

The structures of two new crystal forms of human Cu/Zn superoxide dismutase produced in the eukaryotic expression host L. tarentolae are reported.
Keywords: recombinant proteins, eukaryotic expression systems, Leishmania
Abstract
The rapid and inexpensive production of high-quality eukaryotic proteins in recombinant form still remains a challenge in structural biology. Here, a protein-expression system based on the protozoan Leishmania tarentolae was used to produce human Cu/Zn superoxide dismutase (SOD1) in recombinant form. Sequential integration of the SOD1 expression cassettes was demonstrated to lead to a linear increase in expression levels to up to 30 mg per litre. Chromatographic purification resulted in 90% pure recombinant protein, with a final yield of 6.5 mg per litre of culture. The protein was crystallized and the structures of two new crystal forms were determined. These results demonstrate the suitability of the L. tarentolae expression system for structural research.
1. Introduction
X-ray crystallography is the leading method of protein structure determination at high resolution. Currently, most crystallized proteins are recombinantly produced in Escherichia coli. Despite the obvious advantages, such as rapid growth, well developed methods of protein expression and cheapness of cultivation, E. coli has a range of shortcomings that limit its utility in protein studies. The most prominent problem relates to the inefficiency of E. coli in assisting the folding of eukaryotic proteins. As a result, only ∼15% of eukaryotic proteins can be produced in E. coli in a soluble and active form (Service, 2002 ▶). This has crippled the first generation of structural genomics projects, which have relied heavily on prokaryotic expression. Moreover, prokaryotic expression systems lack the post-translational modifications that are often essential for the functionality of eukaryotic proteins. To circumvent these problems, several in vivo and in vitro eukaryotic expression systems have been developed over the last 30 years. Currently, the mammalian, insect and yeast expression systems represent the main, but not the only, eukaryotic platforms that are used for the production of recombinant proteins for structural studies. Although much progress has been made in adopting these systems to the timelines, quality and cost requirements of structural projects, numerous problems remain. The main shortcomings relate to the cost of insect and mammalian cell cultures, unpredictable and often low expression yields and long development cycles.
We have previously reported several architectures of a new protein-expression system, LEXSY, based on the parasitic protozoan Leishmania tarentolae (reviewed in Basile & Peticca, 2009 ▶). Although it is an intracellular parasite in vivo, this organism can be grown to high cell densities in axenic suspension cultures. Owing to the unique organization of its transcriptional and RNA-processing machinery, Leishmania sp. can efficiently process and translate mRNA generated by RNA polymerase I or foreign polymerases (Kushnir et al., 2005 ▶). In addition, the doubling time of 5 h and the availability of developed methods for microbiological and molecular-genetic manipulation make L. tarentolae a promising protein-expression platform. Furthermore, Leishmania sp. possess eukaryotic protein-folding and post-translational modification machinery that enables them to produce complex proteins in an active form (Zhang et al., 1995 ▶; Breitling et al., 2002 ▶). This work describes the expression of human Cu/Zn superoxide dismutase (SOD1) in the L. tarentolae expression system, its purification, analysis and crystallization, and the X-ray diffraction analysis of two new crystal forms.
2. Materials and methods
2.1. Construction of L. tarentolae expression plasmids
Four expression plasmids were constructed by PCR amplification of a 488 bp fragment encoding the human SOD1 gene, amplified with primers SOD1-for, 5′-TTT AAT TCC ATG GCG ACG AAG GCC GTG-3′, and SOD1-rev, 5′-TTA TTA AGC GGC CGC TTA TTG GGC GAT CCC AAT TAC ACC-3′, using a cDNA clone of human SOD1 as template. The PCR product was digested with NcoI and NotI and subcloned into the Leishmania expression vectors pLEXSY-sat1, pLEXSY-ble1, pLEXSY-hyg1 and pLEXSY-neo1 from the pLEXSYcon1 vector suite (Jena Bioscience) opened with the same enzymes.
