

Abstract
Alzheimer’s disease (AD) is a progressive, neurodegenerative disease histochemically characterized by extracellular deposits of amyloid beta (Aβ) protein and intracellular neurofibrillary tangles of hyperphosphorylated tau protein. AD is considered to be a complex, multifactorial syndrome, with numerous causal factors contributing to its pathogenesis. Thus, for any novel therapeutic molecule to have a “disease-modifying” effect on AD, it must be able to modulate multiple, synergistic targets simultaneously. In this context, we have studied two compounds of plant origin [withanolide A (1) and asiatic acid (2)] for their potential activities against multiple targets associated with Aβ pathways (BACE1, ADAM10, IDE, and NEP). BACE1 is a rate-limiting enzyme in the production of Aβ from amyloid-β precursor protein (AβPP), while ADAM10 is involved in non-amyloidogenic processing of AβPP. IDE and NEP are two of the prominent enzymes involved in effectively degrading Aβ. It was found that both 1 and 2 significantly down-regulated BACE1 and also up-regulated ADAM10 in primary rat cortical neurons. In addition, 1 significantly up-regulated IDE levels, which may help in degrading excess Aβ from the AD brain. Based on the data obtained, the two multi-functional compounds may prove valuable in developing novel, effective therapeutics for the prevention and treatment of AD-associated amyloid pathology.
Alzheimer’s disease (AD) is a very complex, multifactorial, age-related neurodegenerative disease, characterized clinically by severe memory loss and impairment of various cognitive functions.1 Pathologically, AD is characterized by extracellular deposits of amyloid beta (Aβ) protein and intracellular accumulation of neurofibrillary tangles (NFTs) that are composed of hyperphosphorylated tau (τ) protein.2
AD is classified into two categories, viz., familial Alzheimer’s disease (FAD) and sporadic Alzheimer’s disease (SAD). FAD has been shown to be associated with mutations in AβPP, presenilin 1 and 2 (PS1 and PS2) genes on chromosomes 21, 14 and 1, respectively.3–5 Of all AD cases, only 5–10% are due to FAD mutations and the mutations in PS1 are the most frequent of FAD causes.6 Furthermore, the apolipoprotein E4 (ApoE4) gene has been shown to cause a slight predisposition to AD.7 On the other hand, SAD is the major form of AD and comprises 90–95% of all cases.8 Unlike FAD, the etiology of SAD is not well understood9 and several possible risk factors in the development of SAD have been identified. Age is the most significant risk factor for the development of AD.9 Additional risk factors based upon epidemiological studies are, a high fat diet, gender, head trauma, and vascular risk factors such as diabetes, ischemia and hypertension.10 Thus, the lack of any cure for AD potentially may be attributed to this complex etiology. The currently available treatments for AD approved by the United States Food and Drug Administration (FDA) comprise donepezil, tacrine, rivastigmine, and memantine. The first three drugs inhibit acetylcholine esterase, either selectively or non-selectively, and thus help in improving memory in AD patients. However, their use is associated with various adverse side effects.11 In contrast, memantine is a non-competitive inhibitor of N-methyl-D-aspartate (NMDA) receptors, which prevents glutamate excitotoxicity and has relatively fewer adverse drug effects.11 All of these approved drugs have beneficial, but short-lived effects in mediating the symptoms of AD.
Thus, there is a significant need for the development of novel drugs that will not only affect the cholinergic and glutamatergic pathways (symptomatic therapies) but also target other cellular pathways and have lasting, disease-modifying effects against AD. In this regard, investigation of the Aβ pathway may be the most appropriate. Various molecular, cellular, and animal model studies have been used to establish the Aβ protein as a central factor in the development and progression of AD.9 The increased production and deposition of Aβ in the AD brain initiates a pathological cascade leading to the formation of NFTs, gliosis, inflammatory changes, synaptic damage, and neurotransmitter loss.12 Thus, there is a major research focus on finding drugs that may decrease Aβ levels in the AD brain, by lowering its production and/or enhancing its degradation and clearance. These pathways offer multiple molecular therapeutic targets, such as BACE1, the presenilins and ADAM10 (involved in amyloidogenic and non-amyloidogenic processing of AβPP), and insulin degrading enzyme (IDE), neprilysin (NEP) and matrix metalloproteinases (MMPs) (involved in Aβ degradation). The major drawback of the present drug development strategies, however, is the “one-drug-one-target” approach, rendering them limited in their ability to modify the apparent complex pathology of AD.13 Thus, there is an immediate need to develop novel therapeutic molecules that may be able to modulate multiple Aβ-related targets simultaneously, thereby providing disease-modifying therapeutic efficacy against this devastating disease.
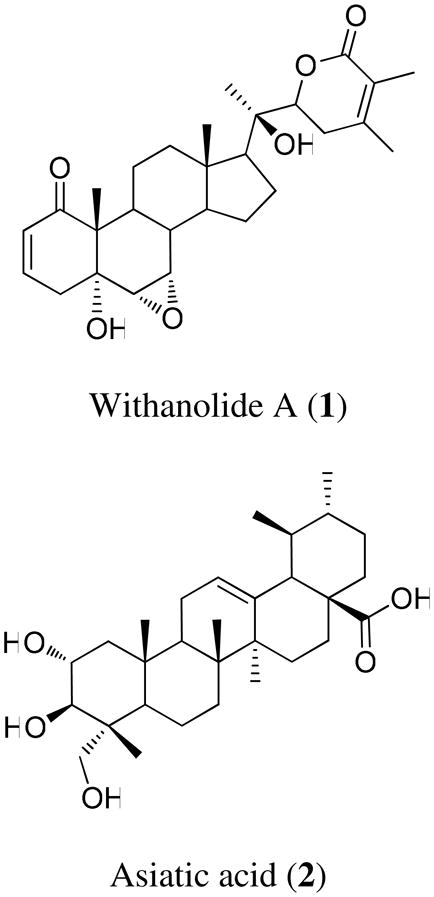
The aim of the present study was to examine two pure natural products (withanolide A, 1 and asiatic acid, 2) isolated from two medicinally important plants (Withania somnifera and Centella asiatica, respectively), for their potential activities against multiple targets associated with AβPP processing and Aβ clearance (BACE1, ADAM10, IDE, and NEP).
