

Abstract
OBJECTIVE:
Human papillomavirus (HPV) 16 is causally associated with over 50% of head and neck cancers arising in the tonsillar area and oropharynx. Most treatment failures result from our inability to treat widely invasive/metastatic growth. This study provides a manipulatable system to study mechanism of HPV16 E6 related transformation of an epithelial cell type affected by HPV16 in humans.
Design:
Biochemical and physiological studies of mouse tonsil epithelial cells (MTEC's) transformed with HPV16 oncogenes as well as H-Ras in vitro and in vivo.
INTERVENTIONS:
Transduction of HPV16 oncogenes, E6 and E7, in retroviral vectors into MTEC's with isolation of multiple individual clones that expressed E6, E7 or both, alone or in conjunction with H-RasGly12.
RESULTS:
MTEC's expressing E6 degraded p53 by a mechanism that is inhibited by proteasomal blockade. While normal MTEC's senesced after 20 population doublings (PDs), E6 alone or in combination with E7 was sufficient to immortalize MTEC's beyond 25 PDs, lower their population-doubling time and permit anchorage independent growth. However, only MTEC's expressing E6+H-Ras or E6/E7+H-Ras formed invasive tumors in immune competent, syngeneic mice at orthotopic intraoral and subdermal sites.
CONCLUSIONS:
HPV16 E6 and E7 alone are not sufficient for invasive growth. However, the synergistic activity of H-Ras and E6 was sufficient to result in invasive growth.
Introduction
The incidence of head and neck squamous cell carcinoma (HNSCCA) has increased worldwide1. Despite advances in surgical and combined chemo/radiation therapies, the overall survival for HNSCCA has not improved during the last thirty year period2. Staging of HNSCCA is based on local invasion and nodal/distant spread. Current treatment of early stage (stage 1 and 2) HNSCCA has very good 5-year disease specific survival (81%), while late stage tumors have poor survival (50%)3. A significant reason for unchanged survival is a lack of effective treatments for late stage cancers that exhibit aggressive local invasion or distant metastasis at the time of presentation2. The mechanisms that control metastasis and aggressive invasion are not understood. In order to design better therapies to improve survival, further investigation into mechanisms of malignant transformation are essential.
One of the obstacles in developing treatments for metastatic disease will be determining the particular pathways that transform a cell and result in metastatic growth. Once a mechanism is defined it would be possible to develop a treatment based on a specific cellular alteration. For any individual cancer it is likely that many different combinations of cellular pathways could cause a malignant phenotype, therefore, a successful mechanism based treatment would be needed to identify susceptible tumors with a particular altered cellular pathway prior to treatment. Treatment of chronic myelogenous leukemia containing the bcr-abl translocation with imatinib mesylate is a successful example of such a strategy4. For many HNSCCA's pre-treatment identification of a malignant mechanism is currently not possible; however, human papilloma virus (HPV) related HNSCCA represents a subset of cancers where many of the mechanisms will be predictable. Significant associative evidence supports a role for HPV16 in the carcinogenic progression of at least 25% of HNSCCA5, 6. A subsite, tonsil cancer, has a higher proportion (60%) of cases that are HPV+, and these cases present with more metastatic and advanced disease5, 7. It is now possible to test for HPV using a simple PCR based test. If one could determine a HPV related mechanism for tumorigenic growth and base a treatment on that HPV mechanism, the potential benefit would be large.
Study of HPV has taught us a great deal about how human cells become transformed. For example, the demonstration that the E6 and E7 genes from high-risk HPV types can immortalize human cells along with the finding that E6 and E7 proteins inactivate the p53 and pRb pathways gave significant insight into the involvement of these pathways in carcinogenesis. Recent advances in the development of prophylactic vaccines to HPV infection were a direct result of studies on HPV biology. However, the hundreds of millions of people who are already infected with HPV worldwide still run the risk of developing HPV related HNSCCA for at least the next several decades.
Understanding HPV related mechanisms of malignant transformation will require in vitro and in vivo studies in tonsil epithelial keratinocytes. Currently, the role HPV 16 plays in malignant transformation has predominantly been investigated in anogenital keratinocytes8 and some of the mechanisms of transformation are known. While some of these mechanisms regarding transformation of anogenital keratinocytes will likely also relate to tonsillar keratinocytes, there will also be differences. Therefore, to best understand the mechanisms of HPV related transformation in HNSCCA, it will be necessary to complete studies in keratinocytes from the most affected area (tonsil).
In addition to studying transformation in tonsil cells in vitro, a mechanistic based therapy will also be aided by an in vivo model. Such a model would not only allow for the exploration of HPV related mechanisms responsible for in vivo invasion and metastasis but would also be a valuable pre-clinical testing site for future therapies. Many different strategies can be pursued to develop such a model. Although it may be possible to test xenografted human tonsil cells with viral genes in immune incompetent mice, such a model has significant limitations (aberrant immune response, species differences in receptors/ligands related to metastatic growth, etc.). The lack of an immune response in this model is a significant problem because several lines of evidence suggest that HPV subverts the immune response to its advantage to encourage growth9. A transgenic mouse model of HPV16 related skin cancer does exist. In this transgenic mouse model the human cytokeratin 14 (K14) promoter drives express of HPV16 E6/E7. The HPV mice develop squamous cancers of the skin (14% in 2 years)10-12. We present a new non-transgenic approach to study HPV related tonsil cancer for several reasons: 1) the current transgenic model does not develop tonsil cell cancer. 2) The development of transgenic mice requires substantial cost and prolonged time to observe results with a low frequency of tumor formation. 3) Multiple genetic changes in a mouse can be difficult to study due to embryonic lethality. 4) The immune reaction to HPV antigens may aid the development of HPV-related malignancy9. In the current transgenic mouse model the mice would be tolerant to viral antigens because they are present throughout development. In our approach, we can rapidly test many hypotheses in mice with many genetic alterations (e.g. knock out mice, mice with conditional expression of genes, immune system variants). Our model also exposes the mice to HPV genes only after implantation in a syngeneic immune competent animal. This method will allow us to understand invasive neoplastic growth in the context of an immune competent animal.
