

The complex between human α-thrombin and a modified thrombin-binding aptamer has been crystallized. Diffraction data were collected to 2.15 Å resolution, the structure was solved by molecular replacement and refinement of the model is in progress.
Keywords: quadruplexes, thrombin-binding aptamers, thrombin, protein–nucleic acid interactions
Abstract
The thrombin-binding aptamer (TBA) is a consensus DNA 15-mer that binds specifically to human α-thrombin at nanomolar concentrations and inhibits its procoagulant functions. Recently, a modified TBA (mTBA) containing a 5′–5′ inversion-of-polarity site has been shown to be more stable and to possess a higher thrombin affinity than its unmodified counterpart. The structure of the thrombin–TBA complex has previously been determined at low resolution, but did not provide a detailed picture of the aptamer conformation or of the protein–DNA assembly, while that of the complex with mTBA is unknown. Crystallographic analysis of the thrombin–mTBA complex has been attempted. The crystals diffracted to 2.15 Å resolution and belonged to space group I222.
1. Introduction
Aptamers are nucleic acid macromolecules that bind to molecular targets, including proteins, with high affinity and specificity. The thrombin-binding aptamer (TBA) is a consensus DNA 15-mer, 5′-GGTTGGTGTGGTTGG-3′, that binds specifically to thrombin at nanomolar concentrations (Bock et al., 1992 ▶); for this reason, it has interesting anticoagulant properties and is in development for use as an anticoagulant during coronary artery bypass graft procedures. The NMR solution structure of TBA revealed that it adopts a unimolecular chair-like quadruplex structure consisting of two G-tetrads connected by two TT loops and a single TGT loop (Macaya et al., 1993 ▶; Schultze et al., 1994 ▶). The X-ray structure of the complex between thrombin and TBA was determined at 2.9 Å resolution in 1993 (Padmanabhan et al., 1993 ▶). Although the central core of TBA is the same in the NMR and crystallographic models, some structural differences exist in the way that the central bases are connected. Some years later, a new model of the complex was proposed in which the NMR structure of TBA was fitted into the electron-density maps. The dominant interactions between thrombin and the aptamer are located in the fibrinogen-recognition site and involve the TGT loop in the crystallographic model and the TT loops in the NMR–crystallographic mixed model. Both of these structures refined to the same R value, presenting a disconcerting ambiguity (Padmanabhan & Tulinsky, 1996 ▶). A subsequent analysis of these structures suggested that the NMR model of TBA is consistent with the X-ray data and identifies the most likely model of the thrombin–TBA complex to be the NMR–crystallographic mixed model (Kelly et al., 1996 ▶). However, a detailed characterization of the complex is still lacking as better quality crystals of the thrombin–TBA complex were never obtained.
In order to improve the properties of TBA, several modified TBAs have been synthesized (Lancellotti & De Cristofaro, 2009 ▶). An interesting modification is the insertion of 5′–5′ inversion sites, since it has been reported that the presence of inversion-of-polarity sites confers an increased resistance to endogenous nucleases. Therefore, a modified TBA (mTBA), 3′-GGT-5′–5′-TGGTGTGGTTGG-3′, has recently been produced and characterized (Martino et al., 2006 ▶). Notably, mTBA is more stable than its unmodified counterpart and possesses a higher thrombin affinity, but a lower inhibitory activity, with respect to its natural counterpart (Pagano et al., 2007 ▶). The structure of the complex between thrombin and mTBA has not yet been determined.
In this article, we describe the crystallization and preliminary X-ray diffraction analysis of the complex of thrombin with mTBA. These data could help to shed light on the molecular details of thrombin–aptamer recognition, which remain ambiguous, and could be a good starting point for the rational design of new aptamers with improved pharmacological properties with respect to TBA.
2. Materials and methods
2.1. Thrombin–mTBA complex preparation
Human d-Phe-Pro-Arg-chloromethylketone (PPACK) inhibited α-thrombin was purchased from Haemtech and mTBA was synthesized as described previously (Martino et al., 2006 ▶). mTBA was dissolved in 10 mM potassium phosphate buffer pH 7.1 to a concentration of 2 mM, heated for 10 min at 360 K and slowly cooled to room temperature to induce folding into the quadruplex structure. Circular-dichroism measurements have shown that the aptamer adopts an antiparallel quadruplex structure in solution.
