

In order to investigate its function in transcriptional gene silencing, the highly conserved motif 2 from A. thaliana Morpheus’ molecule 1 protein was expressed, purified and crystallized. X-ray diffraction analysis is reported to a resolution of 3.2 Å.
Keywords: Morpheus’ molecule 1, conserved MOM1 motif 2, coiled-coil domain, epigenetic, transcriptional gene silencing
Abstract
Of the known epigenetic control regulators found in plants, the Morpheus’ molecule 1 (MOM1) protein is atypical in that the deletion of MOM1 does not affect the level of epigenetic marks controlling the transcriptional status of the genome. A short 197-amino-acid fragment of the MOM1 protein sequence can complement MOM1 deletion when coupled to a nuclear localization signal, suggesting that this region contains a functional domain that compensates for the loss of the full-length protein. Numerous constructs centred on the highly conserved MOM1 motif 2 (CMM2) present in these 197 residues have been generated and expressed in Escherichia coli. Following purification and crystallization screening, diamond-shaped single crystals were obtained that diffracted to ∼3.2 Å resolution. They belonged to the trigonal space group P3121 (or P3221), with unit-cell parameters a = 85.64, c = 292.74 Å. Structure determination is ongoing.
1. Introduction
The Arabidopsis thaliana Morpheus’ molecule 1 (MOM1) is an epigenetic regulator that is involved in transcriptional gene silencing (TGS). However, unlike other epigenetic regulators, which induce TGS by altering epigenetic marks, MOM1 induces TGS without major changes in the covalent modifications of DNA and histones (Amedeo et al., 2000 ▶; Habu et al., 2006 ▶; Vaillant et al., 2006 ▶). These exceptional features of MOM1 suggest that it acts downstream or independently of the classical epigenetic marks. In fact, recent studies revealed that at some loci MOM1 functions downstream of RNA-directed DNA methylation (RdDM), while at other loci MOM1 alters TGS independently of RdDM (Numa et al., 2010 ▶; Yokthongwattana et al., 2010 ▶).
The MOM1 gene encodes a large nuclear protein of 2001 amino acids with homology to part of the helicase domain present in SNF2 chromatin-remodelling factors (Amedeo et al., 2000 ▶). Bioinformatics predictions suggested the presence of an actin-binding domain, nuclear localization signals (NLS) and a putative transmembrane domain (Amedeo et al., 2000 ▶). Surprisingly, functional studies of MOM1 deletions revealed that a small part of the protein, comprising less than 13% of the entire sequence, retains TGS activity when linked to an NLS (Čaikovski et al., 2008 ▶). This active 197-amino-acid polypeptide contains a novel conserved plant-specific protein motif named CMM2 (conserved MOM1 motif 2). The CMM2 sequence corresponds to residues 1734–1814 and can be found in the MOM1 homologues of all vascular plants for which complete genome sequences are available (Čaikovski et al., 2008 ▶). Moreover, the CMM2 from poplar (Populus trichocarpa) can initiate TGS in Arabidopsis thaliana, suggesting structural and functional conservation. Currently, the molecular mechanisms by which CMM2 regulates TGS remain unknown (Čaikovski et al., 2008 ▶; Numa et al., 2010 ▶; Yokthongwattana et al., 2010 ▶).
Structure-prediction programs indicate that the CMM2 sequence could fold into a long α-helix, possibly forming a degenerate coiled-coil domain. A high-resolution X-ray crystal structure of CMM2 would provide a framework to investigate the mechanisms by which MOM1 functions in TGS. In the following, we describe the crystallization and X-ray diffraction analysis of CMM2.
2. Materials and methods
2.1. Protein cloning
The conserved CMM2 sequence encoding the predicted coiled-coil domain was amplified from the miniMOM1 clone (Čaikovski et al., 2008 ▶) using KOD Hot Start DNA polymerase according to the manufacturer’s instructions (Toyobo, Osaka, Japan) and cloned into a pT5T expression vector downstream of a T7 promoter. Multiple constructs with a variety of N-terminal and C-terminal boundaries around this conserved region were created to facilitate protein solubility and, at later stages, crystal optimization. Every clone was sequenced to verify the correct nucleotide sequence.
