

Abstract
A detailed understanding of chemical and biological function and the mechanisms underlying the activities ultimately requires atomic-resolution structural data. Diffraction-based techniques such as single-crystal X-ray crystallography, electron microscopy and neutron diffraction are well established and have paved the road to the stunning successes of modern-day structural biology. The major advances achieved in the last 20 years in all aspects of structural research, including sample preparation, crystallization, the construction of synchrotron and spallation sources, phasing approaches and high-speed computing and visualization, now provide specialists and non-specialists alike with a steady flow of molecular images of unprecedented detail. The present chapter combines a general overview of diffraction methods with a step-by-step description of the process of a single-crystal X-ray structure determination experiment, from chemical synthesis or expression to phasing and refinement, analysis and quality control. For novices it may serve as a stepping-stone to more in-depth treatises of the individual topics. Readers relying on structural information for interpreting functional data may find it a useful consumer guide.
1. Introduction
There are numerous approaches that furnish insight into the conformational properties of biopolymers such as proteins and nucleic acids. Among these, diffraction-based techniques occupy a unique place due to the atomic-resolution picture that they can reveal. Thus, provided a single crystal of a receptor, virus or RNA diffracts X-rays to very high resolution, conformation, molecular interactions and water structure can be visualized in stunning detail. A few selected examples of recent successes in the crystallographic structure determination of macromolecular assemblies, receptors, molecular machines and viruses are depicted in Figure 1. In the last decade we have witnessed an unprecedented increase in the number of new crystal structures. On-line databases such as the Research Collaboratory for Structural Biology/Protein Data Bank [RCSB/PDB; http://www.rcsb.org; 62,119 structures as of December 15, 2009 (Berman et al., 2000)] and the Nucleic Acid Database [NDB; http://ndbserver.rutgers.edu; 4,581 structures deposited as of December 16, 2009 (Berman et al., 1992)] now boast large numbers of entries. Before long the number of new PDB entries per year may surpass 10,000. Indeed, with the advent of structural genomics, the old adage that structure determination is preceded by a thorough understanding of function has given way to structure-driven initiatives that promise insights into function from structure [i.e. the Protein Structure Initiative funded by the US National Institutes of Health: http://www.nigms.nih.gov/Initiatives/PSI/(Chandonia and Brenner, 2006; Terwilliger et al., 2009)].
Figure 1.

Recent Triumphs of Structural Biology. (A) The ribosome (large subunit; PDB entry code 1ffk); (B) Adrenergic receptor (GPCR; PDB entry code 2rh1); (C) Poliovirus (PDB entry code 2plv); (D) Photosystem II (PDB entry code 1s5l); (E) Cyanobacterial master clock protein KaiC (PDB entry code 2gbl); (F) Fatty acid synthase (PDB entry codes 2uvb and 2uvc). Reprinted with permission from David S. Goodsell, RCSB/PDB (http://www.rcsb.org/pdb/static.do?p=education_discussion/molecule_of_the_month/index.html; Molecule of the Month Illustrations; info@rcsb.org).
For some 100 hundred years diffraction techniques have shaped our perception of the structure of condensed matter: An overview of the Nobel prizes awarded to scientists behind discoveries related to diffraction and their application to physics, chemistry, biology and medicine provides evidence for the wide-ranging scientific impact of diffraction phenomena (Table 1). The explosive growth in the number of crystal structures during the last years followed dramatic advances in practically all areas of X-ray crystallography, including crystallization [sparse matrix screens and robotics (Jancarik and Kim, 1991; Doudna et al., 1993; Scott et al., 1995)], crystal handling [flash freezing (Garman and Owen, 2006)], data collection and resolution [synchrotron sources and fast CCD detectors (Hendrickson, 2000)], phasing [single- and multi-wavelength anomalous dispersion (Terwilliger and Berendzen, 1999; Weeks et al., 2003)], electron density map interpretation and model building [automatic chain tracing (CCP4, 1994; Abola et al., 2000)], and structure refinement [increased computer power, simulated annealing and maximum likelihood refinement (Murshudov et al., 1999; Brunger and Adams, 2002)]. It is now feasible to mount a protein crystal in the morning and end up with a preliminary, partially refined structure in the afternoon.
Table 1.
Nobel Prizes Related to Diffraction and Crystallography
| Year | Nobel laureate(s) | Field | Discovery |
|---|---|---|---|
| 1901 | Wilhelm C. Roentgen | Physics | Recognition of the extraordinary services he has rendered by the discovery of the remarkable rays subsequently named after him |
| 1914 | Max von Laue | Physics | Discovery of the diffraction of X-rays by crystals |
| 1915 | William H. Bragg, William L. Bragg | Physics | Their services in the analysis of crystal structure by means of X-rays |
| 1927 | Arthur H. Compton Charles T. R. Wilson |
Physics | Discovery of the effect named after him, method of making the paths of electrically charged particles visible by condensation of vapor |
| 1936 | Peter J. W. Debye | Chemistry | Contributions to our knowledge of molecular structure through his investigations on dipole moments and on the diffraction of X- rays and electrons in gases |
| 1937 | Clinton J. Davisson, George P. Thompson | Physics | Experimental discovery of the diffraction of electrons by crystals |
| 1954 | Linus C. Pauling | Chemistry | Research into the nature of the chemical bond and its application to the elucidation of the structure of complex substances |
| 1962 | John Kendrew, Max Perutz | Chemistry | Studies of the structures of globular proteins |
| 1962 | James D. Watson, Francis H. C. Crick, Maurice H. F. Wilkins | Medicine | Discoveries concerning the molecular structure of nucleic acids and its significance for information transfer in living material |
| 1964 | Dorothy Hodgkin | Chemistry | Determinations by X-ray techniques of the structures of important biochemical substances |
| 1976 | William N. Lipscomb | Chemistry | Studies on the structure of boranes illuminating problems of chemical bonding |
| 1982 | Aaron Klug | Chemistry | Development of crystallographic electron microscopy and his structural elucidation of biologically important nucleic acid-protein complexes |
| 1985 | Herbert A. Hauptman, Jerome Karle | Chemistry | Outstanding achievements in the development of direct methods for the determination of crystal structures |
| 1987 | Robert Huber, Johann Deisenhofer, Hartmut Michel | Chemistry | Determination of the three-dimensional structure of a photosynthetic reaction center |
| 1994 | Betram Brockhouse, Clifford Shull | Physics | Development of neutron spectroscopy, development of the neutron diffraction technique |
| 2003 | Peter Agre, Roderick MacKinnon | Chemistry | Discovery of water channels, structural and mechanistic studies of ion channels |
| 2006 | Roger Kornberg | Chemistry | Studies of the molecular basis of eukaryotic transcription |
| 2009 | Thomas A. Steitz Venkatraman Ramakrishnan Ada E. Yonath |
Chemistry | For studies of the structure and function of the ribosome |
However, all these breakthroughs don’t change the fact that crystallography can be a tedious business. Crystallization and phasing represent common bottlenecks on the way to a structure and what is many times a straightforward exercise can become a make-or-break effort that lasts months or years in some cases. Although it is impossible a priori to identify problem cases, empirical evidence exists supporting the notion that membrane proteins are hard to crystallize, that sampling proteins from various organisms increases the chances of obtaining diffraction-quality crystals, and that derivatization and phasing approaches ideally suited for proteins in the 15 to 50 kDa range are frequently inadequate to crack the structures of large macromolecular assemblies. Particularly as far as the latter are concerned, electron microscopy (EM) represents a powerful approach for structure and function studies at the intermediate 10 to 30 Å resolution range. In favorable cases and with averaging of ≥ 1 million subunits, near-atomic resolution can be achieved (Figure 2) (Baumeister and Steven, 2000; Zhou, 2008). Moreover, hybrid structural approaches, marrying EM and X-ray crystallography or crystallography and solution NMR are becoming ever more popular.
Figure 2.

From the Visible to the Invisible. The diagram depicts the rough sizes of cells and their components on a logarithmic scale and illustrates the range of objects that can be visualized with different techniques.
This chapter gives an overview of some of the major techniques in structural biology, particularly those that rely on diffraction, by briefly summarizing the benefits and limitations of individual methods and comparing them to each other. It will then describe in some detail the main stages of structure determinations by single-crystal X-ray crystallography, from crystallization to structure refinement, analysis and quality control. It is by no means the intent of the author to provide an exhaustive account of the topic of X-ray diffraction and macromolecular structure determination. The interested reader may turn to some of the additional reading material listed at the end for a more in-depth treatment of the individual topics touched upon in this brief review.
2. Major techniques in structural biology
The following methods are considered to be of primary importance for experimental, three-dimensional structure determination: X-ray crystallography, X-ray fiber diffraction, electron diffraction, electron microscopy, neutron diffraction and nuclear magnetic resonance (NMR). There are additional techniques that can provide insight into the shape of macromolecules, such as for example small angle X-ray scattering [SAXS (Putnam et al., 2007)] and fluorescence resonance energy transfer [FRET (Lilley and Wilson, 2000; Schuler and Eaton, 2008)]. Although these and others are very useful in combination with any of the above approaches and can shed light on the dynamic behavior of molecular systems, they will not be considered further here. A key difference between optical or electron microscopy and X-ray diffraction is that, unlike light or electron beams, X-rays cannot be focused (Figure 3). The X-ray crystallographic visualization of a molecule requires a ‘mathematical’ lens – Fourier transformation – that generates a 3D structure from the amplitudes of the scattered radiation (the structure factors) and the phases. The phase information is lost in the diffraction experiment, but several methods allow one to recover the phases and we will get back to the so-called phase problem in X-ray crystallography in section 3.5.).
Figure 3.
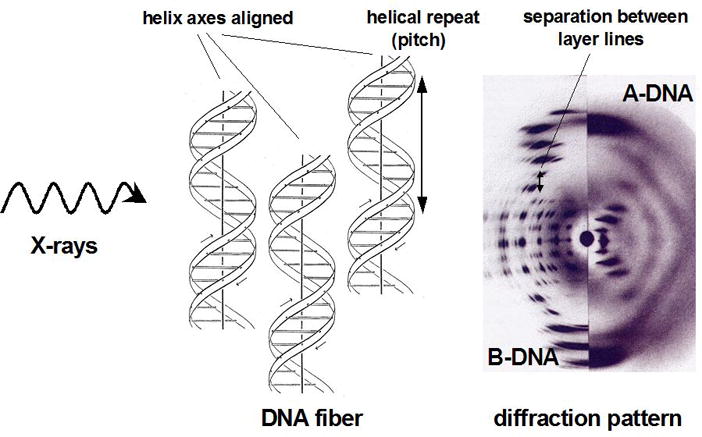
Principles of Fiber Diffraction. The diffraction pattern resulting from aligned helical structures in fibers exposed to X-rays exhibit characteristic cross-like shapes. The drawing of the DNA duplex was originally created by Odile Crick and is adapted from (Kemp, 2003).
2.1. X-ray fiber diffraction
Fiber diffraction can give key insights into the geometry of nucleic acids or fibrous proteins (i.e. collagen) and its golden era coincides with the discovery of the structure of DNA. Very long double-helical DNA molecules tend to be packed side by side in an ordered manner inside fibers. The helical structure gives rise to cross-shaped diffraction patterns with various separations between layer lines (Figure 4). The spacing of layer lines is determined by the helical repeat and as the repeat distance increases the layer lines move closer together. The DNA diffraction pattern depicted in Figure 4 show different numbers of spots and the pattern from A-DNA indicates a higher degree of regularity in the packing arrangement of fibers (there are more spots). The B-form and A-form DNA duplexes differ in their helical repeats (34 and 28 Å, respectively). The larger separation of stacked bases along the helical direction in B-DNA compared with A-DNA can be deduced from the smaller separation of diffraction spots in the B-DNA fiber diffraction pattern. From the helical repeat and the inclination of the arms in the cross, it is possible to derive an approximate radius for the double helix. Moreover, the orientation of the dyad in the diffraction pattern allowed Watson and Crick to conclude that the two strands in the DNA duplex run in opposite directions. X-ray fiber diffraction is still used today but has gradually given way to single crystal studies (Tsuruta and Irving, 2008). For further information, please see the official website for small angle scattering and fiber diffraction studies: http://www.small-angle.ac.uk/.
Figure 4.
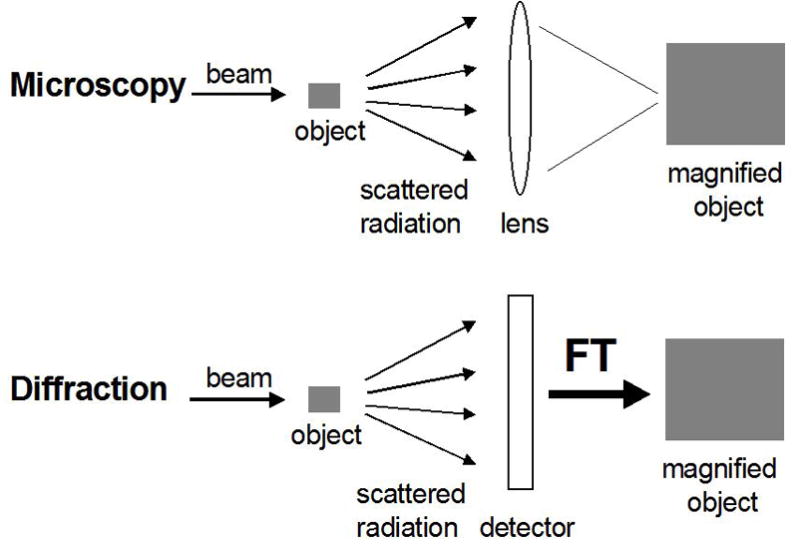
Light Microscopy versus Diffraction. Structure determination by X-ray diffraction entails the use of a mathematical lens, Fourier Transformation (FT), to ‘focus’ the scattered radiation.
2.2. Electron diffraction
In terms of the theoretical framework electron diffraction is similar to X-ray diffraction. However, there are a number of differences that have a significant impact on the practical aspects. Electrons interact strongly with matter and cause serious radiation damage. Thus, the method is typically only applicable to thin layers (2D crystals). Therefore, electron diffraction is useful for certain membrane proteins that may easily form 2D but not 3D crystals. An electron’s wavelength decreases as its velocity increases; in a typical electron microscope the wavelength is around 0.04 Å and thus much lower than the X-ray wavelength used for single crystal diffraction experiments (1–2 Å). However, the damage to biological samples caused by the electron beam is such that the effective resolution is often reduced to 10 to 20 Å.
2.3. Electron microscopy
Unlike with X-rays electromagnetic lenses can be used with electrons to reconstruct the image as in a traditional light microscope. Hence there is no phase problem. A comparison between a standard light microscope and transmission and scanning electron microscopes (TEM and SEM, respectively) is depicted in Figure 5. Samples for EM have to be carefully prepared: (i) they need to be exposed to high vacuum and therefore fixed with special chemicals or frozen; (ii) extremely thin sections are required as electrons have limited penetrating power; and (iii) samples are often exposed to heavy metals (staining) because the contrast depends on the atomic number.
Figure 5.

