

Chronic rejection is a major cause of late liver allograft failure. Generally accepted pathologic features of this condition include chronic obliterative arteriopathy (COA) and bile duct loss. The mechanisms involved in graft damage are poorly understood.
Bile duct loss has been attributed to direct lymphocytotoxic attack, which seems reasonable considering the histologic appearance of rejection. However, ischemia due to COA has been largely overlooked as a cofactor in the pathogenesis of bile duct loss. It is known that the hepatic artery is the only blood supply to the intra-hepatic biliary tree.
In this study, we studied the effect of ischemia on bile duct loss by studying the relationship between the degree of COA and intensity of bile duct loss. Furthermore, in order to get a greater understanding of the pathogenesis of COA, we analyzed the lineage of cells comprising COA lesion in heart, liver and kidney allografts. Special attention was focused on detecting cells with phenotypic characteristics of dendritic cell (DC) lineage. DC play a pivotal role in the initiation of immunological reactions.
MATERIALS AND METHODS
Source of Materials and Selection of Cases
Liver allografts which failed because of chronic rejection were reviewed (N = 28). The diagnosis of chronic rejection was based on a characteristic clinical course and confirmed by histologic analysis of the removed allografts. Normal livers were selected from autopsy cases in which there was no clinical or pathological evidence of liver disease.
Tissue sampling for histologic sections from the failed allografts and control livers was performed according to a predefined protocol. Three sections were taken from the hilum, two sections from the left lobe, and three sections from the right lobe.1
The three sections taken from the hilum of each liver were examined blindly and independently by two of the authors for the degree of narrowing of hepatic arterial branches and the percentage of vessels affected. If greater than 70% of the arteries demonstrated more than 75% stenosis, the case was labeled as “severe COA”. Conversely, in “mild COA”, greater than 70% of arteries showed less than 35% narrowing (n = 10). The intermediate cases (n = 10) were excluded to accentuate any possible differences between the two extreme groups, which may result in recognition of the role of ischemia in bile duct loss. General patient data is shown in Table 1.
Table 1.
Demographic Data and Original Hepatic Disease
| Cases | Graft Survival (months) | Age Sex |
Original Hepatic Disease |
||||||
|---|---|---|---|---|---|---|---|---|---|
| PBC | CAH | HCC | SMN | PSC | AAD | O/C | |||
| Mild | |||||||||
| (n = 10) | 1.3–140.5 (33.7) | 14–47 (31.9) F:M = 8:2 |
4 | 3 | 1 | 0 | 1 | 0 | 1 |
| Severe | |||||||||
| (n=8) | 2.0–17.0 (7.7) | 2–32 (12.6) F:M = 2:6 |
0 | 0 | 1 | 2 | 1 | 2 | 2 |
PBC = Primary Biliary Cirrhosis, CAH = Chronic Active Hepatitis (Non A, Non B), HCC = Hepatocellular Carcinoma, SMN = Submassive Necrosis, PSC = Primary Sclerosing Cholangitis, AAD = Alpha-1-Antitrypsin Deficiency, O/C = Other/Congenital.
The average age of allograft patients with mild COA was 31.9 years compared to 12.6 for the patients with severe COA (P > 0.05). The average graft survival was much shorter in those with severe COA (7.7 months) compared to those with mild COA (33.7 months) (P < 0.05). Complete MHC typing of both donor and recipient was available in only 8 of the 18 cases analyzed. No particular trend was noted in this small sample. All patients were maintained on baseline immunosuppression of cyclosporine and steroids. Episodes of acute cellular rejection were treated with a steroid bolus or recycling doses. If steroids were unsuccessful, OKT3 (Ortho pharmoceuticals, Raritan, NJ) was used.
Histometrical Study for Arterial Loss
The presence or absence of any artery within the confines of the portal tract and the size of the only or largest artery in the portal tract was recorded. This assessment was repeated for all portal tracts in every section of the selected cases (average 200–300 portal tracts/case). The size of the artery was determined by measuring the shortest diameter of the vessel to the outer extent of the media using an ocular micrometer. The percentage of portal tracts with arteries and the size distribution of the only or largest artery was calculated according to a predefined range of arterial diameter (<35, 35–54, 55–74, ≥ 75 microns).