2.2. Protein expression and purification
The same expression and purification procedures were used for all of the CMM2 fragments (Table 1 ▶). Within the MOM1 functional polypeptide that complements MOM1 deletion (residues 1662–1860), the region containing the CMM2 (residues 1699–1814) reproducibly yielded soluble protein, while the construct containing residues 1700–1814 led to the highest diffracting crystals.
Table 1. Relative CMM2 expression levels.
Levels are indicated by ‘+’ signs: a single ‘+’ denotes a low level of expression and ‘+++’ indicates a high level. A ‘–’ sign indicates no expression or insoluble protein.
| Construct boundaries | Expression level | Purification | Crystals obtained | Maximum resolution (Å) |
|---|---|---|---|---|
| 1675–2001 | − | |||
| 1675–1823 | ++ | Yes | ||
| 1692–1805 | ++ | Yes | ||
| 1692–1817 | ++ | |||
| 1692–1823 | ++ | Yes | ||
| 1692–2001 | ||||
| 1699–1805 | + | Yes | ||
| 1699–1813 | +++ | Yes | ||
| 1699–1814 | +++ | Yes | Yes | 3.6–3.8 |
| 1699–1817 | +++ | |||
| 1699–1823 | ++ | |||
| 1700–1813 | +++ | |||
| 1700–1814 | +++ | Yes | Yes | 3.2–3.5 |
| 1715–1805 | + |
The various constructs were expressed in Escherichia coli BL21 (DE3) cells (Stratagene) using LB medium. Cell cultures were grown at 310 K to an OD600 of ∼0.6–0.8, at which point cell cultures were harvested and transferred to fresh LB medium equilibrated at 303 K. Protein expression was performed at 303 K for 4 h by induction with 0.15 mM isopropyl β-d-1-thiogalactopyranoside (IPTG).
The cells were lysed by pressure disruption at 277 K in buffer consisting of 25 mM 2-(N-morpholino)ethanesulfonic acid (MES) pH 6.0, 100 mM NaCl, 5 mM DTT and protease inhibitors, which was followed by ultracentrifugation for 1 h at 277 K to remove cell debris. The CMM2 polypeptides were purified at 277 K by cation exchange (Sepharose SP column; Pharmacia Biotech) in a buffer consisting of 25 mM MES pH 6.0, 5 mM DTT and protease inhibitors using a gradient of 50 mM to 1 M NaCl. A second purification step was performed at 277 K via gel filtration (Superdex 200 column; Pharmacia Biotech) in MES buffer (25 mM MES pH 6.0, 200 mM NaCl, 5 mM DTT) and the fractions containing the CMM2 fragment were concentrated (Centriprep YM-10, Millipore) to 15–18 mg ml−1. The protein solution was either directly used in crystal screening or flash-frozen in liquid nitrogen and stored at 193 K.
2.3. Protein crystallization
The purified MOM1 CMM2 protein fragments were screened for crystallization at 277 K with custom sparse-matrix screens as well as commercial screens (PEG/Ion, Index and Crystal Screens, Hampton Research) in 48-well plates (type VDX48 with sealant from Hampton Research) using the hanging-drop vapour-diffusion technique or by the microbatch method using a novel microfluidics crystallization system (Emamzadah et al., 2009 ▶). Crystallization experiments were performed by mixing equal amounts of protein solution (protein concentration of 15 mg ml−1 in MES buffer) and reservoir solution containing 0.1 M Tris pH 8.5 and 0.3 M magnesium formate dihydrate. The volume of the drops and of the reservoir used in the hanging-drop experiments were 2 and 250 µl, respectively. Crystals of CMM2 (residues 1699–1814 and 1700–1814) formed using both methods and the diamond-shaped crystals reached 200 × 100 × 50 nm in size on average in 3–5 d (Fig. 1 ▶).
Figure 1.

Representative crystals of MOM1 CMM2 (residues 1700–1814) crystallized using 0.1 M Tris pH 8.5, 0.3 M magnesium formate dihydrate that diffracted to 3.2–3.5 Å resolution. This image was captured under polarized light. The crystals are colourless.