Results and Discussion
Aβ PP Processing: Both Withanolide A (1) and Asiatic Acid (2) Enhance Non-amyloidogenic Processing of Aβ PP by Down-regulating BACE1 as well as Up-regulating ADAM10 Activation in Primary Rat Cortical Neurons
To determine whether 1 and 2 affect AβPP processing, primary rat cortical neurons were treated with various doses of these compounds for 24 h. Compound 1 was non-toxic to neurons at a concentration as high as 100 μM, while 2 affected cell viability at 20 μM and caused complete cell death at 100 μM (data not shown). Therefore, the highest concentrations of 1 and 2 used in the present study were 100 and 10 μM, respectively. The morphologies of neurons treated with 100 μM of compound 1 and 10 μM of compound 2 are shown using MAP-2 immunostaining (Figure 1). Both 1 and 2 at these concentrations had no significant effect on cell morphology and viability, as compared to controls.
Figure 1.

MAP-2 immunostaining. Primary rat cortical neurons were treated for 24 h with 100 μM of withanolide A (1), 10 μM of asiatic acid (2), or 0.1% DMSO (control). Images were obtained with Leica DM IL inverted fluorescence microscope (objective lens magnification, 40X).
After 24 h of treatment with 1 and 2, the expression levels of various proteins (cellular and secreted) associated with AβPP processing were measured; the conditioned media were collected for evaluation of sAPPα levels, and the neurons were washed, lysed, and the total cellular protein was used for western blot analysis of different cellular proteins. BACE1 levels decreased dose-dependently in response to both 1 and 2 treatment (Figures 2 and 3, p < 0.05). Also, both 1 and 2 dose-dependently enhanced ADAM10 activation in primary rat cortical neurons as compared to controls; levels of mature ADAM10 (~60 kDa isoform) dose-dependently increased in response to both 1 and 2 treatment (Figures 2 and 3, p < 0.05). BACE1 is involved in amyloidogenic processing of AβPP, whereby it cleaves AβPP forming the smaller, membrane bound C-terminal fragment of APP (C99), which is further cleaved by γ-secretase leading to the formation of Aβ proteins.14 On the other hand, cleavage of AβPP by ADAM10 constitutes the non-amyloidogenic pathway, in which ADAM10 cleaves AβPP within its Aβ region releasing a membrane-bound, ~10-kDa C-terminal fragment (C83) and a soluble, ~120-kDa N-terminal fragment (sAPPα), thus precluding Aβ formation.15 Therefore, the observed down-regulation of BACE1 and up-regulation of ADAM10 activation due to treatment with 1 and 2, may suggest a strong bias towards non-amyloidogenic processing of AβPP, thus producing elevated levels of C83 and sAPPα. Consistent with this, both 1 and 2 dose-dependently increased C83 and sAPPα levels in cortical neurons (Figure 4, p < 0.05).
Figure 2.

Withanolide A (1) down-regulates BACE1 and up-regulates ADAM10. Cortical neurons were treated with 0, 5, 20 and 100 μM of 1 for 24 h. Immunoblots show significant down-regulation in BACE1 levels and also up-regulation of active ADAM10 levels in neurons treated with 1 as compared to respective controls (0.1% DMSO). Histograms corresponding to BACE1 and ADAM10 blots represent quantitative determinations of intensities of the relevant bands normalized to actin. Data represent the mean ± S.D. of three independent experiments. The Student’s t-test was used for analyzing the differences between the two treatment groups (*, p <0.05 compared with respective control).
Figure 3.

Asiatic acid (2) down-regulates BACE1 and up-regulates ADAM10. Cortical neurons were treated with 0, 1, 5 and 10 μM of 2 for 24 h. Immunoblots show significant down-regulation in BACE1 levels as well as up-regulation of mature ADAM10 levels in neurons treated with 2 as compared to respective controls (0.01% DMSO). Histograms corresponding to BACE1 and ADAM10 blots represent quantitative determinations of intensities of the relevant bands normalized to actin. Data represent the mean ± S.D. of three independent experiments. The Student’s t-test was used for analyzing the differences between the two treatment groups (*, p <0.05 compared with respective control).
Figure 4.

Effects of 1 and 2 on AβPP processing. Immunoblots show a dose-dependent increase in the levels of C83 and sAPPα (α-secreatase cleavage products of AβPP) in neurons treated with 1 and 2 as compared to their respective controls (0.1% and 0.01% DMSO, respectively). Histograms corresponding to sAPPα blot represent quantitative determinations of intensities of the relevant bands normalized to actin. Data represent mean ± S.D. of three independent experiments. The Student’s t-test was used for analyzing the differences between the two treatment groups (*, p <0.05 compared with respective control).
