

Abstract
Purpose
The purpose of the study was to characterize signaling intermediates involved in angiogenic responses of retinal endothelial cells (RECs) to the extracellular matrix and growth factors, by using specific inhibitors.
Methods
Tubelike structure formation and the development of secondary sprouts on a basement membrane (BM) matrix, cell proliferation, and cell migration were studied in cultures of bovine and human RECs. Specific inhibitors were tested for inhibition of retinal neovascularization in a mouse model of oxygen-induced retinopathy (OIR).
Results
In initial experiments, the broad-spectrum protein kinase inhibitors, H7 and H89, stabilized REC tubes on BM matrix and inhibited secondary sprouting, cell migration, and cell proliferation. Among more specific kinase inhibitors tested, only inhibitors of protein kinase CK2 (formerly, casein kinase II), such as emodin and DRB, were able to duplicate the effects of H7 and H89. Actinomycin D caused only minor changes in angiogenic assays, suggesting that CK2’s effects on REC did not involve its known impact on transcription. The extent of retinal neovascularization in a mouse OIR model was reduced >70% (versus untreated or vehicle-treated groups) after treatment with emodin (6 days at 60 mg/kg per day) and by approximately 60% after treatment at the same dose with TBB, the most specific CK2 inhibitor known. In the treated retinas, the main vascular tree had minimal changes, but the neovascular tufts were greatly reduced in number or absent.
Conclusions
This is the first demonstration of the involvement of ubiquitous protein kinase CK2 in angiogenesis. Naturally derived CK2 inhibitors may be useful for treatment of proliferative retinopathies.
Retinal neovascularization, as observed in proliferative diabetic retinopathy (PDR), represents an abnormal angiogenic process that occurs in response to insufficient tissue oxygenation.1 In the retinas of diabetic patients, homeostatic abnormalities result in retinal nonperfusion and subsequent ischemia, which leads to characteristic PDR neovascularization and disruption of normal retinal vasculature.2 Retinal neovascularization in PDR appears to involve alterations of a variety of angiogenic growth factors, such as vascular endothelial growth factor (VEGF); its homologue, placenta growth factor (PlGF); basic fibroblast growth factor (FGF)-2; insulin-like growth factor (IGF)-I; hepatocyte growth factor (HGF); and platelet-derived growth factor (PDGF).2–14
Hypoxia-inducible VEGF is usually considered to be the main mediator of PDR neovascularization, probably in concert with IGF-I.15 It may also be the main mediator of choroidal neovascularization in age-related macular degeneration.16,17 VEGF may promote both neovascularization and vascular permeability, resulting in fluid leakage and hemorrhages.6,7,18–20 However, VEGF may not be the sole causative factor of retinal neovascularization in patients with PDR or macular degeneration. Recent data, including ours, suggest that angiogenic growth factors synergize to induce full response of endothelial cells in vitro and in vivo.21–24 Growth factor synergy may be critical for the development of PDR neovascularization, as vitreous of patients with PDR has significantly increased concentrations of various angiogenic growth factors.5–14 A similar situation apparently exists in macular degeneration.17
Angiogenic growth factors exert their action through cell surface tyrosine kinase receptors that bind to and phosphorylate signaling molecules. Considerable progress has been made in dissecting the respective signaling pathways. Different growth factors use the same pathways for migratory, mitogenic, or antiapoptotic effects on cells.25 Depending on the conditions, cell type, and receptor involved, the actual pathway used and hence, the outcome, may vary greatly.26 Such peculiarities of signaling by different growth factors may form the basis of their synergy observed both in vitro and in vivo.
For a successful antiangiogenic therapy, it is important to first understand the mechanisms of angiogenesis. In molecular terms, it means to identify which signaling pathways govern complex angiogenic cell reactions such as migration, stromal invasion, capillary-like tube formation, and pericyte recruitment. In this report, we investigated these pathways using inhibitors of various key signaling molecules. As a result, we have identified the ubiquitous and pleiotropic protein kinase CK2 as a major modifier of key signaling pathways. Previously, there was no evidence for a role of CK2 in angiogenesis despite its known links to angiogenic growth factors. We show herein that specific CK2 inhibitors can stabilize retinal endothelial cell tubes on basement membrane (BM) matrix, inhibit growth-factor–stimulated endothelial cell migration, proliferation, and secondary sprouting on this matrix, and significantly decrease oxygen-induced retinal neovascularization in mice.
Methods
Cell Culture
Retinal endothelial cells (RECs) were isolated from fresh bovine eyes provided by Sierra for Medical Science (Santa Fe Springs, CA) by a modified method of Grant and Guay.27 Cultures of human RECs from healthy and diabetic donor eyes obtained from the National Disease Research Interchange (NDRI, Philadelphia, PA) were also used. NDRI has a human tissue collection protocol approved by a managerial committee and subject to National Institutes of Health oversight. It is managed according to the provisions of the Declaration of Helsinki. Cedars-Sinai Medical Center judged this tissue to be exempt from the institutional review board review (exemption 4). Primary cell cultures were established as published in detail previously.21,28 Briefly, dissected retinas were passed through a sterile 45-μm nylon mesh, digested with collagenase (Worthington Biochemical Corp., Lakewood, NJ) in Dulbecco’s phosphate-buffered saline (PBS) for 30 minutes, resuspended in incomplete REC medium (50% Ham’s F-12, 50% low-glucose DMEM with antibiotics/antimycotics [Invitrogen, Carlsbad, CA] and 10% fetal calf serum [FCS]) and centrifuged at 400g for 5 minutes. The pellet was resuspended in high serum, complete REC medium (incomplete medium plus insulin/transferrin/selenium, endothelial cell growth supplement [Sigma-Aldrich Co., St. Louis, MO] and 20% FCS). After the first passage the serum was reduced to 10%. Only passages 3 to 5 were used for experiments. Cultures were checked for purity by immunostaining with rabbit antibodies to von Willebrand factor (Sigma-Aldrich).
Cell Viability and Proliferation Assay
Bovine RECs (5 × 103 REC per well of triplicate 96-well plates) were incubated in complete REC medium with 10% FCS (proliferation) or 0.5% FCS (viability in low serum). Inhibitors were added at the time of cell seeding. Control cultures received medium with the inhibitor vehicle, dimethylsulfoxide (DMSO), at ≤0.1%. The number of cells was determined on days 4 to 7 using the MTS cell-proliferation assay (Promega Corp., Madison, WI) according to the manufacturer’s instructions.
Tube Formation Assay on a Reconstituted Basement Membrane Matrix
This assay has been published in detail previously.21 Briefly, 50 μL of reconstituted basement membrane (BM) matrix from mouse EHS tumor (Matrigel; BD Biosciences Labware, Bedford, MA) was added into each well of cold 96-well plates and allowed to solidify for 1 hour at 37°C. The 5 × 104 bovine RECs per 100 μL were seeded into triplicate wells in 0.5% FCS-incomplete REC medium. Appropriate inhibitors were added at the time of cell seeding (effect on tube formation) or 24 hours later (effect on tube collapse). Capillary-like tube structures formed by REC on reconstituted basement membrane matrix were photographed at various intervals ranging from 12 to 72 hours. Pictures were scanned, digitized, and analyzed using image-processing software.21
Secondary Sprouting Assay on Matrigel
Our group has recently developed an assay,21 which, unlike many other assays, replicates not one but several steps of the angiogenic process. Briefly, endothelial cells on basement membrane matrix (Matrigel; Invitrogen) form capillary-like tubes within 24 hours, as described earlier. Tubes then start shortening, and the cells aggregate into clumps and are thought to all die by apoptosis in 48 to 72 hours. Because of a longer examination time (up to 2 weeks), we found that the seemingly dead clumps actually contain living cells. By day 5, these von Willebrand factor–positive cells start to proliferate, sprout out of the clumps (migrate), and invade Matrigel forming three-dimensional spheres. Secondary sprouts are usually well developed by day 9. Some spheres form connections between each other that resemble larger capillaries and contain lumens. Growth factors such as FGF-2 and PDGF greatly enhance the sprouting.