2.4. Data collection and analysis
The crystals were gently stabilized for radiation exposure via multiple buffer exchanges at 277 K from the above-mentioned crystallization solution to a final solution composed of 0.1 M Tris pH 8.5, 0.32 M magnesium formate dihydrate and 20% ethylene glycol. The crystals appeared to be quite sensitive and an incubation period of 5–10 min had to be respected between each buffer exchange. Furthermore, each change in the concentrations of the crystallization solution components during buffer exchange had to remain below 15% in order to avoid reduced crystal diffraction properties. At the end of the stabilization procedure, the crystals were left overnight at 277 K to equilibrate against the final cryoprotectant solution. The equilibrated crystals were then mounted in loops and plunged into liquid nitrogen for storage, transport and data collection. Despite the use of the above-mentioned cryostabilization procedure and the delicate treatment of the crystals, several of them showed medium- to low-resolution diffraction properties. All data were collected at the European Synchrotron Radiation Facility (ESRF, Grenoble, France) on beamlines ID23-1, ID29 and ID14-4. A complete 3.2 Å resolution data set was collected from a single CMM2 crystal (residues 1700–1814) at an X-ray wavelength of 1.27 Å on ID14-4 and the images were indexed and integrated using the XDS software (Kabsch, 2010 ▶). Crystal parameters and diffraction statistics are summarized in Table 2 ▶.
Table 2. Data-collection and processing statistics.
Values in parentheses are for the outer shell.
| Crystal system | Trigonal |
| Space group | P3121 or P3221 |
| Unit-cell parameters (Å) | a = 85.64, c = 292.74 |
| Matthews coefficient† (Å3 Da−1) | 1.67–6.25 |
| Corresponding solvent content† (%) | 26–80 |
| Possible No. of molecules per asymmetric unit† | 4–15 |
| Diffraction source | ESRF ID14-4 |
| Wavelength (Å) | 1.27 |
| Resolution range (Å) | 50.0–3.2 (3.28–3.2) |
| No. of observed reflections | 463255 (19098) |
| No. of unique reflections | 21342 (1368) |
| Completeness (%) | 99.5 (96.6) |
| Redundancy | 21.7 |
| I/σ(I) | 20.56 (2.15) |
| Rmrgd-F‡ (%) | 0.072 (0.893) |
Values were calculated using the Matthews Probability Calculator (Kantardjieff & Rupp, 2003 ▶; http://ruppweb.dyndns.org/mattprob/). More extreme values were excluded on the basis of their lower probability (below 0.0011).
R
mrgd-F is the quality of the reduced structure-factor amplitudes according to Diederichs & Karplus (1997 ▶): R
mrgd-F = 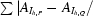
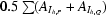 , where A
I = I
1/2 if I ≥ 0 and A
I = −I
1/2 if I < 0, I
h,P =
, where A
I = I
1/2 if I ≥ 0 and A
I = −I
1/2 if I < 0, I
h,P = 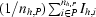 and Ih,Q =
and Ih,Q = 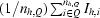 .
.
3. Results and discussion
We generated constructs of various lengths spanning the putative coiled-coil region formed by the CMM2 motif in order to identify N-terminal and C-terminal boundaries that allowed the expression of soluble recombinant protein in E. coli (Table 1 ▶). However, purification of the soluble region had to be performed with particular attention to the sample temperature given that the purified protein immediately precipitated when removed from ice (at a temperature of >280 K). A slight reduction in the lysis buffer pH (from 6.5 to 6.0) helped to increase CMM2 stability. The protein was purified using standard chromatographic techniques (ion-exchange and size-exclusion chromatography), concentrated to ∼15 mg ml−1 and crystallized at 277 K.
