

Abstract
Each heartbeat requires precisely orchestrated action potential propagation through the myocardium achieved by coordination of about a million ion channels on the surface of each cardiomyocyte. Specific ion channels must occur within discrete subdomains of the sarcolemma in order to exert their electrophysiological effects with highest efficiency (e.g. voltage-gated Ca2+ channels at T-tubules and gap junctions at intercalated discs). Regulation of ion channel movement to their appropriate membrane subdomain is an exciting research frontier with opportunity for novel therapeutic manipulation of ion channels in the treatment of heart disease. While much research has generally focused upon internalization and subsequent degradation of ion channels, the field of forward trafficking of de novo ion channels from the cell interior to the sarcolemma has now emerged as a key regulatory step in cardiac electrophysiological function. In this brief review, we provide an overview of the current understanding of the cellular biology governing the forward trafficking of ion channels.
Keywords: Trafficking, ion channel, cytoskeleton, targeted delivery, membrane subdomain
Introduction
Ion channels that occur within the cardiomyocyte plasma membrane (sarcolemma) exist with specific densities and in specific subdomains. Specificity of population and location are important to effect efficient action potential generation and excitation-contraction (EC) coupling. Precise forward trafficking of ion channels to their respective membrane subdomain is an important aspect of ion channel regulation. Cardiac disease can also alter ion channel trafficking, with dangerous consequence to both the electrical and mechanical function of the heart1.
Given the highly complex nature of cardiomyocyte architecture, it is unsurprising that an increasing number of studies are reporting direct and targeted delivery of ion channels to their specific destinations2–4. Although cardiomyocytes generally last the entire human lifetime, ion channels have reported half lives at the membrane on the order of hours2,5,6. The intracellular forward trafficking of ion channels is therefore a key regulatory step in controlling the current density of specific ion channels. In fact, we recently found that perturbed forward trafficking of connexons contributes to losses in cell-cell coupling in stressed human and mouse myocardium7. As the mechanisms governing delivery of ion channels to their destinations at the cell surface are elucidated, the potential for therapeutically manipulating ion channel function by altering their forward trafficking can be realized.
Subcellular Organization of the Cardiomyocyte
Figure 1 contains a schematic representation of the adult cardiomyocyte cellular architecture. Adult ventricular cardiomyocytes are large, usually bi-nucleated contractile cells with a highly polarized phenotype. Cardiac muscle contracts over three billion times during the average human’s lifetime and therefore, cardiomyocytes have a high energy need, provided by high numbers of mitochondria throughout the cytoplasm. Contractility is achieved through contraction of cytoplasmic myofibrils composed of specialized mechanical structures known as sarcomeres. All myocytes contain a form of smooth endoplasmic reticulum (ER) termed the sarcoplasmic reticulum (SR) which houses large stores of calcium ions. For each heartbeat to occur, calcium must be released from intracellular SR stores, precipitating contraction of the sarcomeric machinery. More typical components of the cardiomyocyte include the rough ER and Golgi apparatus, near which microtubules are anchored and extend throughout the cell. The microtubules form an intricate and dynamic network capable of shuttling ion channel containing vesicles to their destinations (Figure 2).
Figure 1. Subcellular Organization of the Ventricular Cardiomyocyte.

Schematic representation of cardiomyocyte subcellular architecture. Intercalated discs couple cardiomyocytes electrically and mechanically to each other. T-tubules proximate sarcolemmal CaV1.2 channels to intracellular ryanodine release channels on the sarcoplasmic reticulum. The sarcomeres of the cardiomyocyte are responsible for contractility in response to Ca2+ release from the sarcoplasmic reticulum. Microtubules provide a means by which newly-synthesized ion channels can navigate the complex cell interior and reach their specific destinations.
Figure 2. Immunofluorescence Detection of Microtubules and the Plus-End Binding Protein EB1 in a Primary Mouse Neonatal Ventricular Cardiomyocte.
