibrogenesis. Chemokine (C-C motif) receptor 5 (CCR5) and cysteine-X-cysteine receptor 4 (CXCR4), the two major coreceptors required for HIV entry into cells, are expressed on activated hepatic stellate cells (HSCs), the principle fibrogenic cell type in the liver. We therefore examined whether HIV can infect HSCs, explored the potential mechanisms of viral entry, and assessed the impact of infection as reflected by the ability of HSCs to transfer virus to T lymphocytes and elicit a proinflammatory and profibrogenic response. We report that the laboratory-adapted viruses HIV-IIIB (CXCR4-tropic or X4) and HIV-BaL (CCR5-tropic or R5) and primary HIV isolates can infect both a human stellate cell line, LX-2, and primary human HSCs. HIV entry and gene expression in HSCs was confirmed using HIV–green fluorescent protein (GFP) expression viral constructs in the presence or absence of the reverse-transcriptase inhibitor azidothymidine. CD4 expression on a subset of primary HSCs was demonstrated using fluorescence-activated cell sorting and immunofluorescence staining. Blocking experiments in the presence of anti-CD4, anti-CXCR4, and anti-CCR5 revealed that HIV entry into HSCs is predominantly CD4/chemokine coreceptor-independent. HIV infection promoted HSC collagen I expression and secretion of the proinflammatory cytokine monocyte chemoattractant protein-1. Furthermore, infected LX-2 cells were capable of transferring GFP-expressing virus to T lymphocytes in a coculture system.
Conclusion
Taken together, our results suggest a potential role of HIV in liver fibrosis/inflammation mediated through effects on HSCs. The role of early highly active antiretroviral therapy initiation in patients with HIV/HCV coinfection warrants further investigation.
Over 40 million people are infected with human immunodeficiency virus (HIV) worldwide. Because of their shared routes of transmission, infection with hepatitis C virus (HCV) is common among patients with HIV infection, and approximately 30% of HIV-infected persons in the United States are also infected with HCV.1 The introduction of highly active antiretroviral therapy (HAART) in 1996 changed the natural history of HIV infection and resulted in a dramatic decline in most opportunistic infections. Given their increased survival, HCV-related liver disease has emerged as a major cause of morbidity and mortality among patients infected with HIV.
Although the effects of HCV on HIV disease progression are less clear, several studies have demonstrated that HIV infection adversely impacts every stage in the natural history of HCV infection. Infection with HIV enhances HCV transmission, decreases the rates of spontaneous HCV clearance leading to higher rates of chronic hepatitis C infection,2 and is associated with higher HCV RNA loads.3 Once chronic infection is established, HIV/HCV-coinfected patients have higher necro-inflammatory activity on liver biopsies, faster rates of fibrosis progression, and earlier development of end-stage liver disease.4,5
Despite the significant adverse clinical consequences of HIV/HCV coinfection, the underlying molecular mechanisms by which HIV infection impacts HCV disease progression have not been clearly defined. While the host T cell responses appear to be crucial in promoting HCV clearance in acute infection and controlling viral replication and recurrence,6 ultimately these responses fail and the majority of patients become chronically infected. Epidemiological studies support a correlation between lower CD4 cell counts, HCV persistence, and progression of liver disease,4 suggesting that the immunosuppression associated with HIV infection partially contributes to the pathogenesis of chronic liver disease. Whereas higher HCV RNA loads in coinfected patients do not correlate with progression of liver disease, HIV RNA levels correlate with fibrosis progression rates in a dose-dependent fashion.7 Moreover, effective suppression of HIV replication by HAART has been associated with better outcomes.7 Taken together, these findings suggest a direct role for HIV on liver disease progression in coinfected patients.
The hepatic stellate cell (HSC) is a central mediator in liver fibrosis. In its quiescent state, it is a vitamin A–rich cell that produces type IV collagen, the collagen characteristic of a normal basement membrane. With injury, such as in chronic hepatitis C, HSCs undergo a process of activation, rendering them susceptible to a variety of stimuli that yield a highly proliferative, contractile, and fibrogenic cell producing predominantly type I collagen, the collagen characteristic of the cirrhotic liver. Activated HSCs express both HIV chemokine coreceptors, chemokine (C-C motif) receptor 5 (CCR5)8 and cysteine-X-cysteine receptor 4 (CXCR4),9 and recent studies suggest effects of HIV envelope protein on HSC responses.10,11 To date there has been no evidence that activated HSCs are a cellular target for HIV infection to account for the accelerated fibrosis observed in coinfected patients.
We report that activated HSCs are infectable by HIV, support viral gene expression, and are capable of transmitting infectious virus to susceptible lymphocytes through cell–cell contact. Furthermore, HIV infection of HSCs induces collagen I expression and secretion of the proinflammatory cytokine, monocyte chemoattractant protein 1 (MCP-1). These findings support direct profibrogenic and proinflammatory effects of HIV on stellate cells.
Materials and Methods
Cell Lines
LX-2 cells, an immortalized human HSC line, were cultured as described.9 TZM cells, a HeLa cell line that stably expresses CD4, CXCR4, and CCR5, have been generated by introducing separate integrated copies of the luciferase and β-galactosidase genes under control of the HIV-1 promoter. MT4 cells are a human T cell line (obtained from the National Institutes of Health AIDS Research & Reference Reagent Program).
Isolation of Primary Human HSCs
Primary HSCs were isolated from wedge sections of normal liver as described.9 Passage #3–activated HSCs from at least three different donors were used for all experiments.
Isolation of Primary CD4 T Cells
Peripheral blood mononuclear cells from healthy donors were isolated by way of Ficoll-Hypaque gradient centrifugation and CD4+ T cells were isolated by negative selection using a CD4+ T cell isolation kit (Miltenyi Biotech, Germany). Cells were cultured in RPMI medium (10% fetal bovine serum) with interleukin-2 (50 U/mL), and stimulated with phytohemagglutinin (5 μg/mL) for 2 days at 37°C.