For this assay, RECs were seeded on BM in duplicate or triplicate and incubated in 0.5% FCS-incomplete REC medium without growth factors for 2 to 3 days, allowing for tube formation and collapse. On days 2 to 3, human recombinant or purified growth factors (VEGF+IGF-I+FGF-2+PlGF21) were added to a final concentration of 10 ng/mL each and incubated for another 5 to 6 days. This combination of growth factors synergistically enhanced secondary sprout formation.21 Inhibitors were added with growth factors. Control cultures received medium with or without inhibitor vehicle (DMSO at ≤0.1%). Digital photographs were obtained with a camera (MDS 100; Eastman Kodak, Rochester, NY) attached to an inverted microscope (DM IL; Ernst Leitz, Wetzlar, Germany). The extent of secondary sprouting was determined by measuring the number of living cells in the sprouting colonies with the MTS cell-proliferation assay.
Wound Migration Assay
Cells were seeded in 24-well plates and allowed to reach confluence. Before growth factor treatment, cells were serum starved overnight in incomplete REC medium with 0.5% FCS. All monolayers within an experiment were wounded with a single sterile wood stick of constant diameter to ensure uniformity in the wound areas. Wounded monolayers were rinsed with low-serum (0.5%) medium to remove detached cells and treated with a combination of four growth factors (VEGF+IGF-I+FGF-2+PlGF)21 at 10 ng/mL each. As with secondary sprouting, the combination of these factors greatly increased cell migration in a synergistic way.21 Inhibitors were added with growth factors. Control cultures received medium with inhibitor vehicle (DMSO at ≤0.1%). On day 7, cells were rinsed three times with PBS, fixed with methanol for 15 minutes, and stained with Mayer hematoxylin for 5 minutes. All wells were photographed with a 4× or 10× objective digital camera (MDS 100; Eastman Kodak). The original wound area at 0 hours was used as a baseline for comparison to the treated wells at the end of the experiment. Migrating cell counting was automated on computer (AAB software; Advanced American Biotechnology, Fullerton, CA).
Inhibitor Treatments
Because various inhibitor doses and assays were used, a large number of cells was required. Therefore, most of the studies were performed using cultured bovine RECs. They were very similar to human RECs in response to growth factors. For each angiogenic assay, cells were treated with growth factor combinations, with or without signaling inhibitors, as described. The inhibitors were as follows: H7 (blocks various protein kinases; Seikagaku America, East Falmouth, MA), H89 (blocks protein kinase [PK]A and PKG, also affects CK229; Seikagaku America), calphostin C (blocks PKC; BioMol, Plymouth Meeting, PA), LY379196 (blocks PKC-β; from author MBG), KN-93 (blocks Ca2+/calmodulin kinase II; Seikagaku America), CKI-7 (blocks casein kinase (CK)1, also affects CK2 at high doses; Seikagaku America), wortmannin (blocks phosphoinositide 3 [P13]) kinase; Sigma-Aldrich), LY294002 (CK2+PI3 kinase; Sigma-Aldrich), emodin (1,3,8-trihydroxy-6-methyl-anthraquinone; Sigma-Aldrich), DRB (5,6-dichloro-1-β-o-ribofuranosyl benzimidazole; BioMol), and TBB (4,5,6,7-tetrabromobenzotriazole; synthesized according to protocol described by Büchel30; all block CK2 with TBB being the most specific to date31–36), PD98059 (blocks mitogen-activated protein [MAP] kinase kinase [MEK])/extracellular signal-regulated kinase [ERK], SB202190 (blocks p38 MAP kinase), SB203580 (blocks p38 MAP kinase and MAP kinase-activated protein [MAPKAP] kinase), and SB202474 (an inactive compound used as a negative control for MAP kinase inhibitors). The last four inhibitors were part of MAP kinase inhibitor set I (Calbiochem, San Diego, CA). In preliminary experiments, inhibitors of CK2 and some other kinases, apigenin (4′,5,7-trihydroxyflavone), and quercetin (3,5,7,3′,4′-penta-hydroxyflavone, both from Sigma-Aldrich), were also used. Quercetin was further used in pilot animal experiments because of its lack of toxicity to cultured cells, animals, and humans.37,38 In some experiments, the matrix metalloproteinase (MMP) inhibitor galardin (GM6001; BioMol) and a transcription inhibitor, actinomycin D (Sigma-Aldrich), were used. Working concentrations of all inhibitors and assays in which they were tested are summarized in Table 1.
Table 1.
Characteristics of Tested Signaling Inhibitors
| Inhibitor (target) | Range Tested | Optimal Concentration | Assays |
|---|---|---|---|
| H7 (broad spectrum) | 0–100 μM | 10–20 μM | SSP, M, T, P, V |
| H89 (broad spectrum) | 0–100 μM | 25 μM | SSP, M, T, P, V |
| galardin (MMPs) | 0.01–10 μM | 2.5 μM | SSP, M, T, P, V |
| KN-93 (Ca2+/calmodulin kinase II) | 0–1 μM | 500 nM | SSP, M, T, P, V |
| CKI-7 (CK-1) | 0–50 μM | 50 μM | SSP, M, T, P, V |
| LY379196 (PKC-β) | 0–5 μM | 50 nM | SSP, M, T, P, V |
| calphostin C (PKC) | 0–5 nM | 2.5 μM | SSP, M, T, P, V |
| wortmannin (PI3K) | 0–500 nM | 100 nM | SSP, M, T, P, V |
| PD98059 (MEK-ERK) | 0–10 μM | 10 μM | SSP, M, T, P, V |
| SB202190 (p38 MAPK) | 0–10 μM | 10 μM | SSP, M, T, P, V |
| SB203580 (p38 MAPK/MAPKAP) | 0–10 μM | 10 μM | SSP, M, T, P, V |
| SB202474 (MAPK negative control) | 0–10 μM | 10 μM | SSP, M, T, P, V |
| LY294002 (CK2, PI3K) | 1–50 μM | 20 μM | SSP, T |
| quercetin (CK2 + others) | 0.001–100 μM | 50 μM | SSP, M, T, P, V |
| chrysin (CK2 + others) | 0.001–100 μM | 50 μM | SSP, M, T, P, V |
| apigenin (CK2 + others) | 0.001–100 μM | 10–25 μM | SSP, M, T, P, V |
| DRB (CK2 + others) | 0.001–100 μM | 10–25 μM | SSP, M, T, P, V |
| emodin (CK2 + a few others) | 0.001–100 μM | 10–25 μM | SSP, M, T, P, V |
| actinomycin D (transcription) | 0–10 μg/mL | 2 μg/mL | SSP, T, V |
SSP, secondary sprouting; M, migration; T, tube formation and collapse; P, proliferation in 10% serum; V, viability in 0.5% serum.
Mouse Oxygen-Induced Retinopathy Model
Proliferative retinopathy in neonatal mice was induced in heterozygous C57BL/6J mice (Jackson Laboratory, Bar Harbor, ME).39,40 All procedures adhered to the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research and were conducted under amended Institutional Animal Care and Use Committee (IACUC) protocol 0524 from the University of Florida. Mice from the retinopathy group were placed in 75% oxygen at postnatal day 7 and maintained in these conditions with their nursing mothers for 5 days. These mice were then returned to normal air and maintained for another 5 days. Nor-moxic control mice were maintained in normal air for the same duration as test mice and under the same conditions of light cycle and temperature.
