

Abstract
Improved techniques to non-invasively image protein-protein interactions (PPIs) are essential. We molecularly engineered a positron emission tomography (PET)-based split reporter (herpes simplex virus type 1 thymidine kinase [TK]), split between Thr265 and Ala266, and used this in a protein-fragment complementation assay (PCA) to quantitatively measure PPIs in mammalian cells and to microPET image them in living mice. An introduced point mutation (V119C) significantly enhanced TK complementation in PCAs based on rapamycin modulation of FRB (FKBP12-rapamycin-binding domain) and FKBP12 (FK506 binding protein), on interaction of hypoxia-inducible factor-1α and the von Hippel-Lindau tumor suppressor, and in an estrogen receptor intramolecular protein folding assay. Applications of this novel split TK are potentially far-reaching, including for example considerably more accurate monitoring of immune and stem cell therapies, allowing unprecedented fully quantitative and tomographic PET localization of PPIs in pre-clinical small and large animal models of disease.
Protein-protein interactions (PPIs) are vital to most cellular functions, being associated with processes as diverse as enzymatic activity, signal transduction, immunological recognition, and DNA repair and replication1. A multitude of experimental qualitative and quantitative techniques have been developed with the common goal of understanding these ubiquitous interactions2. Herein we describe the construction and validation of a new split reporter for use in a protein-fragment complementation assay (PCA) strategy to quantitatively image and define PPIs in living subjects using positron emission tomography (PET). We had previously reviewed the many noteworthy advantages of molecular imaging of biological processes in living subjects3. The non-invasive visual representation, characterization, quantification, and timing of PPIs could create unprecedented opportunities to complement available in vitro or cell culture methodologies, in order to accelerate the evaluation in living subjects of novel drugs that promote or inhibit active homodimeric or heterodimeric protein assembly, and to characterize more fully known PPIs (e.g. the reasons for, and the factors that drive their association) in the context of whole-body physiologically-authentic environments.
In all PCAs, splitting a specific reporter protein into two distinct fragments abolishes its function. Bringing the two fragments back together in a controlled manner, as might occur when driven by a particular PPI under scrutiny, then restores partial functional activity of the reporter, and thus reflects on the occurrence and nature of the PPI4 (Fig. 1a). Selected fragments of many proteins can associate to produce functional bimolecular complexes5. Split reporter systems have been used to date in several model organisms, including E. coli6, yeast7, C. elegans8 and mice9,10.
Figure 1.
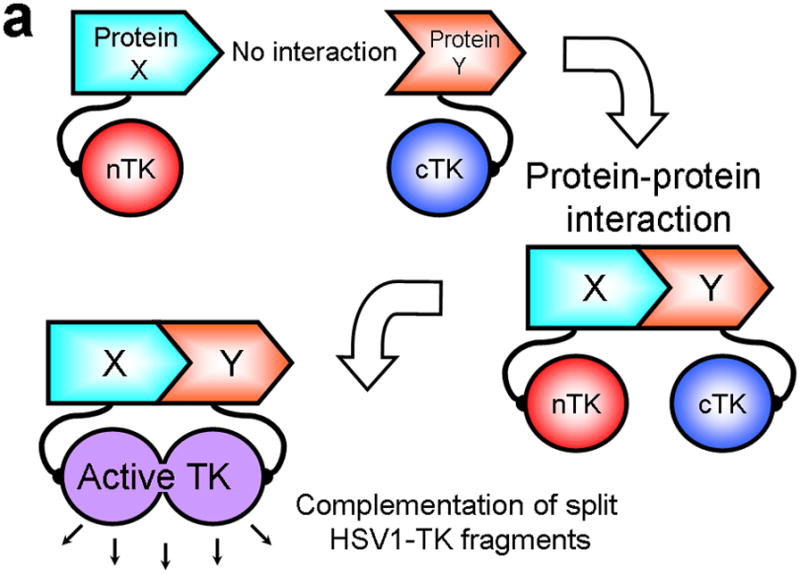
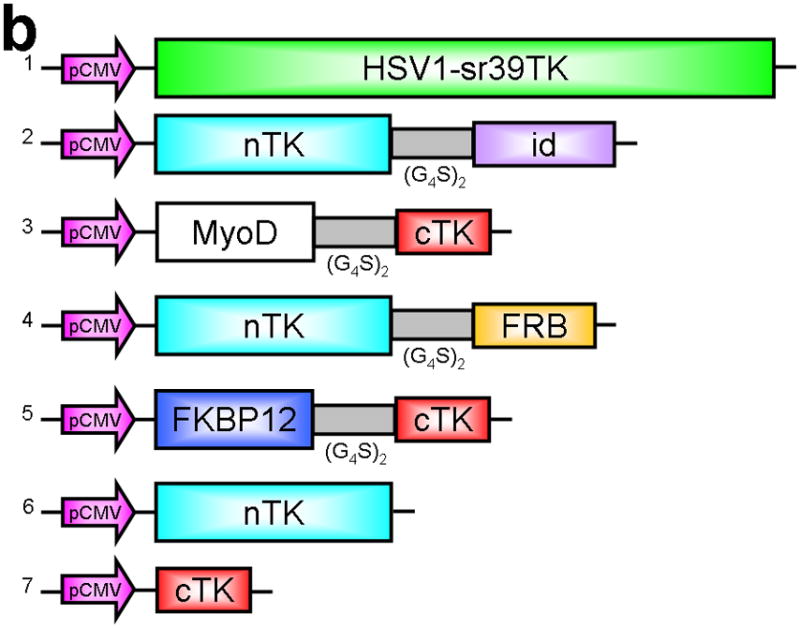
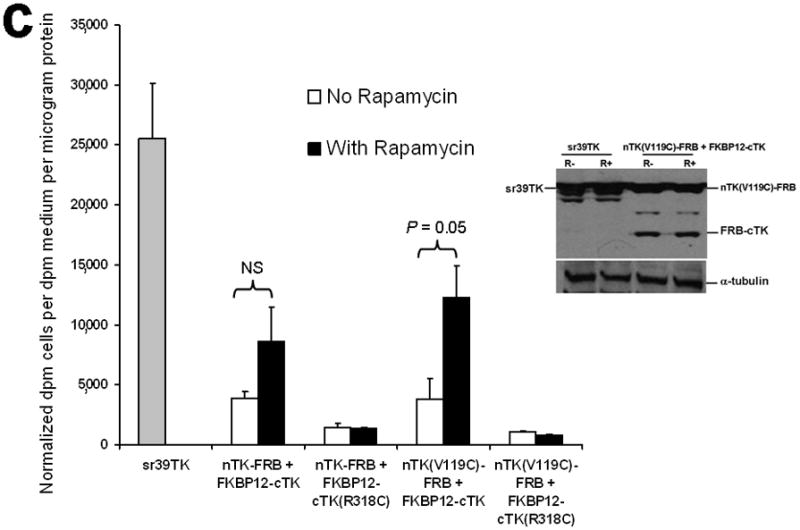
Principle of the TK PCA, expression vectors used, and evaluation of plasmid vector constructs containing the V119C mutation. (a) Schematic diagram showing the PCA strategy using split HSV1-TK (here abbreviated as TK) to monitor the hypothetical X–Y heterodimeric protein-protein interaction. This is accomplished by fusing each of the reporter fragments to heterologous X–Y protein domains to generate two chimeric proteins that have the capacity to interact with one another. If the interaction of the two heterologous protein domains (first and foremost) restores the activity of the reporter by bringing the two reporter fragments into close spatial proximity (as a secondary consequence), then this restoration of reporter activity can be used to monitor the interaction of the two heterologous protein domains. Dimerization of the two proteins restores TK activity through protein complementation and produces a PET imaging signal in the presence of radiolabeled TK substrate. If the reporter protein is an enzyme, then an additional strength of this PCA approach is the capacity to amplify the signal associated with each protein-protein interaction event. Note that this is a simplified schematic diagram representing the forward or ‘folding’ mechanism underlying a PCA. The reverse or ‘unfolding’ mechanism is not depicted here for the sake of simplification. A more realistic and detailed graphic depiction of the interplay between these two mechanisms within a PCA has been published previously by Michnick et al.26. (b) Schematic representation of the plasmid vector constructs made for transient expression of the seven genes transfected individually or in combinations described in the text and Supplementary Methods, for evaluation of the PCA strategy. Each vector was cloned into a pcDNA3.1 (+) plasmid backbone, under control of a CMV promoter. (c) Graph to show comparison of coexpressed chimeras carrying nTK or cTK with FRB/FKBP12 (with and without rapamycin), and chimeras containing the TK point mutations V119C and R318C on enzyme activity in a PCA, measured by TK enzyme uptake (expressed as normalized dpm of cells/dpm in medium/microgram of protein) in transiently transfected 293T cells, with mock (negative) and full length HSV1-sr39TK (positive) controls. The error bar is the standard error of the mean for three samples. Introducing the point mutation V119C to the nTK fragment resulted in an increase (at limit of statistical significance) in measured TK activity upon addition of rapamycin, after co-transfecting nTK(V119C)-FRB and FKBP12-cTK. NS: not significant. Western blot analysis using antibody to TK in the presence or absence of Rapamycin shows adequate expression levels in 293T cells.