2.2. Construction of L. tarentolae SOD1-overexpressing strains
L. tarentolae laboratory strain P10 (Jena Bioscience) was cultivated as a static suspension culture in LEXSY BHI Medium (Jena Bioscience) at 297 K. For electroporation, approximately 5 µg of the expression plasmids was linearized with SwaI. Transfections were carried out by electroporation of in vitro-cultivated promastigotes, as described in Kushnir et al. (2005 ▶) and in the LEXSY manual (available at http://www.jenabioscience.com/cms/en/1/catalog/1013_lexsycon2_kit.html). Recombinant clones were selected as single colonies on solidified growth medium containing 10% FCS and 40 mM HEPES pH 7.4. The selective antibiotics were nourseothricin (LEXSY NTC) at 100 µg ml−1, hygromycin B (LEXSY Hygro) at 100 µg ml−1, bleomycin (LEXSY Bleo) at 100 µg ml−1 and geneticin (LEXSY Neo) at 50 µg ml−1 (Jena Bioscience). Chromosomal integration of the expression constructs at the 18S rRNA locus was confirmed by PCR of genomic DNA with specific primers. In all cases, the analysis confirmed integration of the expression cassettes into the 18S RNA genes. In order to obtain strains that co-expressed multiple copies of the SOD1 gene, the expression cassettes were introduced sequentially in the order of antibiotic resistance: nourseothricin, geneticin, hygromycin and bleomycin. This order of integration ensured the best success rate in the construction of expression strains carrying multiple expression cassettes. The expression levels of SOD1 were assessed by SDS–PAGE and Western blotting analysis of cell lysates with rabbit anti-SOD1 antibodies (Rockland). The blots were developed with alkaline phosphatase-conjugated anti-rabbit antibodies (Sigma).
2.3. Cultivation of recombinant clones and large-scale protein expression
The resulting recombinant clones were maintained on the corresponding antibiotics at 297 K in static suspension cultures in 10 ml LEXSY BHI medium. Culture of the clone harbouring four copies of the SOD1 gene was expanded in two Fernbach flasks containing 500 ml LEXSY BHI medium each and cultivated for 48 h at 297 K with agitation at 75 rev min−1 on a rotary-shaker platform under omission of selection antibiotics. At a density of 108 cells ml−1, the culture was used to inoculate a Biostat C fermenter containing 10 l LEXSY YS medium (Jena Bioscience) supplemented with 1 g l−1 glucose, 20 ml 500× Hemin stock solution (Jena Bioscience) to give a final concentration of 5 mg l−1 and 10 ml of both 0.1 M CuSO4 and ZnSO4. Fermentation was carried out for 53 h at 297 K with aeration at 24 l min−1 without stirring. The pH was maintained in the range 7.2–7.4 and anti-foam M-30 (SERVA) was titrated at a 1:15 dilution to prevent foaming. The cells were harvested by centrifugation (30 min at 5000g), washed once in PBS (50 mM sodium phosphate pH 7.5, 100 mM NaCl) and stored frozen at 193 K in aliquots from 1 l of culture.
2.4. Purification of recombinant SOD1
The cell pellets from 5 l of culture were resuspended in buffer A (50 mM sodium phosphate pH 7.5, 100 mM NaCl, 100 µM CuSO4 and 100 µM ZnSO4) and disintegrated in a fluidizer (Microfluidics). The homogenate was clarified by ultracentrifugation at 60 000g for 1 h at 277 K before ammonium sulfate was slowly added to the supernatant to 70% saturation. The mixture was incubated for 1 h at 277 K with constant mixing and precipitated protein was then separated by centrifugation at 60 000g for 30 min at 277 K. The supernatant was loaded onto a 50 ml phenyl Sepharose column equilibrated with buffer B (70% ammonium sulfate, 50 mM sodium phosphate pH 7.5, 100 mM NaCl, 100 µM CuSO4 and 100 µM ZnSO4). The column was developed with buffer A and fractions containing pure SOD1 were pooled and dialyzed against buffer C (50 mM sodium citrate pH 5.5, 1 mM DTT, 100 µM CuSO4 and 100 µM ZnSO4). The protein was then concentrated to ∼10 mg ml−1, aliquoted, flash-frozen in liquid nitrogen and stored at 193 K.
2.5. Mass spectrometry
For peptide analysis, 500 µg SOD1 was incubated for 4 h at 323 K in 500 µl buffer (0.5 M Tris–HCl pH 8.0, 5 M guanidinium hydrochloride, 2 mM EDTA and 100 µg DTT). The reaction mixture was dialyzed overnight against distilled water, followed by trypsin digestion with 20 µM TPCK trypsin (proteomics grade, Sigma–Aldrich) for 16 h at 310 K. The tryptic peptides were separated on a C18 column (Nucleodur C18 Pyramid, particle size 3 µm, length 125 mm, internal diameter 4 mm; Macherey-Nagel, Germany). The peptides were eluted by a gradient from methanol–H2O (0:100%) containing 0.1% formic acid to methanol–H2O (65:35%) containing 0.1% formic acid over 47 min at a flow rate of 1.0 ml min−1 at room temperature. The peptides were analyzed on an LCQ Advantage Max mass spectrometer.