A limitation of using mice to study human disease is that species differences are present. A mouse model that accurately reflects human disease would provide an adequate testing ground for studies designed to discover mechanism and for testing pre-clinical therapies. In the studies below we describe a method to culture mouse tonsil epithelial cells (MTEC's). We deliver HPV viral oncogenes to MTEC's to determine what viral genes are required for immortalization, anchorage independent growth and in vivo malignant growth. To begin to determine the human relevance of studying HPV transformation in MTEC's, we also examine whether the HPV viral oncogenes cause similar cellular changes in mouse and human cells.
Methods
Isolation and establishment of mouse tonsil epithelial cells
C57BL/6 mice were euthanized and oral epithelium overlying the tonsillar fossa was harvested. Institutional guidelines regarding animal experimentation were followed. The epithelium was dissociated from the underlying dermis with overnight dispase II (Roche, 0.25 gm/10ml) treatment, manual tissue separation, brief trypsinization with 0.25% trypsin (Invitrogen), and subsequent outgrowth in keratinocyte serum free media (K-SFM) (Invitrogen) containing 0.2 ng/mL EGF and 25 μg/mL bovine pituitary extract (Invitrogen, USA), 1% pen/strep, and 25μg/ml Fungizone (Gibco). After the first passages, the cells were grown in Dulbeco's modified Eagle's medium containing 10% fetal bovine serum, 22% Ham's F12, 1% pen/strep, 25 μg/mL hydrocortisone, 8.4 ng/mL cholera toxin, 5 μg/mL transferrin, 5 μg/mL insulin, 1.36 ng/mL tri-iodo-thyronine, and 5 ng/mL epithelial growth factor. Cells in serum containing media required the addition of irradiated murine J2-3T3 feeder cells for maximal growth and viability.
Retroviral production, transduction and generation of stable lines
Replication incompetent retrovirus was produced by plasmid transfection in packaging cell lines using previously reported techniques13. Early passage MTEC's were transduced at passage 2. Retroviruses expressing E6, E7, H-Ras, and a control empty vector (LXSN) were used to transduce ~40% confluent normal mouse tonsillar cells for 12 hours at 37°C, 5% CO2. Following media change, cells were cultured for 16 hours, split 1:4, and subjected to initial antibiotic selection. Transduced cells were selected by the addition of 150 μg/mL G418 (Invitrogen Corporation, Grand Island, NY) for E6 and LXSN, 15 μg/mL Hygromycin B (Invitrogen) for E7, and 1 μg/mL puromycin for H-Ras beginning 24 hours post retroviral transduction. Cells were maintained at this high antibiotic concentration until 100% untransduced control cells had died, approximately 5-10 days. Stable expressing cell lines were maintained in 50 μg/mL G418, 1 μg/mL hygromycin, and 0.2 ug/mL puromycin respectively. During in vitro culture the cells were split 1:4, which represents two population doubling events.
Clonal cell lines from these pooled transductions were generated by splitting pooled populations 1:100 or 1:1000 onto 10 cm tissue culture plates and isolating single colony clones. These clonal populations were ring cloned using 8mm rings (Fisher Scientific). Over 6 clones were initially isolated from each transduction condition (E6, E6E7, H-Ras). In experiments with LXSN and E7 transduced cells, assays were performed on pooled populations because the cells underwent senescence before cloning could be completed. Each transduction condition was repeated multiple times.
Cell lysis and immunoblot
Whole cell lysates were harvested at 4°C with protein lysis buffer consisting of 50mM Tris-HCl pH 7.5, 150mM NaCl, 5mM EDTA, 2mM NaVO4, 10mM NaPPí, 100mM NaF, 10% glycerol, 1% Triton X-100, 10 μg/mL pepstatin, 20 μg/mL leupeptin, and 20 μg/mL aprotinin. Lysates were drawn through a 25-gauge needle ten times to ensure complete protein dissociation from other cellular components. Cell pellets were snap-frozen at −80°C until use. Expression of p53 and actin was measured by immunoblotting 25 μg total cellular proteins with the p53 antibody (Cell Signaling #2524) and actin antibody (Santa Cruz #1616) using a standard western blot technique. Blots were developed by ECL chemiluminescent detection (Pierce).
MG132 inhibitor assay
MTEC's transduced with LXSN or HPV16 E6/E7 were grown in culture at 70% confluency then treated with 50 uM of a proteasomal inhibitor MG132 (Sigma #2211) for 3 hours. Controls received only a media change. Total cell lysate was harvested for protein and a western blot for p53 and actin was performed as described above.
Anchorage independent growth
Cells were grown to 70% confluency then harvested with trypsin. 0.66% noble agar was seeded onto 12 well plates and allowed to congeal. A mixture of 0.33% noble agar and 1×10^4 cells was applied onto the 0.66% agar and set for 15 minutes at 4°C. Cells were grown at 37°C, 5% CO2 for 10 days with feeding of 0.33% agar every 2 days. Colonies greater than 50 cells were counted in quadrants on day 10 with triplicate wells for each cell line.