At the end of the experiment, mice were euthanatized by intraperitoneal injection with tribromoethanol (0.1 mL/g body weight). This method is consistent with the recommendations of the Panel on Euthanasia of the American Veterinary Association and was selected for its rapid action and lack of effect on tissue (especially vascular) ultra-structure. Animals were perfused through the left ventricle with 4% paraformaldehyde in 0.1 M phosphate buffer (pH 7.4) with 50 mg/mL of 2 × 103-kDa fluorescein-dextran (Sigma-Aldrich). The eyes were enucleated and fixed in 4% paraformaldehyde in 0.1 M phosphate buffer (pH 7.4) for 18 hours. Retinas from one eye of each mouse were dissected, the peripheral retinas cut in five places and flat-mounted with glycerol-gelatin, and the specimens photographed under a fluorescence microscope. Fellow eyes from all mice were embedded in paraffin and serial 6-μm sections of whole eyes were cut sagittally through the cornea parallel to the optic nerve. Every 30th section was mounted on a slide. Cell nuclei were counted under a light microscope on 5 to 10 hematoxylin- and eosin-stained sections. Individuals blinded to the treatment identity counted all nuclei above the inner limiting membrane (preretinal nuclei) in each eye. Sections with the optic nerve were excluded, because normal vessels emanating from the optic nerve fulfilled the counting criterion and would have increased the error. Vascular cell nuclei were considered to be associated with new vessels if found on the vitreal side of the inner limiting membrane. Pericytes were not identified in the neovascular tufts and have not been documented in neovasculature. Nevertheless, pericytes or their precursors may have been included in our cell counts.
For in vivo administration, the CK2 inhibitors emodin and TBB were dissolved in PBS (pH 7.2) with 20% polyethylene glycol 400 (PEG 400) and 2% Tween-8041 (both from Sigma-Aldrich). This solvent did not elicit any animal tolerance problems, unlike ethanol or DMSO. Inhibitors were injected intraperitoneally in volumes of 50 μL or less per mouse at doses of 15 to 30 mg/kg body weight, twice daily, starting from day 11. These doses were based on available pharmacokinetic data42 and previously established concentrations of emodin that did not show general toxicity, developmental toxicity, or genotoxicity when administered intraperitoneally to mice.43–45 Control mice were injected with PEG-Tween vehicle alone.
Image and Statistical Analysis
Inhibitor treatment data sets for cultured RECs were individually compared to their respective controls by the paired Student’s t-test (Prism 3.0 program; GraphPad Software, San Diego, CA). In the experiments in which one treatment was compared with several others, a nonparametric one-way ANOVA test was used (GraphPad Software). In animal studies, all data among various groups were compared using ANOVA. Tube formation images were processed by background subtraction, thresholding and measurement of total length of tubes by computer (Photoshop ver. 5.0; Adobe Systems Inc., Mountain View, CA; and the Image Processing Toolkit ver. 3.0, Reindeer Games, Inc., Gainesville, FL), as previously described.21,28
Results
In initial experiments, the broad-spectrum protein kinase inhibitors, H7 and H89, stabilized REC tubes on BM matrix (when added to formed tubes, 24 hours after cell seeding on Matrigel; Invitrogen), inhibited secondary sprouting (see the Methods section), cell migration, and proliferation (Table 2, Fig. 1 for tube stabilization). When added to the cells at the time of their seeding on the substrate, both inhibitors decreased REC tube formation (Table 2). We then tried to identify specific kinase(s) that were inhibited by H7 and H89 and played a role in these events. Most inhibitors of major signaling intermediates caused only minor effects in all assays over the range of tested concentrations (Table 2). Only molecules inhibiting protein kinase CK2 mimicked H-7 and H-89 in their effects on REC proliferation, secondary sprouting, migration, tube formation, and collapse (Table 2).
Table 2.
Effects of Various Inhibitors on Cultured RECs
| Inhibitor | Tube Formation* | Tube Collapse† | Secondary Sprouting | Migration | Proliferation/Viability |
|---|---|---|---|---|---|
| H7 | ↓↓↓ | ↓↓↓ | ↓↓ | ↓–↓↓ | ↓↓ |
| H89 | ↓↓↓ | ↓↓↓ | ↓↓ | ↓–↓↓ | ↓↓ |
| Galardin | ↓ | – | – | ↓ | ↓ |
| KN-93 | ↓ | – | – | No data | ↓ |
| CKI-7 | ↓ | – | – | No data | ↓ |
| LY379196 | ↓ | – | – | ↓ | ↓ |
| Calphostin C | ↓ | – | – | ↓ | ↓ |
| Wortmannin | ↓ | – | – | ↓ | ↓ |
| PD98059 | – | – | – | ↓ | ↓ |
| SB202190 | – | – | ↓↓ | ↓ | ↓ |
| SB203580 | – | – | – | ↓ | ↓ |
| SB202474 | – | – | – | – | – |
| LY294002 | ↓↓↓ | ↓↓↓ | ↓↓ | ↓↓↓ | ↓↓ |
| Quercetin | ↓↓ | ↓↓↓ | ↓↓ | ↓↓↓ | ↓↓ |
| Chrysin | ↓↓ | ↓↓↓ | ↓↓ | ↓↓↓ | ↓↓ |
| Apigenin | ↓↓↓ | ↓↓↓ | ↓↓ | ↓↓↓ | ↓↓ |
| DRB | ↓↓↓ | ↓↓↓ | ↓↓ | ↓↓↓ | ↓↓ |
| Emodin | ↓↓↓ | ↓↓↓ | ↓↓ | ↓↓↓ | ↓↓ |
| Actinomycin D | – | – | Toxic | Toxic | ↓↓ |
–, no effect; ↓, slight decrease; ↓↓, decrease; ↓↓↓, significant decrease.
Inhibitors added at the time of cell seeding on basement membrane.
inhibitors added 24 hours after cell seeding on basement membrane.
Figure 1.
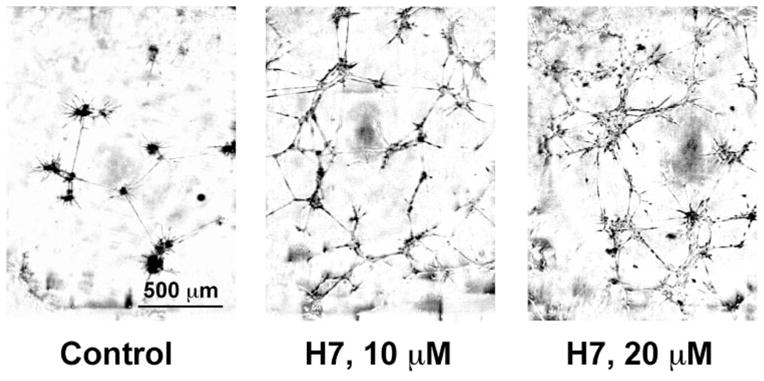
H7 stabilized REC tubes. Cells were seeded on basement membrane and incubated for 48 hours. In control cultures (left), tubes largely collapsed and clumps formed. In H7-treated cultures (middle, right) no tube collapse occurred. Phase-contrast microscopy.