Viral Preparations
The X4-tropic (HIV-IIIB) and R5-tropic (HIV-BaL) laboratory-adapted viruses were obtained from Advanced Biotechnologies Incorporated (ABI, Columbia, MD). Primary X-tropic (92UG021) and R5-tropic (92TH007) isolates were obtained from the National Institutes of Health AIDS Research & Reference Reagent Program.
Green Fluorescent Protein–Expressing Constructs
HIV-1 Gag–interdomain green fluorescent protein (iGFP) is an NL4-3–based HIV-1 molecular clone that carries GFP inserted internally into Gag between the MA and CA domains, and HIV-NL-GI (GFP-IRES) is an NL4-3–based HIV-1 molecular clone that carries GFP in place of the nef start codon, with nef expression restored by inserting an internal ribosome entry site.12
Infectivity Assays
Enzyme-Linked Immunosorbent Assay for HIV-1 p24
Primary HSCs and LX-2 cells were plated (1–1.5 × 105 cells per well) and exposed to HIV-IIIB (X4-tropic) or HIV-BaL (R5-tropic) at a multiplicity of infection (moi) of 0.5 for 4 hours at 37°C. After viral incubation, cells were washed to remove unbound virus, and overlaid with Dulbecco's modified Eagle's medium (1% fetal bovine serum). Supernatants were collected up to 7 days after infection, starting at day 0 (30 minutes postwash). Infectivity was determined by quantification of p24 antigen (HIV-1 viral capsid protein) on culture supernatant by way of enzyme-linked immunosorbent assay (ELISA) according to the manufacturer's protocol (National Cancer Institute, Frederick, MD). The detection limit of the assay is 80 pg/mL of p24.
To determine whether HSC-derived supernatant contained infectious virus, supernatants from HIV-infected HSCs and primary CD4 lymphocytes as controls, were used to infect CD4 lymphocytes and TZM cells using p24 assay and Luciferase Reporter Assay, respectively. The volume of supernatant used to infect TZM cells varied, and was calculated to reach a final p24 concentration of 0.04 pg/cell. For the experiments using primary CD4 T cells, the final p24 concentration used was 0.001 pg/cell.
Luciferase Reporter Assay
Luciferase activity in TZM cells, where the luciferase gene is under control of the HIV-1 promoter, was assessed in both cell-free infection studies as well as coculture studies according to the manufacturer's protocol (Promega, Madison, WI).
GFP Experiments
For experiments using GFP constructs, primary HSCs and LX-2 cells were infected with either HIV-iGFP or HIV-NL-GI (0.4 pg/cell of p24) for 24 hours in serum-free media with or without 100 μM zidovudine (azidothymidine [AZT]; Sigma), washed to remove unbound virus, overlaid with Dulbecco's modified Eagle's medium (1% fetal bovine serum), and monitored daily for GFP expression under a fluorescence microscope (Nikon Eclipse TE 2000-u).
For receptor-blocking experiments, primary HSCs or T lymphocytes were incubated with anti-CD4 (BD Biosciences, clone Leu 3a), anti-CXCR4 (R&D systems, clone 12G5), anti-CCR5 (R&D systems, clone 45523) or respective isotype controls (10 mg/mL) for 1 hour prior to viral challenge.
Fluorescence-Activated Cell Sorting
In order to determine the percentage of HIV-GFP–infected cells, 72 hours after viral exposure with or without AZT, cells were trypsinized, fixed with paraformaldehyde, and analyzed by way of fluorescence-activated cell sorting (FACS) (Becton Dickinson).
Coculture Experiments A total of 5 × 104 TZM cells/well were plated onto 12-well culture plates 1 day prior to the infection. HSCs were either mock-infected or infected with HIV-IIIB at a moi of 0.5 for 4 hours at 37°C. Following infection, cells were washed to remove unbound virus, trypsinized, and plated onto TZM cells in a 1:1 ratio. Cells were cocultured for 72 hours, lysed, and analyzed for luciferase activity according to the manufacturer's protocol (Promega).
To examine whether HSCs are capable of transferring infectious virus to lymphocytes, HSCs were cocultured with MT4 cells. LX-2 cells were infected with the HIV NL-GI GFP viral construct at 0.4 pg/cell as described above. Twenty-four hours after washing of the viral inoculum, cells were trypsinized, replated, allowed to attach, and subsequently cocultured with MT4 cells (2 × 105 cells/well) with or without 100 μM AZT. GFP expression was monitored daily under a fluorescence microscope.
Immunoblots
For detection of collagen I expression, HSCs were exposed to HIV-IIIB at an moi of 0.5 for 4 hours at 37°C, washed, and cultured with serum-free media. Cell lysates were pooled from three wells, and 50 mg of protein was subjected to sodium dodecyl sulfate–polyacrylamide gel electrophoresis. Membrane probed for collagen-I (1:1,000; Rockland), and β-tubulin (Sigma) as a loading control. Blots were developed and analyzed by way of scanning densitometry as described.9 All values were normalized to housekeeping protein and expressed as fold changes relative to control.
ELISA for MCP-1
HSCs were exposed to HIV-IIIB as described above, and supernatants were collected and subjected to ELISA for MCP-1 according to the manufacturer's protocol (R&D Systems, Minneapolis, MN). The lower limit of detection of the assay is of 5 pg/mL.
Cell Viability Assay
HSC viability was assayed 24 hours after 4-hour exposure to AZT and after HIV exposure at all time points used for p24 by means of the CellTiter 96 Aqueous Non-Radioactive Cell Proliferation Assay (Promega).