Histometrical Study for Bile Duct Loss
An analysis similar to that done by Nakanuma was also performed to determine the extent of duct loss and size of vanished ducts.2 In brief, it is known that arteries and bile ducts of a similar diameter occur in parallel in the portal tracts of the normal liver. Precise measurement of the diameter of the only or largest artery in each portal tract was recorded as described above. Tracing an imaginary circle 3 times the recorded arterial diameter from the external media of the vessel, circumscribed the area in which the only or largest accompanying bile duct should be detected. The presence or absence of a bile duct within this area was then recorded for each portal tract of every section. The result was labeled according to the predefined range of arterial diameter for each liver and expressed as the rate of bile duct artery (BD-A) parallelism. It must be noted that the rate of parallelism was determined only for those portal tracts which contained arteries. Portal tracts without arteries and bile ducts were excluded from the determinations of BD-A parallelism.
Study of Chronic Obliterative Vasculopathy (COA)
Heart (n = 7), kidney (n = 9) and liver (n = 7) allografts which failed from chronic rejection were reviewed. Formalin fixed and paraffin embedded blocks were cut at 4 microns and routinely stained with hematoxylin and eosin. Masson’s trichrome and Verhoff/Van Gieson for elastic tissue. Paraffin embedded sections were also subjected to staining with various antibodies using the avidin-biotin—complex (ABC) method.
Antibodies for lysozyme, S100 protein and T-lymphocytes (UCHL-1, DAKO, Santa Barbara, CA) and for actin (Biogenex, San Ramon, CA) were used. Immunofluorescent staining was also performed on frozen tissue embedded in OCT compound using a simple indirect technique with appropriate fluoresceinated secondary antibodies. Primary antibodies included HLA-DR and Leu 4 (CD3) (Becton Dickinson, Mountain View, CA).
RESULTS
Analysis of Bile Duct Loss in Chronic Liver Rejection
Distribution and Size of Portal Tract Arteries
The percentage of portal tracts containing arteries or arterioles were compared for each of the 14 subjects with no COA (controls), the 10 subjects with mild COA and 8 subjects with severe COA (Figure 1). Overall, 90.9% of the portal tracts in controls, 61.3% of the portal tracts in cases with mild COA and 25.3% of the portal tracts in cases with severe COA had at least one artery. The logistic regression model was used to determine whether there was an association between severity of COA and the presence of arteries in the portal tract.3 A statistically significant difference between each pair of groups was found.
Fig 1.
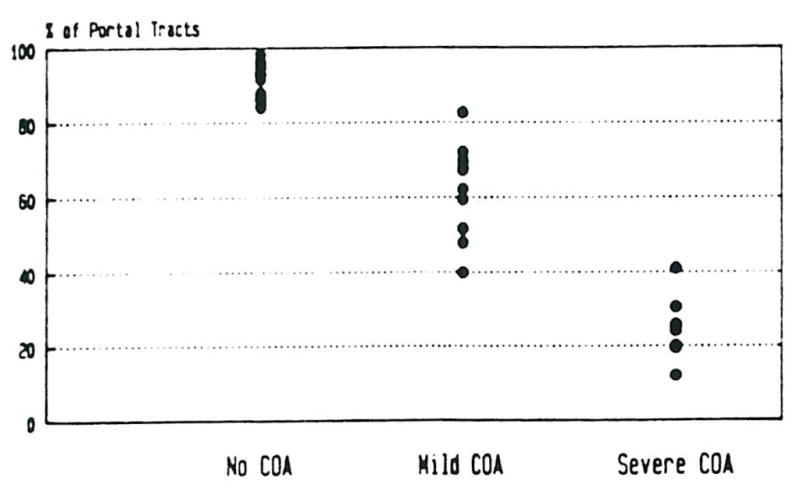
The percentage of portal tracts from normal control and failed allograft livers containing at least one identifiable artery is shown. Note the absolute loss of identifiable portal arteries in failed allografts.
Figure 2 shows the percentage of portal tracts in which the largest or only artery was <35, 35–54, 55–74, ≥ 75 microns in diameter for each subject classified by the degree of COA. Overall, the distribution of the size of the largest or only artery in portal tracts with at least one artery was significantly (P < 0.001) different among groups, using the chi-square test of association for independent samples.
Fig 2.