In vivo tumor growth
Cells were harvested at 70% confluence with trypsin and resuspended in normal growth media with a concentration of 1×10^6 cells/ml. Using an 18-gauge needle, C57BL/6 mice were injected with either 5×10^5 cells in the tongue or 1×10^6 cells in the subcutaneous tissue in the upper dorsal quadrant near the spine under halothane anesthesia. Animals were euthanized when tumor size was >20mm in its greatest dimension, the animal was substantially emaciated, or there appeared to be pain or functional impairment (jaw or leg use). Tissue was preserved in formalin (2%) and sectioned for hematoxylin and eosin staining per standard protocol. Histology was analyzed by a pathology specialist in head and neck cancer.
Results
Establish a method to isolate and culture mouse tonsil epithelial cells
The highest percentage of HPV positive HNSCCA's originate from tonsillar epithelial cells. To establish a relevant cell culture model, we developed a method to isolate and culture tonsil epithelial cells from the mouse tonsillar fossa. After the first passage in serum free media to eliminate contamination with fibroblasts, the cells were plated with irradiated feeders and the media was switched to a basal media with serum. Under these conditions, the cells grow in epithelial colonies (Figure 1). The cells under these conditions can be propagated for up to approximately 15-20 population doublings (PD). However, in rare instances clonal populations of morphologically normal MTEC's can be cultured for up to 30-40 PD's prior to undergoing cell death. This extremely prolonged life of rare clones has also been described for other mouse cells in culture14, 15. Therefore in the subsequent studies that determined immortality related to a particular viral oncogene, we found it necessary to not only follow pooled populations of transduced cells but also quantitate the number of clones that obtained immortality after a retroviral transduction. In subsequent experiments with retroviral insertions of HPV16 oncogenes, MTEC's were not considered immortal until they had exceeded 80 PD. We did not consider an event related to a transgene unless it occurred in greater than 90% of the clones isolated after a transduction or repeated on multiple pooled transductions.
Figure 1.
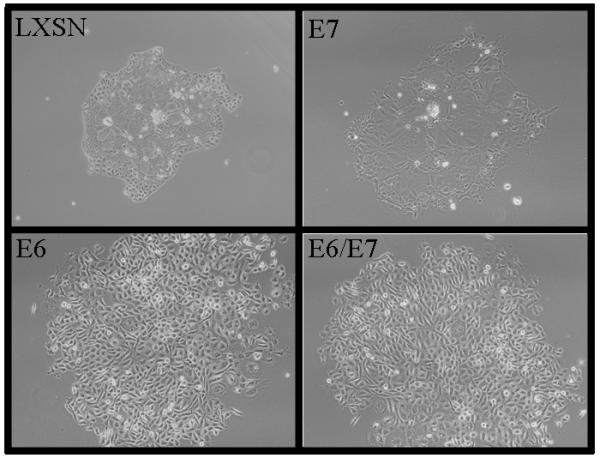
Mouse tonsil epithelial cells infected with LXSN (control vector) or HPV16 E6,E7,E6/E7 grown in cell culture. Phase contrast light microscopy at 20X. Phenotypic change with E6 and E6/E7 show tightly packed cells versus larger senescent cells with LXSN and E7.
E6 alone results in MTEC immortalization and altered morphology
In culture, epithelial cells have a limited lifespan with eventual apoptosis, cell cycle arrest or senescent cell death16, 17. Expression of E6 and E7 oncogenes provide mechanisms that result in efficient immortalization of human tonsil cells (unpublished data) and other human keratinocytes8. The requirements for immortalization of MTEC's have not been previously explored. However, other mouse cells apparently require fewer altered cellular pathways to result in immortalization18. We used retroviral transduction of HPV16 oncogenes into MTEC's in culture to determine what viral oncogenes are required for immortalization.
HPV16 E6 alone was sufficient to alter morphology and immortalize MTEC's. Immediately after selection it became apparent that clones containing E6 alone and E6/E7 grew in small tight colonies (Figure 1). The cells continued to grow as epithelial colonies displacing the irradiated fibroblasts. In contrast, primary mouse tonsil cells transduced with the empty vector LXSN formed colonies with large senescent cells at the center (Figure 1). We continued to passage these cells to determine which genes were required for immortalization in vitro. Expression of E6 by itself resulted in MTEC's immortality (Figure 2), while control cells (LXSN) and cells expressing E7 alone did not immortalize. Compared to E6 alone, clones expressing both E6 and E7 showed a slightly faster overall growth rate in vitro. Thus, unlike human tonsil cells (unpublished data), mouse tonsil cells are immortalized with E6 alone with no added requirement of the degradation of pRb by E7.
Figure 2.
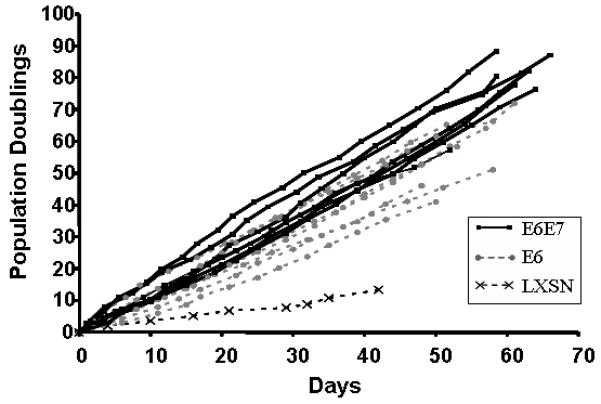
Growth of mouse tonsil epithelial cells infected with LXSN, E7 alone, E6 alone, and E6/E7 retroviruses. E6 and E6/E7 (clonal populations) are still growing while LXSN cells (pooled population) have undergone senescence. E7 cells (pooled population) underwent senescence during selection.