Immunofluorescence Staining
For the detection of CD4, primary HSCs grown on glass coverslips were fixed with cold acetone, rehydrated, permeabilized, blocked, incubated with mouse monoclonal anti-human CD4 or isotype control (anti-IgG1) at 1:20 dilution (BD biosciences), washed, incubated with Alexa Fluor 594 goat anti-mouse at 1:1,000 dilution, and subsequently mounted with Vectashield mounting media for fluorescence (Vector Laboratories, UK). Images were acquired with Nikon Eclipse E600 fluorescence microscope (Nikon, Tokyo, Japan).
Statistical Analysis
All results are expressed as the mean ± standard deviation. Statistical significance was tested using unpaired Student t test, and P < 0.05 was considered significant.
Results
Human HSCs Support HIV Entry, Infection, and Gene Expression
To determine whether HSCs can be infected by HIV, both an immortalized HSC line, LX-2, as well as primary HSCs were challenged with X4-tropic (HIV-IIIB) and R5-tropic (HIV-BaL) laboratory-adapted strains of HIV-1 at an moi of 0.5. Primary HSCs were isolated from normal liver as described9 and activated passage #3 cells from three distinct patients were used for assays. Infectivity was determined through quantification of HIV-1 p24, an HIV capsid protein, in culture supernatants after infection. With progressive time in culture, there was a significant increase in the p24 concentration in culture supernatants of HSCs challenged with both strains of HIV-1, suggesting that HSCs are permissive to HIV infection in vitro (Fig. 1). HIV-IIIB infection of primary HSCs resulted in a four- to eight-fold increase in p24 levels compared with infection with HIV-BaL. Although this observed difference may result from a specific donor susceptibility to X4, HIV-IIIB is a laboratory X4 isolate recognized by its increased efficiency relative to BaL and other R5 strains in vitro. Therefore, primary HSCs were also challenged with HIV-IIIB at a lower moi of 0.1, with resulting p24 levels comparable to HIV-BaL at a moi of 0.5 (data not shown). To support the physiologic relevance of these findings, primary HSCs were also challenged with HIV primary isolates, both X4-tropic and R5-tropic (Fig. 1E,F). Although a higher p24 level was observed with the X4-tropic primary isolate compared with the R5-tropic virus, a much larger panel of isolates would be needed to determine whether primary HSCs are more permissive to infection by X4-tropic viruses.
Fig. 1.

HSCs are permissive to HIV infection in vitro. LX-2 cells and primary HSCs (passage #3) were infected with HIV-IIIB (X4-tropic), HIV-BaL (R5-tropic), or primary X4-tropic (92UG021) and R5-tropic (92TH007) isolates and culture supernatant collected sequentially for p24 ELISA. Significant p24 was noted up to day #7 after infection in both (A,B) LX-2 and primary (C–F) HSCs. Representative graphs of at least three independent experiments shown. Values are expressed as the mean p24 (pg/mL) ± standard deviation in treated versus mock-infected cells (P < 0.05).
To further confirm HIV entry and explore whether HSCs could support HIV gene expression, LX-2 and primary HSCs were exposed to HIV-X4 and HIV-R5 expressing GFP.12 GFP expression indicates different stages of the infectious viral cycle (Fig. 2A). HIV NL-GI is a replication-competent virus carrying an enhanced GFP gene in place of the nef start codon that reflects early gene expression. HIV Gag-iGFP is an infectious clone that carries the GFP gene in subdomains of Gag; expression of its Gag-GFP fusion during later stages of the HIV replication cycle are indicative of late viral gene expression.12 GFP expression was observed in primary HSCs 48–72 hours after exposure to both viral constructs, indicating viral entry and early and late gene expression. Furthermore, preincubation with AZT, a reverse-transcriptase inhibitor, blocked HIV-GFP gene expression in HSCs (Fig. 2B). R5-tropic GFP viruses did not show efficient viral entry into HSCs though the available clones are less infectious, making direct comparisons difficult (data not shown). To quantify the percentage of HSCs infected by HIV NL-GI GFP, LX-2 and primary HSCs were analyzed by way of flow cytometry 72 hours after viral exposure. In three independent experiments, 22%–28% of infected cells were positive for GFP expression (Fig. 2C). AZT almost completely abolished GFP expression in HSCs, with levels comparable to noninfected cells by both fluorescence microscopy and flow cytometry (0%–2% positive cells). Cell viability in the presence of AZT was confirmed by bright field microscopy (Fig. 2E) and cell viability assay (Fig. 2F).
Fig. 2.

Human HSCs support HIV entry and gene expression. (A) HIV-1 Gag-iGFP is an NL4-3-based HIV-1 molecular clone that carries GFP inserted internally into Gag between the MA and CA domains, and HIV-NL-GI (GFP-IRES) is an NL4-3-based HIV-1 molecular clone that carries GFP in place of the nef start codon, with nef expression restored by inserting an internal ribosome entry site as described.12 Primary HSCs were infected with HIV-1 GFP constructs with or without preincubation with the reverse-transcriptase inhibitor AZT and monitored daily for GFP expression using fluorescence microscopy. (B) GFP expression was noted 48–72 hours after exposure to both X4-tropic HIV NL-GI and HIV Gag-iGFP viral constructs indicating viral entry and both early and late gene expression. Furthermore, preincubation with AZT, a reverse-transcriptase inhibitor, blocked HIV-GFP gene expression in stellate cells (magnification ×40). (C) FACS confirmed a reduction in GFP+ cells in the presence of AZT. (D) Results from three independent experiments are shown graphically. **P < 0.008. (E,F) Lack of toxicity from AZT confirmed by Brightfield Microscopy (E) and MTS assay (F).