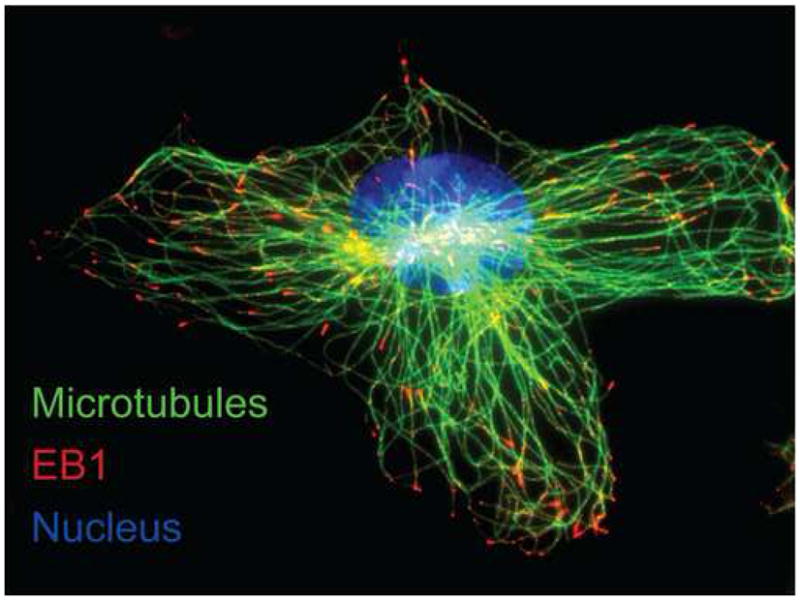
100× confocal immunofluorescence detection of α-tubulin (green) and EB1 (red) in primary mouse neonatal ventricular cardiomyocytes with nuclei counterstained using Hoechst (blue). Microtubules emanate from the centrosome at the cell nucleus close to the Golgi apparatus where vesicular cargo can be loaded from the TGN. Note EB1 ‘comets’ at the plus-ends of microtubules approaching the cell cortex.
Actin, like microtubules, is a major component of the myocyte cytoskeleton, but also occurs in the sarcomere. Essentially, sarcomeres are comprised of thin filaments of actin and thick filaments of myosin, both anchored at Z-discs that occur perpendicular to the ‘actomyosin’ filaments. Release of Ca2+ from the SR causes the sarcomere to contract by revealing actin binding sites to the myosin motor head, pulling actin thin filaments towards the center of the sarcomere, resulting in contraction of the entire cardiomyocyte8. The sarcolemma is connected to myofibrils at the adherens junctions (fascia adherens), of the intercalated disc, along with cytoskeletal actin microfilaments (F-actin) that provide structural support9. Formation and maintenance of specialized sarcolemmal domains such as intercalated discs, caveolae, and T-tubules are dependent on F-actin9,10. Such cortical F-actin is highly dynamic and, in addition to maintaining cell shape, has been shown to participate in vesicular trafficking as well as vesicular fusion and exocytic events in other cell types11. The role of actin in cardiomyocyte ion channel trafficking remains to be explored.
Individual cardiomyocytes couple mechanically and electrically to each other through specialized structures termed intercalated discs which occur at the longitudinal ends of cardiomyocytes, perpendicular to myofibrils. Mechanical coupling of cardiomyocytes is achieved through desmosome and fascia adherens complexes connecting the intercalated disc to structural components of the cytoskeleton such as intermediate filaments and F-actin. Gap junctions are primarily responsible for electrical coupling of the cardiomyocytes as they allow direct communication between the cytoplasms of adjacent cells, permitting free and voltage driven diffusion of ions9.
The polarized localization of gap junctions at cardiomyocyte intercalated discs is essential for appropriately orchestrated propagation of action potentials throughout working myocardium. Altered localization and losses in cell-cell gap junction coupling occur during cardiac disease12, and contribute to arrhythmogenic substrates13. Gap junctions exist in plaques that are comprised of hundreds to thousands of connexon hemichannels, each of which is made up of six connexin molecules. Connexons on the surfaces of opposing cells must coalesce to form continuous conduits spanning both lipid bilayers. Over 20 isoforms of connexin have been identified in humans, of which Connexin 43 (Cx43) is the primary connexin expressed in ventricular myocardium14. Other ion channels, such as voltage-gated sodium channels, are also enriched at the intercalated disc and their loss may contribute to arrhythmogenesis in acquired and inherited diseases4.
Another sarcolemmal domain of the cardiomyocyte, which unlike the intercalated disc is shared with skeletal myocytes, is T-tubule invaginations. Cardiac T-tubules occur at regular intervals along the lateral sides of the cell, closely coincident with the sarcomeric Z-discs. Enrichment of the voltage-gated L-type calcium channel (CaV1.2) at cardiac T-tubules localizes these channels in close proximity (~15 nm) to intracellular SR-based ryanodine receptor (RyR) release channels. Upon membrane depolarization, initial calcium influx occurs through CaV1.2 channels and the close association between CaV1.2 and RyR permits efficient calcium-induced calcium-release (CICR) and subsequent sarcomeric contraction15.T-tubules also include enriched populations of voltage-gated sodium channels (NaV) as well as the Na+/Ca2+ exchanger (NCX) and Na+/K+ ATPase (NKA) channels to help maintain ionic homeostasis16.