E6 degrades mouse p53 via proteasomal-mediated degradation
HPV16 E6 has been shown to decrease apoptosis in anogenital keratinocytes by proteasomal mediated degradation of p5319. We investigated whether a human viral protein E6 will inactivate mouse p53 by similar mechanisms. Indirect evidence showing lack of p53 induction in the K14-16E6 mouse following radiation suggested that E6 degrades p53 in the mouse11. After retroviral transduction we assayed for p53 degradation using western blot analysis. The results (Figure 3) show that in the presence of E6 mouse p53 is almost undetectable. p53 rescue using proteasome inhibition also suggests that targeted degradation occurs by a similar pathway in mouse and human tonsil cells (Figure 4). These findings suggest that the mouse may be a suitable model to study the mechanisms of E6 mediated transformation.
Figure 3.
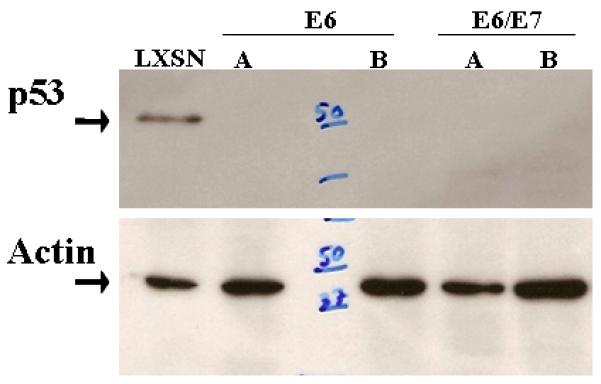
Selective degradation of p53 by HPV16 E6. Mouse tonsil epithelial cells transduced with either LXSN, E6, or E6/E7 were analyzed by western blot probed with p53 and actin (loading control). Two representative clones of E6 and E6/E7 are shown (A,B). “50” and “37” kilodalton molecular weight markers are shown.
Figure 4.
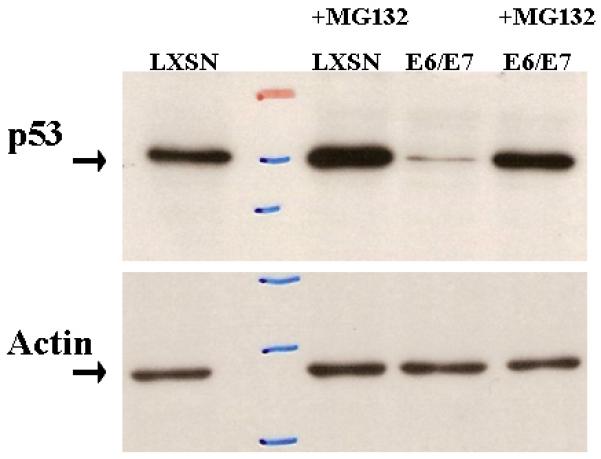
Proteasomal dependence of E6 related p53 degradation. Mouse tonsil epithelial cells transduced with LXSN or E6/E7 were treated with 50 uM of proteasomal inhibitor MG132 (Sigma #2211) for 3 hours. The western blot was probed with antibodies to p53 and actin (loading control).
E6 alone induces anchorage independent growth but not tumor growth in vivo
In the progression to neoplasia, an immortal cell must acquire the ability to grow in the absence of regulatory message from the extracellular environment, such as the basement membrane. To determine the transformed state of the HPV immortalized MTEC's we tested their ability to grow anchorage independent in vitro or form tumors in immune competent syngeneic mice. Anchorage independent growth (AIG) in many circumstances predicts tumor growth in vivo20. As shown in Figure 5, E6 transformed cells were able to grow in an anchorage independent manner. The addition of E7 to E6 increased the efficiency of AIG in vitro. Although AIG was observed for the E6 transformed cells as well as E6/E7 cells, tumor growth in vivo was not seen (Table 1). These data suggest HPV viral oncogenes are sufficient to induce AIG of MTEC's in vitro but alone are not sufficient for in vivo tumor formation.
Fig 5.
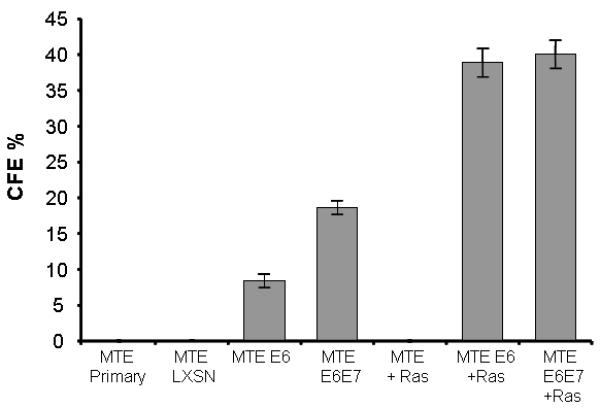
Colony Forming Efficiency in Agar (CFE). Transduced mouse tonsil epithelial cells were suspended in semisolid agar. Colonies greater than 1 mm were counted under 4x magnification on day 10 after seeding. The experiment was done in triplicate and repeated.