2.6. SOD1 crystallization
Since initial experiments using established crystallization buffers from the literature were not successful, a search for new crystallization conditions was carried out at 293 K using the sitting-drop vapour-diffusion method with the Classics, PEGs and PACT suites from Qiagen. For this screen, the protein concentration was adjusted to 8 and 12 mg ml−1 in 50 mM sodium citrate pH 5.5, 1 mM DTT, 100 µM CuSO4 and 100 µM ZnSO4. Drops consisting of 0.1 µl protein solution mixed with 0.1 µl reservoir solution were prepared in 96-well plates by a Mosquito nanodispensing robot (Molecular Dimensions Ltd) and were equilibrated against a 75 µl reservoir. Conditions that produced crystals were optimized with the hanging-drop method, varying the precipitant concentration and pH to increase the size of the crystals and to reduce the amount of nucleation. Diffraction-quality crystals were obtained by mixing equal volumes (1 µl) of protein solution (10 mg ml−1) and reservoir solution containing 21–25%(w/v) PEG 4000, 0.1 M sodium acetate pH 4.2–5.2. The drops were equilibrated against 500 µl reservoir solution. These crystallization conditions differed from those that have been published previously and gave crystals with two different morphologies within the same drop. One crystal form (crystal form 1) consisted of very small, thin and fragile colourless plates that grew on top of each other after 5 d, while the other manifested itself as small and slightly more three-dimensional crystals of oblique shape that appeared after approximately four weeks (crystal form 2; Fig. 2a).
2.7. Data collection
In order to achieve cryoprotection, SOD1 crystals were washed in reservoir solution supplemented with 5%(w/v) PEG 400 or 5%(w/v) glycerol and flash-frozen in liquid nitrogen. Crystals were tested using a Rigaku rotating-anode X-ray generator equipped with a MAR Research imaging-plate detector. Crystal form 1 belonged to space group P212121, with unit-cell parameters a = 75.5, b = 166.6, c = 175.0 Å, α = β = γ = 90°, whereas crystal form 2 belonged to space group P6522, with unit-cell parameters a = b = 112, c = 428 Å, α = β = 90, γ = 120°.
A data set for crystal form 1 was collected at 100 K as 150 non-overlapping 1° oscillation images, whereas 180 images of 0.5° oscillation were collected for crystal form 2. Data collection was performed on beamline X10SA of the Swiss Light Source (SLS) at the Paul Scherrer Institut (Villigen, Switzerland) using a MAR 225 detector with a 1 s exposure time. All data were indexed, integrated and scaled with the XDS package (Kabsch, 2010 ▶).
2.8. Structure solution
Since both crystal forms obtained here have not been described in the literature, phasing was achieved by molecular replacement using the online molecular-replacement pipeline BALBES from the York Structural Biology Laboratory (http://www.ysbl.york.ac.uk/YSBLPrograms/index.jsp; Long et al., 2008 ▶).
For crystal form 1, the program picked an atomic resolution structure of human SOD as a search model (PDB entry 2v0a; Strange et al., 2003 ▶) and placed five SOD dimers in the asymmetric unit. However, visual inspection revealed additional electron density for one further dimer, which was then positioned using MOLREP (Vagin & Teplyakov, 2010 ▶) from the CCP4 software package (Collaborative Computational Project, Number 4, 1994 ▶). Accordingly, the solvent content of the crystal form was 58%, corresponding to a Matthews parameter of 2.9 Å3 Da−1.
Similarly, BALBES placed five protein chains in the asymmetric unit of crystal form 2. Again, the electron density revealed the presence of one further monomer. In addition, the chain neighbouring the missing monomer was clearly misplaced by the molecular-replacement software. This was corrected by first locating the missing monomer with MOLREP, followed by superimposition of the SOD1 dimer from PDB entry 2v0a. The solvent content of crystal form 2 was 67%, which corresponded to a Matthews parameter of 3.72 Å3 Da−1.
2.9. Refinement
Prior to refinement, 5% of the reflections contained in each data set were chosen at random for the calculation of R free. Both structures were refined in REFMAC5 (Murshudov et al., 1997 ▶) using tight noncrystallographic symmetry restraints for both main and side chains owing to the low resolution of the data sets. The structures were subsequently inspected in Coot (Emsley & Cowtan, 2004 ▶) and minor adjustments were introduced by hand.
