

The structure of human carbonic anhydrase II in the monoclinic P21 space group with a doubled a axis from that of the usually observed unit cell has been re-determined and shown that the choice for how the four molecules in the unit cell are grouped (based on their coordinates) into pairs that represent a single asymmetric unit determines whether or not rotational disorder is observed/created during refinement.
Keywords: doubled axis, systematically weak data, pseudo-translational symmetry, redetermination
Abstract
The crystal structure of human carbonic anhydrase II in the monoclinic P21 space group with a doubled a axis from that of the usually observed unit cell has recently been reported, with one of the two molecules in the asymmetric unit demonstrating rotational disorder [Robbins et al. (2010 ▶), Acta Cryst. D66, 628–634]. The structure has been redetermined, with the coordinates of both pseudo-symmetrically related molecules in the crystallographic asymmetric unit translated by x′ = x ± 1/4, and no rotational disorder is observed. This corresponds to a different choice of how the four molecules in the unit cell should be grouped into pairs that represent a single asymmetric unit.
1. Introduction
A crystallographic dilemma in the choice of molecule positions can occur when pseudo-centering is present in a crystal system. Recently, we reported the crystal structure of human carbonic anhydrase II in the monoclinic P21 space group with a doubled a axis from that of the usually observed unit cell. The diffraction data contained alternating strong (h = 2n) and weak (h = 2n + 1) data and a Patterson map of the normalized structure factors showed a strong peak at u = 1/2, v = 0, w = 0. Both the rotation and translation functions gave a single solution and it was only when one molecule was fixed that the position of the second translated molecule became apparent. Our final refined model contained two nearly identical molecules in the asymmetric unit at positions (x, y, z) and (x + 1/2, y, z), with the (x + 1/2, y, z) molecule demonstrating rotational disorder (PDB entry 3ks1; Robbins et al., 2010 ▶). Zwart et al. (2008 ▶) described a crystallographic scenario that can occur when such an observation is made. In the monoclinic P21 case with a pseudo-translational operator (x + 1/2, y, z), the unit cell consists of four molecules [A (x, y, z), B (x + 1/2, y, z), C (−x + 1/2, y + 1/2, −z) and D (−x, y + 1/2, −z)]. Hence, if operators A and D are designated crystallographic symmetry, B and C are translational noncrystallographic symmetry (NCS) operators (Fig. 1 ▶ a). However, a second choice can be made: if operators A and C are incorrectly designated to be crystallographic [corresponding to a shift in reference frame by (1/4, 0, 0)] then B and D are the NCS translational operators (Fig. 1 ▶ b). When the second choice was used in 3ks1, wrongly enforced crystallographic symmetry caused the B and D molecules to appear disordered. In contrast, when the coordinates of both molecules A and B in 3ks1 [Fig. 1 ▶ b; originally termed A and (B′ and B′′) in Robbins et al., 2010 ▶] are translated by x′ = x ± 1/4 the structure can be refined to 1.4 Å resolution with no indication of rotational disorder, more reasonable temperature factors and better agreement with the observed diffraction data (Fig. 1 ▶ a). By comparing the two arrangements, the difference can be seen as an exchange between which 21 screw axes are crystallographic and which are local NCS (Fig. 1 ▶).
Figure 1.

Packing diagrams showing the positions of the molecules of (a) 3mwo (with no observed rotational disorder) and (b) 3ks1 (showing rotational disorder). The crystallographic 21 screw axis is located at (0, 0, 0) and the pseudo-NCS 21 screw axis is located at (1/4, 0, 0). Hence, in the 3ks1 structure solution this arrangement forces the pseudo-NCS 21 screw that relates molecules A and C to be crystallographic and therefore creates the appearance of rotational disorder between the molecules. The axes are as labeled; crystallographic and pseudo-NCS 21 screw [generated by the (1/2, 0, 0) translational symmetry] axes are depicted as solid and open symbols, respectively. The horizontal unit-cell axis is labeled ‘2a’ to indicate that the cell is effectively a doubling of the smaller cell typically observed for this enzyme.