Light microscopy versus electron microscopy. Lenses allow reconstruction of the image in both techniques, but to focus electron beams electromagnetic lenses are required. Standard light microscope (LM, left), transmission electron microscope (TEM, center), and scanning electron microscope (SEM, right). Source: http://www.vcbio.science.ru.nl/images/fesem beam zoom.jpg. Original illustration: Jeol Instruments. Redrawn by vcbio.science.ru.nl, Radboud University Nijmegen. Used with permission.
In SEM the specimen is dried and coated with a thin layer of heavy metal. The technique allows visualization of secondary electrons that are scattered or emitted from the specimen surface. SEM provides great depth of focus but only surface features can be examined and the resolution is not very high (around 100 Å). An example of an SEM image is shown in Figure 6.
Figure 6.
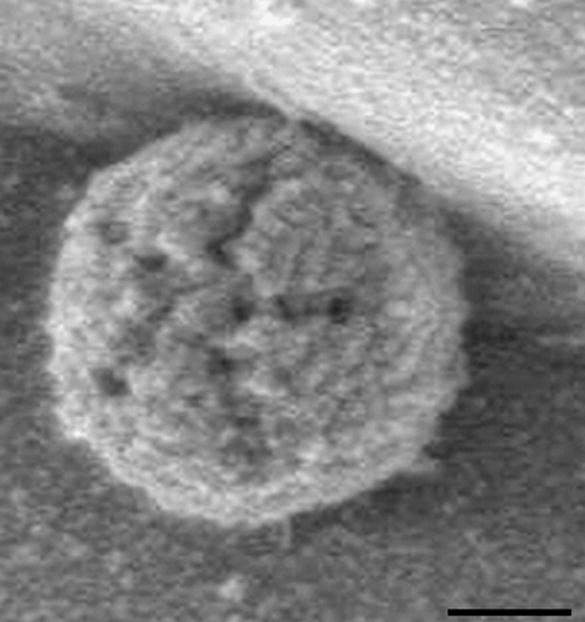
Example of an SEM image. The star-shaped structure in a mature extracellular Acanthamoeba polyphaga mimivirus, an icosahedral double-stranded DNA virus. The scale bar measures 200 nm. Reproduced from Zauberman et al. (2008).
TEM uses electrons that have passed through a specimen to form an image. Specimens are usually fixed, embedded, sectioned, and stained with an electron-dense material. Various techniques can be differentiated, one of them being metal shadowing that allows visualization of surface structures or cell components. Another technique is freeze fracture or freeze etch, used for studying membranes and the cell interior. Finally, negative staining and cryo-electron microscopy (Figure 7) can be applied to unfixed biological samples. Thus, these techniques are useful to visualize large macromolecular assemblies such as viruses or ribosomes.
Figure 7.
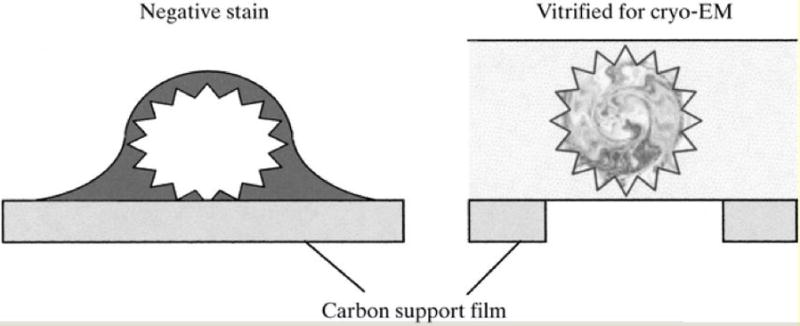
Negative-Stain and Cryo-EM. Left: A virus particle is outlined with good contrast by heavy-metal stain but is somewhat flattened due to dehydration. Right: By comparison it is preserved in the native state in the cryo-EM sample, but the protein-ice contrast is very low. The particle is therefore imaged over holes in the carbon support to maximize the contrast. Reprinted with permission from (Saibil, 2000).
A single protein molecule gives only a weak and ill-defined image in the electron microscope. Increasing the signal by using higher intensity beams or longer exposure only increases the radiation damage. Therefore, it is necessary to combine the information from many molecules so as to average out random errors in the single images. This is more easily achieved when the molecule or particle features high symmetry, a key property of many viruses (Chiu et al., 1997). It is possible to apply averaging techniques and reconstruction analysis also to non-symmetric molecules (Saibil, 2000). Images of randomly oriented molecules are collected and classes of similar particles are generated (Figure 8). Angles are then assigned to each class and a 3D averaging procedure is carried out. The process can be further refined by projecting the image obtained, and using the projections to break the original classes into smaller ones and then assigning more precise angles (Figure 9).
Figure 8.
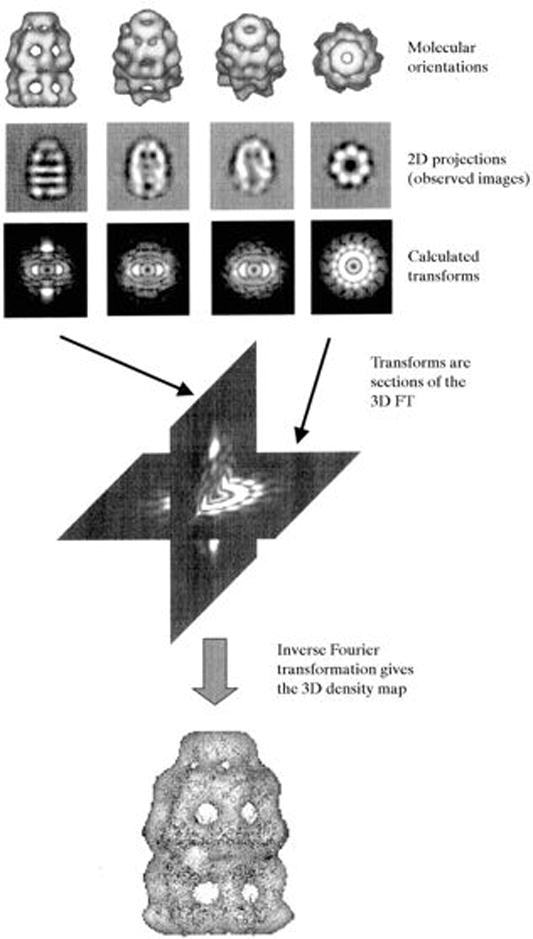
Single-Particle EM 3D-Reconstruction from 2D-Projections. A set of 2D-projections (four in this case) is depicted along rendered iso-surfaces. The Fourier transform of a 2D-projection is equivalent to a central section in the 3D-FT of a molecule. Once a sufficient number of sections are available, the complete 3D-transform can be generated and inverse-transformed into a 3D-density map (bottom). Reprinted with permission from (Saibil, 2000).
Figure 9.
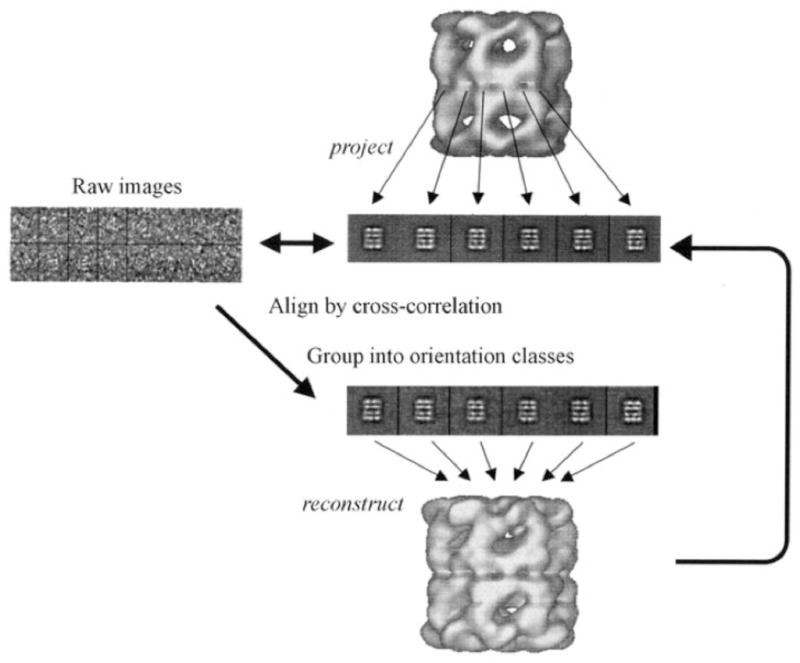
Refinement by Projection Matching. Reference images are created by projecting a 3D-map into a set of different orientations (center). Each raw image from the data set (left) is then rotationally and translationally aligned to individual reference images and given the orientation with the highest correlation coefficient. Images aligned in this fashion are grouped and averaged once again to create an improved 3D-map (bottom). Reprinted with permission from (Saibil, 2000).
In favorable cases cryo-EM can reach near-atomic resolution and if more detailed structures of components of a particle are available from X-ray crystallography or solution NMR, these can be built into the cryo-EM molecular envelope (Zhou, 2008) (Figure 10). Therefore EM and X-ray crystallography are complementary techniques. When they compete directly, crystallography delivers far more detailed information (i.e. ribosome, RNA polymerase). Nevertheless, EM is an extremely useful technique for studying macromolecule assemblies that are difficult to crystallize or in cases where the production of large amounts of materials is problematic. A more detailed comparison of the similarities and differences between EM and X-ray crystallography is provided in Table 2.
Figure 10.
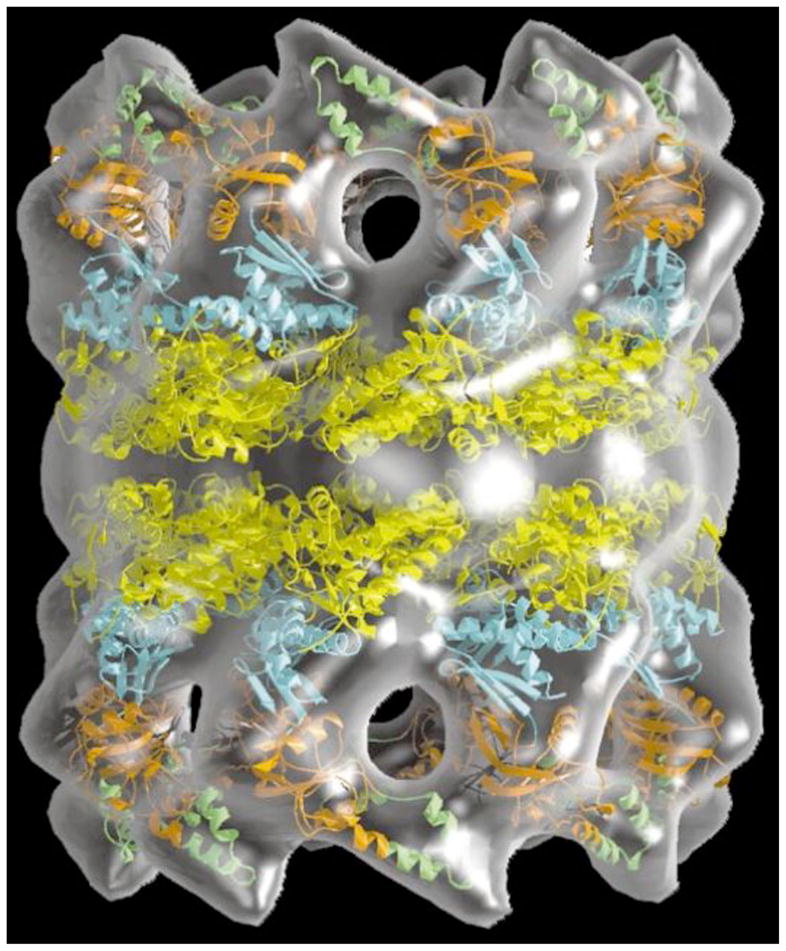
3D-Model of the Archaeal Thermosome Holoenzyme. Crystal structures of the subunits (in color) are modeled into the EM-molecular envelope of the hexadecameric chaperone. Reprinted with permission from (Baumeister and Steven, 2000).
Table 2.
Electron Microscopy versus X-ray Crystallography
| Variable | EM | Crystallography |
|---|---|---|
| Sample size | Relatively low amounts of material needed | Milligram quantities required |
| Crystals | No need for single crystals | Crystallization constitutes a bottleneck on the way to structure determination |
| Molecular size | Typically applied to large-size macromolecules or assemblies, (> 300 kDa), but the technique has been successfully applied to a 78kDa DNA nanostructure (Kato et al., 2009) | No intrinsic size limitation (structures up to the MDa size range have been determined); however large molecules can be difficult to crystallize |
| Resolution | Typically 10 Å or less; in favorable cases near-atomic resolution is possible (Zhou, 2008) | Near-atomic resolution can be achieved even with very large molecules, permitting detailed insights into recognition and mechanistic aspects |
2.4. Neutron diffraction
A fundamental difference between diffraction of X-rays (photons) and neutrons is that the former are scattered by electrons and the latter by protons. Neutrons are highly penetrating and unlike X-rays they are non-destructive, and crystals of macromolecules do not decay in neutron beams even after lengthy exposure times. X-rays are typically blind to hydrogen atoms in crystals of macromolecules, unless diffraction data are available to extremely high resolution (≪1Å). Even in those cases, the hydrogen atoms of water molecules in well ordered solvent networks (first and second shell hydration) normally remain invisible. The atomic form factor f in X-ray scattering (a measure of the scattering intensity of a wave by an isolated atom) is replaced by the scattering length b in neutron diffraction. The scattering length varies randomly across the periodic table and its magnitude can differ significantly even with isotopes of the same element, as in the case for hydrogen (1H) and deuterium (2H). The atomic form factors (f≡Z) and scattering lengths (unit 10−15 m, fm) for selected elements and isotopes are: hydrogen (f=1; b= −3.8), deuterium (f=1; b=6.5), carbon (f=6, b=6.6), nitrogen (f=7, b=9.4), oxygen (f=8, b=5.8), sulfur (f=16, b=3.1) and iron (f=26, b=9.6); for a full list, please see http://www.ncnr.nist.gov/resources/n-lengths/). Thus, deuterium and carbon exhibit very similar scattering lengths and the light element can be observed in the presence of the heavier carbon, oxygen, nitrogen, and sulfur atoms (Figure 11). Deuterium also displays much weaker incoherent scattering than hydrogen. Therefore, visualization of the positions of hydrogen atoms in neutron crystallographic experiments requires perdeuteration of proteins.
Figure 11.
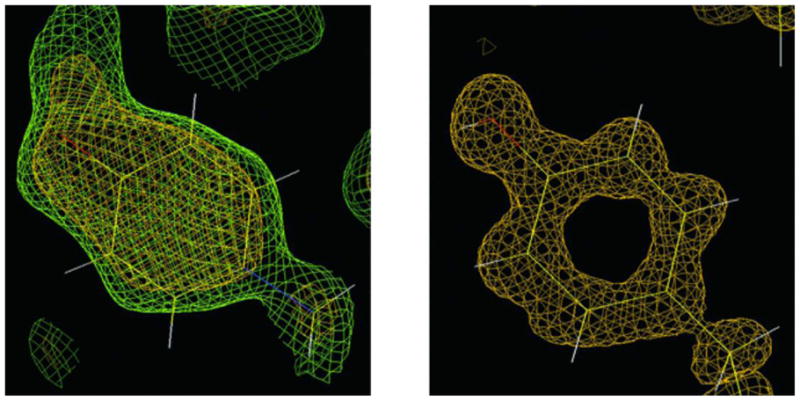
Neutron versus X-ray Macromolecular Crystallography. Left: The neutron density for Tyr137 in the structure of D-xylose isomerase contoured at 1.5σ(green) and 2.0σ(yellow) clearly reveals the orientation of the deuteron on the O atom of tyrosine. Right: The protonation state of Tyr254 remains unclear from electron density maps in the X-ray crystal structure of the same enzyme determined to 0.94-Å resolution at −170°C. Reprinted with permission from (Hanson et al., 2004).
There are a number of advantages of neutron macromolecular crystallography (NMC) for structural biology (Blakeley et al., 2008). The positions of hydrogen atoms can be located even at resolutions of around 2 Å. Thus, NMC is complementary to ultrahigh resolution X-ray macromolecular crystallography (XMC). The protonation and ionization states of atoms can be determined, thus yielding atomic charges and pKa’s. Insights can be gained into hydrogen bonding pattern because NMC allows one to determine the orientation of hydroxyl and amide groups (Hanson et al, 2004). Similarly, the conformations of methyl groups and side chains can be established in neutron density maps, thus providing details on packing arrangements. Because it is possible to observe hydrogen atoms in neutron structures, the orientations of water molecules can be determined, effectively revealing donor and acceptor patterns in water networks. This will contribute to a better understanding of the role of water molecules at active sites and the effects on conformation and stability of solvation shells. Further advantages of NMC concern the monitoring of hydrogen/deuterium (H/D) exchange, permitting insight into solvent accessibility, dynamics and folding patterns. Finally, NMC allows one to discriminate between metals at active sites due to unique neutron scattering cross sections, i.e. Mn(25)= −3.6 fm, Fe(26)=9.5 fm, and Zn(30)=5.6 fm.
More widespread applications of NMC have traditionally suffered from the high cost of the instrumentation required (either a nuclear reactor or a spallation neutron source, SNS; the complexity and cost of neutron detectors also exceed by far those of state-of-the-art X-ray CCDs) and the need for large crystals (ca. 1 mm3). However, the availability of SNSs in Europe, Japan and the USA (some of these are still under construction; Figure 12), that produce high intensity beams has sparked a renewed interest in applications of neutron scattering and promises a renaissance of NMC. The design criteria for the Macromolecular Neutron Diffractometer (MaNDi) on the SNS at Oak Ridge National Laboratory (ORNL, Oak Ridge, Tennessee, USA) anticipate resolution limits of between 1.5 and 2.0 Å for crystals with a lattice constant of up to 150 Å (2.5 – 3.0 Å for constants of 150 – 300 Å). Moreover, the time spent to collect data from a crystal with a volume of 0.125 mm3 and unit cell constants of max. 100 Å is expected to be 24 hours for a resolution of ca. 2 Å.
Figure 12.
The Spallation Neutron Source (SNS) at Oak Ridge National Laboratory (ORNL, Oak Ridge, Tennessee, USA). The SNS instrument hall currently under construction will eventually contain 24 instruments on 18 beam lines. The Macromolecular Neutron Diffractometer (MaNDi, BL-11B) and the Single-Crystal Diffractometer (TOPAZ, BL-12), to be completed in 2012 and 2009, respectively, are of particular interest for neutron macromolecular crystallography research. Source: http://neutrons.ornl.gov/instrument_systems/beamline.shtml; please visit the original website and click on the individual boxes for details of the instrument layout and capabilities.
2.5. Nuclear magnetic resonance spectroscopy
Certain nuclei, such as for example 1H, 13C, 15N and 31P possess an angular momentum. The energy levels associated with nuclei of different spin angular momentums can be separated in high magnetic fields. The spin will align along the field and absorption of electromagnetic radiation of the appropriate frequency (radio waves) then induces a transition. When the nuclei revert to their equilibrium state they emit radiation that can be measured. Most importantly, the precise frequency of the emitted radiation is dependent on the environment of the individual nuclei. The environment of a particular nucleus affects the frequency of the emitted radiation. These different frequencies are referred to as chemical shifts. NMR spectra are further complicated by scalar coupling between neighboring nuclei that is apparent from the splitting of individual signals (Figure 13) (Keeler, 2005).
Figure 13.
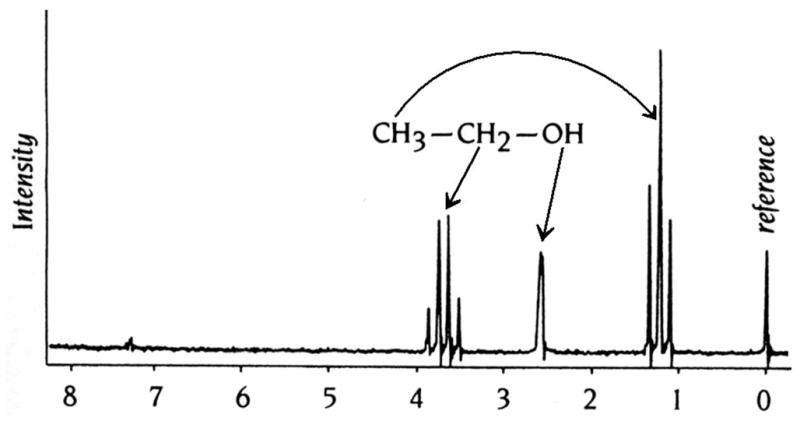
One-Dimensional 1H-NMR Spectrum of Ethanol. The three groups of protons in this small molecule, (C)H3, (C)H2, and (O)H, all exhibit different chemical shifts relative to the protons in reference molecule, tetramethylsilane (TMS). The characteristic splitting of the signals arising from the methyl (1:2:1) and methylene (1:3:3:1) protons is the result of through-bond coupling between neighboring nuclei.
Protein NMR spectra contain a large number of overlapping peaks and it is impossible to interpret a one-dimensional (1D) spectrum. But it is possible to design 2D NMR experiments and to plot the results into a xy-diagram, i.e. a so-called 2D homonuclear COSY (correlation spectroscopy) experiment. In this 2D representation, the diagonal corresponds to the common 1D spectrum. Off-diagonal peaks arise from the interactions between hydrogen atoms that are relatively closely spaced. Another common type of NMR experiments with proteins concerns the heteronuclear single quantum correlation (HSQC), i.e. between the nitrogen atom of an NHx group with the attached proton. Therefore, each signal in a 15N-HSQC spectrum represents a signal from a single amino acid. In addition to the signals from the HN protons in the backbone, the HSQC spectrum also contains signals from the amino groups of the side chains of Asn and Gln and the aromatic N-H groups in the His and Trp side chains. However, unlike a 2D homonuclear spectrum, a heteronuclear 13C- or 15N-HSQC spectrum does not contain a diagonal (Figure 14) (Wüthrich, 1986).
Figure 14.