Aβ Degradation: Withanolide A (1), but not Asiatic Acid (2), Enhances IDE Levels, While NEP is Unaffected by Both WL-A and AS-A in Primary Rat Cortical Neurons
In addition to the observed effects of 1 and 2 on AβPP processing in primary rat cortical neurons, it was intended also to study their possible effects in terms of degradation of Aβ. In this regard, the expression levels of IDE and NEP, two major proteins involved in the degradation of Aβ, were examined.16 The activity as well as mRNA and protein levels of IDE are decreased in the AD brain and this decrease is associated with elevated levels of Aβ as compared to healthy controls.17 Similarly, NEP mRNA and protein levels are reduced significantly in AD brains as compared to controls and this decrease is specific to brain regions that are selectively affected in AD pathology.18 Thus, it has been hypothesized that the increased expression of these enzymes (IDE and NEP) may confer a protective effect against AD-associated Aβ etiology.19 In the present study, it was found that withanolide A dose-dependently enhanced IDE levels in cortical neurons (Figure 5, p < 0.05). In contrast, there was no change in the levels of IDE in neurons treated with asiatic acid at all concentration as compared to untreated ones (Figure S1, Supporting Information). In the case of NEP, 1 had no effect on NEP levels at all concentrations as compared to controls (Figure 5). Furthermore, 2 also had no effect on NEP levels at 1 and 10 μM, but at 5 μM there was a statistically significant, but nevertheless slight increase (~ 32%) in NEP levels (Figure S1, Supporting Information).
Figure 5.

Dose-dependent effects of withanolide A (1) on targets involved in Aβ degradation (IDE and NEP). Cortical neurons were treated with 0, 5, 20 and 100 μM of 1 for 24 h. Immunoblots show significant up-regulation of IDE, while NEP remained unaffected, in neurons treated with 1 in a dose-dependent manner as compared to controls. Histograms corresponding to IDE and NEP blots represent quantitative determinations of intensities of the relevant bands normalized to actin. Data represent mean ± S.D. of three independent experiments. The Student’s t-test was used for analyzing the differences between the two treatment groups (*, p <0.05 compared with respective control).
The “amyloid cascade hypothesis”, which suggests the accumulation of Aβ in the brain as a main trigger for AD, has been studied extensively since the first characterization of Aβ deposits in 1984.20 According to this hypothesis, a chronic imbalance between the production and clearance of Aβ results in the formation of Aβ plaques and plays a major role in the etiopathogenesis of AD.21 Many studies support the amyloid cascade hypothesis. The brains of AD patients are characterized by the presence of Aβ plaques and their number far exceeds that found in the brains of age-matched healthy controls.22 Furthermore, the amount of Aβ plaques is correlated highly with the degree of cognitive impairment.23 In addition, all three genes associated with FAD have been shown to be involved in increased production of Aβ (AβPP, PS1 and PS2).24 Down’s syndrome patients who produce significantly higher amounts of Aβ from birth and deposit Aβ plaques in their brains as early as age 12, consistently develop AD by age the of 50.25 This further emphasizes the central role of Aβ in the pathogenesis of AD. Thus, a major focus of current AD drug discovery efforts is on developing novel therapeutics that may effectively decrease Aβ production and deposition in the AD brain.21
The proteolytic processing of AβPP takes place by sequential cleavage by various proteases named α-, β-, and γ-secretase. α-Secretase is a member of the ADAM (a disintegrin and metalloprotease) family, such as ADAM17 or TACE (tumor necrosis factor-α converting enzyme), ADAM 9, ADAM10, MDC9, and an aspartyl protease, BACE2.26 The α-secretase enzyme cleaves AβPP within the Aβ domain between residues Lys16 and Leu17, thus avoiding the generation of intact Aβ peptides. This leads to the formation of a soluble domain (sAPPα), released into extracellular space, and a 10-kDa C-terminal fragment (C83), which remains within the cellular membrane and serves as substrate for further cleavage by γ-secretase.27 Both sAPPβ and C83 have been shown not to contribute directly to Aβ plaques observed in AD brains.9 In fact, both α-secretase (ADAM10) and sAPPα have been shown to be reduced in AD patients as compared to healthy controls.28 On the contrary, β-secretase (BACE1) is up-regulated significantly in the AD brain.29 BACE1 is a major β-secretase involved in the amyloidogenic processing of APP in neurons.30 BACE1 cleaves APP at the Asp+1 residue of the Aβ region and leads to the generation of a secreted soluble fragment (sAPPβ) and a membrane-bound C-terminal fragment (C99). The γ-secretase cleavage of C99 constitutes an amyloidogenic pathway, leading to the generation of a spectrum of Aβ peptides. The Aβ peptides containing 40 or 42 amino acids (Aβ40/42) are the two most common amyloidogenic Aβ peptides and are involved in the formation of mature, neuritic plaques observed in the AD brain.31 In the present study, it was found that both withanolide A (1) and asiatic acid (2) dose-dependently and significantly down-regulated BACE1 levels in primary rat cortical neurons. BACE1 is a rate-limiting enzyme in the production of Aβ; our group and others have shown previously that a slight increase in BACE1 levels leads to a dramatic increase in the production of Aβ 40/42.32,33 A corollary to this is that even a slight decrease in BACE1 levels may lead to a considerable decrease in the production of Aβ. Thus, 1 and 2, with their significant activity against BACE1, represent potentially effective lead compounds for AD aimed at decreasing Aβ generation and deposition. Furthermore, it has been established recently that BACE1 and α-secretase compete for AβPP processing, whereby BACE1 cleavage of AβPP precludes its processing by α-secretase and vice versa.34,35 Thus, BACE1 down-regulation induced by 1 and 2, in itself, may indirectly lead to the increased processing of AβPP by α-secretase (non-amyloidogenic processing). In the current work, it is encouraging that both 1 and 2 also had direct effects on α-secretase activity, which was evident by significantly enhanced ADAM10 maturation (increased ratio of mature to pro ADAM10 levels). This increased α-secretase activity further affects non-amyloidogenic processing of AβPP (a positive gain of function). It was found that levels of both C83 and sAPPα, non-amyloidogenic products of AβPP, were elevated by treatment with both 1 and 2 as compared to the respective controls. The secreted, α-secretase product of AβPP (sAPPα) has been shown to protect neurons against various insults such as excitotoxic, metabolic, and oxidative.36–38 Thus, 1 and 2, with their dual activities against BACE1 and ADAM10, may prove highly beneficial against AD in terms of lowering Aβ levels directly and also increasing sAPPα levels, thus being neuroprotective indirectly.