Table 1.
Summary population data. MTEC's transduced with retroviruses containing oncogenes with clonal populations obtained in lines that survived subcloning. E6 and E6/E7 conferred immortality; however, H-Ras was necessary for tumor growth in vivo.
| Retrovirus | PD time (days) |
Immortal | % clones immortal |
AI Growth | Growth in vivo |
|
|---|---|---|---|---|---|---|
| oral | flank | |||||
| none | 54 | - | 0/10 | - | nt | 0/6 |
| lxsn | 53 | - | 0/10 | - | nt | 0/6 |
| ras | 52 | - | 0/10 | - | nt | 0/6 |
| E6 | 12 | + | 10/10 | + | nt | 0/6 |
| E7 | 50 | - | * | nt | nt | 0/6 |
| E6/E7 | 12 | + | 10/10 | + | nt | 0/6 |
| E6+ras | 12 | + | nt | + | nt | 7/10 |
| E6/E7+ras | 12 | + | nt | + | 10/12 | 8/10 |
nt = “not tested.”
= cells did not survive selection.
H-Ras synergizes with E6 to induce tumor growth in vivo
In our HPV transformed cells, we chose to express an additional active form oncogene (H-Rasg12) and test for in vivo growth for several reasons: 1) Ras is over-expressed and is implicated in HNSCCA21, 22. 2) It has been shown to synergize with E6/E7 tumor formation in mice23, 24.
We transduced H-Ras or a control retroviral vector into E6 alone and E6/E7 MTEC cells. Pure cultures of transduced cells were obtained through selection with puromycin. H-Ras transduction of these cells greatly increased their ability to grow in an anchorage independent manner; however, anchorage independent growth did not occur in the presence of H-Ras alone (Figure 5). These cells were also transferred either under the epithelial lining of the mouth or subcutaneously in the upper dorsal quadrant near the spine. Aggressive, invading tumors developed at both sites within 1 week (Figure 6). Histologic analysis of the tumors showed they are poorly differentiated squamous cell cancer (Figure 6). Aggressive local invasion of muscle and fat, as well as capillary lymphatic space invasion is present in all specimens. Vector control cells, and cells transduced with H-Ras alone did not grow in mice (Table 1). Although overall survival was similar, survival time was substantially decreased in mice with orthotopic oral, as opposed to flank, tumors (Figure 7A). This likely represents the associated effects of local tumor volume on oral intake. Evaluation of tumor growth using either direct caliper measurement or indirect luciferase mediated light expression shows logarithmic growth (Figure 7C). The cellular changes associated with individual expression of H-Ras, E6, or E6/E7 alone are not sufficient for invasive growth; however, the complement of the cellular effects of E6 and H-Ras results in malignant growth in vivo. The addition of E7 to E6 adds no obvious advantage to tumor formation in the mouse.
Figure 6.
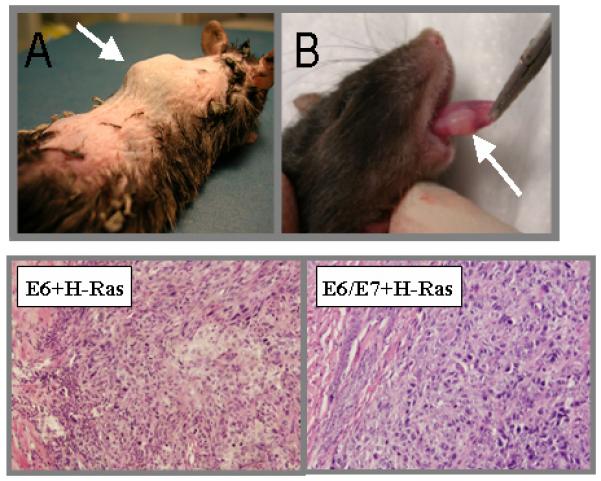
Mice were injected with mouse tonsil epithelial cells expressing HPV16 E6 +Ras developed tumors at two weeks at sites of injection (A) subdermal or (B) tongue. H&E sections of oral tumors at 10X show poorly differentiated SCCA in both specimens.
Figure 7.
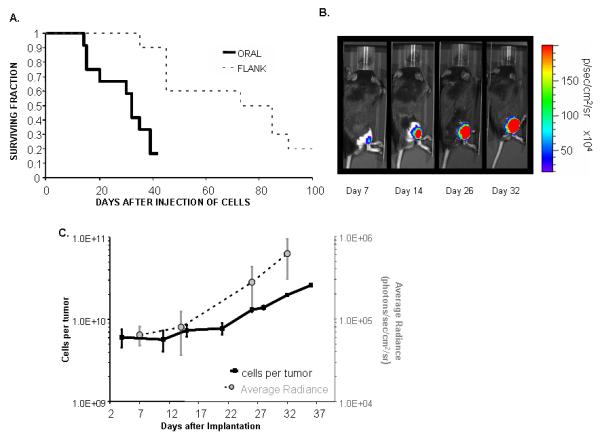
A. Survival. Mice were injected with either 5×10^5 (tongue) or 1×10^6 (flank) tumor-forming cells. Animals were euthanized when tumor size was >20mm in its greatest dimension, the animal was substantially emaciated, or there appeared to be pain or functional impairment (jaw or leg use). B. Representative in vivo tumor growth of E6/E7/H-Ras/Luc cells in C57Bl/6 mice. The mice were injected with 10^6 tumor cells and imaged on the indicated day. Shown are a representative mouse and the tumor growth that occurs at the subcutaneous sites over 32 days. C. Comparison between tumor size and photons. Logarithmic growth of tumors correlates with logarithmic increases in light-emitting tumor radiance.