For crystal form 1, the TLS refinement option in REFMAC5 was then employed (Winn et al., 2001 ▶), defining each dimer as a separate TLS body since this produced lower R factors than using separate TLS bodies for each of the individual monomers. Because one chain consistently only produced very poor electron density, it was excluded from the group of chains defining the noncrystallographic symmetry restraints and the respective SOD dimer was also split into two TLS bodies. The structure was refined to an R factor of 24.3% and an R free of 27.7%.
TLS refinement was also employed for crystal form 2, defining each monomer as a single TLS body. Again, R factors were used as an indicator to determine the optimal strategy for TLS refinement. Here, the refinement converged with R = 20.1% and R free = 20.8%.
Structure factors and coordinates have been deposited in the Protein Data Bank (Berman et al., 2000 ▶) under PDB codes 3kh3 and 3kh4. Table 1 ▶ summarizes the data-collection and refinement statistics.
Table 1. Data-collection and refinement statistics.
Both data sets were collected from one single crystal. Values in parentheses are for the highest resolution shell.
| Crystal form 1 | Crystal form 2 | |
|---|---|---|
| Data collection | ||
| Space group | P212121 | P6522 |
| Unit-cell parameters (Å, °) | a = 75.5, b = 166.6, c = 175.0, α = β = γ = 90 | a = b = 112.2, c = 428.3, α = β = 90, γ = 120 |
| Wavelength | 0.9797 | 0.9797 |
| Resolution (Å) | 50–3.5 (3.6–3.5) | 50–3.5 (3.6–3.5) |
| Rmerge(I)† | 18.9 (57.5) | 17.2 (58.6) |
| Rmrgd-F‡ | 16.0 (41.3) | 13.9 (40.5) |
| Mean I/σ(I) | 11.2 (4.3) | 13.1 (4.2) |
| Completeness (%) | 99.9 (100) | 99.9 (100) |
| Redundancy | 6.1 (6.2) | 10.5 (10.9) |
| Refinement | ||
| Resolution (Å) | 50–3.5 (3.59–3.50) | 50–3.5 (3.59–3.50) |
| No. of reflections | 21151 (2100) | 20080 (1528) |
| Rwork | 24.3 (28.1) | 20.1 (25.7) |
| Rfree | 27.7 (31.1) | 20.8 (28.2) |
| No. of atoms | ||
| Protein | 13312 | 6660 |
| Ligand/ion | 39 | 12 |
| Water | — | — |
| B factors | ||
| Protein | 76 | 90 |
| Ligand/ion | 87 | 105 |
| Water | — | — |
| R.m.s. deviations | ||
| Bond lengths (Å) | 0.013 | 0.017 |
| Bond angles (°) | 1.137 | 1.434 |
| PDB code | 3kh3 | 3kh4 |
R
merge(I) = 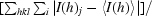
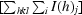 , where I(h)j is the measured diffraction intensity, 〈I(h)〉 is its average and the summation includes all observations.
, where I(h)j is the measured diffraction intensity, 〈I(h)〉 is its average and the summation includes all observations.
R
mrgd-F = 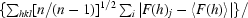
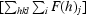 is a redundancy-independent merging R factor of structure-factor amplitudes. Symbols and indices are analogous to those in the calculation of R
merge and n is the number of observations of reflection h (Diederichs & Karplus, 1997 ▶).
is a redundancy-independent merging R factor of structure-factor amplitudes. Symbols and indices are analogous to those in the calculation of R
merge and n is the number of observations of reflection h (Diederichs & Karplus, 1997 ▶).
3. Results and discussion
3.1. Overexpression of human Cu/Zn superoxide dismutase (SOD1) in L. tarentolae
To date, the L. tarentolae-based expression system has been used for the production of intracellular and secreted proteins for various applications, including isotopic labelling of recombinant proteins for NMR studies (Foldynova-Trantirkova et al., 2009 ▶; Niculae et al., 2006 ▶). Although the system is clearly suitable for crystallographic applications, published examples of diffracting crystals obtained from recombinant proteins expressed in L. tarentolae are still lacking. To this end, we chose to produce the antioxidant Cu/Zn superoxide dismutase (EC 1.15.1.1) in L. tarentolae in order to use the recombinant protein for crystallization and structure determination. This enzyme catalyzes the dismutation of superoxide into oxygen and hydrogen peroxide (2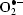 + 2H+ → H2O2 + O2). We chose a well established constitutive L. tarentolae expression system (LEXSY; Breitling et al., 2002 ▶) in which the gene-expression cassette is stably integrated into a chromosomal copy of the 18S rRNA gene. This architecture ensures that the target genes are co-transcribed with ribosomal genes by the host RNA polymerase I. The latter produces approximately ten times more RNA than RNA polymerase II in Leishmania (Clayton, 1999 ▶).