Compounds 1 and 2 are constituents of Withania somnifera (L.) Dunal (Solanaceae) and Centella asiatica Urb. (Apiaceae), respectively. Both species are recommended as “Medhya-Rasayana” (memory and intellect enhancers) in the ayurvedic traditional Indian medicinal system.39 Various modern scientific studies support the memory enhancing role of W. somnifera and C. asiatica, as has been reported.40,41 Thus, both W. somnifera and C. asiatica, may prove beneficial against AD, where memory and other cognitive functions are severely impaired. Moreover, a crude extract of C. asiatica has been shown to decrease Aβ levels in a transgenic mouse model of AD.42 The present study, however, is the first to assess the effects of pure active constituents of these two plants (1 and 2, respectively) on AβPP processing pathways and the underlying molecular mechanisms associated with the increased bias towards non-amyloidogenic processing of AβPP.
In addition to the increased amyloidogenic processing and/or decreased non-amyloidogenic processing of AβPP, the levels of Aβ may also be increased in the AD brain due to its decreased degradation. IDE, NEP, MMPs, plasmin, and endothelin-converting enzymes (ECEs) are some of the major proteolytic enzymes involved in Aβ degradation.43 Growing evidence suggests that defective Aβ degradation may be a central causative factor in the pathogenesis of AD. The genetic deletion or pharmacological inhibition of the Aβ-degrading enzymes has been shown to elevate Aβ levels in animal brains significantly.43 Furthermore, the levels of NEP and IDE proteins are decreased in an age- and brain region-dependent manner.43,44 Thus, modulation of one or more Aβ-degrading enzymes may prove vital in the prevention and treatment of AD. This hypothesis is supported by a recent study, whereby a novel small molecule inhibitor of plasminogen activator inhibitor-1 discovered by Wyeth (PAZ-417), which enhances activity of an Aβ-degrading enzyme (plasmin), has been shown to significantly lower plasma/brain Aβ levels and also reverses cognitive deficits in transgenic mouse models of AD.45 In the present study, it was found that 1, but not 2, significantly increased IDE levels in primary rat cortical neurons. As indicated earlier, both 1 and 2 had no significant effects on NEP levels. The significance of 1 in the up-regulation in IDE levels against AD is emphasized by the fact that over-expression of IDE by 100% decreases Aβ levels, plaque burden, and associated neuronal death by more than 50%.19 Similarly, a seven-fold over-expression of NEP is associated with more than a 90% decrease in Aβ levels.19
At present, the underlying mechanism by which 1 and 2 affect the levels of BACE1, ADAM10, and IDE is unclear. The AD brain is characterized by increased oxidative stress46 and the enzymes involved in AβPP processing and Aβ degradation (BACE1, ADAM10, IDE, and NEP) have been shown to be dependent upon the cellular redox state. Oxidative stress has been demonstrated to increase the expression and activity of BACE1 in NT2 neurons and primary rat cortical neurons, which was accompanied by a proportional elevation of the carboxy-terminal fragments of AβPP.47,48 Furthermore, both ADAM10 promoter activity and transcription of endogenous ADAM10 have been shown to be increased by treatment with retinoic acid.49 Also, (−)-epigallocatechin-3-gallate (EGCG), from green tea, has been shown to significantly increase ADAM10 maturation.50 EGCG has also been shown to increase the expression levels of both NEP and IDE.51 These data, taken together with the realization that both 1 and 2 possess excellent anti-oxidative and anti-inflammatory properties52,53, may explain, in part, their effects on BACE1, ADAM10, and IDE levels. However, the lack of an effect of either 1 or 2 on NEP levels and of 2 on IDE levels suggest other potentially important molecular mechanisms underlying the observed effects of these compounds that remain to be further elucidated. Recently, PPARγ has been shown to regulate IDE expression levels in rat primary neurons.54 This, taken together with the current data, suggests that 1 may have some effect on PPARγ leading to the up-regulation in IDE protein levels. Furthermore, it remains to be seen if compounds 1 and 2 have any affect on activities of one or more of the Aβ-related targets studied here. In this context, the computational docking studies that were conducted indicate the occurrence of favorable interaction complexes between the natural product 1 and the active sites of all four targets indicating its possible direct effect on the levels and the activities of these enzymes (Figure S2, Supporting Information).