Crystals of CMM2 (residues 1699–1814) appeared after 3–4 d but only showed X-ray diffraction to ∼7 Å resolution. After extensive optimization of the cryo-preservation protocol, the diffraction limit could be increased to ∼3.6–3.8 Å (see §2.4). At this stage, slightly smaller constructs were designed in an attempt to improve crystal contacts and possibly further increase the resolution limit. The CMM2 construct encompassing residues 1700–1814 yielded crystals that reproducibly diffracted to ∼3.2–3.5 Å resolution with a reasonable signal-to-noise ratio [I/σ(I) > 2 in the highest resolution shell]. A complete data set was collected with the statistics reported in Table 2 ▶ and after indexing the crystals were found to belong to space group P3121 or P3221 (space group No. 152 or 154, respectively). Our major concern now lies in determining the exact number of molecules in the asymmetric unit. Given that the CMM2 monomer is only 114 amino acids in length (∼13 kDa), the possible number of polypeptides in the asymmetric unit ranges from four to 15 monomers, with corresponding solvent contents of 80 and 26%, respectively (Matthews, 1968 ▶). We expect that the number of molecules per asymmetric unit will be small (4–6 molecules). Indeed, CMM2-containing crystals are very sensitive to variation of the crystallization solution, indicating a large solvent-content value, which corresponds to a small number of molecules. Currently, we are pursuing molecular-replacement strategies using long α-helix structures, as well as de novo heavy-atom location with selenomethionine substitution and heavy-atom derivatives, in order to obtain the missing phase information. We expect that having a model of the CMM2-domain structure will facilitate our understanding of the function of MOM1 in the regulation of transcriptional gene-silencing mechanisms.
Acknowledgments
We thank Daniel Hess of the Friedrich Miescher Institute for Biomedical Research, Basel, Switzerland for LC-MS/MS analysis of the CMM2 protein fragments (residues 1699–1823 and 1692–1805). We acknowledge the support from the ESRF beamline scientists in charge of ID23-1, ID29 and ID14-4 at ESRF (Grenoble, France). Additionally, we thank Scarlet Versini for generously providing materials. This work was supported by grants from the Swiss National Fund (31003A_125005 to JP, 3100A_112434 to TDH and 31003A_124909 to ST).
References
- Amedeo, P., Habu, Y., Afsar, K., Mittelsten Scheid, O. & Paszkowski, J. (2000). Nature (London), 405, 203–206. [DOI] [PubMed]
- Čaikovski, M., Yokthongwattana, C., Habu, Y., Nishimura, T., Mathieu, O. & Paszkowski, J. (2008). PLoS Genet.4, e1000165. [DOI] [PMC free article] [PubMed]
- Diederichs, K. & Karplus, P. A. (1997). Nature Struct. Biol.4, 269–275. [DOI] [PubMed]
- Emamzadah, S., Petty, T. J., De Almeida, V., Nishimura, T., Joly, J., Ferrer, J.-L. & Halazonetis, T. D. (2009). Acta Cryst. D65, 913–920. [DOI] [PMC free article] [PubMed]
- Habu, Y., Mathieu, O., Tariq, M., Probst, A. V., Smathajitt, C., Zhu, T. & Paszkowski, J. (2006). EMBO Rep.7, 1279–12784. [DOI] [PMC free article] [PubMed]
- Kabsch, W. (2010). Acta Cryst. D66, 125–132. [DOI] [PMC free article] [PubMed]
- Kantardjieff, K. A. & Rupp, B. (2003). Protein Sci.12, 1865–1871. [DOI] [PMC free article] [PubMed]
- Matthews, B. W. (1968). J. Mol. Biol.33, 491–497. [DOI] [PubMed]
- Numa, H., Kim, J. M., Matsui, A., Kurihara, Y., Morosawa, T., Ishida, J., Mochizuki, Y., Kimura, H., Shinozaki, K., Toyoda, T., Seki, M., Yoshikawa, M. & Habu, Y. (2010). EMBO J.29, 352–362. [DOI] [PMC free article] [PubMed]
- Vaillant, I., Schubert, I., Tourmente, S. & Mathieu, O. (2006). EMBO Rep.7, 1273–1278. [DOI] [PMC free article] [PubMed]
- Yokthongwattana, C., Bucher, E., Čaikovski, M., Vaillant, I., Nicolet, J., Mittelsten Scheid, O. & Paszkowski, J. (2010). EMBO J.29, 340–351. [DOI] [PMC free article] [PubMed]