The complex with thrombin was prepared by placing a twofold molar excess of the aptamer onto a frozen sample of inhibited thrombin at a protein concentration of 0.9 mg ml−1 in 0.75 M KCl and leaving the sample for 3 h at 277 K. The sample was then diluted and the buffer was changed to 50 mM potassium phosphate pH 7.1 and 0.1 M KCl. The thrombin–mTBA complex was extensively washed and finally concentrated to 8 mg ml−1 using a Centricon mini-concentrator and a refrigerated centrifuge.
2.2. Crystallization
Initial crystallization trials were performed at 293 K using the sparse-matrix sampling method. Screening was carried out using commercially available kits (Crystal Screen, Crystal Screen 2 and Index kits from Hampton Research). ‘Bushes’ of thin and damaged crystals grew from a crystallization condition consisting of 25%(w/v) polyethylene glycol 4000, 0.2 M ammonium sulfate, 100 mM sodium acetate pH 4.5 (Fig. 1 ▶).
Figure 1.

Initial crystals of the thrombin–mTBA complex.
In order to obtain crystals suitable for diffraction data collection, it was necessary to perform an optimization of the crystallization conditions. Initially, we fine-tuned the concentrations of the protein and precipitant, the pH value and the nature of the buffer and performed the crystallization trials at different temperatures (277 and 293 K). Since none of these trials were successful, a screen of polyethylene glycols and methyl-substituted polyethylene glycols with different molecular weights was also performed. An improvement in protein crystal quality was obtained using PEG 20 000, although the crystals were still too fragile and broke when touched with the loop. To avoid this problem, the use of several additives was tested. In particular, alcohols (ethanol, 2-methyl-2,4-pentandiol, 2-propanol, n-propanol, n-butanol, t-butanol and glycerol), sugars (trehalose, sucrose and xylitol) and organic liquids (dimethylformamide, acetonitrile and dimethylsulfoxide) were added to the crystallization mixture at concentrations ranging from 0.5 to 10%(v/v). Eventually, good-quality crystals of the complex were obtained (Fig. 2 ▶) from a crystallization solution consisting of 20%(v/v) polyethylene glycol 20 000, 0.2 M ammonium sulfate, 3%(v/v) n-propanol, 100 mM sodium acetate pH 5.8. Crystals grew in a few days using the sitting-drop vapour-diffusion method at 293 K. The protein:reservoir solution ratio was 1:1 in 1 µl drops and the protein concentration was 8 mg ml−1.
Figure 2.

The best crystals of the thrombin–mTBA complex.
The experience gained in the crystallization of the thrombin–mTBA complex led us to attempt crystallization of the thrombin–TBA complex, with the aim of obtaining better quality crystals, since the diffraction data reported were poor. Preliminary tests have been performed and the results are particularly promising.
2.3. Data collection and processing
Diffraction data were collected in-house on a Saturn 944 CCD detector. The X-ray radiation used was Cu Kα radiation from a Rigaku MicroMax-007 HF generator. After the addition of 25% glycerol to the harvesting solution, the crystal was flash-cooled at 100 K in supercooled N2 gas produced by an Oxford Cryosystem and was maintained at 100 K during data collection. Data were indexed, processed and scaled with HKL-2000 (Otwinowski & Minor, 1997 ▶; Table 1 ▶).
Table 1. Summary of crystallographic data.
Values in parentheses are for the highest resolution shell.
| Space group | I222 |
| Unit-cell parameters | |
| a (Å) | 68.21 |
| b (Å) | 110.57 |
| c (Å) | 120.92 |
| α (°) | 90.00 |
| β (°) | 90.00 |
| γ (°) | 90.00 |
| Resolution limits (Å) | 50.00–2.15 (2.23–2.15) |
| No. of observations | 817402 |
| No. of unique reflections | 25096 |
| Completeness (%) | 99.1 (93.2) |
| 〈I/σ(I)〉 | 9.0 (3.0) |
| Average multiplicity | 5.2 |
| Rmerge† (%) | 13.7 (42.7) |
| Mosaicity | 0.6 |
| VM (Å3 Da−1) | 2.92 |
| Solvent content (%) | 57.9 |
R
merge = 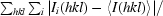
 , where Ii(hkl) is the intensity of the ith measurement of reflection hkl and 〈I(hkl)〉 is the mean value of Ii(hkl) for all i measurements.
, where Ii(hkl) is the intensity of the ith measurement of reflection hkl and 〈I(hkl)〉 is the mean value of Ii(hkl) for all i measurements.