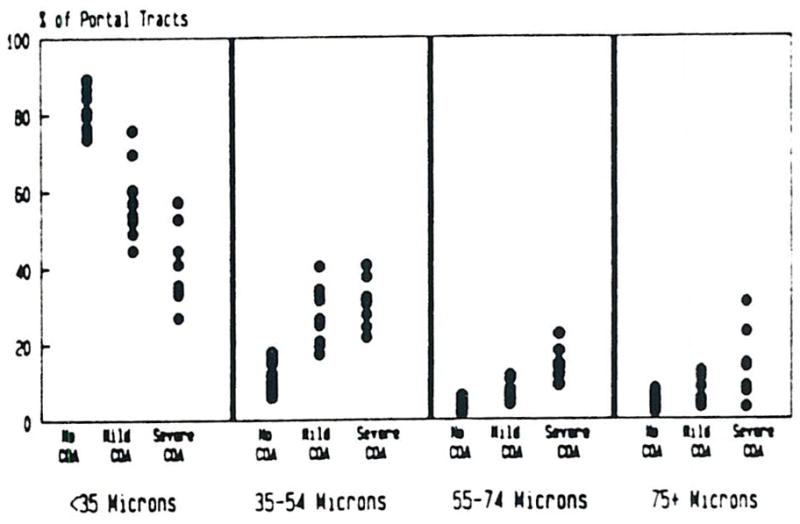
The percentage of portal tracts according to the size of the largest or only portal artery is represented. Note the relative decrease in small portal arterioles and relative Increase in larger arteries.
Pairwise comparison of the three groups using the same statistical technique demonstrated that the distribution of the size of the largest or only artery was significantly different (P < 0.001) between each pair. Among 81% of the portal tracts of controls, the largest or only artery was less than 35 microns, compared to about 58% of the portal tracts in cases with mild COA and 41% of the portal tracts in cases with severe COA. At the other extreme, only 4% of the portal tracts in controls contained a single or largest artery, 75 microns or greater in diameter, ranging from 1.9% to 7.9% among the 14 subjects. Six percent of portal tracts in cases with mild COA (range 3.1% to 12.6%) and 14% of portal tracts in cases with severe COA (range 3.1% to 30.8%) these differences are also statistically significant (P < 0.001).
Bile Duct-Arterial Parallelism
The mean rate of bile duct-artery (BD-A) parallelism versus arterial size, classified by the degree of COA is shown in Figure 3. There is a statistically significant (P < 0.001) trend toward decreasing parallelism with decreasing arterial size in both the mild and severe COA, using a test for linear trend.4 Using the chi-square test for independent samples, statistically significant differences (p < 0.01) for BD-A parallelism between groups (controls vs. mild COA, controls vs. severe COA, and mild COA vs. severe COA) were found in all size categories, except between controls and mild COA for arteries greater than or equal to 75 microns in diameter.
Fig 3.
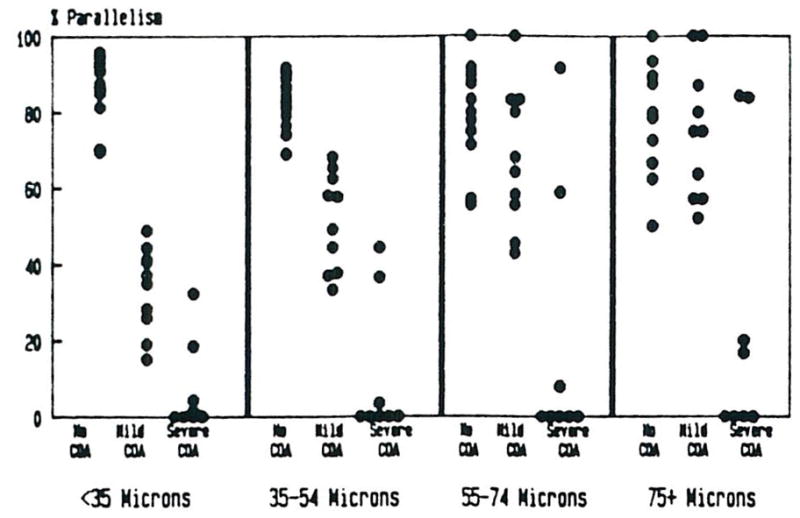
The percentage of portal tracts in which a bile duct is identified along with the corresponding artery is shown. Note the severe loss of small bile ducts in the failed allografts. Duct loss corresponding to larger arteries is seen only in the allografts with severe COA.