HIV Entry Into HSCs Is Predominantly CD4-Independent
Classically, HIV entry requires the interaction of the envelope glycoprotein, gp120, with CD4 and either CCR5 or CXCR4 as coreceptors, but CD4-independent pathways13 have been reported. CD4 binding facilitates viral attachment and mediates conformational changes in gp120 that allow a high-affinity interaction with the respective chemokine receptor. HSCs express both functional CXCR49 and CCR5.8 Therefore, we examined whether HSCs express CD4. FACS analysis revealed that 4% of passage #3 HSCs express CD4 (data not shown). Because CD4 receptors can be disrupted by trypsinization, immunofluorescent staining for CD4 on primary HSCs was performed (Fig. 3A). Although a subset of primary HSCs expressed CD4, the expression level was low. To determine whether HIV entry into HSCs is CD4- and/or CXCR4-dependent, primary HSCs were preincubated with anti-CD4, anti-CXCR4, or isotype control, challenged with HIV-IIIB (X4-tropic), and ELISA for p24 performed on culture supernatants (Fig. 3B). Neither blocking antibody inhibited HIV infection of HSCs. Efficacy of blocking antibodies was simultaneously confirmed in primary CD4 cells where HIV infection was inhibited by both antibodies (Fig. 3C). As additional confirmation, HSCs were incubated with anti-CD4 and anti-CXCR4 prior to challenge with HIV-GFP and FACS analysis (Fig. 3D). Similar to p24 results, GFP expression was not significantly blocked by anti-CXCR4 or anti-CD4 antibodies. Whereas baseline efficiency of viral entry by R5-tropic virus (HIV-BaL) into HSCs was low, infection was not blocked using CCR5 blocking antibodies (data not shown). Taken together, these results indicate that the major pathway of viral entry into HSCs is independent of CD4 and chemokine coreceptor binding. Although alternative HIV receptors such as C-type lectins have been shown to mediate HIV entry into dendritic cells (DCs)14 and astrocytes,15 this mechanism of entry will have to be further explored for HSCs.
Fig. 3.

HIV entry into HSCs is predominantly CD4-independent. (A) Expression of CD4 by primary HSCs was examined by way of immunostaining, revealing a subset of CD4+ cells. A representative image is shown (magnification ×45). (B) To determine whether HIV infection of HSCs is dependent on CD4 and the chemokine coreceptor CXCR4, primary HSCs were preincubated with anti-CD4 (clone Leu 3a, BD Biosciences), anti-CXCR4 (clone 12G5, R&D Systems), or respective isotype control antibodies (all at 10 mg/mL) 30 minutes prior to exposure to the X4-tropic HIV-IIIB, and supernatant was collected for p24 up to 7 days after infection. (C) Efficiency of blocking antibodies was confirmed through simultaneous infection of primary T cells. *P < 0.002. **P < 0.001. ***P < 0.0004. ****P < 0.0006. Representative data from at least three independent experiments performed in triplicate are shown. (D) Primary HSCs were also pretreated with anti-CXCR4 and anti-CD4 antibody prior to exposure to HIV-GI GFP, and FACS for GFP expression performed approximately 3 days after infection. GFP expression was not significantly blocked by anti-CXCR4 or anti-CD4. Although anti-CD4 slightly reduced GFP expression, it was not different than the effect of isotype control. Similarly, infection of HSCs by R5-GFP virus was not blocked by anti-CCR5 antibodies (data not shown). Representative data from three independent experiments are shown.
HSCs Can Transfer Infectious HIV to Lymphocytes Through Direct Cell Contact
To determine whether HSCs can produce infectious virus, culture supernatants from HSCs previously infected with HIV-IIIB were incubated with primary CD4 lymphocytes and TZM cells. There was no detectable p24 in culture supernatant from CD4 cells (Fig. 4A) or luciferase activity in TZM cells (Fig. 4B) exposed to culture supernatants from HIV-infected HSCs, respectively. In contrast, both purified HIV as well as culture supernatants derived from primary CD4 lymphocytes previously infected with HIV led to infection of both CD4 cells as indicated by p24 ELISA and luciferase activity for TZM cells (Fig. 4A). These findings indicate that most of the viral particles released into culture supernatants from HSCs are noninfectious.
Fig. 4.

HIV released in culture supernatant from HIV-infected HSCs is noninfectious to TZM cells and CD4 T lymphocytes. Primary HSCs were infected with HIV-IIIB (moi of 0.5) for 4 hours at 37°C, washed extensively, and overlaid with fresh media. (A,B) Culture supernatants collected up to day 7 were incubated with CD4 cells for p24 assay (A) and TZM cells for luciferase activity assay (B). Both purified HIV-IIIB and culture supernatant from infected CD4 cells served as positive controls where increased p24 over time reflects active ongoing replication in CD4 cells (A) (*P < 0.0003, **P < 0.01) and increased luciferase activity in TZM cells reflects activation of the HIV-1 promoter (B) (***P < 0.004, #P < 0.02). No significant p24 or luciferase activity was observed using supernatant derived from infected HSCs. Mock-infected cells served as negative controls.
Transmission of HIV through points of cell contact has been demonstrated between DCs and T cells16 as well as between T cells.17 Because HSCs share features with DCs,18,19 we examined whether HSCs could transfer infectious virus to lymphocytes in a coculture system. HSCs infected with HIV-IIIB were washed, trypsinized to remove any cell surface–associated virus, and subsequently cocultured with TZM cells. As shown in Fig. 5A, TZM cells cocultured with HIV-infected HSCs showed a significant increase in luciferase activity versus coculture with mock-infected HSCs. To further confirm this finding, HSCs were infected with the HIV-GFP–expressing constructs, washed, trypsinized, and subsequently cultured with MT4 lymphocytes (Fig. 5B). Over time, MT4 cells became infected with HIV, which was blocked by AZT. These results indicate that HSCs are able to capture HIV and transfer viable virus to surrounding lymphocytes. Given that most of the viral particles released in culture supernatants were defective and unable to infect cells, this phenomena appears to require cell–cell contact and potentially occurs through a virological synapse, as demonstrated for other cells,20 and thus requires further investigation.