Comment
HPV plays a critical role in malignant transformation of tonsil epithelial cells. Our data show we have the ability to alter tonsil cells in culture using viral oncogenes and correlate phenotypic changes in their growth both in vitro and in vivo with a syngeneic, immune competent mouse. Previous studies in other mouse cell types not associated with HPV related disease have used HPV viral oncogenes and Ras to obtain invasive growth25-28. However, our data expands on these previous studies in the following ways: 1) the use of an epithelial cell type harvested from a common site of HPV related cancers 2) individual examination of which viral oncogenes are required for in vivo invasive growth 3) use of a quantitative detection mechanism to monitor neoplastic growth 4) examination of multiple in vitro characteristics associated with transformation (anchorage independent growth, immortality, and growth rates).
For a mouse model to be helpful in understanding human disease, the cellular mechanisms and disease process should be similar. Our findings show that in mice E6 expression results in loss of p53. Furthermore, we show that this E6 induced loss is likely mediated by proteasomal degradation because it is reversible with MG132 (proteasome inhibitor). These findings are consistent with E6 mediated loss of p53 in human epithelium 19. Therefore, it would appear that the mouse could be an excellent model to study the role of E6 in tonsil cell transformation. Although our findings on E6 function in mouse cells show similarity in the mechanism of p53 degradation, E6 may have additional functions in mouse cells not present in a human cell.
Our data also suggests that mice are different from humans in the HPV genes required for immortalization. Human tonsil cells and anogenital keratinocytes require the expression of both E6 and E7 for immortalization29-31; therefore, we were initially surprised to find that E6 alone results immortalization of mouse tonsil cells. Other studies examining the requirements for mouse cell immortalization have also found that pRb inactivation is not required18, 32. Transgenic E6 mice develop tumors in the absence of E7 expression12. It is possible that E7 mediated pRb inactivation still occurs in the cells with both E6 and E7. We did notice a slight increase in growth rate as well as increased anchorage independent growth in the cells with both E6/E7 compared to E6 alone. However, preliminary studies in our lab have shown that, unlike human cells, E7 does not induce p16 expression in MTEC's (data not shown). It is possible that the viral genes mechanistically alter mouse cells differently than they do human cells Future studies are currently planned to correlate other known interactions of HPV16 E6 and E7 with cellular targets in human cells with mechanistically similar functions in mouse cells.
This study determines the requirements of transformation of mouse tonsil cells in the context of the major transforming oncogenes of HPV16, E6 and E7. We did not investigate the effects, possibly syngergistic or antagonistic, of other HPV16 genes. Interestingly, other known HPV early genes may also play a role in transformation. For instance, E5 has been shown to alter EGFR processing33. The absence of all the HPV16 genes could limit interpretation of investigations of immunity or tumor clearance. Using our methods, it would be possible in future studies to systematically examine the addition of other viral genes with regards to transformation and invasive growth.
Our findings also suggest that HPV related carcinogenesis is a multi-step process. We have found that expression of HPV viral oncogenes alone is not sufficient to permit metastatic growth. This result supports the clinical finding that not all patients with persistent HPV infection develop cancer. It is likely that at least one additional cellular mutation in the presence of HPV oncogenes is required for metastatic growth. E6 transgenic mouse data also support these findings. In one study, only 14% of mice that overexpressed E6 developed malignant tumors, suggesting that E6 is not sufficient to induce tumor growth by itself. Chromosomal analysis of the tumors that developed in these mice indicated that they exhibited other genetic alterations compared to the surrounding tissue10. The fact that H-Ras synergizes with E6 to result in invasive growth may shed light into what other cellular pathways need to be altered to result in invasive tumorigenic growth. H-Ras activation is a downstream event of growth factor signaling34. Growth factor receptor overexpression is common in HNSCCA35. Although H-Ras mutations have not been reported in HPV related HSCCA, it is possible that alterations like growth factor overexpression or other components of the growth factor signaling cascade could synergize with HPV oncogenes to allow invasive growth.
Other than testing what mechanisms are critical for HPV related carcinogenesis, this model has several other practical applications and advantages. From a cost and time perspective, an in vivo approach takes months to test a hypothesis and is relatively cost effective compared to completing the same study by developing a transgenic or knockout animal. It is also possible to test cellular alterations using this approach that are not possible using a genetic approach. Cellular changes that would cause embryonic lethality in a transgenic mouse could be introduced into the mature mouse. This approach also mimics cancer development, because the cancer is introduced into an immune competent animal. For the current HPV transgenic mouse, the mice become tolerant to the viral proteins because the genes are present throughout development. Therefore, in our model, a mouse may be induced to immunologically clear the tumor by strategies targeting the viral antigens, although a limitation is the presence of only two HPV16 oncogenes out of the full HPV16 genome. We chose retroviral transduction to insert oncogenes because it offers stable expression and selection over time. Also, multiple retroviruses containing various genes can be made inexpensively with little time and resources compared to adenovirus production. Transduction of oncogenes using retroviruses can lead to suppression of cellular genes due to insertional events 36, 37; however, multiple clonal cell lines showing similar results as well as the use of empty vector controls confirm the effects seen are due to the oncogenes and not from random insertions. These methods provide a means to reproducibly observe gene induced changes in cells that will best be confirmed by validations of human tumor specimens.