The Anterograde Trafficking Pathway of de novo Ion Channels
1) Through the Endoplasmic Reticulum
Newly synthesized plasma membrane proteins, such as ion channels, must pass from the cytosol through the ER-Golgi trafficking machinery and be packaged in vesicles to be transported to their final destination (Figure 3). The mRNA transcripts of ion channel genes are translated to protein by ribosomes in the cytosol initially, but amino acid signal sequences within the nascent forming polypeptide effect the localization of the ribosome complex to the rough ER, where translocation of the newly forming ion channel within the ER lipid bilayer can occur. Processes such as folding and posttranslational modifications including phosphorylation and core-glycosylation occur in the ER and once completed correctly, ion channel proteins are transported to the Golgi apparatus, from where they are further modified and sorted for delivery.
Figure 3. The Anterograde Vesicular Trafficking Pathway.
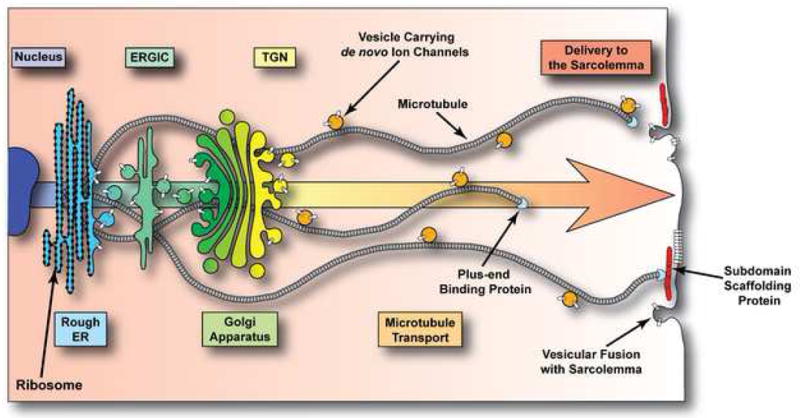
Schematic representation of the forward trafficking pathway that de novo ion channels (white cylinders) traverse to reach their specific destinations. Nascent ion channel polypeptides are translocated to the membrane of the rough ER after which they are modified and move through the endoplasmic reticulum-Golgi intermediate compartment (ERGIC), Golgi apparatus, and trans-Golgi network (TGN). From the TGN, vesicles containing ion channels are loaded onto microtubules and are transported to their destinations. Microtubule plus-end binding proteins interact with scaffolding proteins specific to particular membrane subdomains to achieve targeted delivery of vesicular cargo.
Individual ion channel pore-forming (α) subunit proteins often must associate (oligomerize) with other (β) channel subunits to traffic and function. Pore-forming K+ channel α-subunits are typically comprised of four polypeptides while the voltage gated Ca2+ and Na+ channels are translated as a single large full length protein. β subunit associations predominantly occur within the ER and often aid the α-subunits exit the ER17,18. A common mechanism of ER release is through masking of ER-retention motifs within the ion channel polypeptide following binding with another subunit protein, as is the case with CaV18, or cellular chaperones such as 14-3-3 proteins, as is the case with some K+ channels19. Six connexin proteins oligomerize to form hemichannels in an event which, for different connexin subtypes, occurs at different points in the trafficking pathway14.
Regulation of channel formation can be achieved through posttranslational modifications. For example, several protein-protein interaction motifs (including 14-3-3 binding domains19) are phosphorylation dependent. Substantial evidence exists for regulation of anterograde vesicular traffic by protein kinase A (PKA). Indeed, increases in PKA activity have been associated with enhancing gap junction coupling14. Such effects may not be direct, and most likely involve cooperation from other signaling cascades within the cardiomyocyte.
Importantly, the ER-Golgi checkpoint regulates both exit of ion channels from the ER and channel degradation, as subunits failing to traffic (e.g. due to misfolding) may be redirected to an ER-associated degradation pathway14. As such, following transcription and translation, transport of de novo ion channel proteins from the ER to the Golgi apparatus represents a major rate-limiting step in their trafficking to the sarcolemma. Once exit from the ER is approved, ion channels are packaged into COPII-coated vesicles and transported along microtubules to the ER-Golgi intermediate compartment (ERGIC) by motor proteins20. Further sorting occurs within the ERGIC from where vesicular traffic continues on to the cis-face of the Golgi apparatus. The Golgi apparatus is comprised of several discs, or cisternae, of which the cis-face is most internal and close to the ERGIC and the trans-face is most external and represents the point of vesicular exit.
