

Xylulose-5-phosphate/fructose-6-phosphate phosphoketolase from B. breve was overexpressed and crystallized. The crystals belonged to the tetragonal space group I422 and diffracted to beyond 1.7 Å resolution.
Keywords: xylulose-5-phosphate/fructose-6-phosphate phosphoketolase, Bifidobacterium breve, bifid shunt, heterofermentative lactic acid bacteria
Abstract
The xylulose-5-phosphate/fructose-6-phosphate phosphoketolase gene from Bifidobacterium breve was cloned and overexpressed in Escherichia coli. The enzyme was purified to homogeneity and crystallized by the sitting-drop vapour-diffusion method. Crystals were obtained at 293 K using 0.05 mM thiamine diphosphate, 0.25 mM MgCl2, 24%(w/v) PEG 6000 and 0.1 M Bicine pH 9.0. The crystals belonged to the tetragonal space group I422, with unit-cell parameters a = b = 174.8, c = 163.8 Å, and diffracted to beyond 1.7 Å resolution.
1. Introduction
Heterofermentative lactic acid bacteria (HLAB) ferment sugars using the phosphoketolase (PK) pathway to produce lactic and acetic acids as the major end products (Zaunmüller et al., 2006 ▶). PK, which requires thiamine diphosphate (ThDP) and Mg2+ as cofactors, is a key enzyme in this pathway. PKs have been classified into two types based on substrate preference: xylulose-5-phosphate (X5P) phosphoketolases (XPKs; EC 4.1.2.9), which only act on X5P (Heath et al., 1958 ▶; Hurwitz, 1958 ▶), and X5P/fructose-6-phosphate (F6P) phosphoketolases (XFPKs; EC 4.1.2.22), which act on both X5P and F6P with comparable activities (Schramm et al., 1958 ▶; Goldberg & Racker, 1962 ▶; Sgorbati et al., 1976 ▶; Meile et al., 2001 ▶). PKs (XPK and XFPK) catalyze the cleavage of X5P or F6P utilizing inorganic phosphate (Pi) to produce acetyl phosphate (acetyl-P), H2O and glyceraldehyde 3-phosphate or erythrose 4-phosphate. The high-energy metabolite acetyl-P is subsequently converted to acetic acid by acetate kinase to produce ATP from ADP in the pathway.
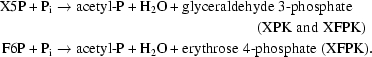 |
Bifidobacteria are strictly anaerobic Gram-positive HLAB. As major components of human intestinal flora, bifidobacteria are beneficial and commensal because they exhibit health-promoting effects such as protection against pathogens, treatment of diarrhoea, immunostimulatory activity and anticarcinogenic activity (Picard et al., 2005 ▶). In contrast to many HLAB that use the pentose monophosphate pathway involving X5P as the only substrate for PK, bifidobacteria use a distinctive bifid shunt as a sugar-fermentation pathway. Both F6P and X5P are phosphorolytically cleaved in this pathway (Scardovi & Trovatelli, 1965 ▶; Sela et al., 2008 ▶). PK activity against F6P thus serves as a tool for identifying bifidobacteria (Scardovi, 1986 ▶).
To date, XFPK-type genes from Bifidobacterium lactis (Meile et al., 2001 ▶) and B. animalis (Chinen et al., 2007 ▶) and XPK-type genes from Lactobacillus pentosus (Posthuma et al., 2002 ▶), L. paraplantarum (Jeong et al., 2007 ▶), L. plantarum (Yevenes & Frey, 2008 ▶) and Leuconostoc mesenteroides (Lee et al., 2005 ▶) have been cloned and characterized. The amino-acid sequence identities between the XPK and XFPK groups are about 50%, but they are phylogenetically distinct. Recently, a detailed kinetic study of XPK from Lactobacillus plantarum has been reported (Yevenes & Frey, 2008 ▶).
PK is a member of a large superfamily of ThDP-dependent enzymes and is classified into the transketolase family (Duggleby, 2006 ▶). In the transketolase family, crystal structures of transketolases from several organisms and 1-deoxy-d-xylulose 5-phosphate synthase have been determined to date (Lindqvist et al., 1992 ▶; Xiang et al., 2007 ▶). However, PKs show only a slight sequence similarity to these structurally known proteins within a short region forming the ThDP-binding site. Here, we report the cloning of the XFPK gene (xfp) from B. breve and the characterization, crystallization and preliminary X-ray studies of the recombinant enzyme (BbXFPK).