In conclusion, this data presents a method to study the HPV viral genes required for specific aspects of transformation in primary mouse tonsil keratinocytes. Although the HPV viral gene requirements for transformation are different between mouse and human tonsil keratinocytes, the E6 dependent loss of p53 appears similar. This system may provide a relevant pre-clinical model for testing potential therapeutic strategies.
Acknowledgements
ACH is a Howard Hughes Medical Institute Medical Research Training Fellow Manuscript Preparation: Andrea Van Gelder
Funding Support:
Funded by Veterans Administration Medical Center and National Institute of Health K08 grant
Footnotes
Presented at AHNS 2006
References
- 1.Johnson N. Tobacco use and oral cancer: a global perspective. J Dent Educ. 2001;65:328. [PubMed] [Google Scholar]
- 2.Buckley JG, Ferlito A, Shaha AR, et al. The treatment of distant metastases in head and neck cancer--present and future. ORL J Otorhinolaryngol Relat Spec. 2001;63:259. doi: 10.1159/000055753. [DOI] [PubMed] [Google Scholar]
- 3.Hoffman HT, Karnell LH, Funk GF, et al. The National Cancer Data Base report on cancer of the head and neck. Arch Otolaryngol Head Neck Surg. 1998;124:951. doi: 10.1001/archotol.124.9.951. [DOI] [PubMed] [Google Scholar]
- 4.O'Brien SG, Guilhot F, Larson RA, et al. Imatinib compared with interferon and low-dose cytarabine for newly diagnosed chronic-phase chronic myeloid leukemia. N Engl J Med. 2003;348:994. doi: 10.1056/NEJMoa022457. [DOI] [PubMed] [Google Scholar]
- 5.Smith EM, Ritchie JM, Summersgill KF, et al. Age, sexual behavior and human papillomavirus infection in oral cavity and oropharyngeal cancers. Int J Cancer. 2004;108:766. doi: 10.1002/ijc.11633. [DOI] [PubMed] [Google Scholar]
- 6.Kreimer AR, Clifford GM, Boyle P, et al. Human papillomavirus types in head and neck squamous cell carcinomas worldwide: a systematic review. Cancer Epidemiol Biomarkers Prev. 2005;14:467. doi: 10.1158/1055-9965.EPI-04-0551. [DOI] [PubMed] [Google Scholar]
- 7.Ritchie JM, Smith EM, Summersgill KF, et al. Human papillomavirus infection as a prognostic factor in carcinomas of the oral cavity and oropharynx. Int J Cancer. 2003;104:336. doi: 10.1002/ijc.10960. [DOI] [PubMed] [Google Scholar]
- 8.Steenbergen RD, Parker JN, Isern S, et al. Viral E6-E7 transcription in the basal layer of organotypic cultures without apparent p21cip1 protein precedes immortalization of human papillomavirus type 16- and 18-transfected human keratinocytes. J Virol. 1998;72:749. doi: 10.1128/jvi.72.1.749-757.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.de Visser KE, Eichten A, Coussens LM. Paradoxical roles of the immune system during cancer development. Nat Rev Cancer. 2006;6:24. doi: 10.1038/nrc1782. [DOI] [PubMed] [Google Scholar]
- 10.Simonson SJ, Difilippantonio MJ, Lambert PF. Two distinct activities contribute to human papillomavirus 16 E6's oncogenic potential. Cancer Res. 2005;65:8266. doi: 10.1158/0008-5472.CAN-05-1651. [DOI] [PubMed] [Google Scholar]
- 11.Nguyen ML, Nguyen MM, Lee D, et al. The PDZ ligand domain of the human papillomavirus type 16 E6 protein is required for E6's induction of epithelial hyperplasia in vivo. J Virol. 2003;77:6957. doi: 10.1128/JVI.77.12.6957-6964.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Song S, Pitot HC, Lambert PF. The human papillomavirus type 16 E6 gene alone is sufficient to induce carcinomas in transgenic animals. J Virol. 1999;73:5887. doi: 10.1128/jvi.73.7.5887-5893.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Halbert CL, Demers GW, Galloway DA. The E7 gene of human papillomavirus type 16 is sufficient for immortalization of human epithelial cells. J Virol. 1991;65:473. doi: 10.1128/jvi.65.1.473-478.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Medina D, Kittrell FS. Immortalization phenotype dissociated from the preneoplastic phenotype in mouse mammary epithelial outgrowths in vivo. Carcinogenesis. 1993;14:25. doi: 10.1093/carcin/14.1.25. [DOI] [PubMed] [Google Scholar]
- 15.Tolstonog GV, Shoeman RL, Traub U, et al. Role of the intermediate filament protein vimentin in delaying senescence and in the spontaneous immortalization of mouse embryo fibroblasts. DNA Cell Biol. 2001;20:509. doi: 10.1089/104454901317094945. [DOI] [PubMed] [Google Scholar]
- 16.Hahn WC. Immortalization and transformation of human cells. Mol Cells. 2002;13:351. [PubMed] [Google Scholar]
- 17.Hayflick L, Moorhead PS. The serial cultivation of human diploid cell strains. Exp Cell Res. 1961;25:585. doi: 10.1016/0014-4827(61)90192-6. [DOI] [PubMed] [Google Scholar]