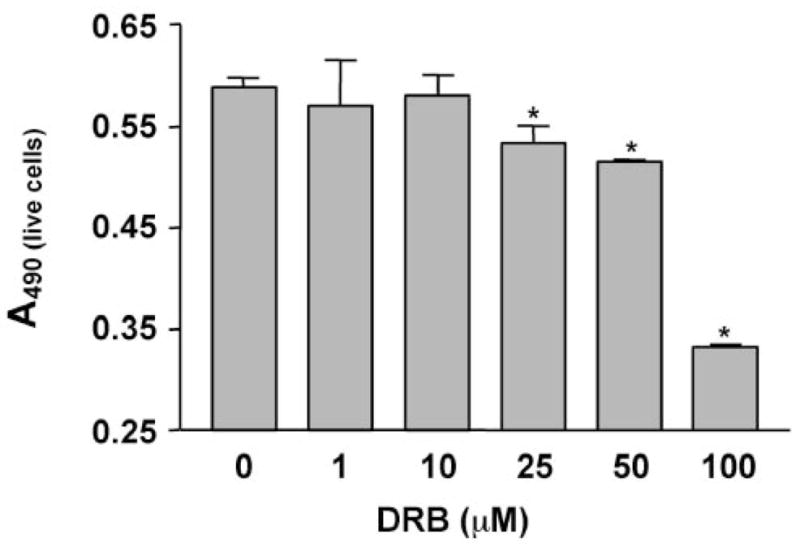
More specific CK2 inhibitors were as effective as H-7 or H-89 in modulating angiogenic behavior of REC. DRB produced a concentration-dependent inhibition of cell proliferation in 10% serum (Fig. 2). In serum-starved cells (0.5% serum), DRB caused a decrease in cell viability (Fig. 2). A similar dose-dependent effect on secondary sprouting was also observed (Fig. 3). Emodin was very similar to DRB (not shown here). These effects of CK2 inhibitors were statistically significant. When cells were treated with a synergistically acting combination of four angiogenic growth factors, VEGF+IGF-I+PlGF+FGF-2,21 secondary sprouting, measured as the number of live cells in cultures, was significantly enhanced (Fig. 4). Emodin completely abrogated this effect of four growth factors, bringing the number of cells to baseline level (Fig. 4). In the wound migration assay, a combination of these four growth factors synergistically increased cell migration more than threefold. Again, emodin was able to reduce this effect significantly (Fig. 5). In separate experiments, we tested whether the inhibitor effects could be due to the known influence of CK2 on transcription.46 To this end, cells were treated with a transcription inhibitor, actinomycin D, over a range of concentrations. This agent caused only minor changes in the multistep secondary sprouting assay (Fig. 6) suggesting that the observed CK2 effects on REC did not involve its impact on transcription.
Figure 2.

Dose-dependent effect of DRB on bovine REC proliferation and viability. Cells were plated on plastic in medium with 0.5% (viability) or 10% (proliferation) serum containing various concentrations of DRB. The number of live cells was measured on day 6 with the MTS assay. Bars, mean ± SEM of results in two experiments in triplicate. DRB significantly lowered the number of cells in both serum concentrations. *P < 0.05 versus vehicle.
Figure 3.
Dose-dependent effect of DRB on bovine REC secondary sprouting. Cells were seeded on basement membrane in medium with 0.5% serum. DRB was added at various concentrations on day 3 after tube collapse.21 The number of live cells was measured on day 9 with the MTS assay. Bars, mean ± SEM of results in two individual experiments in duplicate. DRB significantly decreased the number of cells starting at 25 μM. *P < 0.05 versus vehicle.
Figure 4.

Effect of emodin on growth factor–enhanced REC secondary sprouting. Bovine RECs were seeded on basement membrane, and the tubes were allowed to collapse. Four growth factors (4 GFs), IGF-I+VEGF+FGF-2+PlGF at 1 ng/mL each, were added on day 3 subsequent to emodin at 25 μM. The number of live cells was measured on day 9 by MTS assay. Note the dramatic decrease in the number of cells after treatment with the inhibitor. Bars, mean ± SEM of at least two experiments in duplicate. *P < 0.05 versus 4 GFs.
Figure 5.

Effect of emodin on growth factor–mediated cell migration. Confluent bovine RECs on plastic were wounded and kept for 7 days in medium+0.5% serum, with or without (control) four growth factors (4 GFs), IGF-I+FGF-2+VEGF+PlGF at 10 ng/mL each, with or without emodin (25 μM). Cell migration into the wound was then counted. Note the significant decrease in growth factor–stimulated cell migration by both CK2 inhibitors. Bars, mean ± SEM of at least three experiments. *P < 0.005 versus 4 GFs.
Figure 6.

Effect of actinomycin D on bovine REC secondary sprouting. Cells were seeded on basement membrane in medium with 0.5% serum containing various concentrations of actinomycin D (Act-D). The number of live cells was measured on day 9 with the MTS assay. Act-D only slightly lowered the number of cells, even at 10 μg/mL, with no significant difference from vehicle. Bars, mean ± SEM of results in two individual experiments in duplicate.
We next tested the effects of specific CK2 inhibitors on retinal neovascularization in vivo, using a mouse model of proliferative OIR. In the vehicle-treated retinas, peripheral tufts of newly formed blood vessels were readily visible on retinal angiograms and stained sections (Figs. 7A, 7B, arrows). Emodin treatment reduced the extent of retinopathy by 70% versus untreated group or vehicle-treated group (Fig. 7C). The same results were obtained using 30 or 60 mg/kg per day. The most specific CK2 inhibitor to date, TBB, produced approximately 60% inhibition at 60 mg/kg per day (Fig. 7C). Of note, the main vascular tree had few changes in the treated retinas, but the neovascular tufts were greatly reduced in number or absent. Sections of inhibitor-treated retinas did not show any adverse changes in retinal structure, as in control, normoxic, retinas.
Figure 7.

CK2 inhibitors significantly reduced retinal neovascularization. (A) Fluorescein-dextran perfusion of retinas from mice treated intraperitoneally with solvent (vehicle) or inhibitor (emodin). (B) Hematoxylin-eosin–stained sections of retinas from mice treated with vehicle or TBB. Arrows: neovascular tufts in vehicle-treated retinas. (C) Counts of preretinal nuclei in various groups of mice. Neovascularization is significantly decreased in all inhibitor-treated groups without visibly affecting the main vascular tree. N, number of mice in a group. Five to 10 sections per eye from each mouse were counted. Emodin was used at 30 or 60 mg/kg per day with similar results and TBB at 60 mg/kg per day. *P < 0.001 versus untreated or vehicle.
Discussion
Details and interrelationships of various signaling pathways mediating angiogenic behavior of endothelial cells remain incompletely understood. Using various inhibitors, we examined signaling molecules that may mediate endothelial cell migration, capillary-like tube formation on basement membrane matrix (Matrigel; Invitrogen), and secondary sprouting. Whereas cell migration is an early event in angiogenesis and tube formation is a late one, secondary sprouting recapitulates several steps in the angiogenic process, including tube formation and collapse, sprouting through cell proliferation and migration, and matrix invasion.21,28 Our results show that these diverse angiogenic steps, together with cell proliferation or survival in low serum, could be inhibited by agents blocking the activity of one key cellular enzyme, protein kinase CK2.
Protein kinase CK2 (formerly, casein kinase II) is a tetrameric serine/threonine protein kinase with two catalytic (α and/or α′, 43–45 kDa) and two regulatory (β, 25 kDa) subunits.46 Recently, a variant α chain, α″, with a translated Alu sequence at the C-terminal end, has been described.47 CK2 can be stimulated by tyrosine phosphorylation48 and probably also acts as a tyrosine kinase in some organisms—hence, the term “dual-function kinase.”49 This ubiquitous enzyme plays a role in development, cell cycle, transcription, proliferation, differentiation, apoptosis, DNA repair, and tumor growth.49–59 CK2 is expressed in probably all eukaryotic cells and has more than 300 identified protein substrates in the cytoplasm, nucleus, and extracellular matrix.60 It is found in various intracellular compartments and on the cell surface.46,47,49,54,60–68 CK2 interacts with and/or phosphorylates key signaling molecules including p53, protein phosphatase 2A, tumor suppressor phosphatase PTEN, p38 MAPK, PKC, various transcription factors, A-Raf, Akt/PKB, and S6 kinase46,49,50,60,69,70 (see also Fig. 8). Elevated expression of CK2 is associated with tumor growth, and its downregulation results in tumor cell apoptosis.50,53,57–59 CK2 is involved in growth factor effects. It can be activated by some growth factors, including FGF-2, IGF-I, insulin, and TGF-β.71–73 It can also modulate IGF-I activity by phosphorylating IGF-binding proteins.74,75
Figure 8.