The herpes simplex virus type 1 thymidine kinase (HSV1-TK, here abbreviated as TK) enzyme phosphorylates a wide range of nucleoside analogues, allowing selective anti-herpetic and viral vector-based gene therapies. The two main categories of substrates for TK, uracil nucleoside derivatives labeled with radioactive iodine (e.g. FIAU: 2′fluoro-2′-deoxy-1-beta-D-arabinofuranosyl-5-iodo-uracil)11 or radiolabeled 2′-fluoro-2′-deoxyarabinofuranosyl-5-ethyluracil (FEAU)12, and acycloguanosine derivatives labeled with radioactive 18F-Fluorine (e.g. fluoropenciclovir [FPCV] or 9-(4-[18F]-fluoro-3-hydroxymethylbutyl)-guanine [FHBG])13, have been investigated in the last few years as reporter probes for imaging HSV1-tk reporter gene expression14. These radiolabeled reporter probes are transported into cells, and are trapped as a result of phosphorylation by TK. When used in non-pharmacological tracer doses, these substrates can serve as positron emission tomography (PET) or single photon emission computed tomography (SPECT) targeted reporter probes by their accumulation in just the cells expressing the HSV1-tk gene.
Herein we describe the molecular engineering rationale and construction of a novel split TK reporter, cleaved into two fragments between Thr-265 and Ala-266, for use in a PCA to quantitatively measure real time PPIs in mammalian cells using cell uptake studies, and to PET image these in subcutaneous implants in living mice. We also introduced a point mutation (V119C) in the N-terminal fragment of the split TK reporter to significantly enhance the level of PPI-induced reporter complementation. Importantly, the novel TK PCA approach that we describe has the potential for the first time to provide a sensitive and more accurate means of in vivo fully quantitative imaging and precise tomographic localization of PPIs deeper in the body than currently possible with any other imaging technique. Optical bioluminescence imaging, by contrast, and rather restrictingly, is relatively surface weighted and semi-quantitative in nature. The innovation we report will likely result in a significant benefit when attempting accurate study of more representative animal models of disease based on orthotopic cellular implants, and particularly in transgenic animal applications during the pre-clinical drug discovery and validation process15. Future potential applications in humans may also be possible in theory e.g. when using split reporters to PET image relevant PPIs in cell therapeutic strategies following ex vivo transduction of the cells of interest.
RESULTS
Design of the TK PCA strategy
We first performed a partial circular permutation screen of the TK molecule, and deemed it likely that a split between Thr265 and Ala266 would produce two protein fragments that could be further tested in a PCA strategy for imaging PPIs (Supplementary Discussion and Supplementary Figs. 1 and 7,8). We next determined if removal of the linker joining the native N- and C-terminal ends in CPTK265 (circular permuted TK split at residues 265/266) would yield a pair of TK fragments that could function and complement in a PCA strategy16. We used the two heterologous human proteins FRB (FKBP12-rapamycin-binding domain) and FKBP12 (FK506 binding protein) that are known to strongly interact in the presence of rapamycin17. The use of the FRB/FKBP12/rapamycin system is an example of an intermolecular “three-hybrid” interaction in which a third partner (e.g. a small ligand, rapamycin in this case) mediates a PPI18. The value of this test system had been demonstrated in several previous PCA strategies, including the DHFR fragment complementation assay18, and those using split Firefly luciferase9 and Renilla luciferase10. The N-terminal TK fragment comprising the amino acids Met-1 to Thr-265 (nTK) was fused (via an intervening flexible 10-residue linker [GGGGS]2) proximal to the N-terminal end of FRB, to yield the chimeric protein nTK-FRB (see Methods). The C-terminal TK fragment comprising the amino acids Ala266 to Asn376 (cTK) was fused (via a similar linker) distal to the C-terminal end of FKBP12, to yield the chimeric protein FKBP12-cTK (Fig. 1b). We initially used this particular orientation because it was successful in previously published PCA strategies, e.g. when using split Firefly luciferase9, and Renilla luciferase10, both when used with the same FRB/FKBP12 heterodimerization system. We tested the interaction of these proteins (FRB and FKBP12, modulated by rapamycin) upon transient co-expression of the fusion proteins in 293T cells where there is no endogenous HSV1-tk expression, followed by an in vitro TK enzyme cellular uptake assay to detect dimerization-assisted complementation of the TK fragments.