The crystals of the thrombin–mTBA complex belonged to the orthorhombic space group I222 and diffracted to 2.15 Å resolution.
2.4. Structure determination
The structure of the complex was solved using the program AMoRE (Navaza & Saludjian, 1997 ▶) with PPACK-inhibited thrombin as the search model (PDB code 1ppb; Bode et al., 1989 ▶). To avoid bias, inhibitor and water molecules were removed from the model prior to structure-factor and phase calculation. The correlation for the highest solution was 0.586, with an R factor of 0.359.
3. Results and discussion
More than 15 years after determination of the low-resolution structure of the thrombin–TBA complex (Padmanabhan et al., 1993 ▶), we have been able to crystallize human α-thrombin in complex with the modified DNA aptamer mTBA.
After extensive screening of crystallization conditions, good-quality crystals of the complex between thrombin and mTBA have been obtained. Statistics of the X-ray diffraction data collected from these crystals are given in Table 1 ▶.
Matthews coefficient calculations suggested the presence of a 1:1 complex of thrombin and aptamer in the asymmetric unit. The V M value is 2.92 Å3 Da−1, corresponding to a solvent content of 57.9%. Crystals of the thrombin–mTBA complex diffracted to higher resolution compared with those of the complex of thrombin with TBA (Padmanabhan & Tulinsky, 1996 ▶).
The X-ray diffraction data were used for molecular replacement, as reported in §2. A clear solution was obtained and inspection of the calculated (F o − F c) and (2F o − F c) Fourier difference maps showed the presence of the aptamer. Rebuilding and refinement of the whole structure is in progress. Our model will represent one of the highest resolution structures of a protein–quadruplex complex to be reported to date.
The extensive efforts made in attempting to obtain good diffracting crystals of the complex with the modified TBA prompted us to revisit the crystallization of the complex of thrombin with TBA, the crystallographic structure of which, refined at a rather low resolution, was in partial contradiction with the structure obtained in solution by NMR. The first crystallization trials were very promising and we hope to obtain diffraction data at a sufficiently high resolution to obtain an authoritative model of this structure and to compare it with that of the modified aptamer.
Together, these data will help in the rational design of new aptamers with improved pharmacological properties with respect to TBA.
Acknowledgments
We thank Mr Giosuè Sorrentino and Mr Maurizio Amendola for their technical assistance.
References
- Bock, L. C., Griffin, L. C., Latham, J. A., Vermaas, E. H. & Toole, J. J. (1992). Nature (London), 355, 564–566. [DOI] [PubMed]
- Bode, W., Mayr, I., Baumann, U., Huber, R., Stone, S. R. & Hofsteenge, J. (1989). EMBO J.8, 3467–3475. [DOI] [PMC free article] [PubMed]
- Kelly, J. A., Feigon, J. & Yeates, T. O. (1996). J. Mol. Biol.256, 417–422. [DOI] [PubMed]
- Lancellotti, S. & De Cristofaro, R. (2009). Cardiovasc. Hematol. Agents Med. Chem.7, 19–28. [DOI] [PubMed]
- Macaya, R. F., Schultze, P., Smith, F. W., Roe, J. A. & Feigon, J. (1993). Proc. Natl Acad. Sci. USA, 90, 3745–3749. [DOI] [PMC free article] [PubMed]
- Martino, L., Virno, A., Randazzo, A., Virgilio, A., Esposito, V., Giancola, C., Bucci, M., Cirino, G. & Mayol, L. (2006). Nucleic Acids Res.34, 6653–6662. [DOI] [PMC free article] [PubMed]
- Navaza, J. & Saludjian, P. (1997). Methods Enzymol.276, 581–594. [DOI] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol.276, 307–326. [DOI] [PubMed]
- Padmanabhan, K., Padmanabhan, K. P., Ferrara, J. D., Sadler, J. E. & Tulinsky, A. (1993). J. Biol. Chem.268, 17651–17654. [DOI] [PubMed]
- Padmanabhan, K. & Tulinsky, A. (1996). Acta Cryst. D52, 272–282. [DOI] [PubMed]
- Pagano, B., Martino, L., Randazzo, A. & Giancola, C. (2007). Biophys. J.94, 562–569. [DOI] [PMC free article] [PubMed]
- Schultze, P., Macaya, R. F. & Feigon, J. (1994). J. Mol. Biol.235, 1532–1547. [DOI] [PubMed]