Major signal transduction pathways of angiogenic growth factors and ECM proteins. The most-studied pathways are shown (PKC, PI3K-Akt/PKB, Raf-ERK, and p38 MAPK stress response). Differences in signaling from different receptors (R) of a single growth factor are depicted only for VEGF-PlGF (VEGFR1 and VEGFR2). Major CK2 interaction-phosphorylation points49,59,68,69 that may affect signaling pathways are shown. Up arrows: stimulation; down arrows: inhibition. IRS-1, insulin receptor substrate-1.
Our present data for the first time link this ubiquitous protein kinase with angiogenesis. Inhibitors of CK2 were able to decrease normal and diabetic REC proliferation, migration, and viability in low-serum medium (Figs. 1, 2, 3). It is noteworthy that these inhibitors also readily abrogated potent effects of four combined angiogenic growth factors on all the studied parameters of bovine and human REC. This finding may be explained by the participation of CK2 in several major pathways activated by angiogenic growth factors including Ras-Raf-MEK-ERK, p38 MAPK, PKC, and PI3 kinase-Akt pathways50,60,69,70 (Fig. 8). As TGF-β contributes to REC tube collapse,28 and CK2 inhibitors stabilized these tubes, CK2 could be also involved in TGF-β signaling. Recent findings suggest that such pleiotropic effects of CK2 may be attributed to its ability to phosphorylate a molecular chaperone, Cdc37, which interacts with multiple signaling kinases.76
It may thus be suggested that CK2 enhances cell responses to angiogenic growth factors and protect endothelial cells from apoptosis. This hypothesis is corroborated by recent evidence that CK2 inhibitors promote apoptosis of tumor cells.56,77–80 In case of emodin, it may involve generation of reactive oxygen species and subsequent inhibition of NF-κB and AP1 signaling.78 CK2 activity is generally elevated in actively proliferating cells.77 Therefore, CK2 inhibitors may mostly affect such cells. In our experiments with a mouse model of OIR, specific CK2 inhibitors decreased the extent of retinal neovascularization by up to 70% (Fig. 7). The inhibitory effect primarily concerned neovascular tufts where actively proliferating endothelial cells are concentrated81–84 but was not pronounced in the main vascular tree.
Our data may provide the molecular mechanism for the reported antitumor activity of bioflavonoids such as quercetin, emodin, and apigenin, for example. These compounds among other effects can inhibit CK2, which may reduce tumor growth by adversely affecting tumor neovasculature. In fact, the antiangiogenic effect of quercetin was observed previously,85,86 but it was not attributed to the blocking of CK2, because quercetin is not a specific CK2 inhibitor. In a recent work, tumor growth was significantly reduced by antisense oligonucleotides to CK2,53 but their effects on tumor vascularization were not studied. Distinct antiangiogenic effects of specific CK2 inhibitors, emodin and TBB, were clearly observed in our in vitro and in vivo experiments using retinal endothelial cells and a mouse model of retinal neovascularization. Because of the very high specificity of TBB toward CK2 we can conclude that CK2 is essential for angiogenesis. It is of obvious interest to identify the impact of CK2 inhibition on tumor neovascularization as well.
Both emodin and quercetin are plant-derived compounds found in tea, red wine, lettuce, rhubarb, aloe, and other plants. Moreover, quercetin is part of several over-the-counter dietary supplements. Therefore, these and more specific CK2 inhibitors may be promising tools for developing efficient drug therapy for PDR and other conditions with pathologic neovascularization, including solid tumors.
Emodin’s effects on CK2 activity and downstream expression of c-myc have been fully validated using CK2-specific antisense oligonucleotides.34 However, a small molecule inhibitor approach to CK2 inhibition should be considered with caution. The existing inhibitors may not be completely specific for CK2 and their possible interactions with other known or yet unidentified kinases, some of which may also be expressed only in certain tissues, should be taken into account. Alternative approaches to CK2 inhibition may use powerful RNAi or antisense oligonucleotide technologies50,70 that provide absolute specificity to the target. However, CK2 RNAi has so far achieved only moderate inhibition,70 and antisense oligonucleotides are difficult to use in vivo and may require special carriers. Possibly, a combination of gene-based and pharmacologic CK2 inhibitors would provide in future the magnitude and specificity of action necessary to eliminate pathologic neovascularization without significant side effects.
In summary, we found that a key signaling molecule, protein kinase CK2, was important for many angiogenic characteristics of RECs and their responses to growth factors. Blocking CK2 activity with a variety of inhibitors some of which were quite specific, produced significant changes, especially in tube formation, endothelial cell migration, and experimental retinal neovascularization in mice. CK2 has been shown to impinge on many signaling pathways (Fig. 8). We now provide the first evidence of the participation of CK2 in the angiogenic process. Based on our data, CK2 seems to be a major signaling molecule mediating growth factor effects in normal and pathologic angiogenesis. However, further experimentation is necessary to determine its role in the progression of DR to the vision-threatening proliferative stage.87
Acknowledgments
Supported by National Eye Institute Grants EY07739, EY12601, EY12605, and EY13431 and the Skirball Program in Molecular Ophthalmology.
The authors thank Graziano Fabbrini (Dipartimento di Chimica Biologica, Universita di Padova, Padova, Italy) for synthesizing the TBB.