We introduced a point mutation (V119C) in TK to obtain a variant that permits disulfide bond formation and cross-linking of two TK monomers covalently19. We showed that the use of nTK fragments carrying this point mutation resulted in 41% increased enhancement (which was at the limit of statistical significance) of the degree of PPI-induced complementation of TK fragments, as compared with use of non-mutated nTK, when studied 36 h post transient transfection in 293T cells (Fig. 1c). All experimental results related to further optimization of the TK PCA, before and after constructing this V119C variant, as well as further extensive experiments designed to validate the functional activity of the constructs used are presented in the Supplementary Material and Supplementary Figs. 2–4, 9–19. Of note, we also established that attaching interacting proteins (FRB/FKBP12) to separate nTK and cTK fragments did not hinder the subcellular trafficking of the split reporter, when compared to the localization of the normal full length TK molecule (Fig. 2a, 2b).
Figure 2.
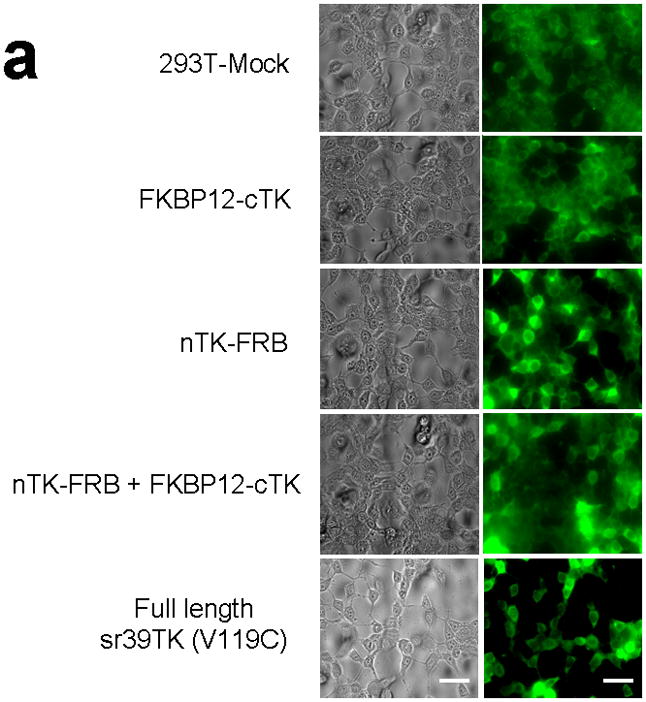
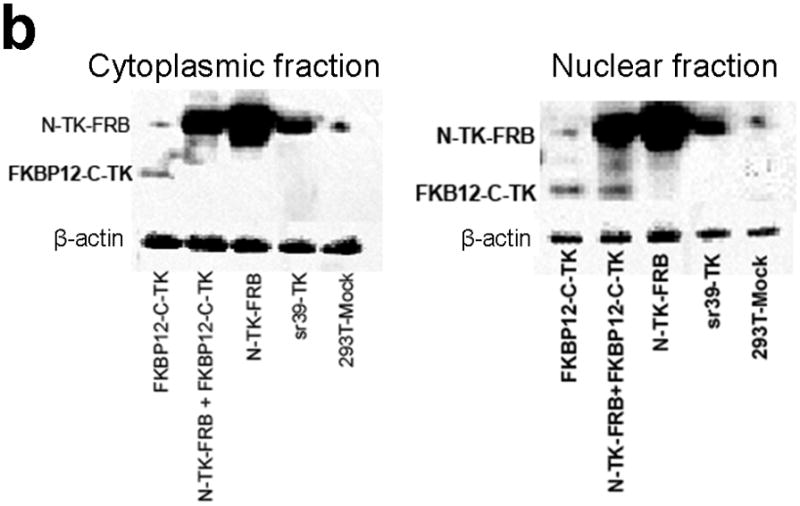
Subcellular localization of components of the PCA. (a) Fluorescence micrographs (×40 magnification) after immunohistochemical staining of negative control (293T-Mock) cells, positive control (Full length sr39TK [V119C] transfected 293T cells) cells, and 293T cells transiently transfected with component vectors of the PCA. These show considerable levels of TK protein expression in both nucleus and cytoplasm regardless of whether the TK reporter was split or intact. Fusion with interacting proteins FRB and FKBP12 did not perceptively hinder normal translocation of functional TK fragments within cells. Scale bar, 50 μm. (b) Western blot analysis reveals expression levels of components of the TK PCA determined in both nuclear and cytoplasmic fractions from lysates of 293T cells transfected accordingly, after immunoblotting with antibody to TK. Antibody to β-actin was used as an internal control for loading. Again, considerable levels of TK protein expression in both nucleus and cytoplasm showed that that there was no perceptible hindrance to normal translocation of functional TK fragments within cells.
Testing the TK PCA in separate cell lines and PPI systems
Since 293T cells are designed for multiepisomal replication and high levels of protein expression, we tested the combination of optimally oriented chimeras in two other cell lines (SKBr3 human breast cancer, and SKOV3 human ovarian cancer) not specifically used for that purpose. True to the features of the developed TK PCA, these too yielded an approximate 3-fold rise in split TK complementation upon PPI (Fig. 3a).
Figure 3.