- 18.Rangarajan A, Hong SJ, Gifford A, et al. Species- and cell type-specific requirements for cellular transformation. Cancer Cell. 2004;6:171. doi: 10.1016/j.ccr.2004.07.009. [DOI] [PubMed] [Google Scholar]
- 19.Hougardy BM, Maduro JH, van der Zee AG, et al. Proteasome inhibitor MG132 sensitizes HPV-positive human cervical cancer cells to rhTRAIL-induced apoptosis. Int J Cancer. 2006;118:1892. doi: 10.1002/ijc.21580. [DOI] [PubMed] [Google Scholar]
- 20.Shin SI, Freedman VH, Risser R, et al. Tumorigenicity of virus-transformed cells in nude mice is correlated specifically with anchorage independent growth in vitro. Proc Natl Acad Sci U S A. 1975;72:4435. doi: 10.1073/pnas.72.11.4435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kiaris H, Spandidos DA, Jones AS, et al. Mutations, expression and genomic instability of the H-ras proto-oncogene in squamous cell carcinomas of the head and neck. Br J Cancer. 1995;72:123. doi: 10.1038/bjc.1995.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ginos MA, P G, Michalowicz BS, Patel KJ, Volker SE, Pambuccian SE, Ondrey FG, Adams GL, Gaffney PM. Identification of a Gene Expression Signature Associated with Recurrent Disease in Squamous Cell Carcinoma of the Head and Neck. Cancer Research. 2004:55. doi: 10.1158/0008-5472.can-03-2144. [DOI] [PubMed] [Google Scholar]
- 23.Eiben GL, Velders MP, Schreiber H, et al. Establishment of an HLA-A*0201 human papillomavirus type 16 tumor model to determine the efficacy of vaccination strategies in HLA-A*0201 transgenic mice. Cancer Res. 2002;62:5792. [PubMed] [Google Scholar]
- 24.Schreiber K, C R, Karrison T, Beck-Engeser G, Huo D, Tennant RW, Jensen H, Kast WM, Krausz T, Meredith SC, Chen L, Schreiber H. Strong synergy between mutant ras and HPV16 E6/E7 in the development of primary tumors. Oncogene. 2004:3974. doi: 10.1038/sj.onc.1207507. [DOI] [PubMed] [Google Scholar]
- 25.Lin KY, Guarnieri FG, Staveley-O'Carroll KF, et al. Treatment of established tumors with a novel vaccine that enhances major histocompatibility class II presentation of tumor antigen. Cancer Res. 1996;56:21. [PubMed] [Google Scholar]
- 26.Liu Z, Ghai J, Ostrow RS, et al. The E6 gene of human papillomavirus type 16 is sufficient for transformation of baby rat kidney cells in cotransfection with activated Ha-ras. Virology. 1994;201:388. doi: 10.1006/viro.1994.1306. [DOI] [PubMed] [Google Scholar]
- 27.Storey A, Banks L. Human papillomavirus type 16 E6 gene cooperates with EJ-ras to immortalize primary mouse cells. Oncogene. 1993;8:919. [PubMed] [Google Scholar]
- 28.Schreiber K, Cannon RE, Karrison T, et al. Strong synergy between mutant ras and HPV16 E6/E7 in the development of primary tumors. Oncogene. 2004;23:3972. doi: 10.1038/sj.onc.1207507. [DOI] [PubMed] [Google Scholar]
- 29.Francis DA, Schmid SI, Howley PM. Repression of the integrated papillomavirus E6/E7 promoter is required for growth suppression of cervical cancer cells. J Virol. 2000;74:2679. doi: 10.1128/jvi.74.6.2679-2686.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Munger K, Howley PM. Human papillomavirus immortalization and transformation functions. Virus Res. 2002;89:213. doi: 10.1016/s0168-1702(02)00190-9. [DOI] [PubMed] [Google Scholar]
- 31.Hawley-Nelson P, Vousden KH, Hubbert NL, et al. HPV16 E6 and E7 proteins cooperate to immortalize human foreskin keratinocytes. Embo J. 1989;8:3905. doi: 10.1002/j.1460-2075.1989.tb08570.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Boehm JS, Hession MT, Bulmer SE, et al. Transformation of human and murine fibroblasts without viral oncoproteins. Mol Cell Biol. 2005;25:6464. doi: 10.1128/MCB.25.15.6464-6474.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim SH, Juhnn YS, Kang S, et al. Human papillomavirus 16 E5 up-regulates the expression of vascular endothelial growth factor through the activation of epidermal growth factor receptor, MEK/ERK1,2 and PI3K/Akt. Cell Mol Life Sci. 2006;63:930. doi: 10.1007/s00018-005-5561-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cox AD, Der CJ. Ras family signaling: therapeutic targeting. Cancer Biol Ther. 2002;1:599. doi: 10.4161/cbt.306. [DOI] [PubMed] [Google Scholar]
- 35.Ford AC, Grandis JR. Targeting epidermal growth factor receptor in head and neck cancer. Head Neck. 2003;25:67. doi: 10.1002/hed.10224. [DOI] [PubMed] [Google Scholar]
- 36.Kohn DB, Sadelain M, Glorioso JC. Occurrence of leukaemia following gene therapy of X-linked SCID. Nat Rev Cancer. 2003;3:477. doi: 10.1038/nrc1122. [DOI] [PubMed] [Google Scholar]
- 37.Lange C, Blankenstein T. Loss of retroviral gene expression in bone marrow reconstituted mice correlates with down-regulation of gene expression in long-term culture initiating cells. Gene Ther. 1997;4:303. doi: 10.1038/sj.gt.3300395. [DOI] [PubMed] [Google Scholar]