The synergistic, multi-target activity demonstrated here by 1 and 2 is in line with a recent shift in the AD drug discovery focus from “one-target molecules” to finding “multi-target ligands”.13,55 This type of multi-functional activity may prove advisable for any novel therapeutic molecule to be effective in modifying the complex pathology of AD. With this in mind, an anti- Aβ multi-target therapeutic index (anti-Aβ MTTI) is proposed, defined simply as the ratio of the fractional up-regulation in anti-amyloid targets to the fractional down-regulation in pro-amyloid targets, as shown below:
| (1) |
This index may serve as a potentially important criterion for determining the effectiveness of a therapeutic molecule in modulating multiple targets that synergistically affect cerebral Aβ levels; the higher the index value, the higher the anti-amyloid, multi-targeting activity of the test compounds. With a minimum number of targets equal to two, at least one anti-amyloid and one pro-amyloid target, and minimum 35% up-regulation and 35% down-regulation, respectively, for biological significance, the following evaluation is obtained:
Thus, a minimum index value for any therapeutic molecule to have a significant, multi-target anti-Aβ activity is 2.10. From the present data, for 1 at the highest concentration of 100 μM, equation (1) can be written as follows by using values from Figures 2 and 5:
Similarly, for 2 at the highest concentration of 10 μM, the following is obtained:
Thus, comparing these anti-Aβ MTTI values for 1 and 2 (6.47 and 6.25, respectively) with the minimum index value of 2.10, both 1 and 2 seem to possess excellent multi-targeted, anti-Aβ activities.
In summary, it has been shown for the first time that withanolide A (1) and asiatic acid (2) positively modulate multiple targets associated with Aβ pathways and thus, may be beneficial in attenuating Aβ levels in the AD brain by both decreasing Aβ production (BACE1 down-regulation and ADAM10 maturation) and also by increasing Aβ degradation (IDE up-regulation). Therapies based on modulating secretases (BACE1 and ADAM10) will act locally to affect Aβ production, while therapies based on increasing Aβ degradation (IDE) may prove essential in acting at sites that are widely separated from the Aβ production sites.43 This kind of “multi-functional” and “multi-level” activity in a given therapeutic molecule may prove highly effective against AD, providing multiple mechanisms to alter amyloid pathology in the AD brain. Finally, in addition to the Aβ-related activities established in the present study, both 1 and 2 have been shown to induce significant regeneration of neurites and dendrites, which may help in reconstructing neuronal networks damaged in AD.56,57 Thus, these two natural products may serve as lead compounds for the development of novel therapeutic molecules with disease-modifying activities, which are urgently needed to tackle the ill-effects of a highly complex, multi-factorial disease like AD.
Experimental Section
Test Compounds
Withanolide A (1) and asiatic acid (2) were purchased from Chromadex Incorporation (Irvine, CA). The purity of 1 and 2 was 99.3% and 93.7%, respectively.
Isolation and Culture of Primary Rat Cortical Neurons
Primary cortical neurons were isolated from 1-day-old Sprague-Dawley rat pups and cultured as described by Chandler et al.58 All procedures were performed according to guidelines developed by the Institutional Animal Care and Use Committee (IACUC) at Michigan State University. The cells were plated on poly-D-lysine coated, 12-well plates at a concentration of 1 × 106 cells per well in fresh cortical medium [Dulbecco’s Modified Eagle’s Medium (DMEM, Invitrogen, Carlsbad, CA) supplemented with 10% horse serum (Sigma, St. Louis, MO), 25 mM glucose, 10 mM HEPES (Sigma), 2 mM glutamine (BioSource International, Camarillo, CA), 100 IU/mL penicillin, and 0.1 mg/mL streptomycin]. The experiments were performed on 3–4 day old neuronal cultures. The cells were treated with 1 and 2 at different doses for 24 h.
Immunostaining of Primary Rat Cortical Neurons
To perform the immunofluorescence microscopic study, neuronal cultures were fixed for 20 min in 4% paraformaldehyde and then incubated for 20 min in blocking solution (0.1% Triton X-100 and 1% bovine serum albumin in phosphate-buffered saline, PBS). After washing 2X with PBS, cells were labeled overnight at 4 °C with primary antibody for neurons [1:50 MAP-2 (Santa Cruz Biotechnology, Santa Cruz, CA)]. After 3X PBS washes, primary antibodies were detected with rhodamine-conjugated secondary antibody (Chemicon, Temecula, CA). The cells were visualized with an inverted fluorescence microscope Leica DM IL (Leica Microsystems, Bannockburn, IL) using a 40X objective lens.
Western Blot Analysis
The following antibodies used for western blotting: anti-BACE1 (Abcam, Cambridge, MA), anti-ADAM10 (Sigma), anti-IDE (Abcam), anti-NEP (Santa Cruz), anti-APP, C-terminal (Sigma), anti-APP, N-terminal (22C11, Millipore) and Actin (Sigma). To detect secreted protein (sAPPα), conditioned media were collected and processed as explained earlier.59 To detect cellular proteins, cells were washed three times with ice-cold TBS (25 mM Tris, pH 8.0, 140 mM NaCl, and 5 mM KCl) and lysed for 30 min by scraping into ice-cold radioimmunoprecipitation assay (RIPA) buffer [1% (v/v) Nonidet P-40, 0.1% (w/v) SDS, 0.5% (w/v) deoxycholate, 50 mM Tris, pH 7.2, 150 mM NaCl, 1 mM Na3VO4 and 1 mM PMSF, all from Sigma].60 The total cell lysate was obtained by centrifugation at 12,000 rpm for 30 min at 4 °C. The total protein concentration was measured by using a BCA protein assay kit from Pierce (Rockford, IL). Equal amounts of total protein from each condition were run at 200 V on 10% Tris–HCl gels (for BACE1, ADAM10, IDE, NEP and actin), 5% gels (for sAPPα), and 10–20% Tris–Tricine gels (for APP-C83). The separated proteins were transferred to polyvinylidene fluoride (PVDF) membranes (for APP-C83 detection) and nitrocellulose membranes (for all other proteins) for 1 h at 100 V and incubated at 4 °C overnight with the appropriate primary antibodies [1:1000 BACE1, 1:1000 ADAM10, 1:1000 IDE, 1:1000 NEP, 1:1000 C-APP, 1:500 N-APP (22C11) and 1:2000 actin]. Blots were washed three times in PBS-Tween (PBS-T) and incubated with appropriate HRP-linked secondary antibodies (Pierce) diluted in PBS-T for 1 h at room temperature. After washing three times in PBS-T, blots were developed with the Pierce SuperSignal West Femto Maximum Sensitivity Substrate (Pierce) and imaged with the BioRad ChemiDoc. Quantity-One software from Bio-Rad was utilized to quantify the signal intensities of the protein bands.