+ 2H+ → H2O2 + O2). We chose a well established constitutive L. tarentolae expression system (LEXSY; Breitling et al., 2002 ▶) in which the gene-expression cassette is stably integrated into a chromosomal copy of the 18S rRNA gene. This architecture ensures that the target genes are co-transcribed with ribosomal genes by the host RNA polymerase I. The latter produces approximately ten times more RNA than RNA polymerase II in Leishmania (Clayton, 1999 ▶).
We cloned the human SOD1 gene into the pLEXSY-sat1 vector, as described in §2, and constructed a recombinant SOD1-expressing L. tarentolae strain. The drug-resistant clones were analyzed by Coomassie Brilliant Blue staining of SDS–polyacrylamide gels and by Western blotting using SOD-specific antibodies (Fig. 1 ▶). Although the specific SOD1 band could be detected on stained SDS–PAGE gels, it was relatively weak, indicating low expression levels. In order to increase the expression levels of SOD1 in this system, we took advantage of the suite of pLEXSYcon1 expression vectors with four alternative antibiotic selection markers, permitting the integration of up to four expression cassettes into the Leishmania genome. The integration of additional SOD1 gene copies resulted in an additive increase in SOD1 expression levels as determined by SDS–PAGE gel staining (Fig. 1 ▶) and Western blotting (not shown). Clones expressing four copies of SOD1 could withstand prolonged cultivation without reductions in expression levels.
Figure 1.

Construction of L. tarentolae strains expressing human SOD and its purification. (a) SDS–PAGE analysis of wild-type and transgenic strains expressing SOD1. 8 × 106 cells from each strain were lysed in SDS–PAGE buffer, loaded onto an 15% SDS–PAGE gel and proteins were visualized by Coomassie staining. (b) Western blot analysis of L. tarentolae expressing four copies of the SOD1 gene in a fermenter using an anti-SOD1 antibody. Lysates from 8 × 106 cells were loaded into each lane. The first lane contains molecular-weight markers (labelled in kDa). (c) Pooled fractions of the main peak of SOD1 eluted from a phenyl Sepharose column analyzed by SDS–PAGE. (d) Analysis of the oligomerization state of recombinant SOD1 using different sample-preparation conditions. An unboiled sample migrates as a tetramer. (e) ESI–MS analysis of purified SOD1. (f) ESI–MS analysis of the presumably acetylated N-terminal SOD1 peptides released by digestion with chemotrypsin.
3.2. Large-scale expression and preparative purification of SOD1 from L. tarentolae
In order to obtain a sufficient amount of recombinant protein for crystallization trials, we cultivated the L. tarentolae strain with four SOD1 gene copies in a 30 l fermentation vessel filled with 10 l cultivation medium. The medium was inoculated with 1 l L. tarentolae pre-culture grown in two agitated Fernbach flasks with 0.5 l culture each. Fermentation was performed for 53 h and the expression levels of SOD1 were monitored by withdrawing samples and analyzing the normalized extracts from 8 × 106 cells by Western blotting. As can be seen in Fig. 1 ▶(b), the SOD1 expression levels per cell remained stable during the entire fermentation period, demonstrating that the expression paralleled the growth of the culture. The expression level reached 30 mg per litre of culture and appeared to be higher than that of small-scale expression. Following fermentation, the cells were collected by centrifugation and stored frozen at 193 K.
To isolate the recombinant SOD1, we disrupted the cells harvested from the fermentation culture using a fluidizer and removed the cell debris by centrifugation. The supernatant was subjected to stepwise ammonium sulfate fractionation. SOD1 remained in the supernatant, even at 70% ammonium sulfate saturation. Next, the supernatant was subjected to hydrophobic chromatography on phenyl Sepharose, which resulted in the elution of a peak containing essentially pure enzyme (Fig. 1 ▶ c). The described purification scheme resulted in a yield of 6.5 mg 90% pure SOD1 from 1 l of culture. The enzyme represented a noncovalent dimer; omission of the boiling step before loading onto the SDS–PAGE gel under nonreducing conditions resulted in a protein band at ∼48 kDa, whereas the boiled protein migrated at ∼24 kDa, a difference from the calculated molecular weight of 15 935.7 Da (Fig. 1 ▶ d). Mass-spectrometric analysis revealed that the molecular weight of the purified protein was 15 898 Da (Fig. 1 ▶ e). This discrepancy could be explained by N-terminal cleavage of methionine followed by acetylation at alanine, as described for human SOD1, and/or post-translational modifications (Kajihara et al., 1988 ▶). N-terminal acetylation would also explain our failure to sequence the N-terminus by Edman degradation (data not shown).