It must be remembered that the rate of BD-A parallelism was determined using only those portal tracts which contained arteries. Bile duct loss with associated artery loss was not specifically tabulated. Therefore, absolute bile duct loss, particularly involving the smaller ducts (<35 microns) is even greater than that expressed as a decrease in the rate of parallelism.
Analysis of Chronic Obliterative Arteriopathy Light Microscopic Features of COA
Many histopathologic features were common to the affected arteries of all of the organ allografts. The vessels most severely affected by COA were those of medium size range, which were often the first and second order branching vessels of the particular arterial trees.
The intima contained a varying degree of mononuclear inflammation, subendothelial foam cells and apparent smooth muscle cell proliferation. Plasmacytic, neutrophilic and eosinophilic inflammation was less commonly present except in the cardiac allografts. The internal elastic lamina was often disrupted in arteries of kidney and heart allografts; in the liver it was mostly intact.
The appearance of the media and periadventitial space was similar in all of the organs. The media was infiltrated to various degrees by macrophages, lymphocytes and foam cells. The inflammatory cells were located in areas of degenerating smooth muscle cells. Cuffs of periadventitial inflammatory cells were seen around affected arteries. These periarterial cells appeared to invade into the media from without, as if the medial smooth muscle cells were the target of cellular mediated immunologic injury. Finally although we did not examine sequential biopsies, there appeared to be a temporal progression of the changes. Early lesions were rich in inflammation and accompanied by smooth muscle cell proliferation. Thereafter, the inflammation gradually subsided and the intima became sclerotic.
Phenotypic Analysis by Immunohistology
Various admixtures of macrophages (lysozyme +) T-lymphocytes (UCHL – 1+), proliferating smooth muscle cells (actin +) and S100 protein positive cells were seen in the thickened intima of involved vessels. The variation, to some extent, was dependent on the “age” of the lesion and organ. A detailed description of the findings present in each of the allograft organs is presented elsewhere. On closer examination, two types of intimal S100 + cells were seen. The first was round to oval with an oval, clefted or angulated nucleus. These cells were uncommon. When present, they were located immediately subjacent to the endothelium. The second type of S100 + cells was dendritic shaped and usually found in contact with nearby T-lymphocytes and/or plasma cells; occasionally macrophages (Figure 4). The S100 + dendritic cells were usually located deeper in the intima, near the internal elastic lamina. Likewise, activated and proliferating smooth muscle cells were seen in two locations, immediately subjacent to hypertrophied endothelial cells and deep intima, near the disrupted elastic lamina. Macrophages, T-lymphocytes and rare S100 + dendritic cells were also seen in the media.
Fig 4.

S100 protein positive dendritic shaped cell in the thickened and inflamed intima in a branch of an allograft coronary artery × 400.
The cuffs of inflammatory cells in the periadventitial space consisted predominantly of macrophages; T-lymphocytes were less common. S100 + dendritic cells and/or terminal nerve twigs were the minority group, but were also easily recognized in the periadventitia.
Serial frozen sections cut at 4 microns consecutively stained for S100 protein and HLA-DR were examined. Round HLA-DR positive cells were seen in the intima. Other HLA-DR positive cells were spindle shaped. However, the morphologic resolution in the frozen section immunofluorescent stains were not of sufficient quality to separate smooth muscle cells from dendritic cells. Nevertheless, the elongated HLA-DR positive cells were in the same general location as the S100 + ones.
DISCUSSION
Analysis of the data reveals that quantifiable bile duct and small arterial loss are features of chronic liver allograft rejection. The degree of damage to both arteries and bile ducts increases as the size of each decreases. Additionally, the severity and size of arterial loss and vanished ducts is directly proportional to the degree of COA.
Bile duct loss was recognized under two circumstances. One occurred when an artery without a corresponding bile duct was found. The other was recognized when there was loss of both the artery and the bile duct. Therefore, the results of this study offer indirect, but supportive evidence that two mechanisms may be responsible for bile duct loss; direct immunologic attack and ischemic injury.