2) The Golgi Apparatus and trans-Golgi Network
As ion channels progress through the cisternae of the Golgi apparatus, further posttranslational modifications including complex-glycosylation, phosphorylation, and cleavage can occur to complete processing and generate a functional ion channel ready for delivery21. The trans-face of the apparatus represents the complex vesicular cargo sorting compartment known as the trans-Golgi network (TGN). Here, clathrin-coated vesicles enriched with membrane proteins form and are loaded onto microtubules and trafficked to specific cellular destinations22. The mechanisms governing membrane protein sorting in the Golgi are better understood in epithelial cells (e.g. apical versus basolateral membrane targets) than in cardiomyocytes23.
Differing connexin isoforms can combine to form heteromeric connexon hemichannels and such isoform composition is understood to influence gap junction conductance properties. An added level of complexity is the possibility of heterotypic gap junctions, whereby isoform-distinct connexons on opposing cells may bind. Interestingly, it is at the TGN that the major ventricular gap junction forming protein Cx43 oligomerizes into hemichannels. This is relatively late for such an event to occur, as other connexins oligomerize in the ER, but it may represent a means of controlling heteromeric hemichannel formation with other connexin isoforms14. Upon exiting the TGN, vesicles containing de novo ion channels must navigate the complex cardiomyocyte intracellular environment, a feat they achieve by trafficking along dynamic microtubules.
3) Vesicular Traffic on the Cytoskeleton Highway
Microtubules act as ‘highways’ within the cardiomyocyte which emanate from the centrosome and provide directionality and specificity to particular compartments within the cell. Vesicles are transported on microtubules by energy-dependent motor proteins such as kinesin which travel towards microtubule plus-ends24. Microtubule plus-ends exist in a state of dynamic instability, whereby tubulin rapidly polymerizes and depolymerizes under the regulation of plus-end binding proteins, altering microtubule velocity25 and binding partners2. Microtubules can therefore respond to stresses or stimuli and regulate delivery of their vesicular cargo to cellular compartments.
Actin is also understood to regulate vesicular transport at early (ER-Golgi) and later (TGN-sarcolemma) stages, and can transport vesicles in a directional manner via myosin motor proteins26,27. Interestingly, vesicles can switch to different microtubules at microtubule-microtubule intersections, or from microtubule to actin filament and vice versa27. The majority of cardiac ion channels have now been shown to traffic in a microtubule and/or actin dependent manner, or at least have their function perturbed through disruption of such cytoskeletal networks. It is likely that a combination of both actin and microtubule based vesicular trafficking are responsible for directing de novo ion channels to the sarcolemma by a process now increasingly understood to occur in a targeted fashion.
4) Targeting of Ion Channels to Specific Sarcolemmal Subdomains Intercalated Discs
The mechanisms by which microtubules exert their specificity in interacting with membrane subdomains are now being elucidated. Dynamic plus-ends of microtubules interact preferentially with certain membrane protein complexes, such as adherens junctions2,25. Plus-end binding proteins have been reported to effect such microtubule interaction with the cell cortex, and can anchor microtubules at membrane protein complexes where they facilitate delivery of vesicular cargo to specific domains of the cardiomyocyte sarcolemma. A major plus-end binding protein, EB1, is known to be necessary for targeted delivery of connexons to adherens junction complexes2. Through interaction with another plus-end protein, p150GLUED, the EB1-tipped microtubule complexes specifically with β-catenin molecules at the fascia adherens of intercalated discs. Vesicular cargo is unloaded and subsequently inserted into the plasma membrane at nearby gap junctions. Other reports propose a less specific paradigm of connexin delivery, whereby connexons are inserted indiscriminately into the lateral membrane of the cell and freely diffuse to gap junction structures28. Both models can exist in parallel. However, the inefficiency of lateral diffusion to a few specific subdomains, the short half life of connexins, and the complex interactions with multiple neighboring cells forming a single cardiomyocyte’s periphery, all suggest directed targeting can be a more effective form of connexon localization to the intercalated disc.