Two-Dimensional Heteronuclear NMR Spectroscopy. 15N-HSQC spectrum of the circadian clock protein KaiB from the cyanobacterium Synechococcus elongatus recorded on an 800 MHz spectrometer.
Relaxation processes are very sensitive to both geometry and motion, but only interactions between atoms that are less than 5 Å apart can typically be detected. Therefore, NMR spectroscopy allows us to map the distances between pairs of atoms and by specifying which pairs are close together in space, NMR spectra contain information about the 3D structure of protein molecules. In reality, it is far from trivial to assign the peaks in a spectrum to a specific H atom in the protein sequence. Kurt Wüthrich worked out a solution to the assignment problem in the 1980s and he was co-awarded the 2002 Nobel Prize in Chemistry for the development of NMR spectroscopy for determining the 3D structure of biological macromolecules in solution. Both solution NMR and X-ray crystallography provide insight into the 3D structures of macromolecules. In many ways the two techniques are complementary with the most significant limitation of NMR and crystallography being size (< 40 kDa) and the need for single crystals, respectively. A more detailed comparison of these two key techniques in structural biology is provided in Table 3.
Table 3.
X-ray Crystallography versus Solution NMR Spectroscopy
| Variable | X-ray crystallography | NMR spectroscopy |
|---|---|---|
| Amount and purity of material (30 kDa protein) | 10 – 50 mg, very pure, stable at room temp. | 10 – 20 mg, 95% pure, stable at room temp.; if >10 kDa must be labeled with 15N and/or 13C |
| Studied sample | Crystals with high content of non-crystalline solvent, (~50% protein) | ~1 mM protein in solution, (~1% protein) |
| Physiological relevance | Artifacts due to crystal packing forces | Artifacts due to use of isolated fragments or domains |
| Experimental variations of sample conditions | Very difficult, crystallization conditions have to be maintained | Straightforward, can change temp., pH and others |
| Size limitations | Virtually none (40 MDa structures determined) | ~50 kDa at present (40 kDa structures solved) |
| Meaning of the single data point | None: one spot has contributions from the whole unit cell | Single inter-atomic interaction - interpreted as distance or angle constraint |
| Meaning of all data points | After FT with proper phases, direct definition of the electron density within one unit cell | All observable inter-atomic interactions - interpreted as distances and torsion angles |
| Interpretation of experimental data | Relatively quantitative | Relatively qualitative |
| Most time consuming | Varies: crystallization, phase generation, model building | Resonance assignments |
| Final result | One model that minimizes R factor; model variations and uncertainties “hidden” in B values | Many models (ensemble) satisfying constraints; variations explicit in multiple models |
| Use of stereo- chemical constraints | Necessary (exception: ultra-high resolution data available) | Necessary |
| Classes of proteins amenable to study | Stable tertiary structure (fold) throughout most of the polypeptide; non-aggregating | Folded and unfolded regions can occur; non-aggregating |
| Observation of dynamic processes | Very difficult, but in principle enzyme reactions can be followed in crystals | Very straightforward |
| Measure of accuracy | Biochemical data, threading calc., ϕ–ψ plots (potential energy) | Biochemical data, threading calculation, ϕ–ψ plots (potential energy) |
| Measure of precision | Resolution, R factors, rms deviations from standard bond lengths and angles | Average root-mean-square deviation (rmsd) among structures in calculated family |
| Comparable resolution | 2 Å resolution, R factor <25% ⇐ | ⇒ rmsd less than 1.0 Å for most of the polypeptide length |
3. Single crystal X-ray crystallography
3.1. Overview
The following sections are dedicated to arguably the most powerful ‘weapon’ in the structural biology arsenal: X-ray crystallography. This technique can provide more detailed models than any of the other approaches available to study macromolecules. In principle there is no limitation as far as size is concerned: the basic principles remain the same independent of whether one is working out the structure of an oligopeptide with a molecular weight of a few kDa or that of a virus a thousand times larger. Individual steps of a structure determination are outlined in Figure 15. Among them crystallization and phasing constitute the biggest hurdles. Despite the fact that impressive advances have been made in recent years to increase the chances of obtaining protein or nucleic acid crystals, crystallization has remained a trial and error approach that frequently fails when only a single construct is available, or can easily escalate into a potentially costly and time-consuming battle when various constructs and/or homologous proteins from different organisms are screened (McPherson, 1998). However, the end – be it a detailed 3-dimensional model of an enzyme, receptor, RNA or protein-DNA complex and the biological insights gained from it – generally justifies the means.
Figure 15.