The absence of a bile duct when an artery is found is supporting evidence for a direct immunologic attack on the biliary epithelium. Other evidence also suggests that a primary immunologic insult to the biliary epithelium occurs. The histologic appearance of acute cellular rejection is characterized by lymphocytic invasion and evidence of degenerative changes in the biliary epithelium.5–9 Although biliary inflammation is less severe in chronic rejection, careful light and electron microscopic examinations of livers undergoing chronic rejection reveals lymphocytes in close contact with degenerating bile duct cells.10
The loss of ducts in conjunction with arterial loss is indirect but suggestive evidence that ischemic injury contributes to bile duct loss. We also found that the severity of duct loss and the size of the “vanished” ducts is directly proportional to the degree of COA. Since the biliary tree in humans receives its blood supply exclusively from the hepatic arterial system.11,12 COA and peripheral arterial loss compromise blood flow to the dependent ducts.
Two mechanisms may be responsible for small arterial loss in the portal tracts. Small portal arteries may be subject to ischemic necrosis because of more proximal vessel occlusion. The other possibility is that small portal arteries may be damaged by direct lymphocytic attack. Regardless of the mechanism, the result is the same: diminished blood flow to dependent bile ducts.13
The results of this study also offer some insight into the puzzling question of whether primary biliary cirrhosis recurs in liver allografts. Nakanuma also discovered that there was a decreased rate of BD-A parallelism for arteries 35–54, 55–74 and 75–94 microns in PBC liver compared to controls.2 In contrast, we found that the loss of bile ducts in the liver allografts was more restricted. Only those ducts with corresponding arteries less than 75 microns in diameter were missing. A decreased rate of BD-A parallelism for larger arteries was seen only when there was coexistant severe COA. However, all of the patients transplanted for PBC in this study fell into the group with mild COA. Therefore, none of the patients with PBC prior to transplantation who were included in this study demonstrated loss of larger ducts as shown by Nakanuma. Furthermore, larger excretory bile duct damage similar to that seen in sclerosing cholangitis is observed in liver allografts with COA but not in PBC.
COA is not only a problem in liver, but all other solid organ allografts.14–16 We therefore investigated the phenotypic characteristics of cells involved in COA lesions. The arteries affected by COA demonstrated a markedly thickened intima, which contained activated smooth muscle cells, macrophages, T-lymphocytes and S100 protein positive dendritic shaped cells (DCs). They were present when ongoing inflammatory destruction was occurring and became less frequent as the inflammation subsided and the organ assumed a “burned out” appearance. The detection of dendritic cells in the inflammed intima might be significant for understanding the development of COA, because they are efficient accessory cells, promoting T-lymphocyte dependent immune reactions.17,18
Information on the role of dendritic cells in the immune cascade is still limited, since they have only recently been studied to any extent. DCs are bone marrow derived and constitute up to 0.5% of peripheral blood mononuclear cells.19–21 They are extremely efficient accessory cells, normally express high density surface HLA-DR antigens and are potent stimulators of mixed lymphocyte reactions; 10–100 times greater than macrophages in these respects.22,23 DCs may also express complement receptors, but are unable to “process” particulate antigens because of the relative lack of endoplasmic and lysozomal activity.19,24 Therefore, DCs may associate with typical macrophages whose phagocytic and lysozomal activities compensate for these relative deficiencies.
Once an effector cascade is directed against some antigens, release of lymphokines and local production of leukotrienes in the inflammed intima may induce smooth muscle cell proliferation and collagen deposition which might be ultimately responsible for luminal narrowing.25 When stimulated, smooth muscle cells express class II antigens which may be a potential target for humoral and/or cellular immunological reactions.26 A very similar scenario may occur during the development of atherosclerosis in the general population.
Finally, these studies demonstrate that obliterative arteriopathy is a major problem in liver transplantation, similar to kidney and heart allografts.14,15 Therefore, since early graft and patient survival has increased with advances in the field of transplantation, attention must be focused on elucidating the pathogenesis of “graft atherosclerosis.” A better understanding of this problem will permit the trial of novel, and perhaps effective therapy for this presently untreatable condition.