Fig. 5.

HSCs are able to transfer infectious viral particles to TZM cells and MT4 lymphocytes in coculture systems. (A) For coculture, 5 × 104 TZM cells/well were plated onto 12-well culture plates 1 day prior to infection. HSCs were either mock-infected or infected with HIV-IIIB at an moi of 0.5 for 4 hours at 37°C. Following infection, cells were washed to remove unbound virus, trypsinized, and plated onto TZM cells in a 1:1 ratio. Cells were cocultured for 72 hours, lysed, and analyzed for luciferase activity. An average eight-fold increase in luciferase activity was observed in TZM cells cocultured with HIV-infected HSCs. ****P < 0.0007. Representative data from at least three independent experiments performed in triplicate are shown. (B) LX-2 cells were mock-infected or infected with HIV NL-GI GFP for 24 hours. After viral incubation, the cells were extensively washed and cultured for another 24 hours. Subsequently, the cells were trypsinized and plated in a separate culture plate. After cell attachment, MT4 cells were added to the culture plate in the presence or absence of AZT. Thirty-six to 48 hours after coculture, MT4 cells cocultured with infected LX-2 cells became progressively positive for GFP expression, which was blocked by AZT.
HIV Promotes Stellate Cell Collagen I Expression and Secretion of MCP-1
HIV/HCV coinfection is associated with rapid fibrosis progression and increased necro-inflammatory activity on biopsy compared with HCV monoinfected patients.4 Therefore, we hypothesized that direct effects of HIV on HSCs may contribute to these clinical observations. Because HSC expression of collagen I is critical to fibrosis, we examined whether HIV infection of HSCs results in increased expression of collagen I. As shown in Fig. 6A, a greater than two-fold increase in collagen I expression by HSCs was observed 48 hours after HIV infection. Chronic inflammation is important for activation of HSCs and fibrogenesis. Because MCP-1 is a potent chemoattractant for monocytes and lymphocytes, is up-regulated during chronic hepatitis, and correlates with the number of cells infiltrating the portal tract,21 we examined whether HIV stimulates the HSC secretion of MCP-1. An average 80-fold increase in MCP-1 secretion was observed 48 hours after HIV infection (Fig. 6B). Therefore, HIV may have both profibrogenic and proinflammatory effects on HSCs.
Fig. 6.
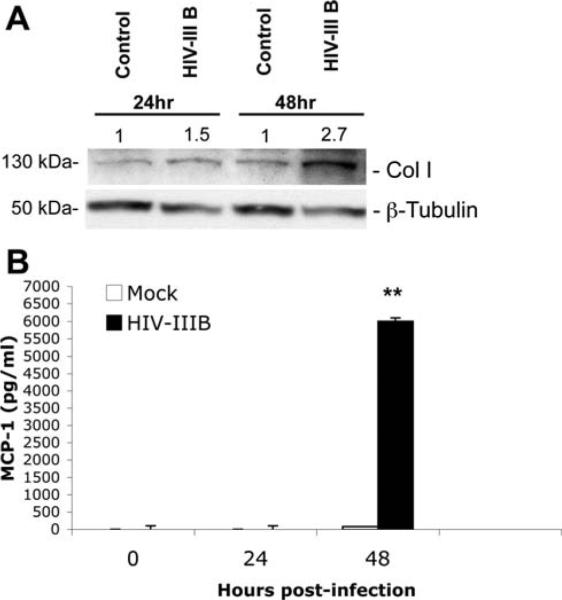
HIV promotes HSC collagen I expression and secretion of MCP-1. Primary HSCs (passage #3) were serum-starved for 24 hours, infected with HIV-IIIB for 4 hours at 37°C, washed extensively, and overlaid with fresh media. (A) Western blotting for collagen I was performed on cell lysates prepared 24 and 48 hours after infection. (A) An average 1.5-fold and 2.7-fold increase in collagen I expression was noted at 24 and 48 hours, respectively. β-Tubulin was used as a protein loading control, and fold increase over control is expressed in arbitrary units. (B) Culture supernatant was collected for MCP-1 by way of ELISA. An average 80-fold increase in MCP-1 secretion was observed 48 hours after HSCs were exposed to HIV-IIIB (**P < 0.0004). Representative data from two independent experiments are shown.
Discussion
HIV/HCV-coinfected patients have accelerated fibrosis progression rates compared with HCV-monoinfected patients with the development of cirrhosis 12–16 years earlier.4,22 Moreover, HIV/HCV-coinfected patients with ongoing HIV viremia have faster rates of HCV-related fibrosis progression,7 suggesting an accelerating effect of HIV on fibrosis progression. When HIV is successfully suppressed with HAART, fibrosis progression rates and necro-inflammatory activity are reduced, closely resembling HCV-monoinfected patients. These observations reinforce the role of HIV in promoting inflammation and fibrosis in HIV/HCV-coinfected livers. The molecular mechanisms by which HIV accelerates fibrosis are not clearly understood, and direct HIV infection of activated HSCs, the main fibrogenic cell in the liver, has not been reported.