2. Materials and methods
2.1. Cloning, expression and purification
B. breve strain 203 was isolated from infant faeces in our laboratory (Nunoura et al., 1996 ▶). The xfp gene was cloned from the genomic DNA of B. breve 203 using a standard colony hybridization method. An xfp-specific probe was prepared by PCR using the primer pair xfp-1 (5′-GTYATHGGNACNCNTGGCA-3′) and xfp-2 (5′-CCRTTRTCNACNGCRAAYTGRAA-3′), which were designed based on the N-terminal (TNPVIGTPWQK) and internal (AFQFAVDNGY) amino-acid sequences of the purified enzyme. The DNA sequence of the insert was determined for both strands by the dideoxy chain-termination method. The nucleotide sequence of the xfp gene was deposited in GenBank under accession No. GU936109. The open reading frame of the xfp gene in the plasmid pSA103 was amplified by PCR using the following primer pair: 5′-GGGAATTCCATATGACAAATCCTGTTATTGG-3′ and 5′-CGCGGATCCTCACTCGTTGTCGCCTGCGGT-3′ (restriction sites are shown in bold). The PCR product was digested with NdeI and BamHI and then inserted into pET28b(+) vector (Novagen, Madison, Wisconsin, USA) to construct the expression plasmid. This plasmid produces protein with a His6 tag at its N-terminus. The expression plasmid was introduced into Escherichia coli BL21 CodonPlus (DE3)-RIL strain (Stratagene, La Jolla, California, USA). The transformants were cultured in Luria–Bertani medium containing 50 mg ml−1 kanamycin at 310 K. Protein expression was induced by treatment with 1 mM isopropyl β-d-1-thiogalactopyranoside for 16 h at 298 K. The cells were harvested by centrifugation at 4000g for 15 min and suspended in buffer A (20 mM HEPES–NaOH pH 7.2, 0.2 mM ThDP and 1 mM MgCl2) supplemented with 0.2 mM phenylmethylsulfonyl fluoride. Cells were lysed using a sonifier and the resulting cell debris was removed by centrifugation at 14 000g for 40 min. The supernatant was subjected to an Ni–NTA column (Qiagen, Hilden, Germany) equilibrated with buffer A. The eluate with 250 mM imidazole pH 7.2 was desalted and concentrated using a Vivaspin (10 000 molecular-weight cutoff; Sartorius Stedim, Göttingen, Germany). The sample was loaded onto a MonoQ HR 10/100 GL column (GE Healthcare, Buckinghamshire, England) and the protein was eluted with a linear gradient (0–1 M) of NaCl in buffer A. The protein was further purified by HiLoad 16/60 Superdex 200 pg gel-filtration chromatography (GE Healthcare) in buffer A supplemented with 0.1 M NaCl. The active fractions were collected individually and concentrated using an Amicon Ultra centrifugal filter device (10 000 molecular-weight cutoff; Millipore, Bedford, Massachusetts, USA). Using the filter device, the sample was desalted and the buffer was changed to 5 mM HEPES–NaOH pH 7.2, 0.05 mM ThDP and 0.25 mM MgCl2. The His6 tag was not removed prior to crystallization.
2.2. Crystallization and data collection
Initial crystallization trials were performed using the sitting-drop vapour-diffusion method at 288 and 277 K using Wizard I, II and III (Emerald BioSystems, Bainbridge Island, Washington, USA) and Crystal Screen and Crystal Screen 2 (Hampton Research, Aliso Viejo, California, USA) crystallization kits. Intelli-Plates (Art Robbins Instruments, Sunnyvale, California, USA) were used for crystallization. Crystals grew in condition No. 11 of the Wizard III kit [20%(w/v) PEG 6000, 0.1 M Bicine pH 9.0]. After refining the crystallization condition, large plate-like crystals were obtained within a few days at 293 K (Fig. 1 ▶). The optimized conditions were obtained by mixing 0.7 µl protein solution containing 25 mg ml−1 BbXFPK, 0.05 mM ThDP and 0.25 mM MgCl2 with an equal volume of reservoir solution [24%(w/v) PEG 6000, 0.1 M Bicine pH 9.0] and equilibrating against 60 µl reservoir solution. Diffraction data were collected on beamline NE3A at Photon Factory-Advanced Ring, High Energy Accelerator Research Organization (KEK, Tsukuba, Japan; λ = 1.000 Å). The crystals were soaked in cryoprotectant solution [20%(v/v) ethylene glycol in precipitant solution] and then flash-frozen in a nitrogen stream at 100 K. Diffraction data were recorded using an ADSC Quantum 270 charge-coupled device camera with an oscillation steps of 0.5° over a range of 180°. The data set was processed and scaled using HKL-2000 (Otwinowski & Minor, 1997 ▶).