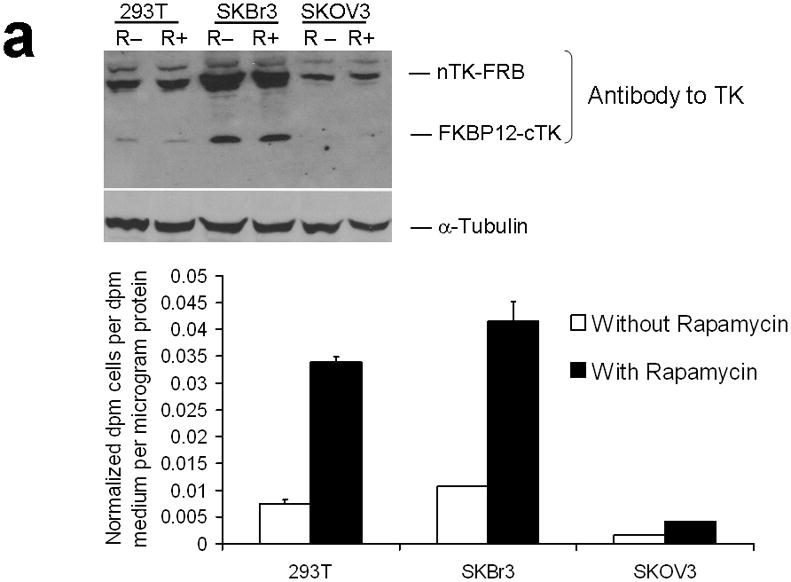
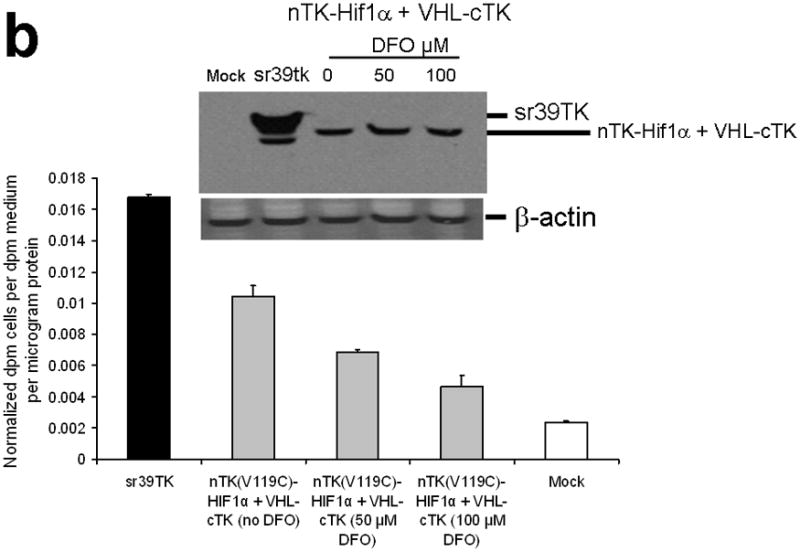
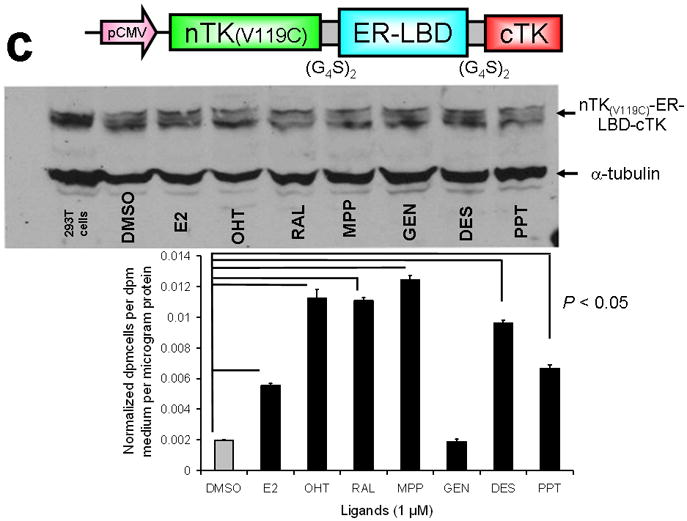
Testing the TK PCA in separate cell lines and PPI systems. (a) Graph to show comparison of coexpressed chimeras carrying nTK or cTK with FRB/FKBP12 (with and without rapamycin) in different cell lines, as measured by TK enzyme uptake (expressed as normalized dpm of cells/dpm in medium/microgram of protein) in transiently transfected 293T cells, SKBr3 cells, and SKOV3 cells. The error bar is the standard error of the mean for three samples. There was a statistically significant increase in measured TK activity upon addition of rapamycin to all 3 cell lines. Despite lack of normalization of transfection efficiency, there was a similar approximate 3-fold rise in split-TK complementation upon PPI regardless of which cell line was used. Western blot analysis of all 3 cells using antibody to TK before and after addition of rapamycin shows adequate expression levels. (b) A vector was constructed that transiently expresses a fusion protein with split TK fragments (containing a V119C point mutation) and substituting HIF1α for FRB, and VHL for FKBP12. Each construct was cloned into a pcDNA3.1 (+) plasmid backbone, under control of a CMV promoter. 293T cells were transiently co-transfected (200 ng DNA each vector per well) to express nTK(V119C)-HIF1α and VHL-cTK, an empty vector, and full length HSV1-sr39TK as a positive control for 48 h, followed by measurement of TK enzyme uptake (expressed as normalized dpm of cells/dpm in medium/microgram of protein). Treatment with increasing doses of desferrioxamine (DFO) resulted in a proportional reduction in interaction of HIF1α and VHL. The error bar is the standard error of the mean for three samples. Western blot analysis using antibody to TK after treatment with the different doses of DFO shows adequate expression levels in 293T cells. (c) Schematic diagram of the intramolecular folding sensor construct with the split TK fragments on either side of the estrogen receptor ligand binding domain (ER–LBD), and each construct was made by cloning into a pcDNA3.1 (+) plasmid backbone, under control of a CMV promoter. 293T cells were transiently transfected (500 ng DNA per well) to express the intramolecular folding sensor and treated with the indicated ER ligands or carrier control (dymethyl sulfoxide, DMSO) for 24 h, followed by measurement of TK enzyme uptake (expressed as normalized dpm of cells/dpm in medium/microgram of protein). Treatment with the ER ligands 17β-estradiol (E2), 4-hydroxytamoxifen (4-OHT), raloxifene (RAL), methyl piperidinylethoxy pyrazole (MPP), genistein (GEN), diethylstilbestrol (DES), and 1,3,5-tris(4-hydroxyphenyl)-4-propyl-1H-pyrazole (PPT) led to levels of intramolecular-folding-assisted complementation that were significantly higher than that of carrier control-treated cells (P < .05) except for genistein. The error bar is the standard error of the mean for three samples. Western blot analysis using antibody to ERα after treatment with the different ligands shows adequate expression levels in 293T cells.
To further demonstrate general applicability of the engineered split TK fragments we also tested their complementation in two other separate systems: First, a system that also employs an intermolecular PPI of hypoxia-inducible factor (its subunit 1α, HIF1α) and the von Hippel-Lindau tumor suppressor (VHL) (Supplementary Discussion)20. Hypoxia-inducible factor (its subunit 1α, HIF1α) is a transcriptional complex that plays a central role in the regulation of gene expression by oxygen, and von Hippel-Lindau tumor suppressor (VHL) serves as the recognition component of a ubiquitin ligase that promotes ubiquitin-dependent proteolysis of HIF1α. In hypoxic cells (desferrioxamine [DFO] is an iron chelator with ‘hypoxia-mimetic’ activity; DFO stabilizes HIF 1α from proteolysis by inhibiting the activity of iron-dependent prolyl hydroxylases) HIF1α degradation is suppressed owing to a diminished interaction with VHL. To test this system we constructed a vector that transiently expresses a fusion protein with split TK fragments (containing the V119C point mutation) and substituting FRB with HIF1α, and FKBP12 with VHL. We studied this in 293T cells after treatment with various doses of DFO. There was a significant level of split TK complementation (62.2% of activity of intact TK) upon interaction of HIF1α and VHL, which diminished with exposure to increasing concentrations of DFO (Fig. 3b, including Western blot showing adequate protein expression levels).