In a recent publication from our group, perturbed forward trafficking of connexons was found to be a significant contributor to the losses of cell-cell coupling during cardiac disease7. During oxidative stress, the microtubule plus-end protein EB1 is displaced from the microtubule, resulting in less dynamic microtubules that fail to interact with the cell cortex (Figure 4). Manipulation of EB1 as well as the upstream regulators of EB1 localization at microtubules could potentially preserve or enhance gap junction coupling during stress. As many ion channels rely on microtubules for their transport, it is likely that such disruption of microtubule trafficking machinery inhibits delivery of many essential channels to the sarcolemma.
Figure 4. Oxidative Stress Limits Connexin 43 Forward Trafficking.
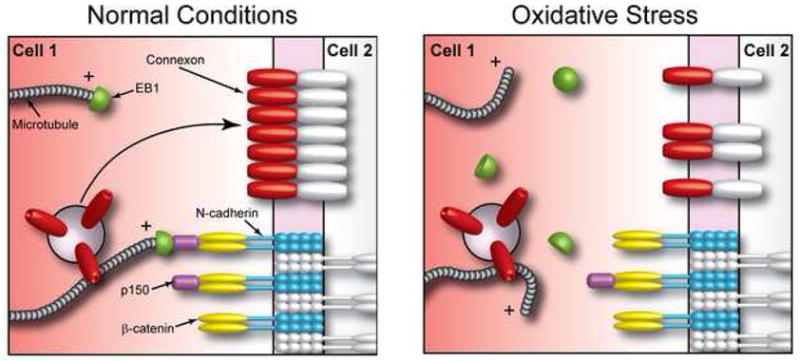
Schematic representation of the Cx43 forward trafficking machinery. Under normal conditions dynamic microtubules interact specifically with adherens junction structures through interaction of the plus-end binding proteins EB1 and p150GLUED and β-catenin (left panel). This interaction permits targeted delivery of vesicles containing Cx43 hemichannels to gap junctions at the intercalated disc. During oxidative stress (right panel) EB1 is displaced from microtubules which subsequently fail to interact with the cell cortex, thus perturbing delivery of their vesicular cargo.
Several membrane scaffolding proteins have recently been uncovered as critical regulators of targeting and maintenance of ion channels at sarcolemmal subdomains of cardiomyocytes. One such protein is ankyrin-G, which has also been shown to target NaV1.2 and NaV1.6 to specialized excitable membrane domains in Purkinje and granule cell neurons. In the heart, ankyrin-G localizes NaV1.5 to the intercalated disc, a model supported by a human Brugada Syndrome SCN5A mutation abolishing the NaV1.5 ankyrin-binding motif (for brevity, Mohler lab publications contained in4). The important role of dynamic microtubules in delivery of NaV1.5 to the sarcolemma was very recently highlighted in a study that found deleterious effects on NaV1.5 function following microtubule stabilization with Taxol29. Further elucidation of this microtubule-based trafficking pathway could uncover exciting mechanistic insight into differential targeting of ion channels to the intercalated disc.
T-Tubules
Enrichment of CaV1.2 channels in the T-tubules is essential for the efficient contractile function of the myocardium. We recently reported that trafficking of CaV1.2 vesicles from the TGN to T-tubules occurs in a microtubule-dependent manner3. Moreover, just as with Cx43 connexons, dynamic microtubules preferentially interact with a specific membrane complex in order to effect targeted delivery of their ion channel cargo. In the case of CaV1.2, the membrane scaffolding protein BIN1 tethers microtubules at T-tubules, permitting delivery of CaV1.2 channels directly to their functional location. BIN1 contains a membrane curvature BAR-domain (which confers the ability to form invaginations in the cell membrane, i.e. T-tubules), a coiled-coil domain, and an SH3 protein-protein interaction domain. Perhaps most compelling is the finding that deletion of the coiled-coil and SH3 domains does not affect membrane invagination, but abrogates CaV1.2 colocalization with these structures. Therefore, it is through interaction specifically with the BIN1 protein, and not T-tubule structures, that targeting of CaV1.2 delivery is achieved.
An additional ankyrin isoform found in ventricular cardiomyocytes, ankyrin-B, is associated with targeting and maintenance of the NCX and NKA channels at T-tubules where they proximate with the InsP3 receptor of the SR and regulate Ca2+ export. Mutations in ankyrin-B ablating its interaction with NCX/NKA/InsP3 result in arrhythmogenic cardiac disorders in humans, including type-4 long-QT syndrome16. Ankyrin-B is also expressed in the pacemaking cells of the sinoatrial node. In addition to NCX/NKA/InsP3, CaV1.3 targeting is perturbed by loss-of-function mutations and thus extracellular calcium entry deregulated, possibly contributing to ‘sick sinus syndrome’4.