In order to assess the ability of Leishmania to N-terminally acetylate heterogeneously expressed proteins, we digested the obtained SOD1 with chymotrypsin and performed mass-spectrometric analysis of the resulting peptides. Using electrospray ionization mass spectrometry, a triple-charged acetylated chymotryptic peptide, Ac-ATKAVCVLKGDGPVQGIINF, with corresponding masses of m/z = 691.40, 691.67 and 691.93 Da was identified (the calculated masses were 691.37, 691.70 and 692.03 Da, respectively). Analysis of the fragmentation patterns demonstrated that there was no other chymotryptic peptide from SOD1 or chymotrypsin that corresponded to a mass of m/z = 691.40 Da.
3.3. Crystallization and X-ray analysis of L. tarentolae-produced recombinant SOD1
The purity and homogeneity of the recombinant SOD1 preparation, together with the fact that the protein could be concentrated without forming a precipitate, encouraged us to screen for new crystallization conditions employing standard methods, since this may indicate the general usefulness of the L. tarentolae expression system for structural studies of otherwise difficult-to-obtain eukaryotic proteins. Indeed, two previously undescribed crystal forms of SOD1 were obtained and it was possible to refine both structures to respectable R factors despite the low resolution of both data sets. This is likely to be a consequence of the availability of high-resolution template structures of SOD1 and is also likely to be a consequence of the presence of a large number of monomers in the asymmetric unit of both crystal forms, allowing the setup of well defined noncrystallographic symmetry restraints.
With respect to the large number of copies, new crystal form 1 is similar to previously published structures of SOD1 containing ten, 12 and 18 chains (Table 2 ▶). Similar to these structures and in contrast to several other PDB entries for SOD1, three SOD1 dimers are arranged in the form of a triangular wheel in which the individual dimers form the spokes. Crystal form 1 contains two such wheels lying side-by-side and stacked face-to-face at their centres in a perpendicular direction. The hub of the wheels contains a sulfate anion, which probably stems from the CuSO4 and ZnSO4 added to stabilize the active site of the enzyme. A sulfate ion was also observed in the corresponding position of PDB entry 1sos (Parge et al., 1992 ▶). In crystal form 1 it is held in place by a cluster of lysine residues (Lys75 and Lys128) that point towards this position from the three chains surrounding the centre of the wheel (Fig. 2 ▶ b).
Table 2. SOD1 crystal structures with high asymmetric unit content.
| No. of chains | PDB entry | Space group | Approximate unit-cell parameters (Å, °) | Reference |
|---|---|---|---|---|
| 10 | 1fun | C2221 | a = 166, b = 204, c = 144 | Fisher et al. (1997 ▶) |
| 1n18 | Cardoso et al. (2002 ▶) | |||
| 1pu0 | DiDonato et al. (2003 ▶) | |||
| 1sos | Parge et al. (1992 ▶) | |||
| 1uxl | Hough et al. (2004 ▶) | |||
| 2gbv | Hornberg et al. (2007 ▶) | |||
| 2zky | S. Yoshikawa, M. Kukimoto-Niino, K. Ito, K. Shirouzu, M. Urushitani, R. Takahashi & S. Yokoyama (unpublished work) | |||
| 3gzo | Galaleldeen et al. (2009 ▶) | |||
| 12 | 1uxm | P21 | a = 112, b = 146, c = 113, β = 120.1 | Hough et al. (2004 ▶) |
| 18 | 1hl5 | P21 | a = 77, b = 172, c = 112, β = 93.5 | Strange et al. (2003 ▶) |
Figure 2.

Crystal structure analysis of two new crystal forms of L. tarentolae-produced recombinant human SOD1. (a) Photograph of crystals. (b) Asymmetric unit of the new P212121 crystal form (crystal form 1). The asymmetric unit contains six SOD dimers arranged as two triangular wheels around sulfate ions. The wheels are arranged in a side-to-side fashion. Cu2+ (orange) and Zn2+ (grey) ions are shown as spheres. The sulfate is held in place by interaction with Lys129 from three different chains (insert). The packing arranges neighbouring wheels in a face-to-face orientation. (c) Honeycomb-like packing of SOD1 in the new P6522 crystals (crystal form 2). The asymmetric unit contains three SOD1 dimers arranged as a triangular wheel, which is shown as a coloured ribbon. For clarity, only one layer of the crystal lattice is shown.