In the present study, we provide some insight into potential molecular mechanisms by which HIV, through its effects on activated HSCs, can accentuate liver injury in chronic liver diseases. Our results show that both laboratory and clinical isolates of HIV-1 can infect activated HSCs. The GFP viral constructs indicate that these cells support viral gene expression, which was blocked by AZT. Despite the low level expression of CD4 on HSCs, and the previously reported expression of the HIV coreceptors, CXCR4 and CCR5, our results suggest that HIV entry occurs by way of mechanisms independent of receptor engagement. Two interesting findings from our study may enhance our understanding of the role of HIV in chronic liver disease: (1) HSCs are able to retain viral particles that can subsequently be transferred to and infect susceptible cells; and (2) exposure to HIV results in increased collagen I expression as well as secretion of the potent proinflammatory chemokine MCP-1, thereby providing a direct link between HIV and fibrosis through effects on HSCs. These findings add a new perspective to our growing understanding of the mechanisms by which HIV promotes inflammation and accelerates fibrosis and must be placed in the context of other important observations.
In vivo studies in seropositive patients support the presence of HIV proviral DNA by polymerase chain reaction in whole liver tissue, as well as HIV RNA in liver cells (particularly Kupffer cells, but also isolated hepatocytes) by way of in situ hybridization. In addition, HIV proteins have been detected in parenchymal and nonparenchymal liver cells by immunohistochemistry.23–25 The specific cell type expressing HIV proteins, however, remains unclear given the lack of coimmunostaining. In vitro, several liver cell types are infectable by HIV, including hepatoma cell lines, Kupffer cells, and sinuosoidal endothelial cells (reviewed in Blackard and Sherman26).
Previously proposed mechanisms by which HIV may promote inflammation and fibrosis include: (1) hepatocyte apoptosis in response to HCV and HIV envelope proteins27,28; (2) induction of hepatocyte-derived transforming growth factor-β1 by HIV and gp12029; and (3) reduced interleukin-10 secretion by intrahepatic CD4+ cells derived from HIV/HCV patients in response to HCV proteins.30 Since interleukin-10 may be both anti-inflammatory and antifibrotic by directly inhibiting HSC apoptosis, reduced interleukin-10 secretion may contribute to accelerated fibrosis in coinfected patients.29 Increased transforming growth factor-β1 may promote fibrosis by way of (1) direct profibrogenic effects on HSCs and; (2) reduction in the IFN-γ response of CD8+ cells to viral infection which could promote HCV persistence.31 Our group as well as others have reported profibrogenic effects of HIV-1 gp120 on HSCs.10,11 Therefore, it is likely that HIV and its proteins promote liver injury, inflammation, and fibrosis by effects on both parenchymal and nonparenchymal cells of the liver.
Upon activation, HSCs exhibit features of professional antigen-presenting cells where they acquire the ability to endocytose external particles and to stimulate T lymphocyte proliferation.18,19 Our observation that infected HSCs are able to transmit infectious virus to susceptible CD4+ lymphocytes by cell–cell contact supports the notion that these cells may be able to function like DCs, which can efficiently transmit HIV to lymphocytes.32 The association of HSCs with lymphocytes in hepatitis, their positioning below the fenestrated sinusoidal endothelium, and recent demonstration of their direct interaction with lymphocytes in vivo by way of confocal microscopy makes this interaction even more likely.33,34 Moreover, studies have suggested that cell-associated HIV-1 may be internalized into CD4+ T cells, resulting in a much more efficient infection than cell-free virus, further highlighting the importance of our findings. Future studies will address whether HIV-infected HSCs may elicit proliferative responses in specific subsets of lymphocytes, in addition to promoting lymphocyte infiltration by secretion of chemokines such as MCP-1. Lastly, HSCs may provide an important intrahepatic source of HIV proteins (such as the envelope protein gp120) that have been shown to elicit biologic responses in neighboring cells such as hepatocytes.27,29 Moreover, HIV gp120, has been shown to modulate HSC responses in vitro in a receptor-dependent manner.10,11 Whether levels used for in vitro studies reflect physiologically relevant tissue levels and whether viral protein is derived from autocrine sources (HSCs) or paracrine sources (Kupffer cells, DCs) is not clear.
Our results suggest that viral entry into HSCs occurs predominantly independent of CD4, CXCR4, and CCR5. Despite the lack of a clear mechanism of entry, HSCs support HIV infection and gene expression. Potential mechanisms of CD4-independent pathways for entry include the use of alternative receptors such as C-type lectins, as has been described for other cell types such as DCs, as well as receptor-independent endocytosis13; each will be explored in future studies.
A compelling question arises from our findings: “Why don't patients monoinfected with HIV develop inflammation and fibrosis if HIV can infect HSCs and stimulate collagen I and MCP-1 expression?” In this study, we used either a moderately activated cell line (LX-2) or passage #3–activated HSCs. The phenotype of these cells changes with activation, including increased expression of cell-surface receptors and changes in the cytoskeleton. The ability for HSCs to act as APCs has only been demonstrated in activated HSCs. Preliminary work in our laboratory suggests that quiescent stellate cells are not infectable by HIV. Therefore, we hypothesize that initial injury from HCV, or from other etiologies, serves as an initiating signal to activate HSCs, which creates a permissive environment for the effects of HIV on HSCs. Typically chronic HCV infection precedes HIV infection in coinfected individuals, further supporting this hypothesis. A recent prospective analysis of paired liver biopsies from HIV/HCV-coinfected patients found increased fibrosis progression rates associated with increased necroinflammatory activity, uncontrolled HIV replication, and untreated HCV.5 The authors suggest earlier antiretroviral therapy initiation in coinfected patients in whom HCV has not been eradicated. Our results provide a potential physiological rationale for this approach.
In conclusion, our study provides a link between HIV and hepatic fibrosis through direct effects on HSCs and broadens our understanding of the mechanisms underlying liver disease in patients coinfected with HIV/HCV. Furthermore, these findings provide a rationale to examine whether HAART should be initiated in coinfected patients earlier than current guidelines recommend.