Caveolae
Within the lateral sarcolemma of ventricular cardiomyocytes there exists high numbers of much smaller invaginations termed caveolae. The lipid bilayers of these flask-shaped structures are enriched with cholesterol, and the cholesterol-binding scaffolding protein Caveolin-3 (Cav-3) is responsible for their formation. Biochemical fractionation and electron microscopy studies have identified a subpopulation of many ion channels at caveolae, and loss of caveolae contributes to arrhythmogenesis. The precise role of ion channels in caveolae is still under investigation, and they may regulate signal transduction, such as Ca2+ dependent signaling through CaV1.230. The mechanisms effecting ion channel enrichment at caveolae are unknown, but reported intimate interactions between caveolae and the cytoskeleton present an appealing possibility of targeted ion channel delivery to these sarcolemmal microdomains31.
5) Insertion of Ion Channels into the Sarcolemma
Regulation of vesicular fusion with the plasma membrane represents a final regulatory step in ion channel forward trafficking. Cortical actin is known to negatively regulate exocytic vesicle fusion, and its reorganization is likely to influence distal ion channel trafficking11. At the cell cortex, unloading of vesicles onto F-actin may occur which would introduce another level of potential targeting specificity. To achieve delivery of its ion channel cargo, a vesicle must fuse with the plasma membrane, an event subject to further cellular regulation. The specifics of these mechanisms in cardiomyocytes, and how they may differ for individual ion channels, are poorly understood. Elucidation of each rate-limiting step in the forward trafficking of cardiac ion channels will facilitate development of specific therapeutics aimed at altering channel densities at the sarcolemma.
Therapeutic Implications of Ion Channel Trafficking
In parallel to the growing understanding of cardiomyocyte biology is an increasing awareness that many drugs that block ion channels may do so by altering trafficking rather than having direct pore blocking effects32,33. In an elegant study, Takemasa et a 34 were able to separate the hERG pore blocking from trafficking effect of ketoconazole by distinguishing time of onset from minutes (pore blocking) to hours (trafficking). While detailed electrophysiology is the gold standard, it remains a challenge to separate pore blocking from trafficking effects of drug action. Equally challenging is the attempt to understand where in the trafficking pathway drugs may have their effect. Interestingly, Delisle et al reported that the sarco/endoplasmic reticulum ATPase inhibitor thapsigargin can rescue trafficking of mutated hERG channels possibly through altered Ca2+-dependent chaperone activity in the ER35. In a separate study, both thapsigargin and the antiarrhythmogenic drug E4031 were found to independently rescue surface expression of trafficking-deficient mutated hERG channels36. Mutations in ion channel genes can result in premature termination codons and therefore encode non-functional truncated protein products which fail to traffic. Aminoglycosides reduce translation accuracy, permitting read-through of such termination codons and rescue function and trafficking of ion channels37. At the opposite end of the trafficking spectrum, quinidine may exert its effect of decreasing already membrane bound Kv1.5 by increasing channel internalization via endocytosis38. Understanding the pathways involved in delivering channels to the plasma membrane will undoubtedly reveal specific mechanisms of both existing and novel anti-arrhythmic agents.
Concluding Remarks
The life cycle of cardiac ion channels extends from DNA transcription to translation into proteins, protein modification, protein oligomerization, channel transport to sarcolemma and targeting to specific subdomains, insertion into the sarcolemma subdomains and, finally, internalization for degradation or recycling. Most current pharmaceutical therapy is believed to be based only on the time channels spend in the sarcolemma. Turnover of channels occurs within hours, and therefore pre-sarcolemmal regulatory mechanisms not only are subject to altered regulation in disease, but present opportunities for novel pharmaceutical regulation of channels. Applying basic cell biological principles to understand cardiomyocyte specific biology is expected to reap many targets for a new generation of ion channel modifying drugs.
Acknowledgments
We thank Ting-Ting Hong, PhD and Samy Y Lamouille, PhD (both at UCSF) for critical review of this manuscript.