Interestingly, new crystal form 2 contains a similar triangular wheel of SOD1 dimers in the asymmetric unit, but here no sulfate ion is observed in the centre despite the fact that both crystal forms originate from the same crystallization drops. In crystal form 2 neighbouring wheels also pack face-to-face, but the contacts in the horizontal direction are toe-to-toe rather than side-by-side as in crystal form 1. This generates a honeycomb-like packing, explaining the high solvent content of 67% (Fig. 2 ▶ c).
While the low resolution of the diffraction data presented here precludes a more detailed analysis of the structure and although such an analysis is also beyond the aim of this study, analysis of the electron density could not confirm the presence of the N-terminal acetylation proposed based on the ESI–MS analysis. This is probably a consequence of the flexibility of the corresponding amino acids, as exemplified by the two different conformations observed in higher resolution structures of SOD1 (e.g. PDB entry 2v0a; Strange et al., 2007 ▶). The electron density was nevertheless clear enough to highlight several smaller deviations with respect to the molecular-replacement search model in crystal contact areas of the two new crystal forms described here. No indications of post-translational modifications, such as the previously described phosphorylation or glutathionylation observed in SOD1 isolated from human erythrocytes (Wilcox et al., 2009 ▶), were obtained. The electron density also revealed that some of the metal-binding sites were not fully occupied by Cu2+ or Zn2+, despite addition of these ions during the course of fermentation and protein purification. This may also explain why attempts to crystallize the protein under the conditions that have been established in the literature were unsuccessful.
4. Conclusions
In summary, our results demonstrate that L. tarentolae is well suited as an expression host for the production of recombinant protein for structural studies. With a large choice of defined complexes and media, some of which are comparable in price to bacterial media, Leishmania provides an inexpensive alternative to mammalian and insect cultures (Fritsche et al., 2008 ▶). The scalability of the system and the availability of vectors for co-expression of multiple genes, as well as its inducible architectures, compare favourably with other eukaryotic expression systems (Basile & Peticca, 2009 ▶). We therefore believe that the L. tarentolae expression system is a system of choice if the protein cannot be produced in E. coli. Development of multihost Leishmania vectors that are capable of mediating protein production in E. coli would be particularly advantageous and would expand the utility of the system to structural proteomics applications.
Supplementary Material
PDB reference: human Cu/Zn superoxide dismutase, 3kh3
PDB reference: 3kh4
Acknowledgments
We wish to thank Anett Geyer and Sandra Thuns for technical assistance during construction of the expression plasmids, Nataliya Lupilova for technical assistance during SOD1 purification and Tim Bergbrede for ESI–MS analysis. This work was supported in part by DFG grant AL 484/8-1 to KA, MSMT LC07032 and the Praemium Academiae award to JL. We thank the X-ray communities of the Max-Planck-Institutes of Molecular Physiology (Dortmund, Germany) and for Medical Research (Heidelberg, Germany) for collecting diffraction data at the Swiss Light Source of the Paul Scherrer Institute (Villigen, Switzerland). The help of the staff at beamline X10SA is gratefully acknowledged.
References
- Basile, G. & Peticca, M. (2009). Mol. Biotechnol.43, 273–278. [DOI] [PubMed]
- Berman, H. M., Westbrook, J., Feng, Z., Gilliland, G., Bhat, T. N., Weissig, H., Shindyalov, I. N. & Bourne, P. E. (2000). Nucleic Acids Res.28, 235–242. [DOI] [PMC free article] [PubMed]
- Breitling, R., Klingner, S., Callewaert, N., Pietrucha, R., Geyer, A., Ehrlich, G., Hartung, R., Muller, A., Contreras, R., Beverley, S. M. & Alexandrov, K. (2002). Protein Expr. Purif.25, 209–218. [DOI] [PubMed]
- Cardoso, R. M., Thayer, M. M., DiDonato, M., Lo, T. P., Bruns, C. K., Getzoff, E. D. & Tainer, J. A. (2002). J. Mol. Biol.324, 247–256. [DOI] [PubMed]
- Clayton, C. E. (1999). Parasitol. Today, 15, 372–378. [DOI] [PubMed]
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50, 760–763.