2. Redetermination of the model
The coordinates of 3ks1 were translated by x ± 1/4 and the coordinates of the lower occupied disordered molecule were removed. Occupancies of all atoms were then changed to 1.0 and re-refinement was initiated using the PHENIX package (Adams et al., 2002 ▶). Changes to the model between cycles of refinement were made using the graphics program Coot (Emsley & Cowtan, 2004 ▶). The refinement protocol consisted of three macrocycles, each comprising refinement of restrained isotropic temperature factors on all atoms with TLS treatment of the protein atoms only, restrained atomic positional refinement and automated solvent searches. Solvent molecules were removed if their B factors exceeded 50 Å2 and riding H atoms were added in the final refinement macrocycle. The final re-refined model contained two monomers in the asymmetric unit, 745 waters and 22 alternate conformations of side chains in the two molecules (PDB code 3mwo). The final R cryst was 16.6% and R free was 18.4%, with r.m.s.d. values for bonds and angles of 0.009 Å and 1.3°, respectively. The re-refinement resulted in an improvement of 5.3% in R free over the 3ks1 model, although some of this improvement was owing to the riding H atoms. PROCHECK (Laskowski et al., 1993 ▶) was used to check the geometry of the final model and indicated that only Lys252 in both molecules is in the generously allowed region of the Ramachandran plot. Complete refinement and model statistics of 3mwo are provided and compared with those of 3ks1 in Table 1 ▶.
Table 1. Data and final model statistics.
Values in parentheses are for the highest resolution bin.
| 3mwo, new solution | 3ks1, original solution | |
|---|---|---|
| Data statistics | ||
| Space group | P21 | |
| Unit-cell parameters (Å, °) | a = 84.0, b = 41.1, c = 73.6, β = 109.3 | |
| No. of unique reflections | 90672 (8910) | |
| Resolution (Å) | 17.3–1.4 (1.45–1.40) | |
| Rmerge† (%) | 6.4 (19.4) | |
| I/σ(I) | 14.5 (6.1) | |
| Completeness (%) | 96.8 (95.7) | |
| Redundancy | 3.1 (3.1) | |
| Final model statistics | ||
| Rcryst‡ (%) | 16.6 | 20.4 |
| Rfree§ (%) | 18.4 | 23.7 |
| Residue Nos. | 4–261 | 4–261 |
| No. of protein atoms (including alternate conformations) | 4196 | 6177 |
| No. of H2O molecules | 745 | 536 |
| R.m.s.d. for bond lengths (Å) | 0.009 | 0.006 |
| R.m.s.d. for bond angles (°) | 1.3 | 1.1 |
| Ramachandran statistics (%) | ||
| Most favored regions | 87.5 | 88.1 |
| Additionally allowed regions | 12.0 | 11.4 |
| Generously allowed regions | 0.5 | 0.5 |
| B factors (Å2) | ||
| Main chain | 11.0 | 19.9 |
| Side chain | 15.0 | 15.5 |
| Solvent | 27.7 | 19.1 |
R
merge = 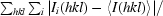
 .
.
R
cryst = 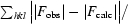
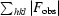 .
.
R free is calculated in the same manner as R cryst, except that it uses 5% of the reflection data that were omitted from refinement.
3. Results and discussion
In contrast to the 3ks1 coordinates, the re-refined structure exhibits no rotational disorder for the second (1/2, 0, 0) translated molecule. When monomer A is translated ½ along a, the differences in Cα coordinates between molecules result in a maximum and minimum displacement of 0.75 and 0.02 Å, respectively, with an overall r.m.s.d. of 0.20 Å in the final model. Several side chains have different conformations between the two molecules and these presumably contribute to the observed doubling of the a axis. In general, the two molecules in the crystallographic asymmetric unit differ in a similar way to the pair of disordered molecules in the 3ks1 model. The largest deviations between pairs of residues occur only on one side of the molecule, with closer agreement on the other side.