The second system used employed an intramolecular protein folding sensor; we designed and validated this system previously by encoding various human estrogen receptor ligand-binding domain (hER-LBD) fusion proteins that could lead to split reporter complementation in the presence of the appropriate ligands21. There was a significant level of ligand-induced split TK complementation for several ligands, in a similar pattern to that documented previously for complementation of split synthetic Renilla luciferase complementation21 (Fig. 3c). All experiments were conducted with comparison to a positive control derived from the activity of intact TK, and a mock transfection acting as a negative control.
MicroPET imaging of PPIs in subcutaneous xenografts in mice
Next, 5 mice subcutaneously injected with 293T cells stably expressing nTK(V119C)-FRB plus FKBP12-cTK from a single vector were imaged using the [18F]-FHBG probe and microPET in the presence of rapamycin. The signal from each implant was quantified directly from the microPET images to determine the %IDg−1 for the FHBG probe. These 5 mice were allowed initially to grow 1-wk-old subcutaneous xenografts of 293T cells stably expressing nTK(V119C)-FRB plus FKBP12-cTK on one shoulder and control tumors (mock transfected 293T cells) on the other shoulder prior to microPET imaging before and 24 h after exposure to 50 μg of rapamycin. The animals were repeatedly imaged on day 1 (before rapamycin), day 2 (one dose post rapamycin) and day 5 (3 doses post rapamycin) of this imaging protocol, i.e. after 1 week of intial xenograft growth. The incremental increases in mean %ID g−1 values for FHBG accumulation in the tumor formed by cells stably expressing nTK(V119C)-FRB plus FKBP12-cTK are shown in (Fig. 4a). Comparison was made with the control tumor showing background signal. Statistically significant differences in imaging signal were seen in tumors expressing split TK plus the interacting proteins when comparing probe accumulation pre- and post-injection of rapamycin (P = 0.02 on imaging Day 5) (Fig. 4b). Only background signal was obtained from both implanted xenografts in a separate cohort of 4 control mice not receiving rapamycin (data not shown). In another group of 4 mice, xenografts expressing nTK(V119C)-FRB plus FKBP12-cTK were compared with control implants stably expressing nTK plus cTK only (also in a single vector). There was minimal FHBG accumulation in the control tumors on the fifth day of a similar imaging protocol (Fig. 4c). Preliminary pilot imaging studies using transiently transfected cells had also been carried out in 3 mice using C6 cells stably transfected with HSV1-sr39tk as positive controls (data not shown).
Figure 4.
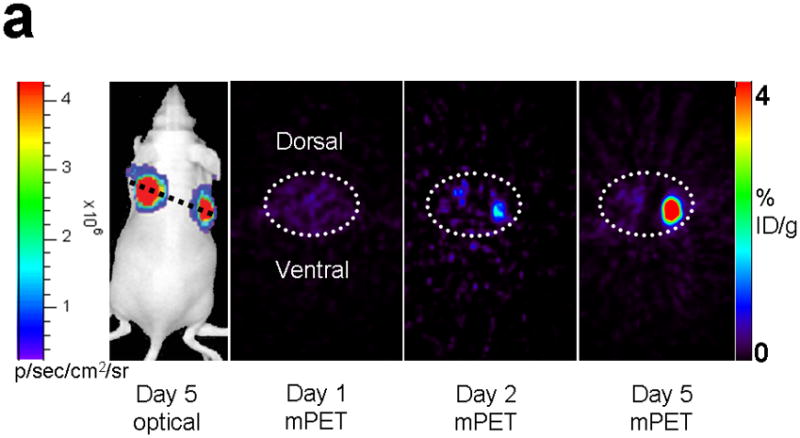
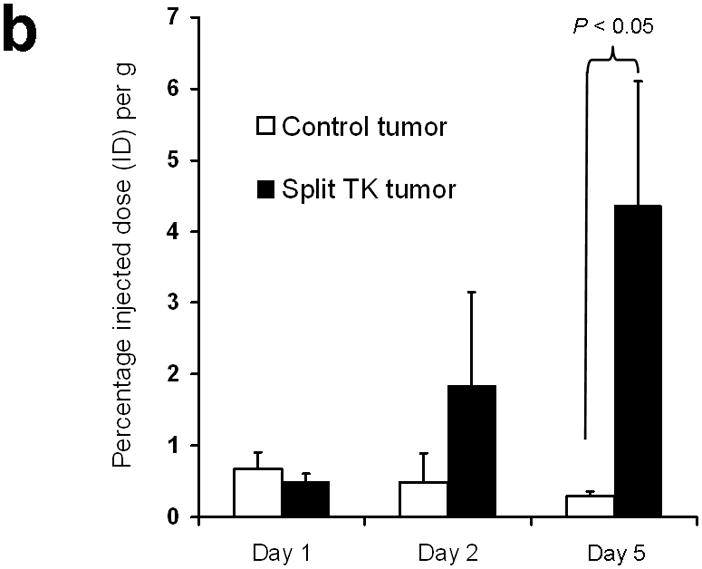
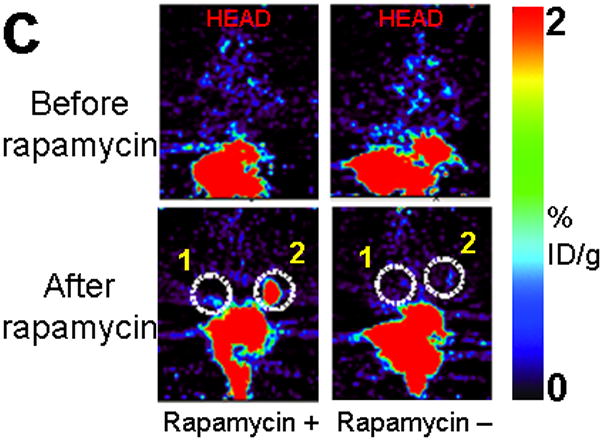
Imaging of tumors containing the split TK constructs. (a) Transaxial tomographic microPET images through a representative prone-positioned mouse implanted subcutaneously over the left shoulder with mock transfected 293T cells, and over the right shoulder with 293T cells stably expressing both nTK(V119C)-FRB and FKBP12-cTK. The mouse was injected with 200 μCi of [18F]-FHBG prior to imaging on days 1, 2, and 5 into the imaging protocol (i.e., after 7 days of initial xenograft growth). Elliptical dotted white line outlines the surface of the mouse’s upper thorax. Color intensity is a reflection on probe accumulation after its phosphorylation by the complemented TK enzyme. Quantitative analysis of this probe accumulation shows a mean %ID g−1 (obtained from 5 tomographic slices through each tumor for all animals) as displayed in accompanying graph (b). The difference between accumulation in tumors exhibiting split TK complementation and control tumors was statistically significant (P = 0.02) on Day 5. Also shown is the optical CCD imaging on day 5 of the imaging protocol of the same mouse immediately before its subsequent microPET imaging of bilateral shoulder region subcutaneous xenografts, and is shown as a visible light image superimposed on the CCD bioluminescence image with a scale in photons/sec/cm2/steradian. Mice were imaged in the prone position after tail-vein injection of 4 mg D-Luciferin per animal. Each mouse was implanted subcutaneously over the left shoulder with 5 × 106 mock transfected 293T cells admixed with 50,000 293T stable cells expressing Firefly luciferase, and subcutaneously over the right shoulder with 5 × 106 293T stable cells expressing both nTK(V119C)-FRB and FKBP12-cTK admixed with 50,000 293T stable cells expressing Firefly luciferase. Bioluminescence imaging shows equivalent viable tumor load in both xenografts. Angeled black dotted line shows the transaxial plane, bisecting each tumor, through which the microPET images were obtained. (c) Coronal tomographic microPET images through two representative prone mice implanted subcutaneously over the left shoulder (circle 1) with control tumors of 293T cells stably expressing nTK plus cTK only (in a single vector), and over the right shoulder (circle 2) with 293T cells expressing nTK(V119C)-FRB plus FKBP12-cTK in a single vector. Unlike tumors containing the complemented TK enzyme, there was minimal FHBG accumulation (%ID g−1) in the control tumors on the fifth day of the imaging protocol upon systemic administration of rapamycin (see text). Intense accumulation in centre of image is due to non-specific probe excretion in the gut.