- DiDonato, M., Craig, L., Huff, M. E., Thayer, M. M., Cardoso, R. M., Kassmann, C. J., Lo, T. P., Bruns, C. K., Powers, E. T., Kelly, J. W., Getzoff, E. D. & Tainer, J. A. (2003). J. Mol. Biol.332, 601–615. [DOI] [PubMed]
- Diederichs, K. & Karplus, P. A. (1997). Nature Struct. Biol.4, 269–275. [DOI] [PubMed]
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed]
- Fisher, C. L., Cabelli, D. E., Hallewell, R. A., Beroza, P., Lo, T. P., Getzoff, E. D. & Tainer, J. A. (1997). Proteins, 29, 103–112. [DOI] [PubMed]
- Foldynova-Trantirkova, S., Matulova, J., Dotsch, V., Lohr, F., Cirstea, I., Alexandov, K., Breitling, R., Lukes, J. & Trantirek, L. (2009). J. Biomol. Struct. Dyn.26, 755–761. [DOI] [PubMed]
- Fritsche, C., Sitz, M., Wolf, M. & Pohl, H. D. (2008). J. Basic Microbiol.48, 488–495. [DOI] [PubMed]
- Galaleldeen, A., Strange, R. W., Whitson, L. J., Antonyuk, S. V., Narayana, N., Taylor, A. B., Schuermann, J. P., Holloway, S. P., Hasnain, S. S. & Hart, P. J. (2009). Arch. Biochem. Biophys.492, 40–47. [DOI] [PMC free article] [PubMed]
- Hornberg, A., Logan, D. T., Marklund, S. L. & Oliveberg, M. (2007). J. Mol. Biol.365, 333–342. [DOI] [PubMed]
- Hough, M. A., Grossmann, J. G., Antonyuk, S. V., Strange, R. W., Doucette, P. A., Rodriguez, J. A., Whitson, L. J., Hart, P. J., Hayward, L. J., Valentine, J. S. & Hasnain, S. S. (2004). Proc. Natl Acad. Sci. USA, 101, 5976–5981. [DOI] [PMC free article] [PubMed]
- Kabsch, W. (2010). Acta Cryst. D66, 125–132. [DOI] [PMC free article] [PubMed]
- Kajihara, J., Enomoto, M., Nishijima, K., Yabuuchi, M. & Katoh, K. (1988). J. Biochem.104, 851–854. [DOI] [PubMed]
- Kushnir, S., Gase, K., Breitling, R. & Alexandrov, K. (2005). Protein Expr. Purif.42, 37–46. [DOI] [PubMed]
- Long, F., Vagin, A. A., Young, P. & Murshudov, G. N. (2008). Acta Cryst. D64, 125–132. [DOI] [PMC free article] [PubMed]
- Murshudov, G. N., Vagin, A. A. & Dodson, E. J. (1997). Acta Cryst. D53, 240–255. [DOI] [PubMed]
- Niculae, A., Bayer, P., Cirstea, I., Bergbrede, T., Pietrucha, R., Gruen, M., Breitling, R. & Alexandrov, K. (2006). Protein Expr. Purif.48, 167–172. [DOI] [PubMed]
- Parge, H. E., Hallewell, R. A. & Tainer, J. A. (1992). Proc. Natl Acad. Sci. USA, 89, 6109–6113. [DOI] [PMC free article] [PubMed]
- Service, R. F. (2002). Science, 298, 948–950. [DOI] [PubMed]
- Strange, R. W., Antonyuk, S., Hough, M. A., Doucette, P. A., Rodriguez, J. A., Hart, P. J., Hayward, L. J., Valentine, J. S. & Hasnain, S. S. (2003). J. Mol. Biol.328, 877–891. [DOI] [PubMed]
- Strange, R. W., Yong, C. W., Smith, W. & Hasnain, S. S. (2007). Proc. Natl Acad. Sci. USA, 104, 10040–10044. [DOI] [PMC free article] [PubMed]
- Vagin, A. & Teplyakov, A. (2010). Acta Cryst. D66, 22–25. [DOI] [PubMed]
- Wilcox, K. C., Zhou, L., Jordon, J. K., Huang, Y., Yu, Y., Redler, R. L., Chen, X., Caplow, M. & Dokholyan, N. V. (2009). J. Biol. Chem.284, 13940–13947. [DOI] [PMC free article] [PubMed]
- Winn, M. D., Isupov, M. N. & Murshudov, G. N. (2001). Acta Cryst. D57, 122–133. [DOI] [PubMed]
- Zhang, W. W., Charest, H. & Matlashewski, G. (1995). Nucleic Acids Res.23, 4073–4080. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: human Cu/Zn superoxide dismutase, 3kh3
PDB reference: 3kh4