Statistical Analysis
Data are shown as means ± S.D. for the indicated number of experiments. The Student’s t-test was used to evaluate statistical significances between different treatment groups. Statistical significance was set at p <0.05.
Supplementary Material
Acknowledgments
We thank L. Liu, N. Tran and H. Geekiyanage for isolating primary rat neuronal cells, and A. Abramczyk for preparing SDS-PAGE gels. We also thank A. Rillorta from Chromadex, Inc. for affording pure test compounds in timely manner. This work was funded in part by the MSU Foundation, the National Institutes of Health (R01 GM079688-01), the National Science Foundation (CBET 0941055), and the Michigan Universities Commercialization Initiative (MUCI) Challenge Fund to C. Chan.
Footnotes
Supporting Information Available. IDE and NEP western blot data for asiatic acid (2) treatment, and computational docking data for withanolide A (1). This information is available free of charge via the Internet at http://pubs.acs.org.
References and Notes
- 1.Wang DC, Chen SS, Lee YC, Chen TJ. Neurosci Lett. 2006;398:78–82. doi: 10.1016/j.neulet.2005.12.057. [DOI] [PubMed] [Google Scholar]
- 2.Mattson MP. Nature. 2004;431:631–639. doi: 10.1038/nature02621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chartier-Harlin MC, Crawford F, Houlden H, Warren A, Hughes D, Fidani L, Goate A, Rossor M, Roques P, Hardy J, Mullan M. Nature. 1991;353:844–846. doi: 10.1038/353844a0. [DOI] [PubMed] [Google Scholar]
- 4.Sherrington R, Rogaev EI, Liang Y, Rogaeva EA, Levesque G, Ikeda M, Chi H, Lin C, Li G, Holman K, Tsuda T, Mar L, Foncin JF, Bruni AC, Montesi MP, Sorbi S, Rainero I, Pinessi L, Nee L, Chumakov I, Pollen D, Brookes A, Sanseau P, Polinsky RJ, Wasco W, Da Silva HAR, Haines JL, Pericak-Vance MA, Tanzi RE, Roses AD, Fraser PE, Rommens JM, St George-Hyslop PH. Nature. 1995;375:754–760. doi: 10.1038/376775a0. [DOI] [PubMed] [Google Scholar]
- 5.Levy-Lahad E, Wasco W, Poorkaj P, Romano DM, Oshima J, Pettingell WH, Yu CE, Jondro PD, Schmidt SD, Wang K, Crowley AC, Fu YH, Guenette SY, Galas D, Nemens E, Wejsman EM, Bird TD, Schellenberg GD, Tanzi RE. Science. 1995;269:973–977. doi: 10.1126/science.7638622. [DOI] [PubMed] [Google Scholar]
- 6.Rogaev EI, Sherrington R, Rogaeva EA, Levesque G, Ikeda M, Liang Y, Chi H, Lin C, Holman K, Tsuda T, Mar L, Sorbi S, Nacrnias B, Piacentim S, Amaducci L, Chminakov I, Cohen D, Lannfelt L, Fraser P, Romnmens J, St George-Hyslop P. Nature. 1995;376:775–778. doi: 10.1038/376775a0. [DOI] [PubMed] [Google Scholar]
- 7.Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, Roses AD, Haines JL, Pericak-Vance MA. Science. 1993;261:921–923. doi: 10.1126/science.8346443. [DOI] [PubMed] [Google Scholar]
- 8.Hoyer S. Exp Gerontol. 2000;35:1363–72. doi: 10.1016/s0531-5565(00)00156-x. [DOI] [PubMed] [Google Scholar]
- 9.Selkoe DJ. Physiol Rev. 2001;81:741–766. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- 10.Haan MN, Wallace R. Annu Rev Public Health. 2004;25:1–24. doi: 10.1146/annurev.publhealth.25.101802.122951. [DOI] [PubMed] [Google Scholar]
- 11.Allain H, Bentue-Ferrer D, Tribut O, Gauthier S, Michel BF, Drieu-La Rochelle C. Fundam Clin Pharmacol. 2003;17:419–428. doi: 10.1046/j.1472-8206.2003.00153.x. [DOI] [PubMed] [Google Scholar]
- 12.Selkoe DJ. Ann N Y Acad Sci. 2000;924:17–25. doi: 10.1111/j.1749-6632.2000.tb05554.x. [DOI] [PubMed] [Google Scholar]
- 13.Li W, Mak M, Jiang H, Wang Q, Pang Y, Chen K, Han Y. Neurotherapeutics. 2009;6:187–201. doi: 10.1016/j.nurt.2008.10.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vassar R, Bennett BD, Babu-Khan S, Kahn S, Mendiaz EA, Denis P, Teplow DB, Ross S, Amarante P, Loeloff R, Luo Y, Fisher S, Fuller J, Edenson S, Lile J, Jarosinski MA, Biere AL, Curran E, Burgess T, Louis JC, Collins F, Treanor J, Rogers G, Citron M. Science. 1999;286:735–741. doi: 10.1126/science.286.5440.735. [DOI] [PubMed] [Google Scholar]