Footnotes
Disclosure: A.V. Ljubimov, (P); S. Caballero, None; A.M. Aoki, None; L.A. Pinna, None; M.B. Grant, (P); R. Castellon, (P)
References
- 1.Dor Y, Eli K. Ischemia-driven angiogenesis. Trends Cardiovasc Med. 1997;7:289–294. doi: 10.1016/S1050-1738(97)00091-1. [DOI] [PubMed] [Google Scholar]
- 2.Frank RN. Diabetic retinopathy. N Engl J Med. 2004;350:48–58. doi: 10.1056/NEJMra021678. [DOI] [PubMed] [Google Scholar]
- 3.Østerby R. Basement membrane morphology in diabetes mellitus. In: Rifkin H, Porte D Jr, editors. Diabetes Mellitus. Theory and Practice. 4. New York: Elsevier; 1990. pp. 220–233. [Google Scholar]
- 4.Grant MB, Spoerri PE, Player DW, et al. Plasminogen activator inhibitor (PAI)-1 overexpression in retinal microvessels of PAI-1 transgenic mice. Invest Ophthalmol Vis Sci. 2000;41:2296–2302. [PubMed] [Google Scholar]
- 5.Paques M, Massin O, Gaudric A. Growth factors and diabetic retinopathy. Diabetes Metab. 1997;23:125–130. [PubMed] [Google Scholar]
- 6.Aiello LP, Hata Y. Molecular mechanisms of growth factor action in diabetic retinopathy. Curr Opin Endocrinol Diabetes. 1999;6:146–156. [Google Scholar]
- 7.Aiello LP, Wong JS. Role of vascular endothelial growth factor in diabetic vascular complications. Kidney Int. 2000;58(suppl 77):113–119. doi: 10.1046/j.1523-1755.2000.07718.x. [DOI] [PubMed] [Google Scholar]
- 8.Boulton M, Gregor Z, McLeod D, et al. Intravitreal growth factors in proliferative diabetic retinopathy: correlation with neovascular activity and glycaemic management. Br J Ophthalmol. 1997;81:228–233. doi: 10.1136/bjo.81.3.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Grant M, Russell B, Fitzgerald C, Merimee TJ. Insulin-like growth factors in vitreous: studies in control and diabetic subjects with neovascularization. Diabetes. 1986;35:416–420. doi: 10.2337/diab.35.4.416. [DOI] [PubMed] [Google Scholar]
- 10.Sivalingam A, Kenney J, Brown GC, Benson WE, Donoso L. Basic fibroblast growth factor levels in the vitreous of patients with proliferative diabetic retinopathy. Arch Ophthalmol. 1990;108:869–872. doi: 10.1001/archopht.1990.01070080113046. [DOI] [PubMed] [Google Scholar]
- 11.Pfeiffer A, Spranger J, Meyer-Schwickerath R, Schatz H. Growth factor alterations in advanced diabetic retinopathy: a possible role of blood retina barrier breakdown. Diabetes. 1997;46(suppl 2):S26–S30. doi: 10.2337/diab.46.2.s26. [DOI] [PubMed] [Google Scholar]
- 12.Canton A, Burgos R, Hernandez C, et al. Hepatocyte growth factor in vitreous and serum from patients with proliferative diabetic retinopathy. Br J Ophthalmol. 2000;84:732–735. doi: 10.1136/bjo.84.7.732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Grierson I, Heathcote L, Hiscott P, Hogg P, Briggs M, Hagan S. Hepatocyte growth factor/scatter factor in the eye. Prog Retin Eye Res. 2000;19:779–802. doi: 10.1016/s1350-9462(00)00015-x. [DOI] [PubMed] [Google Scholar]
- 14.Freyberger H, Brocker M, Yakut H, et al. Increased levels of platelet-derived growth factor in vitreous fluid of patients with proliferative diabetic retinopathy. Exp Clin Endocrinol Diabetes. 2000;108:106–109. doi: 10.1055/s-2000-5803. [DOI] [PubMed] [Google Scholar]
- 15.Smith LE, Shen W, Perruzzi C, et al. Regulation of vascular endothelial growth factor-dependent retinal neovascularization by insulin-like growth factor-1 receptor. Nat Med. 1999;5:1390–1395. doi: 10.1038/70963. [DOI] [PubMed] [Google Scholar]
- 16.Witmer AN, Vrensen GF, Van Noorden CJ, Schlingemann RO. Vascular endothelial growth factors and angiogenesis in eye disease. Prog Retin Eye Res. 2003;22:1–29. doi: 10.1016/s1350-9462(02)00043-5. [DOI] [PubMed] [Google Scholar]
- 17.Schlingemann RO. Role of growth factors and the wound healing response in age-related macular degeneration. Graefes Arch Clin Exp Ophthalmol. 2004;242:91–101. doi: 10.1007/s00417-003-0828-0. [DOI] [PubMed] [Google Scholar]
- 18.Ozaki H, Seo MS, Ozaki K, et al. Blockade of vascular endothelial cell growth factor receptor signaling is sufficient to completely prevent retinal neovascularization. Am J Pathol. 2000;156:697–707. doi: 10.1016/S0002-9440(10)64773-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Aiello LP. Vascular endothelial growth factor and the eye: biochemical mechanisms of action and implications for novel therapies. Ophthalmic Res. 1997;29:354–362. doi: 10.1159/000268033. [DOI] [PubMed] [Google Scholar]
- 20.Miller JW, Adamis AP, Aiello LP. Vascular endothelial growth factor in ocular neovascularization and proliferative diabetic retinopathy. Diabetes Metab Rev. 1997;13:37–50. doi: 10.1002/(sici)1099-0895(199703)13:1<37::aid-dmr174>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 21.Castellon R, Hamdi HK, Sacerio I, Aoki AM, Kenney MC, Ljubimov AV. Effects of angiogenic growth factor combinations on retinal endothelial cells. Exp Eye Res. 2002;74:523–535. doi: 10.1006/exer.2001.1161. [DOI] [PubMed] [Google Scholar]
- 22.Shyu KG, Chang H, Isner JM. Synergistic effect of angiopoietin-1 and vascular endothelial growth factor on neoangiogenesis in hypercholesterolemic rabbit model with acute hindlimb ischemia. Life Sci. 2003;73:563–579. doi: 10.1016/s0024-3205(03)00318-7. [DOI] [PubMed] [Google Scholar]
- 23.Stavri GT, Zachary IC, Baskerville PA, Martin JF, Erusalimsky JD. Basic fibroblast growth factor upregulates the expression of vascular endothelial growth factor in vascular smooth muscle cells: synergistic interaction with hypoxia. Circulation. 1995;92:11–14. doi: 10.1161/01.cir.92.1.11. [DOI] [PubMed] [Google Scholar]
- 24.Gerritsen ME, Tomlinson JE, Zlot C, Ziman M, Hwang S. Using gene expression profiling to identify the molecular basis of the synergistic actions of hepatocyte growth factor and vascular endothelial growth factor in human endothelial cells. Br J Pharmacol. 2003;140:595–610. doi: 10.1038/sj.bjp.0705494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rakhit S, Pyne S, Pyne NJ. The platelet-derived growth factor receptor stimulation of p42/p44 mitogen-activated protein kinase in airway smooth muscle involves a G-protein-mediated tyrosine phosphorylation of Gab1. Mol Pharmacol. 2000;58:413–420. doi: 10.1124/mol.58.2.413. [DOI] [PubMed] [Google Scholar]
- 26.Lawlor MA, Rotwein P. Coordinate control of muscle cell survival by distinct insulin-like growth factor activated signaling pathways. J Cell Biol. 2000;151:1131–1140. doi: 10.1083/jcb.151.6.1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Grant MB, Guay C. Plasminogen activator production by human retinal endothelial cells of nondiabetic and diabetic origin. Invest Ophthalmol Vis Sci. 1991;32:53–64. [PubMed] [Google Scholar]
- 28.Castellon R, Caballero S, Hamdi HK, et al. Effects of tenascin-C on normal and diabetic retinal endothelial cells in culture. Invest Ophthalmol Vis Sci. 2002;43:2758–2766. [PubMed] [Google Scholar]