Abbreviations
- AZT
azidothymidine
- CCR5
chemokine (C-C motif) receptor 5
- CXCR4
cysteine-X-cysteine receptor 4
- DC
dendritic cell
- ELISA
enzyme-linked immunosorbent assay
- FACS
fluorescence-activated cell sorting
- GFP
green fluorescent protein
- HAART
highly active antiretroviral therapy
- HCV
hepatitis C virus
- HIV
human immunodeficiency virus
- HSC
human hepatic stellate cell
- iGFP
interdomain green fluorescent protein
- MCP-1
monocyte chemoattractant protein 1
- moi
multiplicity of infection
Footnotes
Potential conflict of interest: Nothing to report.
References
- 1.Sherman KE, Rouster SD, Chung RT, Rajicic N. Hepatitis C virus prevalence among patients infected with human immunodeficiency virus: a cross-sectional analysis of the US adult AIDS Clinical Trials Group. Clin Infect Dis. 2002;34:831–837. doi: 10.1086/339042. [DOI] [PubMed] [Google Scholar]
- 2.Thomas D, Astemborski J, Rai R, Anania F, Schaeffer M, Galai N, et al. The natural history of hepatitic C infection: host, viral, and environmental factors. JAMA. 2000;284:450–456. doi: 10.1001/jama.284.4.450. [DOI] [PubMed] [Google Scholar]
- 3.Bonacini M, Govindarajan S, Blatt L, Schmid P, Conrad A, Lindsay K. Patients co-infected with human immunodeficiency virus and hepatitis C virus demonstrate higher levels of hepatic HCV RNA. J Viral Hepatitis. 1999;6:203–208. doi: 10.1046/j.1365-2893.1999.00153.x. [DOI] [PubMed] [Google Scholar]
- 4.Benhamou Y, Bochet M, Di Martino V, Charlotte F, Azria F, Coutellier A, et al. Liver fibrosis progression in human immunodeficiency virus and hepatitis C virus coinfected patients. The Multivirc Group. HEPATOLOGY. 1999;30:1054–1058. doi: 10.1002/hep.510300409. [DOI] [PubMed] [Google Scholar]
- 5.Macias J, Berenguer J, Japon M, Giron J, Rivero A, Lopez-Cortes L, et al. Fast fibrosis progression between repeated liver biopsies in patients coinfected with human immunodeficiency virus/hepatitis C virus. HEPATOLOGY. 2009;50:1056–1063. doi: 10.1002/hep.23136. [DOI] [PubMed] [Google Scholar]
- 6.Kim AY, Schulze zur Wiesch J, Kuntzen T, Timm J, Kaufman DE, Duncan JE, et al. Impaired hepatitis C virus-specific T cell responses and recurrent hepatitis C virus in HIV coinfection. PloS Med. 2006;3:e492. doi: 10.1371/journal.pmed.0030492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brau N, Salvatore M, Rios-Bedoya C, Fernandez-Carbia A, Paronetto F, Rodriguez-Orengo J, et al. Slower fibrosis progression in HIV/HCV-coinfected patients with successful HIV suppression using antiretroviral therapy. J Hepatol. 2005;44:47–55. doi: 10.1016/j.jhep.2005.07.006. [DOI] [PubMed] [Google Scholar]
- 8.Schwabe R, Bataller R, Brenner D. Human hepatic stellate cells express CCR5 and RANTES to induce proliferation and migration. Am J Physiol Gastrointest Liver Physiol. 2003;285:G949–G958. doi: 10.1152/ajpgi.00215.2003. [DOI] [PubMed] [Google Scholar]
- 9.Hong F, Tuyama A, Lee T, Loke J, Agarwal R, Cheng X, et al. Hepatic stellate cells express functional CXCR4: role in stromal cell-derived factor 1a mediated stellate cell activation. HEPATOLOGY. 2009;49:2055–2067. doi: 10.1002/hep.22890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bruno R, Galastri S, Sacchi P, Cima S, Caligiuri A, DeFranco R, et al. The HIV envelope protein GP120 modulates the biology of human hepatic stellate cells: a link between HIV infection and liver fibrogenesis. Gut. 2010;59:513–520. doi: 10.1136/gut.2008.163287. [DOI] [PubMed] [Google Scholar]
- 11.Hong F, Bansal MB. HIV gp120(X4) promotes hepatic stellate cell activation, fibrogenesis, and proliferation: a potential mechanism for rapid fibrosis progression in HIV/HCV coinfected patients. HEPATOLOGY. 2009:51. [Google Scholar]
- 12.Hubner W, Chen P, Del Portillo A, Liu Y, Gordon R, Chen B. Sequence of human immunodeficiency virus type I (HIV-1) gag localization and oligomerization monitored with live confocal Imaging of replication-competent flourescently tagged HIV-1. J Virol. 2007;81:12597–12607. doi: 10.1128/JVI.01088-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Clapham P, McKnight A, Talbot S, Wilkinson D. HIV entry into cells by CD4-independent mechanisms. Perspectives in Drug Discovery and Design. 1996;5:83–92. [Google Scholar]
- 14.Turville S, Cameron P, Handley A, Lin G, Pohlmann S, Doms R, et al. Diversity of receptors binding HIV on dendritic cell subsets. Nat Immunol. 2002;3:975–983. doi: 10.1038/ni841. [DOI] [PubMed] [Google Scholar]
- 15.Liu Y, Liu H, Kim BO, Gattone VH, Li J, Nath A, et al. CD4-independent infection of astrocytes by human immunodeficiency virus type 1: requirement for the human mannose receptor. J Virol. 2004;78:4120–4133. doi: 10.1128/JVI.78.8.4120-4133.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cameron P, Freudenthal P, Barker J, Gezelter S, Inaba K, Steinman R. Dendritic cells exposed to human immunodeficiency virus type-1 transmit a vigorous cytopathic infection to CD4+ T cells. Science. 1992;257:383–387. doi: 10.1126/science.1352913. [DOI] [PubMed] [Google Scholar]