Individual Stages of a Macromolecular X-ray Crystal Structure Determination. Selected methods are highlighted on the right. Approaches for refining structures include least squares fitting and simulated annealing. Adapted from (Ringe and Petsko, 1996).
3.2. Sample preparation
Crystallography requires large, milligram, amounts of pure material, precluding in most cases isolation of enzymes or receptors for crystallization from tissues. Instead, proteins based on recombinant DNA technology are used for the structural studies. The DNA is subcloned from a cDNA library or, alternatively the gene is synthesized. A battery of expression vectors is commercially available and, while E. coli still represents the most common organism for over-expression, insect cells, yeast and human cell lines are becoming ever more popular for producing recombinant proteins. In addition, cell-free expression should also be considered as an alternative approach.
Molecules for crystallization need to be reasonably well structured and not floppy. Therefore, it is important to consider possibly unstructured or flexible regions, i.e. at the N- or C-terminus, in the design of the construct. Constructs amenable to crystallization can often be identified by limited proteolysis (Dong et al., 2007). In many cases, only domains can be crystallized or it is necessary to resort to the homologous proteins from a thermophilic organism for successful crystallization. Induced-fit binding of a ligand may render the protein with the ligand bound more likely to crystallize than protein alone. It is also worthwhile to consider whether there are a great many charged residues solvent exposed. This is because reduction of surface entropy by mutation of Lys to Ala or other strategies can dramatically increase the chances of obtaining crystals or of producing higher quality crystals (Czepas et al., 2004). Another important aspect concerns the size of the protein: Is the target a small protein (< ca. 70 amino acids) or a polypeptide? In that case crystallization of the small protein as a fusion with a larger and well characterized protein, such as glutathione-S-transferase (GST) should be tried (Smyth et al., 2003). This often improves solubility and allows for phasing by molecular replacement of the GST.
Fusion with a variety of tags or proteins also facilitates purification via affinity chromatography (Structural Genomics Consortia, 2008). Some popular ones include the (His)6 tag, GST, maltose binding protein (MBP), and small ubiquitin-like modifier (SUMO) protein. Further purification steps may involve gel filtration and/or ion exchange chromatography. Procedures that should be avoided are ammonium sulfate precipitation and lyophilization and care should be applied when combining various fractions following column chromatography or different batches of protein. In general the purification should be carried out quickly and proteins need to be handled gently and maintained at reduced temperature. Turbid samples need to be centrifugated and, for filtrations, cartridges with minimal dead volume should be used and one should check for adsorption (OD/activity) after filtering. As a rule of thumb, the purity of a protein should be 90–95% by SDS- polyacrylamide gel electrophoresis (PAGE) with Coomassie stain. The purified protein can be further characterized with native PAGE, light scattering, isoelectric focusing (to determine the pI), mass spectrometry, circular dichroism (CD) spectroscopy and other techniques. Proteins of low solubility (less than 1 mg/mL) are typically not suitable for crystallization experiments and a search for other constructs or mutation via in vitro directed evolution may be advisable in such cases.
DNA is produced by solid phase chemical synthesis using suitably protected phosphoramidite building blocks (Gait, 1984). Two basic methods exist for producing RNAs of sufficient quality suitable for crystallization and X-ray structure determination. Longer fragments (> 50 nucleotides) can be generated by in vitro transcription using the DNA-dependent T7 RNA polymerase (Milligan and Uhlenbeck, 1989; Wyatt et al., 1991). For shorter RNA oligonucleotides the method of choice is chemical synthesis, usually by the solid phase phosphoramidite technique. Due to the presence of the 2′-hydroxyl group in the furanose sugar, chemical synthesis of RNA is more complicated compared with DNA. Common protection groups for the 2′-OH moiety are the tertiary butyl dimethyl silyl [TBDMS (Scaringe et al., 1990; Wincott et al., 1995)] group, the 2′-acetoxy ethyl orthoester [2′-ACE (Scaringe et al., 1998)], and the triisopropylsilyloxymethyl functionality [TOM (Pitsch et al., 2001)]. The latter approach has allowed production of RNAs as long as 100 residues, a size range that includes many biologically interesting RNA motifs. Once deprotected and cleaved from the solid support, DNA and RNA oligonucleotides are typically purified via trityl-on reverse phase HPLC or ion-exchange chromatography. However, column chromatography is not suitable for the purification of longer fragments. Instead, large RNAs need to be purified by denatured PAGE and desalted following elution from the gel (Wyatt et al., 1991).
3.3. Crystallization
There are a number of crystallization techniques commonly used with proteins or nucleic acids: Hanging-drop and sitting-drop vapor diffusion, batch/microbatch under oil, free interface diffusion employing either integrated fluidic circuits (i.e. the Topaz® crystallization system) or the Zeppezauer tube, and dialysis (Carter and Sweet, 1997a, b; McPherson, 1998; McRee, 1999; Carter, 2003a, b; Rhodes, 2006; Drenth, 2007). The first two techniques are illustrated schematically in Figure 16. Both are fast and easy to setup and versatile for both screening and optimization. The droplets can be viewed through glass (hanging drop) or either a plastic lid or a transparent tape (sitting drop) under a microscope. The drop size can vary but the volume of hanging drops is usually limited to ca. 5 μL. In both cases, the concentration of the particular precipitant in the reservoir exceeds that in the drop. As a result, water will diffuse from the drop to the reservoir, thus increasing the concentration of the precipitant in the drop over time and slowly lowering the solubility of the protein. Ideally, the protein solution will change from the unsaturated region (in terms of a phase diagram) to a labile, supersaturated region, where stable nuclei spontaneously form and grow. The advantage of the sitting drop method is that it can be automated and used in combination with crystallization robots. Microbatch crystallizations using petroleum oil or silicon oil are also easily setup and can be automated to some degree as well. By comparison, crystallizations using dialysis are somewhat more time consuming to setup, but the method allows for a greater control of the individual parameters that affect crystallization. Moreover, dialysis is ideal for replacing the crystallization buffer by a cryo solution, required for flash freezing crystals. Free interface diffusion in a Zeppezauer tube works better in microgravity, but crystallization experiments in space are expensive and not likely to be available in the foreseeable future.
Figure 16.

Two Related Methods for Growing Single Crystals of Biomacromolecules. Schematic depictions of the (A) hanging and (B) sitting drop vapor diffusion techniques. The volume of the droplets is in the nL (Nanodrop setting robots) to μL range.
Crystallization remains a trial and error – mostly error – approach and there is no general recipe for overcoming the nucleation barrier, i.e. a universal nucleant. There are many ways to achieve supersaturation in principle, including adding protein directly to precipitant, altering the temperature, increasing the salt concentration (salt out), decreasing the salt concentration (salt in), adding a ligand that changes the solubility of the protein, altering the dielectric constant of the medium, evaporating water, adding polymer (i.e. polyethylene glycols, PEGs) to produce volume exclusion, adding a cross-linking agent, concentrating the macromolecule and removing a solubilizing agent. Success in crystallization is to a large degree dependent on crystal packing interactions and these remain unpredictable. Lattice contacts are non-covalent and entail various classes of hydrogen bonds (direct bonds between polar, uncharged groups such as OH, NH2, =O; direct bonds between one or more charged groups, so-called salt bridges; two polar or charged groups bridged by a water molecule; bridging of two moieties by a chain of two or more waters) and van der Waals interactions. Optimal packing requires electrostatic and shape complementarity.
It is now common to resort to so-called sparse matrix crystallization screens to increase the chances of obtaining crystals. Such screening kits are commercially available (see, for example http://www.hamptonresearch.com) and they come in a variety of flavors, suitable for proteins, protein-protein complexes, membrane proteins, DNA oligonucleotides, RNA and so forth. The initial set of protein crystallization solutions compiled by Jancarik and Kim in the early 1990s is shown in Figure 17 (Jancarik and Kim, 1991). Individual solutions typically feature a salt, a particular precipitant and a buffer. The pH of the buffers ranges from ca. 4 to 9 and ammonium sulfate figures prominently in the list of salts or precipitants. Similarly, various classes of PEGs are favorites among the precipitants. The recipes for many of these screens are largely based on empirical data that demonstrate, for example, that many proteins can be crystallized from ammonium sulfate solutions. However, not all salts are the same and in the Hofmeister series one can distinguish between stabilizing kosmotropes (weakly hydrated cations such as NH4+ or Cs+ and strongly hydrated anions such as citrate or sulfate) and destabilizing chaotropes [strongly hydrated cations such as Mg2+ or Al3+ and weakly hydrated anions such as nitrate or perchlorate (Collins, 2004)]. The use of PEGs in protein crystallization is based on the tendency of the random coil, water-soluble polymers to reduce protein solubility by volume exclusion (PEG and protein cannot occupy the same space at the same time). This mutual exclusion is mainly dependent on size and shape as well as on concentration.
Figure 17.

Example of a Sparse Matrix Crystallization Screen. Composition of the 50 solutions in the so-called Crystal Screen™ that is commercially available from Hampton Research Inc. (Aliso Viejo, CA). R eprinted with permission from: http://hamptonresearch.com/product_detail.aspx?cid=1&sid=17&pid=1.
Setting up hundreds or perhaps thousands of crystallization trials is a tedious task and the screening process is nowadays facilitated by crystallization robotics. An example of a crystallization robot is depicted in Figure 18. Robotics can be used to generate crystallization screens (so-called liquid handlers), to setup sitting-drop crystallization plates (the 96-well format is quite common), and to barcode, store, retrieve and image at regular intervals of one’s choice the plates. Epifluorescence microscopy can be used to differentiate between crystals of salt and protein; phosphate buffer should be avoided as phosphate tends to crystallize readily and such crystals are then often mistaken for crystals of a macromolecule. Initial leads can be further optimized by manual crystallization setups and the size optimized by seeding. Micro-seeding uses seed beads from crushed crystals in a serial dilution to seed fresh drops in the hope that the introduction of a few seed nuclei into a metastable solution will produce larger crystals. Streak seeding is similar to micro-seeding but quicker in that a whisker is used to pull off seeds from a crystal in order to then streak it through a fresh drop. Finally, macro-seeding consists of partially dissolving the surface layers of a crystal and then placing it into a fresh metastable solution for growth (http://xray.bmc.uu.se/~terese/crystallization/tutorials/tutorial4.html).
Figure 18.