As anticipated, the microPET detected counts from shoulder tumors expressing split TK plus interacting proteins did not differ in significance whether the animals were imaged in the supine or prone positions (probe %ID g−1 of 0.89 and 1.02 respectively, Fig. 5). The optical cooled charge-coupled device (CCD) camera bioluminescence imaging of the co-injected 293T cells stably expressing Firefly luciferase (see Supplementary Methods) showed equivalent signal from both control and experimental tumors in all the animals used for microPET imaging, reflecting on the equivalent cell load and viability in all tumors under study (Fig. 4a). However, these optical imaging findings were evident only upon imaging the mice in the prone position, with a peak light emission of 2.3 × 105 p/sec/cm2/sr (Fig. 5). In contrast, when imaged in the supine position, the light emanating from subcutaneous xenografts on the shoulder/back did not penetrate through the full thickness of the mouse (the mouse torso being > 1 cm thick) (Fig. 5). These findings highlight the relative advantage of PET over optical imaging in imaging sources of signal at depths greater than 1 cm from the exterior which is a well known advantage of PET over optical bioluminescence imaging.
Figure 5.
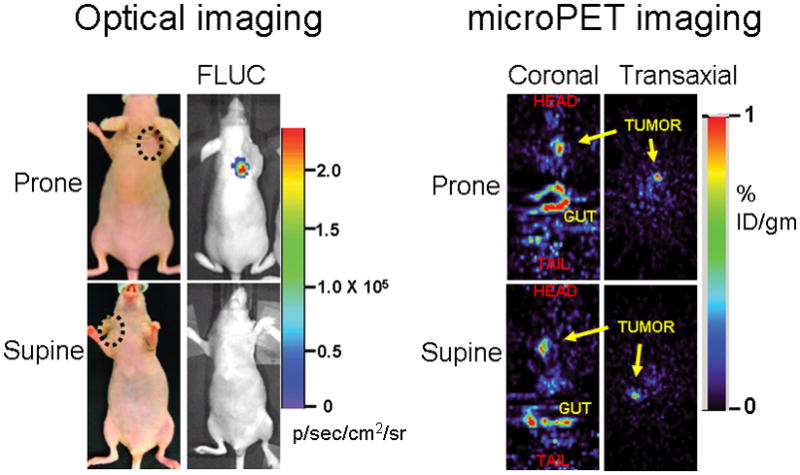
The relative advantage of PET over optical imaging in imaging sources of signal at depths below 1 cm from the exterior is exemplified in this separate experiment. Five mice were imaged 6 days post implantation with 5 × 106 293T cells stably expressing TK or Firefly luciferase (FLUC). The microPET signal return (FHBG accumulation in %ID g−1) from shoulder tumors (circled) expressing TK did not differ in significance whether the animals were imaged in the supine or prone positions (probe %ID g−1 of 0.89 and 1.02 respectively), as demonstrated in the transaxial and coronal tomographic images of a representative animal (intense accumulation in centre and lower aspect of coronal images is due to non-specific probe excretion in the gut). When imaged in the supine position, the light emanating from subcutaneous xenografts on the shoulder or back showed no penetration through the full thickness of the mouse.
DISCUSSION
Typical protein-protein interactions (PPIs) represent low level biological events, and are thus challenging to locate and image in intact living subjects. In part owing to its sensitivity and its fully quantitative and tomographic nature, PET is one of the few noninvasive imaging technologies that currently can be applied in humans for in vivo monitoring of reporter gene expression, and with the potential to image complex biological phenomena such as PPIs22. To create a PET-based PCA using TK, the first step was to identify sites where we could disrupt the primary amino acid sequence of TK to separate the enzyme into two complementary protein fragments. The circularly permuted variant CPTK265 (where the split site was between Thr265 and Ala266) was the only mutation to retain TK enzymatic activity (85.2%, as compared to full length normal HSV1-sr39TK). We next exhaustively studied these engineered TK fragments with respect to their orientation in interacting chimeras, temporal changes in complementation, optimal dosage of a modulating drug (rapamycin) for heterodimerization-induced complementation as well as in a variety of other cell lines and interacting protein systems, issues of self complementation and steric hindrance, testing of a point mutated version of split TK, reversibility of the TK PCA, and microPET imaging in living mice. A more detailed analysis is presented in the Supplementary Discussion.
In essence, the molecular engineering efforts employed in this study, like all protein engineering, are concerned with adapting proteins to function under different regimes23. In this regard, most protein engineering used so far in general, and in attempts to split TK more specifically, have been by ‘rational re-design’23, i.e. preconceived alterations based on a detailed knowledge of protein structure, function and mechanism24. The alternative approach in enzyme engineering is through ‘directed evolution’, whereby libraries of mutated genes (normally by using error-prone PCR to create a library of mutagenized genes24, but in this case the principle would be for these ‘mutations’ to be split genes that express fragments of TK) might be created, and genetic selection or high-throughput screening subsequently identifies the mutants (i.e. the split fragments) that possess retained TK activity. Although either rational re-design or directed evolution can be very effective, a combination of both strategies would probably represent the most successful route in future studies aimed at further refinement of the above strategies to obtain better functioning split TK fragments (exhibiting greater protein-protein interaction-induced complementation, with less self complementation) for use in a split reporter PCA strategy for PET imaging of PPIs24.