The originally incorrect positions of the molecules with respect to the crystallographic symmetry operators should generate two overlapping images (rotational disorder) for each molecule in the unit cell (Fig. 1 ▶ b). To simulate this phenomenon, we split the final correct model 3mwo into A and B chains and removed the solvent molecules. We then translated the A molecule x + 1/2 and the B molecule x − 1/2 and paired these with the correct B and A molecules (i.e. A onto B and B onto A), respectively. After assigning occupancies of 0.5 to all protein atoms, we translated the entire model x + 1/4 (to the location of the incorrect model). Finally, using PHENIX, we refined the two pairs of disordered chains as rigid bodies and allowed the program to complete the solvent structure. The resulting R work of 21.9% and R free of 23.6% were similar to those of the original 3ks1 model. However, in this case the disordered molecule was A. To assure ourselves that we had not made an inadvertent translation error, we verified that the 3ks1 model superposed with the newly refined rigid-body model. This exercise demonstrates that after as few as three macrocycles in phenix.refine the presumed two images in one of the chains coalesce into a single image and the other two diverge such that the distances between atomic positions become even larger. For example, the starting distance between the alternate positions of the Cα atoms of Gly235A was 0.7 Å, but after rigid-body refinement this distance had increased to 1.5 Å. We were not surprised that the electron density of the A molecule was now much worse than that of the B molecule. It should be noted that we started the original 3ks1 refinement at 1.25 Å resolution using SHELXL (Sheldrick, 2008 ▶) and that refinement also produced an apparent disorder, but in this case only in the B chains. Therefore, this unusual result of refinement is not restricted to the use of PHENIX for refinement, nor is it restricted to rigid-body refinement.
In conclusion, the lesson to be learned from our mistake is simple: when faced with translational pseudo-symmetry, explore the effects of all possible symmetry operators, especially the NCS operators. In our case, we should have realised that coordinates translated by x ± 1/4 from our original molecular-replacement solution would have a very different outcome. Fig. 1 ▶(a) shows that the true crystallographic symmetry requires A and C to be different, whereas when the A molecule is misplaced along the a axis as in Fig. 1 ▶(b) the symmetry requires A and C to be identical. This results in the apparent rotational disorder that we observed in one of the molecules of the asymmetric unit of 3ks1.
Supplementary Material
PDB reference: human carbonic anhydrase II, 3mwo
Acknowledgments
We are indebted to Dr Robyn Stanfield for quickly discerning the error in the 3ks1 model and wish to thank Drs Todd Yeates and Zbigniew Dauter for helpful comments and insights. This work was funded by grants from the National Institutes of Health (GM25154 to RM) and the Thomas Maren Foundation (RM) and the MacCHESS grant (US NIH grant RR001646), by US DOE grant DE-FG02-97ER62443 and CHESS, which is supported by the US NSF and NIH–NIGMS through NSF grant DMR-0225180.
References
- Adams, P. D., Grosse-Kunstleve, R. W., Hung, L.-W., Ioerger, T. R., McCoy, A. J., Moriarty, N. W., Read, R. J., Sacchettini, J. C., Sauter, N. K. & Terwilliger, T. C. (2002). Acta Cryst. D58, 1948–1954. [DOI] [PubMed]
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed]
- Laskowski, R. A., MacArthur, M. W., Moss, D. S. & Thornton, J. M. (1993). J. Appl. Cryst.26, 283–291.
- Robbins, A. H., Domsic, J. F., Agbandje-McKenna, M. & McKenna, R. (2010). Acta Cryst. D66, 628–634. [DOI] [PMC free article] [PubMed]
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Zwart, P. H., Grosse-Kunstleve, R. W., Lebedev, A. A., Murshudov, G. N. & Adams, P. D. (2008). Acta Cryst. D64, 99–107. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: human carbonic anhydrase II, 3mwo