Automation of Crystallization Experiments. Crystallization robot in the laboratory of the author, the MaX WorkCell by Thermo Fisher Scientific Inc. The WorkCell integrates nanodrop setting (“Mosquito”, Molecular Dimensions Inc., Apopka, FL; on the left), liquid handling and screen preparation (“Starlet”, Hamilton Company, Reno, NV.; on the right), and sealing of crystallization plates (bottom right). The robot can handle a wide range of crystallization plates and formats and is typically combined with so-called storage hotels and a plate imager (not shown), that permit automated, periodic access to bar-coded plates and digital photography of individual droplets, respectively.
There are some differences between the crystallizations of proteins and nucleic acids, owing to the polyanionic nature of the latter. Thus, many DNA or RNA oligonucleotides can be crystallized in the presence of either magnesium chloride or polyamines (e.g. spermine tetrahydrochloride) (Berger et al., 1996). Other alkaline earth metal ions such as Ca2+, Sr2+ and Ba2+ are also quite widespread, as are Na+, K+ and Rb +. Sodium cacodylate represents a very common buffer and 2-methyl-2,4-pentanediol (MPD), ammonium sulfate and PEGs are probably the most commonly used precipitants (Baeyens et al., 1994). When all attempts to crystallize a protein fail, it is a good idea to resort to a different construct or to try a homologue from a different organism. Similarly, the key to success in nucleic acid crystallization is to try multiple sequences and to include overhanging bases at the 5′- or 3′-termini. Another option in RNA crystallography is helix engineering, for example by incorporating a tetraloop at the end of a stem (double helical) region and a tetraloop receptor elsewhere (Ferré-D’Amaré et al., 1998a). The pairing of such motifs often mediates stabilizing intermolecular contacts. A related approach to potentially generate a stable lattice is the use of mutagenized RNAs with a binding site for a particular protein. An example of this is constituted by a hepatitis delta virus ribozyme that contains the high-affinity binding site for the basic RNA binding domain of the U1A spliceosomal protein (Ferré-D’Amaré et al., 1998b).
A note of caution at the end of this section: although it is exciting to see crystals under a microscope, it turns out that many crystals don’t diffract X-rays at all or only very weakly. Before letting the excitement build up too much, it is therefore a good idea to test the crystals for diffraction on an in-house X-ray setup.
3.4. Data collection and processing
X-rays are high-energy photons and the wavelengths of those used in macromolecular crystallography experiments lie in the 0.5 to 1.8 Å range (Blundell and Johnson, 1976; Woolfson, 1997; Rhodes, 2006). X-rays can, for example, be generated in sealed high-voltage tubes where an anode (Cu, Mb, Fe etc.) is bombarded with electrons from a heated cathode filament. An electron is hitting the anode material and as it passes within proximity of an atom, the electron is attracted to the nucleus by the Coulombic force. This alters the trajectory of the electron and the closer the electron to the nucleus, the greater the change in its trajectory. To conserve momentum a photon is created, whereby the photon’s energy depends on the degree to which the electron’s trajectory was changed. The energy released in the form of photons is referred to as Brems-Strahlung (“braking radiation” or “white radiation”). Every now and then, an electron that hits the anode target is of sufficiently high energy to displace an electron from an inner shell (i.e. the K shell) and an electron from a higher shell (L, M etc.) then takes its place, with the energy difference between them being emitted as monochromatic X-ray radiation. Normally X-rays are polychromatic but monochromatic radiation can be obtained by way of a monochromator, for example a graphite crystal. However, most of the energy is generated as heat and not ‘light’, and X-rays from a sealed-tube setup (Figure 19A) are typically not of high enough intensity for data collection with weakly diffracting macromolecular crystals. By comparison, so-called rotating anode units feature (Figure 19B) an effective increase in the area of the anode target bombarded by accelerated electrons. But the advantage in terms of higher intensity X-rays comes at a cost: rotating anode generators require more maintenance then sealed-tube setups as parts need to be replaced (cathode filament), cleaned (rotating anode) or rebuilt (ferrofluidic seal).
Figure 19.

X-ray Generators and Detectors. Two 4-circle, kappa-geometry X-ray diffraction setups currently used by researchers at Vanderbilt University: (A) The sealed tube Oxford Xcalibur PX2 Ultra (Oxford Diffraction Inc., Blacksburg, VA), and (B) the rotating anode Bruker Microstar (Bruker AXS Inc., Madison, WI). Tube housing (Xcalibur), beam collimator, beam stop, CCD detector, crystal cooler, goniostat, goniometer head and telescope are clearly visible.
Today, most diffraction data collections are conducted at X-ray synchrotrons, where electron or positron beams are circling close to the speed of light in a storage ring (Figure 20). X-rays are emitted in a tangential fashion when the beam is deflected by extremely strong electromagnets, so-called wigglers or undulators (Helliwell, 1992). Unlike the above sealed-tube or rotating anode generators that produce X-rays of a particular wavelength (i.e. CuKα = 1.5418 Å), the wavelength of the X-ray beam at synchrotrons is tunable. The availability of synchrotrons has had a major impact on structural biology and has impacted many other areas of research in a dramatic fashion (Table 4) (Hendrickson, 2000). The higher intensity of X-rays at synchrotrons leads to significant improvements in the resolution of diffraction data (>0.5 Å and more), but also causes radiation damage of crystals. Damage inflicted over the long run on a rotating anode source can occur in minutes on an unattenuated undulator beamline. Primary radiation damage is due to the large absorption cross section of heavier atoms such as sulfur or selenium and secondary damage is caused by free radicals and photoelectrons.
Figure 20.

X-ray Synchrotron. Aerial view of the Advanced Photon Source (APS) at Argonne National Laboratory (Argonne, IL), a so-called 3rd generation X-ray synchrotron.
Table 4.
Impact of X-ray Synchrotron Radiation
|
To preserve crystals in the beam, they need to be flash-frozen and maintained near liquid nitrogen temperature in a cold stream during data collection (Figure 21) (Harp et al., 1998; Garman and Owen, 2006). Crystals mounted in capillaries (possible for neutron data collection; see http://www.mitegen.com/ for rapid room temperature mounting) will not last very long in the beam. For flash-freezing crystals are scooped up from a droplet with a nylon loop and then swiped through a cryoprotectant before being plunged into liquid nitrogen. The choice of cryoprotectant is important as ice inside the loop formed during freezing will lead to diffuse scattering and powder patterns rings in diffraction images. Popular protectants are glycerol, sucrose, ethylene glycol, propylene glycol, low-molecular weight PEGs, MPD and 2,3-butanediol. Very high concentrations of salts such as sodium malonate have also been reported to be suitable for cryoprotection. Crystals are then shipped to the synchrotron source in the frozen state inside so-called dryshippers. Most macromolecular crystallography synchrotron beamlines are now equipped with automatic sample changers and some feature remote access, allowing users to collect data without leaving the office or the laboratory.
Figure 21.