Financial Support: NIH grants HL094414 and HL075449 (RMS)
American Federation for Aging Research grant A112457 (JWS)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Aiba T, Tomaselli GF. Electrical remodeling in the failing heart. Current opinion in cardiology. 2010;25:29–36. doi: 10.1097/HCO.0b013e328333d3d6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shaw RM, et al. Microtubule plus-end-tracking proteins target gap junctions directly from the cell interior to adherens junctions. Cell. 2007;128:547–560. doi: 10.1016/j.cell.2006.12.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hong TT, et al. BIN1 localizes the L-type calcium channel to cardiac T-tubules. PLoS biology. 2010;8:e1000312. doi: 10.1371/journal.pbio.1000312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hashemi SM, Hund TJ, Mohler PJ. Cardiac ankyrins in health and disease. Journal of molecular and cellular cardiology. 2009;47:203–209. doi: 10.1016/j.yjmcc.2009.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.van Ginneken AC. MinK: a double-hearted partner in cardiac repolarization. Cardiovascular research. 2009;82:390–391. doi: 10.1093/cvr/cvp112. [DOI] [PubMed] [Google Scholar]
- 6.Colley BS, Biju KC, Visegrady A, Campbell S, Fadool DA. Neurotrophin B receptor kinase increases Kv subfamily member 1.3 (Kv1.3) ion channel half-life and surface expression. Neuroscience. 2007;144:531–546. doi: 10.1016/j.neuroscience.2006.09.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Smyth JW, et al. Limited forward trafficking of connexin 43 reduces cell-cell coupling in stressed human and mouse myocardium. The Journal of clinical investigation. 2010;120:266–279. doi: 10.1172/JCI39740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boateng SY, Goldspink PH. Assembly and maintenance of the sarcomere night and day. Cardiovascular research. 2008;77:667–675. doi: 10.1093/cvr/cvm048. [DOI] [PubMed] [Google Scholar]
- 9.Noorman M, et al. Cardiac cell-cell junctions in health and disease: Electrical versus mechanical coupling. Journal of molecular and cellular cardiology. 2009;47:23–31. doi: 10.1016/j.yjmcc.2009.03.016. [DOI] [PubMed] [Google Scholar]
- 10.Itoh T, et al. Dynamin and the actin cytoskeleton cooperatively regulate plasma membrane invagination by BAR and F-BAR proteins. Developmental cell. 2005;9:791–804. doi: 10.1016/j.devcel.2005.11.005. [DOI] [PubMed] [Google Scholar]
- 11.Jaiswal JK, Rivera VM, Simon SM. Exocytosis of post-Golgi vesicles is regulated by components of the endocytic machinery. Cell. 2009;137:1308–1319. doi: 10.1016/j.cell.2009.04.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Saffitz JE, Hames KY, Kanno S. Remodeling of Gap Junctions in Ischemic and Nonischemic Forms of Heart Disease. J Membr Biol. 2007;218:65–71. doi: 10.1007/s00232-007-9031-2. [DOI] [PubMed] [Google Scholar]
- 13.Saffitz JE. Arrhythmogenic cardiomyopathy and abnormalities of cell-to-cell coupling. Heart Rhythm. 2009;6:S62–65. doi: 10.1016/j.hrthm.2009.03.003. [DOI] [PubMed] [Google Scholar]
- 14.Hesketh GG, Van Eyk JE, Tomaselli GF. Mechanisms of gap junction traffic in health and disease. Journal of cardiovascular pharmacology. 2009;54:263–272. doi: 10.1097/FJC.0b013e3181ba0811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Orchard C, Brette F. t-Tubules and sarcoplasmic reticulum function in cardiac ventricular myocytes. Cardiovascular research. 2008;77:237–244. doi: 10.1093/cvr/cvm002. [DOI] [PubMed] [Google Scholar]
- 16.Mohler PJ, Davis JQ, Bennett V. Ankyrin-B coordinates the Na/K ATPase, Na/Ca exchanger, and InsP3 receptor in a cardiac T-tubule/SR microdomain. PLoS biology. 2005;3:e423. doi: 10.1371/journal.pbio.0030423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cusdin FS, Clare JJ, Jackson AP. Trafficking and cellular distribution of voltage-gated sodium channels. Traffic. 2008;9:17–26. doi: 10.1111/j.1600-0854.2007.00673.x. [DOI] [PubMed] [Google Scholar]
- 18.Karunasekara Y, Dulhunty AF, Casarotto MG. The voltage-gated calcium-channel beta subunit: more than just an accessory. Eur Biophys J. 2009;39:75–81. doi: 10.1007/s00249-009-0467-4. [DOI] [PubMed] [Google Scholar]