- 15.Kang J, Lemaire HG, Unterbeck A, Salbaum JM, Masters CL, Grzeschik KH, Multhaup G, Beyreuther K, Muller-Hill B. Nature. 1987;325:733–736. doi: 10.1038/325733a0. [DOI] [PubMed] [Google Scholar]
- 16.Dorfman VB, Pasquini L, Riudavets M, Lopez-Costa JJ, Villegas A, Troncoso JC, Lopera F, Castano EM, Morelli L. Neurobiol Aging. 2008 Nov 17; doi: 10.1016/j.neurobiolaging.2008.09.016. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cook DG, Leverenz JB, McMillan PJ, Kulstad JJ, Ericksen S, Roth RA, Schellenberg GD, Jin LW, Kovacina KS, Craft S. Am J Pathol. 2003;162:313–319. doi: 10.1016/s0002-9440(10)63822-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Iijima-Ando K, Hearn SA, Granger L, Shenton C, Gatt A, Chiang HC, Hakker I, Zhong Y, Iijima K. J Biol Chem. 2008;283:19066–19076. doi: 10.1074/jbc.M710509200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Eckman EA, Eckman CB. Biochem Soc Trans. 2005;33:1101–1105. doi: 10.1042/BST20051101. [DOI] [PubMed] [Google Scholar]
- 20.Glenner GG, Wong CW. Biochem Biophys Res Commun. 1984;120:885–890. doi: 10.1016/s0006-291x(84)80190-4. [DOI] [PubMed] [Google Scholar]
- 21.Hardy J, Selkoe DJ. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 22.Perry EK, Tomlinson BE, Blessed G, Bergmann K, Gibson PH, Perry RH. Br Med J. 1978;2:1457–1459. doi: 10.1136/bmj.2.6150.1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cummings BJ, Cotman CW. Lancet. 1995;346:1524–1528. doi: 10.1016/s0140-6736(95)92053-6. [DOI] [PubMed] [Google Scholar]
- 24.Scheuner D, Eckman C, Jensen M, Song X, Citron M, Suzuki N, Bird TD, Hardy J, Hutton M, Kukull W, Larson E, Levy-Lahad E, Viitanen M, Peskind E, Poorkaj P, Schellenberg G, Tanzi R, Wasco W, Lannfelt L, Selkoe D, Younkin S. Nat Med. 1996;2:864–870. doi: 10.1038/nm0896-864. [DOI] [PubMed] [Google Scholar]
- 25.Lemere CA, Blusztajn JK, Yamaguchi H, Wisniewski T, Saido TC, Selkoe DJ. Neurobiol Dis. 1996;3:16–32. doi: 10.1006/nbdi.1996.0003. [DOI] [PubMed] [Google Scholar]
- 26.Allinson TM, Parkin ET, Turner AJ, Hooper NM. J Neurosci Res. 2003;74:342–352. doi: 10.1002/jnr.10737. [DOI] [PubMed] [Google Scholar]
- 27.Weidemann A, Konig G, Bunke D, Fischer P, Salbaum JM, Masters CL, Beyreuther K. Cell. 1989;57:115–126. doi: 10.1016/0092-8674(89)90177-3. [DOI] [PubMed] [Google Scholar]
- 28.Colciaghi F, Borroni B, Pastorino L, Marcello E, Zimmermann M, Cattabeni F, Padovani A, Di Luca M. Mol Med. 2002;8:67–74. [PMC free article] [PubMed] [Google Scholar]
- 29.Gatta LB, Albertini A, Ravid R, Finazzi D. Neuroreport. 2002;13:2031–2033. doi: 10.1097/00001756-200211150-00008. [DOI] [PubMed] [Google Scholar]
- 30.Cai H, Wang Y, McCarthy D, Wen H, Borchelt DR, Price DL, Wong PC. Nat Neurosci. 2001;4:233–234. doi: 10.1038/85064. [DOI] [PubMed] [Google Scholar]
- 31.Burdick D, Soreghan B, Kwon M, Kosmoski J, Knauer M, Henschen A, Yates J, Cotman C, Glabe C. J Biol Chem. 1992;267:546–554. [PubMed] [Google Scholar]
- 32.Patil S, Melrose J, Chan C. Eur J Neurosci. 2007;26:2131–2141. doi: 10.1111/j.1460-9568.2007.05797.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li Y, Zhou W, Tong Y, He G, Song W. Faseb J. 2006;20:285–292. doi: 10.1096/fj.05-4986com. [DOI] [PubMed] [Google Scholar]
- 34.Haass C, Lemere CA, Capell A, Citron M, Seubert P, Schenk D, Lannfelt L, Selkoe DJ. Nat Med. 1995;1:1291–1296. doi: 10.1038/nm1295-1291. [DOI] [PubMed] [Google Scholar]
- 35.Gandhi S, Refolo LM, Sambamurti K. J Mol Neurosci. 2004;24:137–143. doi: 10.1385/JMN:24:1:137. [DOI] [PubMed] [Google Scholar]
- 36.Mattson MP, Guo ZH, Geiger JD. J Neurochem. 1999;73:532–537. doi: 10.1046/j.1471-4159.1999.0730532.x. [DOI] [PubMed] [Google Scholar]
- 37.Furukawa K, Sopher BL, Rydel RE, Begley JG, Pham DG, Martin GM, Fox M, Mattson MP. J Neurochem. 1996;67:1882–1896. doi: 10.1046/j.1471-4159.1996.67051882.x. [DOI] [PubMed] [Google Scholar]