- 29.Cotlin LF, Siddiqui MA, Simpson F, Collawn JF. Casein kinase II activity is required for transferrin receptor endocytosis. J Biol Chem. 1999;274:30550–30556. doi: 10.1074/jbc.274.43.30550. [DOI] [PubMed] [Google Scholar]
- 30.Büchel KH. Inhibitors of photosynthesis. VI. Syntheses of electro-negatively substituting benzimidazoles (in German) Z Naturforsch B. 1970;25:945–953. [PubMed] [Google Scholar]
- 31.Negulescu O, Bognar I, Lei J, Devarajan P, Silbiger S, Neugarten J. Estradiol reverses TGF-β1-induced mesangial cell apoptosis by a casein kinase 2-dependent mechanism. Kidney Int. 2002;62:1989–1998. doi: 10.1046/j.1523-1755.2002.00679.x. [DOI] [PubMed] [Google Scholar]
- 32.Xagorari A, Roussos C, Papapetropoulos A. Inhibition of LPS-stimulated pathways in macrophages by the flavonoid luteolin. Br J Pharmacol. 2002;136:1058–1064. doi: 10.1038/sj.bjp.0704803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yim H, Lee YH, Lee CH, Lee SK. Emodin, an anthraquinone derivative isolated from the rhizomes of Rheum palmatum, selectively inhibits the activity of casein kinase II as a competitive inhibitor. Planta Med. 1999;65:9–13. doi: 10.1055/s-1999-13953. [DOI] [PubMed] [Google Scholar]
- 34.Channavajhala P, Seldin DC. Functional interaction of protein kinase CK2 and c-Myc in lymphomagenesis. Oncogene. 2002;21:5280–5288. doi: 10.1038/sj.onc.1205640. [DOI] [PubMed] [Google Scholar]
- 35.Sarno S, Reddy H, Meggio F, et al. Selectivity of 4,5,6,7-tetrabro-mobenzotriazole, an ATP site-directed inhibitor of protein kinase CK2 (casein kinase-2) FEBS Lett. 2001;496:44–48. doi: 10.1016/s0014-5793(01)02404-8. [DOI] [PubMed] [Google Scholar]
- 36.Battistutta R, De Moliner E, Sarno S, Zanotti G, Pinna LA. Structural features underlying selective inhibition of protein kinase CK2 by ATP site-directed tetrabromo-2-benzotriazole. Protein Sci. 2001;10:2200–2206. doi: 10.1110/ps.19601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hayashi A, Gillen AC, Lott JR. Effects of daily oral administration of quercetin chalcone and modified citrus pectin. Altern Med Rev. 2000;5:546–552. [PubMed] [Google Scholar]
- 38.Ross JA, Kasum CM. Dietary flavonoids: bioavailability, metabolic effects, and safety. Annu Rev Nutr. 2002;22:19–34. doi: 10.1146/annurev.nutr.22.111401.144957. [DOI] [PubMed] [Google Scholar]
- 39.Pierce EA, Avery RL, Foley ED, Aiello LP, Smith LE. Vascular endothelial growth factor/vascular permeability factor expression in a mouse model of retinal neovascularization. Proc Natl Acad Sci USA. 1995;92:905–909. doi: 10.1073/pnas.92.3.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mino RP, Spoerri PE, Caballero S, et al. Adenosine receptor antagonists and retinal neovascularization in vivo. Invest Ophthalmol Vis Sci. 2001;42:3320–3324. [PubMed] [Google Scholar]
- 41.Caltagirone S, Rossi C, Poggi A, et al. Flavonoids apigenin and quercetin inhibit melanoma growth and metastatic potential. Int J Cancer. 2000;87:595–600. doi: 10.1002/1097-0215(20000815)87:4<595::aid-ijc21>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 42.Bachmann M, Schlatter C. Metabolism of [14C]emodin in the rat. Xenobiotica. 1981;11:217–225. doi: 10.3109/00498258109045294. [DOI] [PubMed] [Google Scholar]
- 43.Arosio B, Gagliano N, Fusaro LM, et al. Aloe-emodin quinone pretreatment reduces acute liver injury induced by carbon tetra-chloride. Pharmacol Toxicol. 2000;87:229–233. doi: 10.1034/j.1600-0773.2000.d01-79.x. [DOI] [PubMed] [Google Scholar]
- 44.National Toxicology Program. NTP toxicology and carcinogenesis studies of emodin (CAS No. 518-82-1) feed studies in F344/N rats and B6C3F1 mice. Natl Toxicol Program Tech Rep Ser. 2001;493:1–278. [PubMed] [Google Scholar]
- 45.Jahnke GD, Price CJ, Marr MC, Myers CB, George JD. Developmental toxicity evaluation of emodin in rats and mice. Birth Defects Res B. 2004;71:89–101. doi: 10.1002/bdrb.20002. [DOI] [PubMed] [Google Scholar]
- 46.Guerra B, Issinger OG. Protein kinase CK2 and its role in cellular proliferation, development and pathology. Electrophoresis. 1999;20:391–408. doi: 10.1002/(SICI)1522-2683(19990201)20:2<391::AID-ELPS391>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 47.Hilgard P, Huang T, Wolkoff AW, Stockert RJ. Translated Alu sequence determines nuclear localization of a novel catalytic subunit of casein kinase 2. Am J Physiol Cell Physiol. 2002;283:C472–C483. doi: 10.1152/ajpcell.00070.2002. [DOI] [PubMed] [Google Scholar]
- 48.Donella-Deana A, Cesaro L, Sarno S, et al. Tyrosine phosphorylation of protein kinase CK2 by Src-related tyrosine kinases correlates with increased catalytic activity. Biochem J. 2003;372:841–849. doi: 10.1042/BJ20021905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Litchfield DW. Protein kinase CK2: structure, regulation and role in cellular decisions of life and death. Biochem J. 2003;369:1–15. doi: 10.1042/BJ20021469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ahmed K, Gerber DA, Cochet C. Joining the cell survival squad: an emerging role for protein kinase CK2. Trends Cell Biol. 2002;12:226–230. doi: 10.1016/s0962-8924(02)02279-1. [DOI] [PubMed] [Google Scholar]
- 51.Pinna LA. Protein kinase CK2: a challenge to canons. J Cell Sci. 2002;115:3873–3878. doi: 10.1242/jcs.00074. [DOI] [PubMed] [Google Scholar]
- 52.Pinna LA. The raison d’être of constitutively active protein kinases: the lesson of CK2. Acc Chem Res. 2003;36:378–384. doi: 10.1021/ar020164f. [DOI] [PubMed] [Google Scholar]
- 53.Unger GM, Davis AT, Slaton JW, Ahmed K. Protein kinase CK2 as regulator of cell survival: implications for cancer therapy. Curr Cancer Drug Targets. 2004;4:77–84. doi: 10.2174/1568009043481687. [DOI] [PubMed] [Google Scholar]
- 54.Filhol O, Martiel JL, Cochet C. Protein kinase CK2: a new view of an old molecular complex. EMBO Rep. 2004;5:351–355. doi: 10.1038/sj.embor.7400115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Loizou JI, El-Khamisy SF, Zlatanou A, et al. The protein kinase CK2 facilitates repair of chromosomal DNA single-strand breaks. Cell. 2004;117:17–28. doi: 10.1016/s0092-8674(04)00206-5. [DOI] [PubMed] [Google Scholar]
- 56.Ruzzene M, Penzo D, Pinna LA. Protein kinase CK2 inhibitor 4,5,6,7-tetrabromobenzotriazole (TBB) induces apoptosis and caspase-dependent degradation of haematopoietic lineage cell-specific protein 1 (HS1) in Jurkat cells. Biochem J. 2002;364:41–47. doi: 10.1042/bj3640041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Landesman-Bollag E, Song DH, Romieu-Mourez R, et al. Protein kinase CK2: signaling and tumorigenesis in the mammary gland. Mol Cell Biochem. 2001;227:153–165. [PubMed] [Google Scholar]