Figure 1.

A BbXFPK crystal.
2.3. Enzyme assays
PK activity was detected by measuring the generated acetyl-P (Lipmann & Tuttle, 1945 ▶; Racker, 1962 ▶). A colouring reagent was prepared by mixing 43 µl 15%(v/v) trichloroacetic acid and 152 µl each of 5%(w/v) FeCl3 in 0.1 M HCl, 4 M HCl and Milli-Q water just before use. The standard reaction solution consisted of 27 mM X5P or F6P, 40 mM potassium phosphate pH 6.5, 25 mM MES buffer pH 6.5, 0.02 mM ThDP and 0.1 mM MgCl2. To determine the kinetic parameters, 1–80 mM F6P and 0.5–80 mM potassium phosphate were used. The reaction was initiated by adding 25 µl enzyme solution to a final volume of 250 µl reaction solution. To determine the pH optimum, 100 mM potassium phosphate buffer pH 4.3–9.0 was used. After incubation at 310 K for 15, 30 and 45 min, 250 µl 2 M hydroxylamine solution pH 6.4 was added to stop the reaction. The solution was kept at room temperature for 10 min to allow hydroxamic acid formation. Finally, 500 µl of the colouring reagent was added to the solution, which was then centrifuged at 17 400g for 5 min. The amount of acetyl-P produced was quantified by the absorbance of the supernatant at 505 nm using a DU 7400 spectrometer (Beckman, Palo Alto, California, USA). Acetyl-P (Sigma, St Louis, Missouri, USA) was used as a standard. One unit of activity was defined as the amount of enzyme catalyzing the formation of 1 µmol acetyl-P per minute.
3. Results and discussion
BbXFPK consists of 825 amino acids with a calculated molecular mass of 92.7 kDa and exhibits high amino-acid sequence identity (94%) to XFPK from B. lactis (Meile et al., 2001 ▶). The estimated molecular mass of the recombinant BbXFPK protein on SDS–PAGE was approximately 90 kDa, while three active peaks appeared on gel-filtration chromatography with estimated native molecular masses of 200 kDa (homodimer), 350 kDa (homotetramer) and 600 kDa (homohexamer). From the Coomassie-stained SDS–PAGE, all of the peaks were judged to contain pure BbXFPK. The major peak corresponded to the homohexamer, while the other two peaks were minor. XFPKs from several bifidobacterial strains have been reported to be homohexamers (Meile et al., 2001 ▶; Sgorbati et al., 1976 ▶). The sample from the peak corresponding to the homohexamer was used for subsequent characterization. BbXFPK exhibited typical Michaelis–Menten-type saturation kinetics (data not shown). The specific activities of BbXFPK towards F6P and X5P were 14.5 and 29.0 U mg−1, respectively, demonstrating that it is indeed a dual substrate-specific XFPK. XFPK from B. lactis also exhibits a higher activity towards X5P than F6P (Meile et al., 2001 ▶). The K m values for F6P and Pi and the k cat value were 9.7 ± 0.3 mM, 1.2 ± 0.2 mM and 1540 ± 60 min−1, respectively. The K M values are comparable with previously reported values for PKs from various HLABs (Grill et al., 1995 ▶; Jeong et al., 2007 ▶; Meile et al., 2001 ▶; Schramm et al., 1958 ▶; Sgorbati et al., 1976 ▶; Yevenes & Frey, 2008 ▶). The optimum pH range of BbXFPK was from 5.5 to 6.5 when F6P was used as a substrate.
A sample from the major peak, which consisted of the active homohexamer fraction, was successfully crystallized, whereas those from the minor homodimer and homotetramer peaks failed to crystallize. The BbXFPK crystals diffracted to beyond 1.7 Å resolution and belonged to the tetragonal space group I422, with unit-cell parameters a = b = 174.8, c = 163.8 Å. The data-collection statistics are summarized in Table 1 ▶. Assuming the presence of one molecule in the asymmetric unit, the calculated V M value and solvent content were 3.3 Å3 Da−1 and 62.8%, respectively. The absence of threefold or sixfold symmetry in the crystal appeared to be inconsistent with the biochemical data, since the crystals were obtained from a fraction that was estimated to contain a homohexamer. The biological assembly may have collapsed under the crystallization conditions. Attempts to determine the structure using a selenomethionine derivative are currently under way.
Table 1. Data-collection statistics.