References
- 1.Demetris AJ, Jaffe R, Starzl TE. Pathol Annu. 1987;22(part II):347–386. [PubMed] [Google Scholar]
- 2.Nakanuma Y, Ohta G. Gastroenterology. 1979;76:1328–1332. [PubMed] [Google Scholar]
- 3.Cox DR. The Analysis of Binary Data. London: Metheun; 1970. [Google Scholar]
- 4.Cochran WG. Biometrics. 1954;10:417–441. [Google Scholar]
- 5.Porter K. Pathology of the orthotopic homograft and heterograft. In: Starzl TE, editor. Experience in Hepatic Transplantation. Philadelphia: Saunders; 1969. pp. 422–468. [Google Scholar]
- 6.Fennel RH. Pathol Annu. 1981;16:289–294. [PubMed] [Google Scholar]
- 7.Vierling JM, Fennel RH. Hepatology. 1985;5:1076–1082. doi: 10.1002/hep.1840050603. [DOI] [PubMed] [Google Scholar]
- 8.Demetris AJ, Lasky S, Van Thiel DH, Starzl TE, Dekker A. Am J Pathol. 1985;118:151–161. [PMC free article] [PubMed] [Google Scholar]
- 9.Snover DC, Sibley RK, Freese DK, Sharp HL, Bloomer JR, Najarian JS, Ascher NL. Hepatology. 1984;4:1212–1222. doi: 10.1002/hep.1840040620. [DOI] [PubMed] [Google Scholar]
- 10.Fennel RH, Vierling JM. Hepatology. 1985;5:1083–1087. doi: 10.1002/hep.1840050604. [DOI] [PubMed] [Google Scholar]
- 11.Male AJ. Glasgow Med J. 1951;32:283–301. [PMC free article] [PubMed] [Google Scholar]
- 12.Mitra SK. J Anat. 1966;100:651–663. [PMC free article] [PubMed] [Google Scholar]
- 13.White RM, Zajko AB, Demetris AJ, Bron KM, Dekker A, Starzl TE. Am J Roent. 1987;148:1095–1098. doi: 10.2214/ajr.148.6.1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Porter KA, Thomson WB, Owen K, Kenyon JR, Mowbray JF, Peart W. Br J Med. 1963;2:639–645. doi: 10.1136/bmj.2.5358.639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gao S, Schroeder JS, Alderman EL, Hunt SA, Silverman JF, Wiederhold V, Stimson EB. Circulation. 1987;76(suppl 5):V-56. [PubMed] [Google Scholar]
- 16.Starzl TE, Iwatsuki S, Van Thiel DH, Gartner JC, Zitelli BJ, Malatack J, Schade RR, Shaw BW, Hakala TR, Rosenthal JT, Porter KA. Hepatology. 1982;2:614–636. doi: 10.1002/hep.1840020516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Inaba K, Steinman RM. J Exp Med. 1984;160:1717–1735. doi: 10.1084/jem.160.6.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Inaba K, Steinman RM. Science. 1985;229:475–479. doi: 10.1126/science.3160115. [DOI] [PubMed] [Google Scholar]
- 19.Voorhis WC, Hair LS, Steinman RM, Kaplan G. J Exp Med. 1982;155:1172–1187. doi: 10.1084/jem.155.4.1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Flelinger JG, Hood L, Hill S. Nature. 1979;282:321–323. doi: 10.1038/282321a0. [DOI] [PubMed] [Google Scholar]
- 21.Katz SI, Tamaki K, Snacks DH. Nature. 1979;282:324–326. doi: 10.1038/282324a0. [DOI] [PubMed] [Google Scholar]
- 22.Steinman RM, Witmer MD. Proc Natl Acad Sci USA. 1978;75:5132–5136. doi: 10.1073/pnas.75.10.5132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Steinman RM, Kaplan G, Witmer MD, Cohn ZA. J Exp Med. 1979;149:1–16. doi: 10.1084/jem.149.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kapsenberg ML, Teunissen MB, Stiekena FE, Keizer HG. Eur J Immunol. 1986;16:345–350. doi: 10.1002/eji.1830160405. [DOI] [PubMed] [Google Scholar]
- 25.Palmberg L, Claesson HE, Thyberg J. J Cell Sci. 1987;88:151–159. doi: 10.1242/jcs.88.2.151. [DOI] [PubMed] [Google Scholar]
- 26.Jonasson L, Holm J, Hansson GK. Lab Invest. 1988;58:310–315. [PubMed] [Google Scholar]