Inside a Synchrotron Experimental Station. The Marresearch Charge Coupled Device detector (MARCCD 225; foreground, http://www.marresearch.com/) mounted on the MAR Desktop Beamline® (DTB) at the Insertion Device beamline (5-ID-D hutch) of the DuPont-Northwestern-Dow Collaborative Access Team (DND-CAT), located at sector 5 of the APS (Argonne, IL). The view is into the beam that is transported along the tube visible in the center of the upper half of the photograph. The instrumentation colored light blue in the background is not part of the macromolecular crystallography setup. Work conducted at the DND-CAT now focuses more on surface and interface science, nano-materials, catalysis and environmental science. The macromolecular crystallography efforts have moved to the new Life Sciences (LS-CAT) at sector 21 of the APS that offers four ID lines and is jointly run by Michigan institutions, Northwestern University, the University of Illinois at Urbana-Champaign, the University of Wisconsin and Vanderbilt University. Further consortia that operate ID and/or Bending Magnet (BM) beamlines for macromolecular crystallography at the APS include BioCARS-CAT (sector 14), IMCA-CAT (sector 17), SBC-CAT (sector 19), SER-CAT (sector 22), GM/CA-CAT (sector 23) and NE-CAT (sector 24).
Prior to the actual data collection, a single or multiple test frames (Figure 22) are recorded and indexed and the orientation matrix determined and refined. Once Bravais lattice type and Laue group are assigned, one needs to decide on the best data acquisition protocol. Important parameters are the angle of rotation (around the phi axis in most cases), exposure time and the crystal to detector distance. In terms of the correct rotation angle, fine phi slicing guarantees a reduced background whereas coarse phi slicing is more suitable for rapid data collection. In cases where crystals diffract to very high resolution, it is necessary to collect separate low-, medium- and high-resolution data sets, whereby proper acquisition of low-resolution reflections may require an attenuated beam. In general data collection is now a matter of hours and as long as the crystal survives, it is better to collect too much data than too little. CCD detectors are used to record individual diffraction frames (Figures 19, 21, 22). These detectors offer several advantages over multi-wire proportional counters or image plate area detectors, i.e. a linear response and high dynamic range, rapid readout and high spatial resolution. Unlike standard data collections that use X-rays with a discrete wavelength in the rotation mode, Laue diffraction experiments employ ‘white’ or polychromatic radiation with exposures in as little as 50 psec for time-resolved structural studies. Such experiments are complicated by multiple intensities, variations in the absorption coefficient, an uneven detector response at varying wavelengths and reflection spot overlaps, among others.
Figure 22.
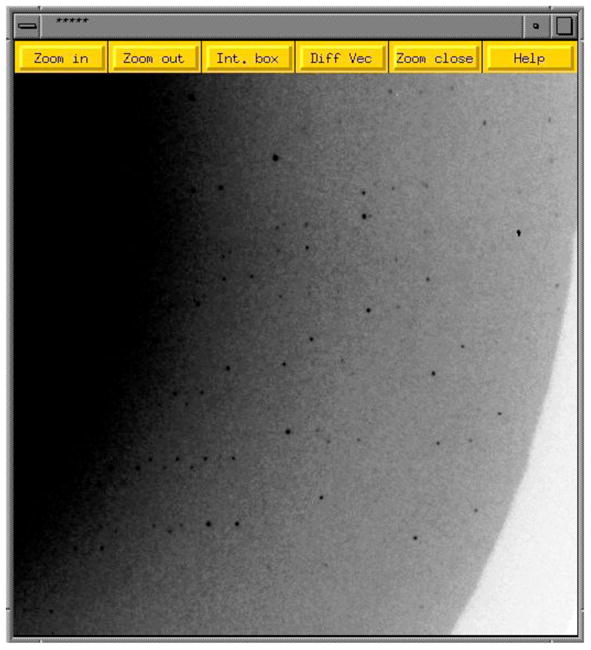
Diffraction Data Collection. Close-up of a 1-degree (ΔPhi) diffraction image obtained from a single crystal of the so-called Dickerson Drew Dodecamer (DDD; B-form DNA of sequence CGCGAATTCGCG). The dark spots represent individual reflections and the diffraction limit is around 1 Å. Data statistics for this particular crystal of the DDD are listed in Table 5.
While the data collection is ongoing, the experimenter starts the data reduction. The reflections (spots) in the individual images or frames are indexed and the crystal and detector parameters are refined before the diffraction peaks are integrated, i.e. their intensities extracted. After establishing the relative scale factors between measurements, these parameters are once more refined using the total data set. Finally the frames are merged and a statistical analysis of reflections based on the space group symmetry is computed. An example of the completeness and quality of a diffraction data set broken down into resolution shells or bins is shown in Table 5. The final product of the diffraction experiment is a file with the amplitudes of individual reflections (the so-called structure factors, Fobs) and their standard deviations σ(Fobs). The Rsym represents the spread of equivalent reflections (the smaller the better) and the resolution limit can be estimated from the mean[I/σ(I)] ratio (the highest resolution shell included should have a mean[I/σ(I)] ≥ 2) and/or the completeness of the data in a higher shell (i.e. > 70% in the outermost shell).
Table 5.
Diffraction Data Quality – Breakdown into Resolution Bins
| Resolution [Å] | N (unique) | Mean[I/σ(I)] | Completeness [%] | Rsym |
|---|---|---|---|---|
| 20.00 – 3.00 | 1,436 | 22.4 | 98.8 | 0.067 |
| 3.00 – 2.50 | 997 | 26.1 | 99.8 | 0.059 |
| 2.50 – 2.00 | 2,218 | 24.5 | 99.5 | 0.063 |
| 2.00 – 1.80 | 1,626 | 18.5 | 97.4 | 0.049 |
| 1.80 – 1.60 | 2,578 | 18.5 | 99.0 | 0.049 |
| 1.60 – 1.40 | 4,282 | 16.0 | 100.0 | 0.064 |
| 1.40 – 1.20 | 7,524 | 13.7 | 100.0 | 0.085 |
| 1.20 – 1.10 | 6,060 | 8.5 | 99.7 | 0.154 |
| All data | 26,721 | 15.5 | 99.5 | 0.064 |
Rsym = ΣhklΣi|I(hkl)i - <I(hkl)>|/ΣhklΣi<I(hkl)i>
The Rsym is a measure for the similarity of the intensities of symmetry-equivalent reflections. It should be a small as possible, typically around 5% or below. Another parameter, Rmerge, is used to characterize the similarity of corresponding reflections in different data sets (i.e. low- and high-resolution data sets) or in data sets from different crystals.
3.5. Phasing approaches
Unfortunately, the measured structure factor amplitudes alone are insufficient for building a structural model. The Fourier transformation of the diffraction pattern that is needed to generate the crystal structure (expressed in terms of an electron density distribution) requires both the amplitudes and the phases of structure factors (Blundell and Johnson, 1976; McRee, 1999; Woolfson, 1997; Rhodes, 2006; Drenth, 2007). However, the phase information is lost in the diffraction experiment. Contrary to data collections that are rapid and more or less automatic, the determination of a structure can therefore still be a time-consuming challenge. There are four basic techniques for solving the phase problem with crystals of macromolecules: Multiple Isomorphous Replacement (MIR), Multi-wavelength Anomalous Dispersion (MAD) and a combination of the two (MIRAS), Molecular Replacement (MR) and Direct Methods (DM). Molecular replacement requires a good model structure and it s the method of choice for complexes of the same enzyme with different ligands (i.e. inhibitors) or multi-domain proteins for which the structure of a domain is available (i.e. fusion proteins). Particularly with crystals of oligonucleotide duplexes, one is often tempted to perform rotation and translation searches using A- or B-form models. However, the failure rate is quite high and relatively small deviations between the conformations of the model and the actual structure are sufficient to derail the search.
Direct Methods are model-independent, but will only work in cases for which diffraction data to very high resolution are available (< 1.0 Å). In addition, there is a size limit and the structure of a 100 kDa protein is unlikely to be phased by DM even with crystals diffracting to atomic resolution. Of the 50,000 or so structures of proteins currently deposited in the Protein Data Bank, less than 0.5% were determined at resolutions of 1 Å and higher. Unlike with crystal structures of small molecules that are mostly solved by DM, the approach is not likely to replace MAD or MIR as the standard phasing techniques for new macromolecular structures in the near future (Terwilliger and Berendzen, 1999; Weeks et al. 2003).
Both MIR and MAD require derivatization of a macromolecule, that is the introduction of heavy atoms into the crystal lattice. Heavy atoms can be bound covalently or by coordination and can be incorporated synthetically (nucleic acids), covalently during protein expression (selenium), by co-crystallization, soaking of native crystals, or in a pressure cell (xenon). A key difference between MAD and MIR is the requirement with the latter that native crystal and derivative crystals (two but better more derivatives are needed for MIR) are closely isomorphous. In this context it is noteworthy that highly similar unit cell constants are not necessarily an indication that the orientations of the protein or nucleic acid in two crystals are identical. The classic approach for introduction of heavy atoms is soaking and a resource for heavy-atom derivatization can be found here: http://www.sbg.bio.ic.ac.uk/had/heavyatom.html (Heavy Atom Databank). Among the favorites for proteins are mercurial compounds (bind to free cysteines or methionine) and platinum compounds (bind mainly methIonine, histidine and cysteine; Pt(CN)2 binds to positively charged residues) (Petsko, 1985; Rould 1997; Garman and Murray, 2003). The heavier the atom the better since the scattering amplitude is proportional to the number of electrons. Several classes of heavy atoms can be differentiated: single metal ions are bound electrostatically, endogenous metal ions such as zinc in zinc fingers and iron in heme that can be used directly for phasing or substituted to obtain a larger signal (i.e. Sr2+ for Ca2+), compounds requiring a chemical reaction, multi-metal complexes for larger molecules (for example the tantalum bromide cluster), xenon and krypton, and anions such as halides or triiodide. For nucleic acids, Rb+, Sr2+, Ba2+ (Tereshko et al., 2001) and Tl+ (Conn et al., 2002) are particularly useful and helix engineering for generating a coordination site for Co (III) hexamine has been used for large RNAs (Keel et al., 2007), as have lanthanides (Holbrook and Kim, 1985; Kim et al., 1985). For soaking it is important to establish a suitable stabilizing solution or artificial mother liquor. The crystal is then transferred to the stabilizing solution that contains the heavy atom at a concentration of typically < 1 to 10 mM. Occasionally, cracks or ragged edges develop and crystals need to be tested for diffraction at various time intervals, whereby it is useful to have a diffraction image prior to soaking for comparison. There are various ways to determine whether the heavy atom is indeed bound. A color change or cracking may be taken as evidence for binding. Mass spectrometry or MicroPIXE (particle induced X-ray emission microprobe) can also be used to confirm successful derivatization. Ultimately, the experimental determination of difference Patterson peaks (there are various means to retrieve the locations of heavy atoms) is the best proof for a useful derivative that paves the way to an interpretable electron density map.
Contrary to MIR, MAD phasing can be accomplished with a single derivative and the technique has gained widespread popularity in the past 15 years and now accounts for the majority of newly determined structures (Hendrickson, 2000). However, since diffraction data need to be collected at the absorption peak of a particular anomalously scattering atom (Figure 23), MAD or the related SAD (single-wavelength anomalous dispersion) experiments have to be performed at a synchrotron source. The most common anomalous scatterer for proteins is selenium that can be incorporated as Se-methionine in E. coli using an auxotrophic strain or metabolic inhibition (Hendrickson et al., 1990; Doublie, 1997). High concentrations of isoleucine, lysine and threonine are known to block methionine synthesis in E. coli by inhibiting aspartokinases. In addition, phenylalanine and leucine act in synergy with lysine. Thus, growth in a medium lacking methionine but supplemented with Se-methionine and plenty of the methionine pathway inhibitors allows for efficient incorporation of the Met analog. Se-Met derivatization does not always work and a number of caveats need to be considered. Selenium is toxic and so cells will not grow as fast. Se-Met derivatized proteins are often less soluble and the altered solubility can affect crystallization. Selenium is also easily oxidized and this may blur the absorption edge or render phasing more difficult. Moreover, it is crucial to precisely determine the peak of the anomalous absorption signal with a particular crystal on the beamline using a fluorescence detector (Figure 23). Very tiny deviations from the maximum may subsequently lead to failure in locating the anomalous scatterers or adversely affect the quality of the MAD electron density map (for a successful example, see Figure 24). Bromine is the most popular anomalous scatterer for derivatization of crystals of nucleic acids or protein-nucleic acid complexes and can be covalently incorporated in the form of Br5U or Br5C. Naturally, many other heavy atoms are not only useful for MIR but can also serve as anomalous scatterers. For example, most crystals of oligonucleotides are grown in the presence of alkaline earth metal ions and it is advisable to always collect MAD data with crystals that contain Sr2+ or Ba2+. This is because the common assumption that structures of oligonucleotide fragments typically yield to phasing by MR is incorrect. Selenium has also been covalently incorporated into nucleic acids for structure determination via SAD or MAD (Pallan and Egli, 2007a, b).
Figure 23.

Multiwavelength Anomalous Dispersion (MAD) Experiment. Example of an X-ray fluorescence spectrum from a protein crystal that contains Se-methionine (Se-Met) in place of Met (Maf protein from Bacillus subtilis; 189 amino acids and 6 Se atoms per protein molecule). The theoretical K absorption edge of selenium lies at 12.6578 keV or 0.9795 Å (an energy of 12.398 keV corresponds to 1.0 Å; http://skuld.bmsc.washington.edu/scatter/). In a typical MAD experiment, diffraction data of high redundancy from the same crystal are collected at three or four wavelengths (i.e. reference below the edge, low, inflection point, inf., peak, max, and reference above the edge, high).
Figure 24.

MAD Phasing. Experimental electron density map based on five Se sites obtained from a Se-Met crystal of the Maf protein (2.7 Å resolution, no solvent flattening), calculated with the program SOLVE (Terwilliger and Berendzen, 1999). The map displays clear boundaries and reveals large solvent-filled channels (black regions).
Accurate phases are very important as they influence the quality of the experimental electron density and without accurate density it is impossible to build a model. MAD phasing has the advantage that the derivative does not have to be isomorphous with the native crystal. Once the model based on the, say, Se-Met protein structure is built and refined, it can be used to solve the native crystal structure via MR if the two are not isomorphous. However, that is not always necessary and one may decide to just use the structure of the Se-Met protein unless the native dataset is of higher resolution. MAD electron density maps are often of excellent quality, making it is possible to automatically trace the protein and build an initial model. Thus, it is not uncommon to end up with a preliminary model of a protein within hours of completing data collection. But in most cases the initial electron density needs to be improved. This is achieved by improving the phases since they are the terms with the largest amount of error in the Fourier transformation. Inaccuracies in the phases dominate those in the amplitudes with regard to the quality of the electron density. The general approach to improve the phase information is to apply constraints in real space; this is referred to as density modification. Density modification methods commonly used are solvent flattening (and flipping), non-crystallographic symmetry averaging (multiple molecules per asymmetric unit that are not related by crystallographic symmetry, i.e. in viruses), histogram matching, phase combination and extension, and the maximum likelihood approach (Çarter and Sweet, 1997a, b; Carter, 2003a, b).
3.6. Refinement and analysis
The model built into the experimental density typically represents just a rough approximation and to arrive at a final structure it is necessary to refine it. Each atom in the model is represented by coordinates x, y and z, an occupancy parameter (q≤1) and a temperature factor (B-factor). The atomic coordinates are stored in a file of a particular format, i.e. the so-called PDB format. The objective of crystallographic refinement is to apply changes to the atomic model such that the difference between model (represented by calculated structure factors Fcalc) and the observed structure factors Fobs are minimized. The R-factor is a measure for the deviations between the calculated and observed amplitudes:
whereby h, k and l represent the Miller indices, the coordinates of reflections in reciprocal space.
Refinement is an iterative process that entails the following basic steps: manual building and (re)fitting, automatic constrained least squares optimization taking into account both X-ray data and geometric constraints of the physical model, and electron density map calculation from the improved model (so-called Fourier sum and difference electron density maps), followed by additional building and so forth. The model will profit from a large excess of reflection data over the number of parameters (x, y, z, q, B) that define the model. A ratio of, say, 10 would be considered excellent and a ratio of 2 represents a poorly over-determined structure. To reduce the total number of parameters that need to be refined, stereochemical restraints are applied (i.e. bond length, bond angle, torsion angle, planarity, chirality, van der Waals distances). The restraints are entered as terms in the refinement target and are weighted so that the deviations from ideal values match those found in databases of high-resolution structures. Thus the target function is an energy that consists of an X-ray (Fobs, Fcalc) and an empirical term (bonds, angles, van der Waals contacts etc.), and optimization algorithms such as steepest descent or conjugate gradient are used to find the nearest minimum in the target function.
To escape local energy minima in the target function and to improve the radius of convergence simulated annealing (molecular dynamics, MD) is used (Brunger and Adams, 2002). Atoms are given random starting velocities and their motion is modeled according to Newton’s laws of motion (bond stretching and angle bending). The temperature of the system is increased (to 2000°C or more) with periodic cooling (annealing), followed by energy minimization. The MD equations are modified through addition of crystallographic residual to the empirical potential energy. Overall, the random element and the thermal motion help to overcome local minima in the target function. Another variant of the least squares optimization is maximum likelihood (Murshudov et al., 1999). Its basic premise is that refinement is not just a matter of making Fcalc equal to Fobs but also needs to consider the phases. To decide how to move an atom we need to take into account the overall accuracy of the model and the best model is consistent with all observations. Consistency is measured statistically by the probability that these observations would be made given the current model. The probabilities include all sources of error (including the model) and as the model gets better, errors get smaller and probabilities become sharper which in turn increases the likelihood.
The R-factor serves as one guide for the status of the refinement. An R-factor of around 60% is consistent with a random relation between the observed and calculated amplitudes. A good starting model will have an R-factor of 40 to 45% and a final model of a macromolecular structure may exhibit an R-factor of around 20%. During the refinement (2Fobs-Fcalc) sum electron density maps should look like the corrected model although they can be biased by incorrect phases/models. On the other hand, (Fobs-Fcalc) difference electron density maps will indicate missing or incorrectly placed atoms. So-called omit maps can be used to remove phase bias that results from least-squares refinement using wrong coordinates. These are difference electron density maps calculated after removing a part of the model from the calculation of Fcalc amplitudes. Because nearby atoms have been influenced by the incorrect portions, the ‘memory’ associated with the omitted atoms needs to be removed. This is achieved by annealed omit maps that are calculated after removal of specific portions of the structure and additional MD. A composite omit map can be generated by placing a 3D grid over the entire unit cell and removing one grid box at the time, calculating the Fcalc, and then repeating this for all grid boxes and summing over all grid points.
An independent measure of the quality of the fit is provided by the R-free, an R-factor that is based on a test data set, reflections (typically amounting to 5% of the total diffraction data) that are set aside and are not included in the refinement (Brunger, 1992). The R-free will be higher than the R-factor (i.e. by up to 5%) and an R-free of 30% with an R-factor (also called R-work) of 20% may indicate errors or over-refinement. Obviously, model building and refinement are easier with high-resolution data. Figure 25 depicts sum electron densities around an aromatic moiety at different resolutions and it is obvious that a map at 3 Å offers some challenges to the model builder. Other parameters beyond R-factors and resolution that need to be considered for judging quality and correctness of a structure are the root mean square deviations (r.m.s.d.’s) of bond lengths and angles from standard values (should be less than 0.02 Å and 3°, respectively) and the B-factors (portions of a structure with atoms displaying B-factors > 50 Å 2 indicate weak electron density). With crystal structures of proteins, the so-called Ramachandran plot (Figure 26) can be used to pinpoint problematic areas in a structure based on deviations of the backbone torsion angles from commonly encountered values.
Figure 25.