- 17.Hubner W, McNerney G, Chen P, Dale B, Gordon R, Chuang F, et al. Quantitative 3D video microscopy of HIV transfer across T cell virological synapses. Science. 2009;323:1743–1747. doi: 10.1126/science.1167525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Winau F, Hegasy G, Weiskirchen R, Weber S, Cassan C, Sieling P, et al. Ito cells are liver-resident antigen-presenting cells for activating T-cell responses. Immunity. 2007;26:117–129. doi: 10.1016/j.immuni.2006.11.011. [DOI] [PubMed] [Google Scholar]
- 19.Vinas O, Bataller R, Sancho-Bru P, Gines P, Berenguer C, Enrich C, et al. Human hepatic stellate cells show features of antigen-presenting cells and stimulate lymphocyte proliferation. HEPATOLOGY. 2003;38:919–929. doi: 10.1053/jhep.2003.50392. [DOI] [PubMed] [Google Scholar]
- 20.Nguyen D, HIldreth J. Involvement of macrophage mannose receptor in the binding and transmission of HIV by macrophages. Eur J Immunol. 2003;33:483–493. doi: 10.1002/immu.200310024. [DOI] [PubMed] [Google Scholar]
- 21.Marra F, DeFranco R, Grappone C, Milani S, Pastacaldi S, Pinzani M, et al. Increased expression of monocyte chemotactic protein-1 during active fibrogenesis: correlation with monocyte infiltration. Am J Pathol. 1998;152:423–430. [PMC free article] [PubMed] [Google Scholar]
- 22.Soto B, Sánchez-Quijano A, Rodrigo L, del Olmo JL, García-Bengoechea M, Hernández-Quero J, et al. Human immunodeficiency virus infection modifies the natural history of chronic parentally-acquired hepatitis C with an unusually rapid progression to cirrhosis. J Hepatol. 1997;26:1–5. doi: 10.1016/s0168-8278(97)80001-3. [DOI] [PubMed] [Google Scholar]
- 23.Housset C, Lamas E, Brechot C. Detection of HIV-1 RNA and p24 antigen in HIV-1 infected human liver. Res Virol. 1990;141:153–159. doi: 10.1016/0923-2516(90)90017-d. [DOI] [PubMed] [Google Scholar]
- 24.Housset C, Boucher O, Girard P, Leibowitch J, Saimot A, Brechot C, et al. Immunohistochemical evidence for human immunodeficiency virus-1 infection of liver Kupffer cells. Hum Pathol. 1990;21:404–408. doi: 10.1016/0046-8177(90)90202-g. [DOI] [PubMed] [Google Scholar]
- 25.Cao YZ, Dieterich D, Thomas PA, Huang YX, Mirabile M, Ho DD. Identification and quantitation of HIV-1 in the liver of patients with AIDS. AIDS. 1992;6:65–70. doi: 10.1097/00002030-199201000-00008. [DOI] [PubMed] [Google Scholar]
- 26.Blackard JT, Sherman KE. HCV/ HIV co-infection: time to reevaluate the role of HIV in the liver? J Viral Hepat. 2008;15:323–330. doi: 10.1111/j.1365-2893.2008.00970.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Munshi N, Balasbramanian A, Koziel M, Ganju R, Groopman J. Hepatitis C and Human immunodeficiency virus envelope proteins cooperatively induce hepatocytic apoptosis via an innocent bystander mechanism. J Infect Dis. 2003;188:1192–1204. doi: 10.1086/378643. [DOI] [PubMed] [Google Scholar]
- 28.Vlahakis S, Villasis-Keever A, Gomez T, Bren GD, Paya CV. Human immunodeficiency virus-induced apoptosis of human hepatocytes via CXCR4. J Infect Dis. 2003;188:1455–1460. doi: 10.1086/379738. [DOI] [PubMed] [Google Scholar]
- 29.Lin W, Weinberg E, Tai A, Peng L, Brockman M, Kim K, et al. HIV increases HCV replication in a TGF-b1-dependent manner. Gastroenterology. 2008;134:803–811. doi: 10.1053/j.gastro.2008.01.005. [DOI] [PubMed] [Google Scholar]
- 30.Graham C, Curry M, He Q, Afdhal N, Nunes D, Fleming C, et al. Comparison of HCV-specific intrahepatic CD4+ T cells in HIV/HCV versus HCV. HEPATOLOGY. 2004;40:125–132. doi: 10.1002/hep.20258. [DOI] [PubMed] [Google Scholar]
- 31.Garba M, Pilcher C, Bingham A, Eron J, Frelinger J. HIV antigens can induce TGFb1-producing immunoregulatory CD8+ T cells. J Immunol. 2002;168:2247–2254. doi: 10.4049/jimmunol.168.5.2247. [DOI] [PubMed] [Google Scholar]
- 32.Piguet V, Steinman R. The interaction of HIV with dendritic cells: outcomes and pathways. Trends Immunol. 2007;28:503–510. doi: 10.1016/j.it.2007.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Holt AP, Haughton EL, Lalor PF, Filer A, Buckley CD, Adams DH. Liver myofibroblasts regulate infiltration and positioning of lymphcytes in human liver. Gastroenterology. 2009;136:705–714. doi: 10.1053/j.gastro.2008.10.020. [DOI] [PubMed] [Google Scholar]
- 34.Muhanna N, Horani A, Doron S, Safadi R. Lymphocyte-hepatic stel-late cell proximity suggests direct interaction. Clin Exp Immunol. 2007;148:338–347. doi: 10.1111/j.1365-2249.2007.03353.x. [DOI] [PMC free article] [PubMed] [Google Scholar]