In conclusion, the development and comprehensive proof-of-principle validation of the novel engineered TK PCA described herein now pave the way for many more research directions to enable improved tomographic localization and quantitative imaging of PPIs, signal transduction cascades, and systems biology networks in transgenic animal models of disease25. The application of this particular innovation to PET imaging also heralds other potential research directions that could lead for the first time to a successful split reporter-based strategy for future clinical imaging of PPIs in humans.
METHODS
Further details can be found in the Supplementary Methods and elsewhere15.
Preparation and testing of 293T stable cells
To make stable cells, we incubated 5 × 106 293T cells plated in a 10 cm dish for 24 h at 37°C with 95% oxygen and 5% CO2. The cells were then transfected with 10 μg of plasmid vector pcDNA-pUbi-FKBP12-cTK-pCMV-nTK(V119C)-FRB using lipofectamine 2000 transfection reagent and 24 h later the medium was replaced with fresh medium containing 10 μg/ml puromycin. Every two days the medium was changed and replaced with fresh containing puromycin. The steps were repeated until we achieved 100% puromycin resistant cells. The cells were checked for the expression of split TK with the interacting proteins using the [8-3H]Penciclovir cell uptake assay before and after exposure of cells to 40 nM rapamycin.
Western immunoblotting
Stably transfected cell samples were also lysed and expression levels of FRB (mTOR) and FKBP12 (either endogenous or in their fusion with split TK fragments) in total lysates were determined by immunoblotting with antibody to TK (1:500, Polyclonal, raised in rabbit) for nTK-FRB and FKBP12-cTK, antibody to FRB (Cell Signaling) for cellular FRB (mTOR), antibody to FKBP12 (Abcam) for cellular FKBP12, and antibody to α-tubulin or β-actin (Sigma) as an internal control for loading.
Nuclear and cytoplasmic extraction
A Thermo Scientific NE-PER® kit was used. Extracts obtained with this product have less than 10% contamination between nuclear and cytoplasmic fractions. These extracts were further analysed by Western blotting for protein expression.
Immunostaining and fluorescence microscopy of cells for subcellular localization of components of the TK PCA
293T cells were transfected with the corresponding vectors shown in Fig. 2a. 48 h later the cells were fixed with acetone for 2 min, after which this was made to evaporate by keeping at room temperature for 10 min. The cells were blocked by incubating with Tris-Buffered Saline with Tween (TBST) containing 2% bovine serum albumin (BSA) for 60 min at room temperature. The cells were further incubated in TBST with 2% BSA containing antibody to TK (1:500, Polyclonal, raised in rabbit) for an additional 60 min. They were washed three times (5 min each) with TBST. The cells were then incubated with TBST containing fluorescein-labeled goat anti-rabbit secondary antibody (1:200; Chemicon, Temicula, CA) for 60 min at room temperature. Fluorescent microscopy (using an excitation filter 365 nm) of washed cells was performed using an Axiovert 25 microscope (Carl Zeiss AG) and micrographs were obtained at ×40 magnification using an AxioCam MRc camera (Bernried).
MicroPET imaging in living mice
All animal handling and care was performed in accordance with Stanford University Animal Research Committee guidelines. For imaging studies using stable cell lines, five 12-week old female nude mice (nu/nu) were implanted subcutaneously over the left shoulder with 5× 106 mock transfected 293T cells admixed with 50,000 293T stable cells expressing Firefly luciferase (see below), and subcutaneously over the right shoulder with 5× 106 293T stable cells expressing both nTK(V119C)-FRB and FKBP12-cTK admixed with 50,000 293T stable cells expressing Firefly luciferase. The cells were allowed to grow as tumors for 7 days (to ~0.3–0.4 cm in diameter). On day 8 mice were tail vein injected with 200 μCi of [18F]-FHBG after undergoing anesthesia with 2% isoflurane in oxygen at 2 L/min. Three hours later, mice were microPET imaged in a spread prone position in a FOCUS microPET scanner (Concorde Microsystems). Images were reconstructed using a three-dimensional filtered back projection and iterative maximum a posteriori algorithm, and no partial volume correction27. Immediately after imaging the animals were intraperitoneally injected with 50 μg of rapamycin; 24 h later all the animals were re-subjected to microPET imaging as mentioned above. The animals were further injected with intraperitoneal 50 μg rapamycin for two more days at 24 h intervals and subjected to repeat microPET imaging. In a separate cohort of 4 control mice, the above temporal imaging studies were performed in an identical fashion except that no rapamycin was administered. The counts from regions of interest (ROI) were converted to the percentage injected dose per gram (%ID g−1) of tumor using filtered back projection as described previously27. This %ID g−1 is a measure of the amount of tracer accumulated in a given tissue site normalized to the injected amount and to the mass of the tissue examined. The FHBG accumulation (%ID g−1) in the 293T tumor cells reflects their TK activity. For statistical analysis, the 2-tailed Student t-test was used. Differences were considered significant at P < 0.05.
For imaging in these 5 mice, 293T cells stably expressing Firefly luciferase enzyme were used as secondary gene marked cells to report on overall tumor viability when admixed with 293T cells stably expressing split TK plus the interacting proteins FRB and FKBP12 under study. We therefore used Firefly luciferase activity measured by optical CCD camera imaging as an indirect crude measure of the viability and adequacy of cell number of admixed 293T cells acting as the source of the microPET images. Details of optical imaging are as reported previously9. We removed the tumors from the animals after microPET imaging for immunostaining to confirm the expression of split TK protein within the tumors.
Supplementary Material
Acknowledgments
We thank S. Gobalakrishnan, J. Willmann, and O. Gheysens for their assistance with the microPET imaging. T. Massoud was supported in part by US National Cancer Institute ICMIC grants P50 CA86306, and by a Mid-Career Award for Established Practitioners from The Health Foundation, General Electric Medical Systems, the Palgrave Brown Foundation, the Cancer Prevention Research Trust, a Royal College of Radiologists X-Appeal pump priming grant, the Steel Charitable Trust, the Sir Samuel Scott of Yews Trust, and the National Institute of Health Research Cambridge Biomedical Research Centre, all in the U.K. This work was funded by US National Cancer Institute grant ICMIC P50 CA114747 (S.S.G.) and US National Cancer Institute RO1 CA082214 (S.S.G.).
Footnotes
AUTHOR CONTRIBUTIONS
All authors designed experiments. SSG supervised experiments. TFM and RP performed the experiments. TFM and SSG wrote the manuscript. All authors discussed the results and commented on the manuscript.