Values in parentheses are for the outer shell.
| Resolution range (Å) | 50–1.70 (1.73–1.70) |
| No. of unique reflections | 137538 (6810) |
| No. of observed reflections | 2049689 |
| Completeness (%) | 100.0 (100.0) |
| Redundancy | 14.9 (14.8) |
| 〈I/σ(I)〉 | 49.6 (7.4) |
| Rmerge | 6.5 (32.2) |
Acknowledgments
We thank the staff of the Photon Factory for the X-ray data collection. This work was supported in part by the Program for the Promotion of Basic Research Activities for Innovative Bioscience (PROBRAIN) in Japan.
References
- Chinen, A., Kozlov, Y. I., Hara, Y., Izui, H. & Yasueda, H. (2007). J. Biosci. Bioeng.103, 262–269. [DOI] [PubMed]
- Duggleby, R. G. (2006). Acc. Chem. Res.39, 550–557. [DOI] [PubMed]
- Goldberg, M. L. & Racker, E. (1962). J. Biol. Chem.237, 3841–3842. [PubMed]
- Grill, J. P., Crociani, J. & Ballongue, J. (1995). Curr. Microbiol.31, 49–54. [DOI] [PubMed]
- Heath, E. C., Hurwitz, J., Horecker, B. L. & Ginsburg, A. (1958). J. Biol. Chem.231, 1009–1029. [PubMed]
- Hurwitz, J. (1958). Biochim. Biophys. Acta, 28, 599–602. [DOI] [PubMed]
- Jeong, D.-W., Lee, J. M. & Lee, H. J. (2007). J. Microbiol. Biotechnol.17, 822–829. [PubMed]
- Lee, J. M., Jeong, D.-W., Koo, O. K., Kim, M. J., Lee, J.-H., Chang, H. C., Kim, J. H. & Lee, H. J. (2005). Biotechnol. Lett.27, 853–858. [DOI] [PubMed]
- Lindqvist, Y., Schneider, G., Ermler, U. & Sundstrom, M. (1992). EMBO J.11, 2373–2379. [DOI] [PMC free article] [PubMed]
- Lipmann, F. & Tuttle, L. C. (1945). J. Biol. Chem.159, 21–28. [PubMed]
- Meile, L., Rohr, L. M., Geissmann, T. A., Herensperger, M. & Teuber, M. (2001). J. Bacteriol.183, 2929–2936. [DOI] [PMC free article] [PubMed]
- Nunoura, N., Ohdan, K., Yano, T., Yamamoto, K. & Kumagai, H. (1996). Biosci. Biotechnol. Biochem.60, 188–193. [DOI] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol.276, 307–326. [DOI] [PubMed]
- Picard, C., Fioramonti, J., Francois, A., Robinson, T., Neant, F. & Matuchansky, C. (2005). Aliment. Pharmacol. Ther.22, 495–512. [DOI] [PubMed]
- Posthuma, C. C., Bader, R., Engelmann, R., Postma, P. W., Hengstenberg, W. & Pouwels, P. H. (2002). Appl. Environ. Microbiol.68, 831–837. [DOI] [PMC free article] [PubMed]
- Racker, E. (1962). Methods Enzymol.5, 276–280.
- Scardovi, V. (1986). Bergey’s Manual of Systematic Bacteriology, edited by P. H. A. Sneath, N. S. Mair, M. E. Sharpe & J. G. Holt, pp. 1418–1434. Baltimore: Williams & Wilkins.
- Scardovi, V. & Trovatelli, L. D. (1965). Ann. Microbiol.15, 19–29.
- Schramm, M., Klybas, V. & Racker, E. (1958). J. Biol. Chem.233, 1283–1288. [PubMed]
- Sela, D. A., Chapman, J., Adeuya, A., Kim, J. H., Chen, F., Whitehead, T. R., Lapidus, A., Rokhsar, D. S., Lebrilla, C. B., German, J. B., Price, N. P., Richardson, P. M. & Mills, D. A. (2008). Proc. Natl Acad. Sci. USA, 105, 18964–18969. [DOI] [PMC free article] [PubMed]
- Sgorbati, B., Lenaz, G. & Casalicchio, F. (1976). Antonie Van Leeuwenhoek, 42, 49–57. [DOI] [PubMed]
- Xiang, S., Usunow, G., Lange, G., Busch, M. & Tong, L. (2007). J. Biol. Chem.282, 2676–2682. [DOI] [PubMed]
- Yevenes, A. & Frey, P. A. (2008). Bioorg. Chem.36, 121–127. [DOI] [PubMed]
- Zaunmüller, T., Eichert, M., Richter, H. & Unden, G. (2006). Appl. Microbiol. Biotechnol.72, 421–429. [DOI] [PubMed]