- 38.Gralle M, Botelho MG, Wouters FS. J Biol Chem. 2009;284:15016–15025. doi: 10.1074/jbc.M808755200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kumar V. Phytother Res. 2006;20:1023–1035. doi: 10.1002/ptr.1970. [DOI] [PubMed] [Google Scholar]
- 40.Schliebs R, Liebmann A, Bhattacharya SK, Kumar A, Ghosal S, Bigl V. Neurochem Int. 1997;30:181–190. doi: 10.1016/s0197-0186(96)00025-3. [DOI] [PubMed] [Google Scholar]
- 41.Veerendra Kumar MH, Gupta YK. J Ethnopharmacol. 2002;79:253–260. doi: 10.1016/s0378-8741(01)00394-4. [DOI] [PubMed] [Google Scholar]
- 42.Dhanasekaran M, Holcomb LA, Hitt AR, Tharakan B, Porter JW, Young KA, Manyam BV. Phytother Res. 2009;23:14–19. doi: 10.1002/ptr.2405. [DOI] [PubMed] [Google Scholar]
- 43.Leissring MA. J Biol Chem. 2008;283:29645–29649. doi: 10.1074/jbc.R800022200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Turner AJ, Nalivaeva NN. Int Rev Neurobiol. 2007;82:113–135. doi: 10.1016/S0074-7742(07)82006-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jacobsen JS, Comery TA, Martone RL, Elokdah H, Crandall DL, Oganesian A, Aschmies S, Kirksey Y, Gonzales C, Xu J, Zhou H, Atchison K, Wagner E, Zaleska MM, Das I, Arias RL, Bard J, Riddell D, Gardell SJ, Abou-Gharbia M, Robichaud A, Magolda R, Vlasuk GP, Bjornsson T, Reinhart PH, Pangalos MN. Proc Natl Acad Sci USA. 2008;105:8754–8759. doi: 10.1073/pnas.0710823105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Montine TJ, Neely MD, Quinn JF, Beal MF, Markesbery WR, Roberts LJ, Morrow JD. Free Radic Biol Med. 2002;33:620–626. doi: 10.1016/s0891-5849(02)00807-9. [DOI] [PubMed] [Google Scholar]
- 47.Tamagno E, Bardini P, Obbili A, Vitali A, Borghi R, Zaccheo D, Pronzato MA, Danni O, Smith MA, Perry G, Tabaton M. Neurobiol Dis. 2002;10:279–288. doi: 10.1006/nbdi.2002.0515. [DOI] [PubMed] [Google Scholar]
- 48.Patil S, Sheng L, Masserang A, Chan C. Neurosci Lett. 2006;406:55–59. doi: 10.1016/j.neulet.2006.07.015. [DOI] [PubMed] [Google Scholar]
- 49.Prinzen C, Muller U, Endres K, Fahrenholz F, Postina R. Faseb J. 2005;19:1522–1524. doi: 10.1096/fj.04-3619fje. [DOI] [PubMed] [Google Scholar]
- 50.Obregon DF, Rezai-Zadeh K, Bai Y, Sun N, Hou H, Ehrhart J, Zeng J, Mori T, Arendash GW, Shytle D, Town T, Tan J. J Biol Chem. 2006;281:16419–16427. doi: 10.1074/jbc.M600617200. [DOI] [PubMed] [Google Scholar]
- 51.Kozina LS, Kochkina EG, Nalivaeva NN, Belyaev ND, Turner AJ, Arutjunyan AV. Neurochem J. 2008;2:69–71. [Google Scholar]
- 52.Malik F, Singh J, Khajuria A, Suri KA, Satti NK, Singh S, Kaul MK, Kumar A, Bhatia A, Qazi GN. Life Sci. 2007;80:1525–1538. doi: 10.1016/j.lfs.2007.01.029. [DOI] [PubMed] [Google Scholar]
- 53.Xiong Y, Ding H, Xu M, Gao J. Neurochem Res. 2009;34:746–754. doi: 10.1007/s11064-008-9844-0. [DOI] [PubMed] [Google Scholar]
- 54.Du J, Zhang L, Liu S, Zhang C, Huang X, Li J, Zhao N, Wang Z. Biochem Biophys Res Commun. 2009;383:485–490. doi: 10.1016/j.bbrc.2009.04.047. [DOI] [PubMed] [Google Scholar]
- 55.Buccafusco JJ. Neurotherapeutics. 2009;6:4–13. doi: 10.1016/j.nurt.2008.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kuboyama T, Tohda C, Komatsu K. Br J Pharmacol. 2005;144:961–971. doi: 10.1038/sj.bjp.0706122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Soumyanath A, Zhong YP, Gold SA, Yu X, Koop DR, Bourdette D, Gold BG. J Pharm Pharmacol. 2005;57:1221–1229. doi: 10.1211/jpp.57.9.0018. [DOI] [PubMed] [Google Scholar]
- 58.Chandler LJ, Newsom H, Sumners C, Crews F. J Neurochem. 1993;60:1578–1581. doi: 10.1111/j.1471-4159.1993.tb03326.x. [DOI] [PubMed] [Google Scholar]
- 59.Chen M, Fernandez HL. Biochem Biophys Res Commun. 2004;316:332–340. doi: 10.1016/j.bbrc.2004.02.052. [DOI] [PubMed] [Google Scholar]
- 60.Wen Y, Onyewuchi O, Yang S, Liu R, Simpkins JW. Brain Res. 2004;1009:1–8. doi: 10.1016/j.brainres.2003.09.086. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.