References
- 1.Valdar WS, Thornton JM. Protein-protein interfaces: analysis of amino acid conservation in homodimers. Proteins. 2001;42:108–124. [PubMed] [Google Scholar]
- 2.Shoemaker BA, Panchenko AR. Deciphering protein-protein interactions. Part I Experimental techniques and databases. PLoS Comput Biol. 2007;3:e42. doi: 10.1371/journal.pcbi.0030042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Massoud TF, Gambhir SS. Molecular imaging in living subjects: seeing fundamental biological processes in a new light. Genes Devel. 2003;17:545–580. doi: 10.1101/gad.1047403. [DOI] [PubMed] [Google Scholar]
- 4.Michnick SW. Exploring protein interactions by interaction-induced folding of proteins from complementary peptide fragments. Curr Opin Struct Biol. 2001;11:472–477. doi: 10.1016/s0959-440x(00)00235-9. [DOI] [PubMed] [Google Scholar]
- 5.Hu CD, Kerppola TK. Simultaneous visualization of multiple protein interactions in living cells using multicolor fluorescence complementation analysis. Nat Biotechnol. 2003;21:539–545. doi: 10.1038/nbt816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Michnick SW, Remy I, Campbell-Valois FX, Vallee-Belisle A, Pelletier JN. Detection of protein-protein interactions by protein fragment complementation strategies. Methods Enzymol. 2000;328:208–230. doi: 10.1016/s0076-6879(00)28399-7. [DOI] [PubMed] [Google Scholar]
- 7.Park K, et al. A split enhanced green fluorescent protein-based reporter in yeast two-hybrid system. Protein J. 2007;26:107–116. doi: 10.1007/s10930-006-9051-2. [DOI] [PubMed] [Google Scholar]
- 8.Zhang S, Ma C, Chalfie M. Combinatorial marking of cells and organelles with reconstituted fluorescent proteins. Cell. 2004;119:137–144. doi: 10.1016/j.cell.2004.09.012. [DOI] [PubMed] [Google Scholar]
- 9.Paulmurugan R, Umezawa Y, Gambhir SS. Noninvasive imaging of protein-protein interactions in living subjects by using reporter protein complementation and reconstitution strategies. Proc Natl Acad Sci USA. 2002;99:15608–15613. doi: 10.1073/pnas.242594299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Paulmurugan R, Gambhir SS. Monitoring protein-protein interactions using split synthetic renilla luciferase protein-fragment-assisted complementation. Anal Chem. 2003;75:1584–1589. doi: 10.1021/ac020731c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tjuvajev JG, et al. Imaging the expression of transfected genes in vivo. Cancer Res. 1995;55:6126–6132. [PubMed] [Google Scholar]
- 12.Kang KW, Min JJ, Chen X, Gambhir SS. Comparison of [14C]FMAU, [3H]FEAU, [14C]FIAU, and [3H]PCV for monitoring reporter gene expression of wild type and mutant herpes simplex virus type 1 thymidine kinase in cell culture. Mol Imaging Biol. 2005;7:296–303. doi: 10.1007/s11307-005-0010-7. [DOI] [PubMed] [Google Scholar]
- 13.Iyer M, et al. 8-[18F]Fluoropenciclovir: an improved reporter probe for imaging HSV1-tk reporter gene expression in vivo using PET. J Nucl Med. 2001;42:96–105. [PubMed] [Google Scholar]
- 14.Gambhir SS, et al. A mutant herpes simplex virus type 1 thymidine kinase reporter gene shows improved sensitivity for imaging reporter gene expression with positron emission tomography. Proc Natl Acad Sci USA. 2000;97:2785–90. doi: 10.1073/pnas.97.6.2785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Massoud TF. PhD thesis. University of Cambridge; 2007. Novel approaches to molecular imaging of protein-protein interactions in living subjects. [Google Scholar]
- 16.Spotts JM, Dolmetsch RE, Greenberg ME. Time-lapse imaging of a dynamic phosphorylation-dependent protein-protein interaction in mammalian cells. Proc Natl Acad Sci USA. 2002;99:15142–15147. doi: 10.1073/pnas.232565699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen J, Schreiber SL. Identification of an 11-kDa FKBP12-rapamycin-binding domain within the 289-kDa FKBP12-rapamycin-associated protein and characterization of a critical serine residue. Proc Natl Acad Sci USA. 1995;92:4947–4951. doi: 10.1073/pnas.92.11.4947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Remy I, Michnick SW. Clonal selection and in vivo quantitation of protein interactions with protein-fragment complementation assays. Proc Natl Acad Sci USA. 1999;96:5394–5399. doi: 10.1073/pnas.96.10.5394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wurth C, Thomas RM, Folkers G, Scapozza L. Folding and self-assembly of herpes simplex virus type 1 thymidine kinase. J Mol Biol. 2001;313:657–670. doi: 10.1006/jmbi.2001.5060. [DOI] [PubMed] [Google Scholar]
- 20.Jaakkola P, et al. Targeting HIF-α to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science. 2001;292:468–472. doi: 10.1126/science.1059796. [DOI] [PubMed] [Google Scholar]
- 21.Paulmurugan R, Gambhir SS. An intramolecular folding sensor for imaging estrogen receptor-ligand interactions. Proc Natl Acad Sci USA. 2006;103:15883–15888. doi: 10.1073/pnas.0607385103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Penuelas I, et al. Positron emission tomography imaging of adenoviral-mediated transgene expression in liver cancer patients. Gastroenterology. 2005;128:1787–1795. doi: 10.1053/j.gastro.2005.03.024. [DOI] [PubMed] [Google Scholar]
- 23.Brannigan JA, Wilkinson AJ. Protein engineering 20 years on. Nat Rev Mol Cell Biol. 2002;3:964–970. doi: 10.1038/nrm975. [DOI] [PubMed] [Google Scholar]
- 24.Chen R. Enzyme engineering: rational redesign versus directed evolution. Trends Biotechnol. 2001;19:13–14. doi: 10.1016/s0167-7799(00)01522-5. [DOI] [PubMed] [Google Scholar]
- 25.Massoud TF, Gambhir SS. Integrating noninvasive molecular imaging into molecular medicine: an evolving paradigm. Trends Mol Med. 2007;13:183–191. doi: 10.1016/j.molmed.2007.03.003. [DOI] [PubMed] [Google Scholar]
- 26.Michnick SW, Ear PH, Manderson EN, Remy I, Stefan E. Universal strategies in research and drug discovery based on protein-fragment complementation assays. Nat Rev Drug Discov. 2007;6:569–82. doi: 10.1038/nrd2311. [DOI] [PubMed] [Google Scholar]
- 27.Qi J, Leahy RM, Cherry SR, Chatziioannou A, Farquhar TH. High-resolution 3D Bayesian image reconstruction using the microPET small-animal scanner. Phys Med Biol. 1998;43:1001–1013. doi: 10.1088/0031-9155/43/4/027. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.