Resolution and Quality of the Electron Density. Comparison of the quality of the Fourier (2Fo-Fc) sum electron density around adenosine monophosphate in crystal structures obtained at various resolutions (1σ threshold): (A) 2.85 Å, ATP in the crystal structure of the KaiC protein from Synechococcus elongatus; (B) 1.80 Å, A residue in the crystal structure of a B-form DNA; (C) 1.10 Å, atomic resolution, A residue in the crystal structure of an A-form DNA.
Figure 26.
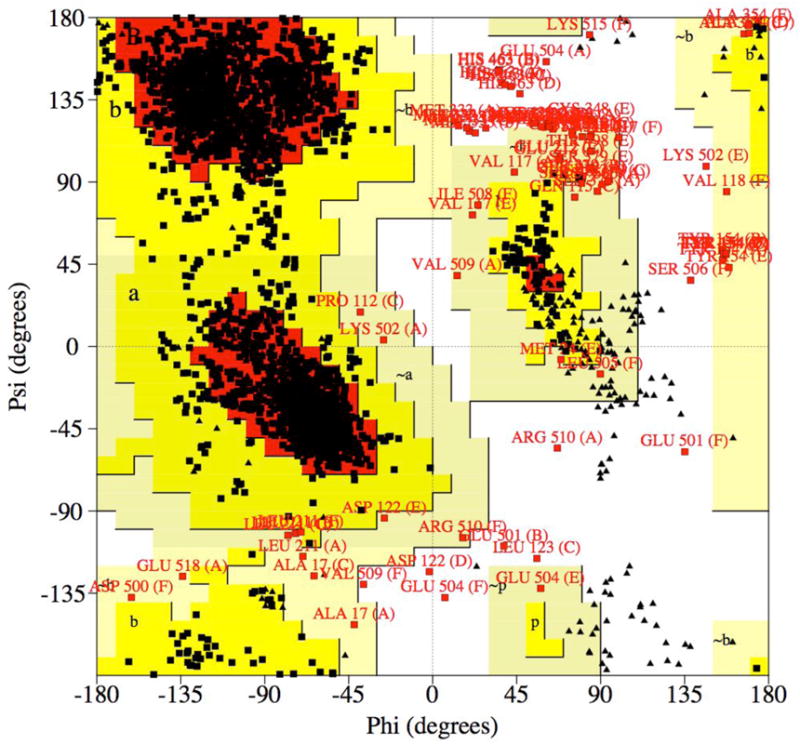
Structure Refinement and Quality Control. Conformations of the Φ and Ψ backbone torsion angle pairs (Ramachandran plot) for amino acids in the crystal structure of the KaiC clock protein from S. elongatus (PDD ID code 2GBL) (CCP4, 1994). Individual angles fall into the most favored regions (A, B, L; red [84%]), additionally allowed regions (a, b, l, p; yellow [13.3%]), generously allowed regions (~a, ~b, ~l, ~p; faint yellow [1.8%]), or disallowed regions (white [1%]). There are 244 glycine (shown as triangles) and 74 proline residues.
It is important to realize that crystallographic models often lack parts of a protein or nucleic acid sequence. The N- and C-terminal portions of a protein are normally more flexible than the core as are the terminal nucleotides in DNA duplexes or single-stranded regions in RNAs. In the crystal structure of E. coli DNA polymerase I (Klenow fragment) about 10% of the amino acids are missing because they could not be seen in the electron density map at ca. 2.5 Å. This indicates that proteins packed into a crystal lattice can still retain considerable flexibility. Indeed, some enzymes are active in the crystalline state and enzymatic reactions have been studied using Laue crystallography. Along with protein or nucleic acid, crystals contain a lot of water (in some cases crystals consist of 70 to 80% water), and the final model consist not just of the coordinates of protein atoms but many first and second shell water molecules, ions and other cosolutes.
Crystal packing forces obviously have an effect on the structure of a macromolecule and need to be considered in the conformational analysis of a protein. Rather than cursing them, lattice forces should be considered a blessing as they can provide valuable information on the deformability of a loop region or particular features of the interface between a protein and its interacting partner. Apart from anisotropic B-factors from data at very high resolution, crystal structures typically provide mostly static information. Occasionally, two or more crystal forms are available, however, allowing one to sample multiple conformations of the same molecule. In such cases, it is possible to determine how packing forces affect the structure of a protein and to identify flexible regions and relative motions of domains.
4. Summary and outlook
Progress in structural biology over the last quarter of a century has been dramatic on all fronts, including instrumentation, mechanistic insights into ever-larger molecules and multi-protein complexes, and the automation of individual steps on the way to a structure determination (for those involved in a crystal structure analysis, see Figure 15). The increasing complexity of the problems being tackled has led to the recognition that one technique alone cannot possibly provide all the answers and has motivated researchers to apply hybrid structural approaches, i.e. combinations of single crystal X-ray crystallography and cryo-EM, crystallography and SAXS, or NMR and computational simulations (computational biology has not been discussed beyond applications in crystallography in this chapter).
Looking into the crystal ball, one can see significant developments in the area of X-ray synchrotron sources in the future, with the emergence of so-called compact light sources (tabletop synchrotron) based on the free-electron laser (FEL) process. In an FEL electrons traveling at nearly the speed of light make their way through an undulator magnet where they are accelerated, resulting in the release of photons. Electrons continue to move in phase with the field of the light emitted and the fields add together in a coherent fashion. The wavelength of the resulting X-ray beam of high brilliance can be tuned by changes in the magnetic field strength of the undulators or the energy of the electron beam. This setup precludes the need for a large storage ring (Figure 20) and the equipment could be housed on university campuses or in medical centers, thus allowing users local access to X-ray synchrotron radiation. Beyond crystallography, applications could include material science, single-molecule X-ray diffraction (Hajdu, 2000), imaging and surgery. For additional information see the following websites: http://www.lynceantech.com/sci_tech_cls.html and http://www.photon-production.co.jp/e/PPL-HomePage.html).
Automation of protein expression and purification, crystallization, data collection and structure determination and model building will continue, driven by the need for high-throughput crystallography as part of structural genomics projects and drug discovery. A decade of large-scale structure determination of proteins has had a major impact on technological advances that have clearly benefited traditional structural biology projects. However, the expectation that one may have had regarding potential outcomes of the PSI, namely that function could be gleaned from structure alone, has not been fulfilled in most cases (Chandonia and Brenner, 2006; Terwilliger et al., 2009). A more likely scenario is that structural information deposited in publicly accessible databases and improved data sharing in combination with biochemical, mutational and genetic studies (that are perhaps initiated by the structural data) will allow the classification of proteins of unknown function at an increased pace.
The achievements made in terms of the structural characterization of soluble proteins, RNA, molecular machines, multi-subunit complexes and others cannot detract from the fact that there are areas where progress has been slower and significant challenges remain. An example that comes to mind is the membrane protein field. Several structures have been determined including photo systems, ion channels and the first G-coupled protein receptors (GPCRs) in 2008. However, expression of stable constructs of membrane proteins in amounts suitable for structural characterization, solubilization and crystallization still constitute formidable obstacles on the way to a more routine generation of structural data. Capturing dynamic systems involving formation of relatively labile protein-protein complexes represents another frontier of structural biology (Radaev and Sun, 2002; Dafforn, 2007). One such system studied in the laboratory of the author is the minimal circadian clock from the cyanobacterium S. elongatus that can be reconstituted in vitro from three proteins in the presence of ATP. The KaiA, KaiB and KaiC proteins interact to form complexes of different compositions throughout the 24-hour cycle, whereby the concentrations of the free proteins and the respective complexes oscillate (Johnson et al., 2008). Clearly, only by using hybrid structural approaches such as those outlined above can one expect to make headway with regard to a structural dissection of the clock and a better understanding of its mechanism.
Acknowledgments
The author would like to thank the US National Institutes of Health for financial support.
Literature cited
- Abola E, Kuhn P, Earnest T, Stevens RC. Automation of X-ray crystallography. Nat Struct Biol. 2000;7(suppl):973–977. doi: 10.1038/80754. [DOI] [PubMed] [Google Scholar]
- Baeyens KJ, Jancarik J, Holbrook SR. Use of low-molecular-weight polyethylene glycol in the crystallization of RNA oligomers. Acta Cryst D. 1994;50:764–767. doi: 10.1107/S0907444994003458. [DOI] [PubMed] [Google Scholar]
- Baumeister W, Steven AC. Macromolecular electron microscopy in the era of structural genomics. Trends Biochem Sci. 2000;25:624–631. doi: 10.1016/s0968-0004(00)01720-5. [DOI] [PubMed] [Google Scholar]
- Berger I, Kang CH, Sinha N, Wolters M, Rich A. A highly efficient 24-condition matrix for the crystallization of nucleic acid fragments. Acta Cryst D. 1996;52:465–468. doi: 10.1107/s0907444995013564. [DOI] [PubMed] [Google Scholar]
- Berman HM, Olson WK, Beveridge DL, Westbrook J, Gelbin A, Demeny T, Hsieh S-H, Srinivasan AR, Schneider B. The nucleic acid database: a comprehensive relational database of three-dimensional structures of nucleic acids. Biophys J. 1992;63:751–759. doi: 10.1016/S0006-3495(92)81649-1. On the web: http://ndbserver.rutgers.edu. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The protein data bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. On the web: http://www.rcsb.org/pdb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blakeley MP, Langan P, Niimura N, Podjarny A. Neutron crystallography: opportunities, challenges and limitations. Curr Opin Struct Biol. 2008;18:593–600. doi: 10.1016/j.sbi.2008.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blundell TL, Johnson LN. Protein Crystallography. Academic Press Inc; New York, NY: 1976. [Google Scholar]
- Brunger AT. The free R value: a novel statistical quantity for assessing the accuracy of crystal structures. Nature. 1992;355:472–474. doi: 10.1038/355472a0. [DOI] [PubMed] [Google Scholar]
- Brunger AT, Adams PD. Molecular dynamics applied to X-ray structure refinement. Acc Chem Res. 2002;35:404–412. doi: 10.1021/ar010034r. [DOI] [PubMed] [Google Scholar]
- Carter CW Jr, Sweet RM, editors. Methods Enzymol. Vol. 276. Academic Press Inc; New York, NY: 1997a. Macromolecular crystallography, part A. [Google Scholar]
- Carter CW Jr, Sweet RM, editors. Methods Enzymol. Vol. 277. Academic Press Inc; New York, NY: 1997b. Macromolecular crystallography, part B. [Google Scholar]
- Carter CW Jr, editor. Methods Enzymol. Vol. 368. Academic Press Inc; New York, NY: 2003a. Macromolecular Crystallography, Part C. [Google Scholar]
- Carter CW Jr, editor. Methods Enzymol. Vol. 374. Academic Press Inc; New York, NY: 2003b. Macromolecular Crystallography, Part D. [Google Scholar]
- CCP4. The CCP4 suite: programs for protein crystallography. Acta Cryst D. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- Chandonia JM, Brenner SE. The impact of structural genomics: expectations and outcomes. Science. 2006;311:347–351. doi: 10.1126/science.1121018. [DOI] [PubMed] [Google Scholar]
- Chiu W, Burnett RM, Garcea RL, editors. Structural Biology of Viruses. Oxford University Press; Oxford, UK: 1997. [Google Scholar]
- Collins KD. Ions from the Hofmeister series and osmolytes: effects on proteins in solution and in the crystallization process. Methods. 2004;34:300–311. doi: 10.1016/j.ymeth.2004.03.021. [DOI] [PubMed] [Google Scholar]
- Conn GL, Gittis AG, Lattman EE, Misra VK, Draper DE. A compact RNA tertiary structure contains a buried backbone – K+ complex. J Mol Biol. 2002;318:963–973. doi: 10.1016/S0022-2836(02)00147-X. [DOI] [PubMed] [Google Scholar]
- Czepas J, Devedjiev Y, Krowarsch D, Derewenda U, Otlewski J, Derewenda ZS. The impact of Lys→Arg surface mutations on the crystallization of the globular domain of RhoGDI. Acta Cryst D. 2004;60:275–280. doi: 10.1107/S0907444903026271. [DOI] [PubMed] [Google Scholar]