- 58.Xu X, Landesman-Bollag E, Channavajhala PL, Seldin DC. Murine protein kinase CK2: gene and oncogene. Mol Cell Biochem. 1999;191:65–74. [PubMed] [Google Scholar]
- 59.Romieu-Mourez R, Landesman-Bollag E, Seldin DC, Sonenshein GE. Protein kinase CK2 promotes aberrant activation of nuclear factor-κB, transformed phenotype, and survival of breast cancer cells. Cancer Res. 2002;62:6770–6778. [PubMed] [Google Scholar]
- 60.Meggio F, Pinna LA. One-thousand-and-one substrates of protein kinase CK2? FASEB J. 2003;17:349–368. doi: 10.1096/fj.02-0473rev. [DOI] [PubMed] [Google Scholar]
- 61.Pinna LA, Meggio F. Protein kinase CK2 (“casein kinase-2”) and its implication in cell division and proliferation. Prog Cell Cycle Res. 1997;3:77–97. doi: 10.1007/978-1-4615-5371-7_7. [DOI] [PubMed] [Google Scholar]
- 62.Bosc DG, Luscher B, Litchfield DW. Expression and regulation of protein kinase CK2 during the cell cycle. Mol Cell Biochem. 1999;191:213–222. [PubMed] [Google Scholar]
- 63.Yu S, Wang H, Davis A, Ahmed K. Consequences of CK2 signaling to the nuclear matrix. Mol Cell Biochem. 2001;227:67–71. [PubMed] [Google Scholar]
- 64.Tawfic S, Yu S, Wang H, Faust R, Davis A, Ahmed K. Protein kinase CK2 signal in neoplasia. Histol Histopathol. 2001;16:573–582. doi: 10.14670/HH-16.573. [DOI] [PubMed] [Google Scholar]
- 65.Stepanova V, Jerke U, Sagach V, et al. Urokinase-dependent human vascular smooth muscle cell adhesion requires selective vitronectin phosphorylation by ectoprotein kinase CK2. J Biol Chem. 2002;277:10265–10272. doi: 10.1074/jbc.M109057200. [DOI] [PubMed] [Google Scholar]
- 66.Faust M, Jung M, Gunther J, Zimmermann R, Montenarh M. Localization of individual subunits of protein kinase CK2 to the endoplasmic reticulum and to the Golgi apparatus. Mol Cell Biochem. 2001;227:73–80. [PubMed] [Google Scholar]
- 67.Ford HL, Landesman-Bollag E, Dacwag CS, Stukenberg PT, Pardee AB, Seldin DC. Cell cycle-regulated phosphorylation of the human SIX1 homeodomain protein. J Biol Chem. 2000;275:22245–22254. doi: 10.1074/jbc.M002446200. [DOI] [PubMed] [Google Scholar]
- 68.Filhol O, Nueda A, Martel V, et al. Live-cell fluorescence imaging reveals the dynamics of protein kinase CK2 individual subunits. Mol Cell Biol. 2003;23:975–987. doi: 10.1128/MCB.23.3.975-987.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kusk M, Ahmed R, Thomsen B, Bendixen C, Issinger OG, Boldyreff B. Interactions of protein kinase CK2β subunit within the holoenzyme and with other proteins. Mol Cell Biochem. 1999;191:51–58. [PubMed] [Google Scholar]
- 70.Ruzzene M, Di Maira G, Salvi M, Arrigoni G, Pinna LA. Signalling events affected by down-regulation of protein kinase CK2. 4th International Conference on Protein Kinase CK2; London, Canada. 2004. Abstract 10. [Google Scholar]
- 71.Bonnet H, Filhol O, Truchet I, et al. Fibroblast growth factor-2 binds to the regulatory β subunit of CK2 and directly stimulates CK2 activity toward nucleolin. J Biol Chem. 1996;271:24781–24787. doi: 10.1074/jbc.271.40.24781. [DOI] [PubMed] [Google Scholar]
- 72.Bailly K, Soulet F, Leroy D, Amalric F, Bouche G. Uncoupling of cell proliferation and differentiation activities of basic fibroblast growth factor. FASEB J. 2000;14:333–344. [PubMed] [Google Scholar]
- 73.Higashi K, Ogawara H. Daidzein inhibits insulin- or insulin-like growth factor-1-mediated signaling in cell cycle progression of Swiss 3T3 cells. Biochim Biophys Acta. 1994;1221:29–35. doi: 10.1016/0167-4889(94)90212-7. [DOI] [PubMed] [Google Scholar]
- 74.Ankrapp DP, Jones JI, Clemmons DR. Characterization of insulin-like growth factor binding protein-1 kinases from human hepatoma cells. J Cell Biochem. 1996;60:387–399. doi: 10.1002/(sici)1097-4644(19960301)60:3<387::aid-jcb10>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 75.Coverley JA, Martin JL, Baxter RC. The effect of phosphorylation by casein kinase 2 on the activity of insulin-like growth factor-binding protein-3. Endocrinology. 2000;141:564–570. doi: 10.1210/endo.141.2.7306. [DOI] [PubMed] [Google Scholar]
- 76.Miyata Y, Nishida E. CK2 controls multiple protein kinases by phosphorylating a kinase-targeting molecular chaperone, Cdc37. Mol Cell Biol. 2004;24:4065–4074. doi: 10.1128/MCB.24.9.4065-4074.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Guo C, Yu S, Davis AT, Wang H, Green JE, Ahmed K. A potential role of nuclear matrix-associated protein kinase CK2 in protection against drug-induced apoptosis in cancer cells. J Biol Chem. 2001;276:5992–5999. doi: 10.1074/jbc.M004862200. [DOI] [PubMed] [Google Scholar]
- 78.Yi J, Yang J, He R, et al. Emodin enhances arsenic trioxide-induced apoptosis via generation of reactive oxygen species and inhibition of survival signaling. Cancer Res. 2004;64:108–116. doi: 10.1158/0008-5472.can-2820-2. [DOI] [PubMed] [Google Scholar]
- 79.Shieh DE, Chen YY, Yen MH, Chiang LC, Lin CC. Emodin-induced apoptosis through p53-dependent pathway in human hepatoma cells. Life Sci. 2004;74:2279–2290. doi: 10.1016/j.lfs.2003.09.060. [DOI] [PubMed] [Google Scholar]
- 80.Chen YC, Shen SC, Lee WR, et al. Emodin induces apoptosis in human promyeloleukemic HL-60 cells accompanied by activation of caspase 3 cascade but independent of reactive oxygen species production. Biochem Pharmacol. 2002;64:1713–1724. doi: 10.1016/s0006-2952(02)01386-2. [DOI] [PubMed] [Google Scholar]
- 81.de Juan E, Jr, Humayun MS, Hatchell DL, Wilson D. Histopathology of experimental preretinal neovascularization. Invest Ophthalmol Vis Sci. 1989;30:1495–1503. [PubMed] [Google Scholar]
- 82.de Juan E, Jr, Stefansson E, Ohira A. Basic fibroblast growth factor stimulates 3H-thymidine uptake in retinal venular and capillary endothelial cells in vivo. Invest Ophthalmol Vis Sci. 1990;31:1238–1244. [PubMed] [Google Scholar]
- 83.Chan-Ling T, Gock B, Stone J. The effect of oxygen on vasoformative cell division. Evidence that “physiological hypoxia” is the stimulus for normal retinal vasculogenesis. Invest Ophthalmol Vis Sci. 1995;36:1201–1214. [PubMed] [Google Scholar]
- 84.McLeod DS, Crone SN, Lutty GA. Vasoproliferation in the neonatal dog model of oxygen-induced retinopathy. Invest Ophthalmol Vis Sci. 1996;37:1322–1333. [PubMed] [Google Scholar]
- 85.Tan WF, Lin LP, Li MH, et al. Quercetin, a dietary-derived flavonoid, possesses antiangiogenic potential. Eur J Pharmacol. 2003;459:255–262. doi: 10.1016/s0014-2999(02)02848-0. [DOI] [PubMed] [Google Scholar]
- 86.Igura K, Ohta T, Kuroda Y, Kaji K. Resveratrol and quercetin inhibit angiogenesis in vitro. Cancer Lett. 2001;171:11–16. doi: 10.1016/s0304-3835(01)00443-8. [DOI] [PubMed] [Google Scholar]
- 87.Aiello LP, Gardner TW, King GL, et al. Diabetic retinopathy. Diabetes Care. 1998;21:143–156. doi: 10.2337/diacare.21.1.143. [DOI] [PubMed] [Google Scholar]