- 19.Shikano S, Coblitz B, Wu M, Li M. 14-3-3 proteins: regulation of endoplasmic reticulum localization and surface expression of membrane proteins. Trends in cell biology. 2006;16:370–375. doi: 10.1016/j.tcb.2006.05.006. [DOI] [PubMed] [Google Scholar]
- 20.Watson P, Forster R, Palmer KJ, Pepperkok R, Stephens DJ. Coupling of ER exit to microtubules through direct interaction of COPII with dynactin. Nature cell biology. 2005;7:48–55. doi: 10.1038/ncb1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Glick BS, Nakano A. Membrane traffic within the Golgi apparatus. Annual review of cell and developmental biology. 2009;25:113–132. doi: 10.1146/annurev.cellbio.24.110707.175421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.De Matteis MA, Luini A. Exiting the Golgi complex. Nature reviews. 2008;9:273–284. doi: 10.1038/nrm2378. [DOI] [PubMed] [Google Scholar]
- 23.Duffield A, Caplan MJ, Muth TR. Protein trafficking in polarized cells. International review of cell and molecular biology. 2008;270:145–179. doi: 10.1016/S1937-6448(08)01404-4. [DOI] [PubMed] [Google Scholar]
- 24.Verhey KJ, Hammond JW. Traffic control: regulation of kinesin motors. Nature reviews. 2009;10:765–777. doi: 10.1038/nrm2782. [DOI] [PubMed] [Google Scholar]
- 25.Lansbergen G, Akhmanova A. Microtubule plus end: a hub of cellular activities. Traffic. 2006;7:499–507. doi: 10.1111/j.1600-0854.2006.00400.x. [DOI] [PubMed] [Google Scholar]
- 26.Lanzetti L. Actin in membrane trafficking. Current opinion in cell biology. 2007;19:453–458. doi: 10.1016/j.ceb.2007.04.017. [DOI] [PubMed] [Google Scholar]
- 27.Ross JL, Ali MY, Warshaw DM. Cargo transport: molecular motors navigate a complex cytoskeleton. Current opinion in cell biology. 2008;20:41–47. doi: 10.1016/j.ceb.2007.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Laird DW. Life cycle of connexins in health and disease. The Biochemical journal. 2006;394:527–543. doi: 10.1042/BJ20051922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Casini S, et al. Tubulin polymerization modifies cardiac sodium channel expression and gating. Cardiovascular research. 2010;85:691–700. doi: 10.1093/cvr/cvp352. [DOI] [PubMed] [Google Scholar]
- 30.Balijepalli RC, Kamp TJ. Caveolae, ion channels and cardiac arrhythmias. Progress in biophysics and molecular biology. 2008;98:149–160. doi: 10.1016/j.pbiomolbio.2009.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Head BP, et al. Microtubules and actin microfilaments regulate lipid raft/caveolae localization of adenylyl cyclase signaling components. The Journal of biological chemistry. 2006;281:26391–26399. doi: 10.1074/jbc.M602577200. [DOI] [PubMed] [Google Scholar]
- 32.van der Heyden MA, Smits ME, Vos MA. Drugs and trafficking of ion channels: a new pro-arrhythmic threat on the horizon? British journal of pharmacology. 2008;153:406–409. doi: 10.1038/sj.bjp.0707618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McEwen DP, Martens JR. Antifibrillatory agents and potassium channels in the atria: pore block versus channel trafficking. Molecular interventions. 2009;9:79–86. doi: 10.1124/mi.9.2.7. [DOI] [PubMed] [Google Scholar]
- 34.Takemasa H, et al. Coexistence of hERG current block and disruption of protein trafficking in ketoconazole-induced long QT syndrome. British journal of pharmacology. 2008;153:439–447. doi: 10.1038/sj.bjp.0707537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Witchel HJ. The hERG potassium channel as a therapeutic target. Expert opinion on therapeutic targets. 2007;11:321–336. doi: 10.1517/14728222.11.3.321. [DOI] [PubMed] [Google Scholar]
- 36.Anderson CL, et al. Most LQT2 mutations reduce Kv11.1 (hERG) current by a class 2 (trafficking-deficient) mechanism. Circulation. 2006;113:365–373. doi: 10.1161/CIRCULATIONAHA.105.570200. [DOI] [PubMed] [Google Scholar]
- 37.Yao Y, et al. Aminoglycoside antibiotics restore functional expression of truncated HERG channels produced by nonsense mutations. Heart Rhythm. 2009;6:553–560. doi: 10.1016/j.hrthm.2009.01.017. [DOI] [PubMed] [Google Scholar]
- 38.Schumacher SM, et al. Antiarrhythmic drug-induced internalization of the atrial-specific k+ channel kv1.5. Circulation research. 2009;104:1390–1398. doi: 10.1161/CIRCRESAHA.108.192773. [DOI] [PMC free article] [PubMed] [Google Scholar]