- Dafforn TR. So how do you know you have a macromolecular complex? Acta Cryst D. 2007;63:17–25. doi: 10.1107/S0907444906047044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong A, Xu X, Edwards AM, et al. In situ proteolysis for protein crystallization and structure determination. Nat Methods. 2007;4:1019–1021. doi: 10.1038/nmeth1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doublié S. Preparation of selenomethionyl proteins for phase determination. Methods Enzymol. 1997;276:523–530. [PubMed] [Google Scholar]
- Doudna JA, Grosshans C, Gooding A, Kundrot CE. Crystallization of ribozymes and small RNA motifs by a sparse matrix approach. Proc Natl Acad Sci USA. 1993;90:7829–7833. doi: 10.1073/pnas.90.16.7829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drenth J. Principles of Protein X-Ray Crystallography. 3. Springer Science and Business Media LLC; New York, NY: 2007. [Google Scholar]
- Ducruix A, Giegé R, editors. Crystallization of Nucleic Acids and Proteins: A Practical Approach. 2. Oxford University Press; Oxford, UK: 1992. [Google Scholar]
- Ferré-D’Amaré AR, Zhou K, Doudna JM. A general module for RNA crystallization. J Mol Biol. 1998a;279:621–631. doi: 10.1006/jmbi.1998.1789. [DOI] [PubMed] [Google Scholar]
- Ferré-D’Amaré AR, Zhou K, Doudna JM. Crystal structure of a hepatitis delta virus ribozyme. Nature. 1998b;395:567–574. doi: 10.1038/26912. [DOI] [PubMed] [Google Scholar]
- Gait MJ, editor. Oligonucleotide synthesis: a practical approach. IRL Press Ltd; Oxford, UK: 1984. [Google Scholar]
- Garman EF, Murray JW. Heavy-atom derivatization. Acta Cryst D. 2003;59:1903–1913. doi: 10.1107/s0907444903012794. [DOI] [PubMed] [Google Scholar]
- Garman EF, Owen RL. Cryocooling and radiation damage in macromolecular crystallography. Acta Cryst D. 2006;62:32–47. doi: 10.1107/S0907444905034207. [DOI] [PubMed] [Google Scholar]
- Hajdu J. Single-molecule X-ray diffraction. Curr Opin Struct Biol. 2000;10:569–573. doi: 10.1016/s0959-440x(00)00133-0. [DOI] [PubMed] [Google Scholar]
- Hanson BL, Langan P, Katz A, Li X, Harp JM, Glusker JP, Schoenborn BP, Bunick GJ. A preliminary time-of-flight neutron diffraction study of S. rubiginosus D-xylose isomerase. Acta Cryst D. 2004;60:241–249. doi: 10.1107/S0907444903025873. [DOI] [PubMed] [Google Scholar]
- Harp JM, Timm DE, Bunick GJ. Macromolecular crystal annealing: overcoming increased mosaicity associated with cryocrystallography. Acta Cryst D. 1998;54:622–628. doi: 10.1107/s0907444997019008. [DOI] [PubMed] [Google Scholar]
- Helliwell JR. Macromolecular crystallography with synchrotron radiation. Cambridge University Press; Cambridge, UK: 1992. [Google Scholar]
- Hendrickson WA, Horton JR, LeMaster DM. Selenomethionyl proteins produced for analysis by multiwavelength anomalous diffraction (MAD): a vehicle for direct determination of three-dimensional structure. EMBO J. 1990;9:1665–1672. doi: 10.1002/j.1460-2075.1990.tb08287.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendrickson WA. Synchrotron crystallography. Trends Biochem Sci. 2000;25:637–643. doi: 10.1016/s0968-0004(00)01721-7. [DOI] [PubMed] [Google Scholar]
- Holbrook SR, Kim SH. Crystallization and heavy-atom derivatives of polynucleotides. Methods Enzymol. 1985;114:167–176. doi: 10.1016/0076-6879(85)14017-6. [DOI] [PubMed] [Google Scholar]
- Jancarik J, Kim SH. Sparse matrix sampling: a screening method for crystallization of proteins. J Appl Cryst. 1991;24:409–411. [Google Scholar]
- Johnson CH, Egli M, Stewart PL. Structural insights into a circadian oscillator. Science (Wash DC) 2008;322:697–701. doi: 10.1126/science.1150451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato T, Goodman RP, Erben CM, Turberfield AJ, Namba K. High-resolution structural analysis of a DNA nanostructure by cryoEM. Nano Lett. 2009;9:2747–2750. doi: 10.1021/nl901265n. [DOI] [PubMed] [Google Scholar]
- Keel AY, Rambo RP, Batey RT, Kieft JS. A general strategy to solve the phase problem in RNA crystallography. Structure. 2007;15:761–772. doi: 10.1016/j.str.2007.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keeler J. Understanding NMR. 1. John Wiley & Sons; 2005. [Google Scholar]
- Kemp M. The Mona Lisa of modern science. Nature. 2003;421:416–420. doi: 10.1038/nature01403. [DOI] [PubMed] [Google Scholar]
- Kim SH, Shin WC, Warrant RW. Heavy metal ion-nucleic acid interaction. Methods Enzymol. 1985;114:156–167. doi: 10.1016/0076-6879(85)14016-4. [DOI] [PubMed] [Google Scholar]
- Lilley DMJ, Wilson TJ. Fluorescence resonance energy transfer as a structural tool for nucleic acids. Curr Opin Chem Biol. 2000;4:507–517. doi: 10.1016/s1367-5931(00)00124-1. [DOI] [PubMed] [Google Scholar]
- McPherson A. Crystallization of Biological Macromolecules. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 1998. [Google Scholar]
- McRee DE. Practical Protein Crystallography. 2. Academic Press Inc; San Diego, CA: 1999. [Google Scholar]
- Milligan JF, Uhlenbeck OC. Synthesis of small RNAs using T7 RNA-polymerase. Methods Enzymol. 1989;180:51–62. doi: 10.1016/0076-6879(89)80091-6. [DOI] [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Cryst D. 1999;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- Pallan PS, Egli M. Selenium modification of nucleic acids. Preparation of phosphoroselenoate derivatives for crystallographic phasing of nucleic acid structures. Nat Protocols. 2007a;2:640–646. doi: 10.1038/nprot.2007.74. [DOI] [PubMed] [Google Scholar]
- Pallan PS, Egli M. Selenium modification of nucleic acids. Preparation of oligonucleotides with incorporated 2′-SeMe-uridine for crystallographic phasing of nucleic acid structures. Nat Protocols. 2007b;2:647–651. doi: 10.1038/nprot.2007.75. [DOI] [PubMed] [Google Scholar]
- Petsko GA. Preparation of isomporphous heavy-atom derivatives. Methods Enzymol. 1985;114:147–156. doi: 10.1016/0076-6879(85)14015-2. [DOI] [PubMed] [Google Scholar]
- Pitsch S, Weiss PA, Jenny L, Stutz A, Wu X. Reliable chemical synthesis of oligoribonucleotides (RNA) with 2′-O-[(triisopropylsilyl)oxy]methyl (2′-O-tom) protected phosphoramidites. Helv Chim Acta. 2001;84:3773–3795. [Google Scholar]
- Putnam CD, Hammel M, Hura GL, Tainer JA. X-ray solution scattering (SAXS) combined with crystallography and computation: defining accurate macromolecular structures, conformations and assemblies in solution. Q Rev Biophys. 2007;40:191–285. doi: 10.1017/S0033583507004635. [DOI] [PubMed] [Google Scholar]
- Radaev S, Sun PD. Crystallization of protein-protein complexes. J Appl Cryst. 2002;35:674–676. [Google Scholar]
- Rhodes G. Crystallography Made Crystal Clear: A Guide for Users of Macromolecular Models. 3. Academic Press Inc; San Diego, CA: 2006. [Google Scholar]
- Ringe D, Petsko GA. A consumer’s guide to protein crystallography. In: Carey PR, editor. Protein Engineering and Design. Academic Press Inc; San Diego, CA: 1996. pp. 205–229. [Google Scholar]
- Rould MA. Screening for heavy-atom derivatives and obtaining accurate isomorphous differences. Methods Enzymol. 1997;276:461–472. doi: 10.1016/S0076-6879(97)76072-5. [DOI] [PubMed] [Google Scholar]
- Saibil HR. Macromolecular structure determination by cryo-electron microscopy. Acta Cryst D. 2000;56:1215–1222. doi: 10.1107/s0907444900010787. [DOI] [PubMed] [Google Scholar]
- Scaringe SA, Francklyn C, Usman N. Chemical synthesis of biologically active oligoribonucleotides using β-cyanoethyl protected ribonucleoside phosphoramidites. Nucleic Acids Res. 1990;18:5433–5441. doi: 10.1093/nar/18.18.5433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scaringe SA, Wincott FE, Caruthers MH. Novel RNA synthesis method using 5′-silyl-2′-orthoester protecting groups. J Am Chem Soc. 1998;120:11820–11821. [Google Scholar]
- Schuler B, Eaton WA. Protein folding studied by single-molecule FRET. Curr Opin Struct Biol. 2008;18:16–26. doi: 10.1016/j.sbi.2007.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott WG, Finch JT, Grenfell R, Fogg J, Smith T, Gait MJ, Klug A. Rapid crystallization of chemically synthesized hammerhead RNAs using a double screening procedure. J Mol Biol. 1995;250:327–332. doi: 10.1006/jmbi.1995.0380. [DOI] [PubMed] [Google Scholar]
- Smyth DR, Mrozkiewicz MK, McGrath WJ, Listwan P, Kobe B. Crystal structures of fusion proteins with large-affinity tags. Protein Sci. 2003;12:1313–1322. doi: 10.1110/ps.0243403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Structural Genomics Consortia. Protein production and purification. Nat Methods. 2008;5:135–146. doi: 10.1038/nmeth.f.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tereshko V, Wilds CJ, Minasov G, Prakash TP, Maier MA, Howard A, Wawrzak Z, Manoharan M, Egli M. Detection of alkali metal ions in DNA crystals using state-of-the-art X-ray diffraction experiments. Nucleic Acids Res. 2001;29:1208–1215. doi: 10.1093/nar/29.5.1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terwilliger TC, Berendzen J. Automated MAD and MIR structure solution. Acta Cryst D. 1999;55:849–861. doi: 10.1107/S0907444999000839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terwilliger TC, Stuart D, Yokoyama S. Lessons from structural genomics. Annu Rev Biophys. 2009;38:371–383. doi: 10.1146/annurev.biophys.050708.133740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuruta H, Irving TC. Experimental approaches for solution X-ray scattering and fiber diffraction. Curr Opin Struct Biol. 2008;18:601–608. doi: 10.1016/j.sbi.2008.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weeks CM, Adams PD, Berendzen J, Brunger AT, Dodson EJ, Grosse-Kunstleve RW, Schneider TR, Sheldrick GM, Terwilliger TC, Turkenburg MG, Uson I. Automatic solution of heavy-atom substructures. Methods Enzymol. 2003;374:37–83. doi: 10.1016/S0076-6879(03)74003-8. [DOI] [PubMed] [Google Scholar]
- Wilds CJ, Pattanayek R, Pan C, Wawrzak Z, Egli M. Selenium-assisted nucleic acid crystallography: use of DNA phosphoroselenoates for MAD phasing. J Am Chem Soc. 124:14910–14916. doi: 10.1021/ja021058b. [DOI] [PubMed] [Google Scholar]
- Wincott F, DiRenzo A, Shaffer C, Grimm S, Tracz D, Workman C, Sweedler D, Gonzalez C, Scaringe S, Usman N. Synthesis, deprotection, analysis and purification of RNA and ribozymes. Nucleic Acids Res. 1995;23:2677–2684. doi: 10.1093/nar/23.14.2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolfson MM. An Introduction to X-ray Crystallography. 2. Cambridge University Press; Cambridge, UK: 1997. [Google Scholar]
- Wüthrich K. NMR of Proteins and Nucleic Acids. 1. John Wiley & Sons; 1986. [Google Scholar]
- Wyatt JR, Chastain M, Puglisi JD. Synthesis and purification of large amounts of RNA oligonucleotides. BioTechniques. 1991;11:764–769. [PubMed] [Google Scholar]
- Zauberman N, Mutsafi Y, Ben Halevy D, Shimoni E, Klein E, Xiao C, Sun S, Minsky A. Distinct DNA exit and packaging portals in the virus Acanthamoeba polyphaga mimivirus. PLoS Biol. 2008;6:e114. doi: 10.1371/journal.pbio.0060114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou ZH. Towards atomic resolution structural determination by single-particle cryo-electron microscopy. Curr Opin Struct Biol. 2008;18:218–228. doi: 10.1016/j.sbi.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]