

Abstract
Transglutaminase 2 (TG2) is a multifunctional protein that modulates cell survival and death pathways. It is upregulated in numerous ischemic models, and protects primary neurons from oxygen and glucose deprivation. TG2 binds to the hypoxia inducible factor (HIF) 1β and decreases the upregulation of hypoxic-induced proapoptotic genes. To investigate the role of TG2 in ischemic stroke in vivo, we used the murine, permanent middle cerebral artery (MCA) ligation model. TG2 mRNA levels are increased after MCA ligations, and transgenic mice that express human TG2 in neurons had significantly smaller infarct volumes than wild type littermates. Further, TG2 translocates into the nucleus within 2 hours post ligation. Nuclear-localized TG2 is also apparent in human stroke cases. TG2 suppressed the up regulation of the HIF-induced, proapoptotic gene, Noxa. The findings of this study indicate that TG2 plays a role in attenuating ischemic-induced cell death possibly by modulating hypoxic-induced transcriptional processes.
Keywords: Transglutaminase 2, Stroke, Ischemia, Hypoxia, Hypoxia inducible factor, Neuron, Noxa
Introduction
Stroke is a major cause of adult acquired disability. Inadequate delivery of oxygen and glucose to cerebral tissue initiates numerous processes including excitotoxic and apoptotic cell death, as well as adaptive responses in the penumbra (tissue surrounding the core of the stroke; (Aarts and Tymianski, 2004; Sharp et al., 2000)). Molecular mechanisms in response to stroke that dictate cell survival or eventual death in the penumbra are incompletely understood. It is clear, however, that transcriptional processes play key roles.
A decrease in oxygen delivery activates hypoxic signaling, with hypoxia inducible factors (HIFs; (Wang et al., 1995)) being responsible for the majority of changes in gene expression. The extent and cell type specificity of HIF activation results in alternative outcomes regarding cell fate (Baranova et al., 2007; Chen et al., 2008; Vangeison et al., 2008) with the differential upregulation of both pro-survival and pro-death genes (Aminova et al., 2005; Dayan et al., 2006; Hu et al., 2007). One protein upregulated in ischemia that has previously been shown to regulate cell death and survival pathways is Transglutaminase 2 (TG2) (Ientile et al., 2004; Tolentino et al., 2004). Although the mechanism of TG2 upregulation in hypoxia remain unclear, TG2 has recently been shown to be directly regulated by HIF (Jang et al., 2009). TG2 binds the constitutively expressed subunit of HIF, HIF1β, attenuates HIF signaling, and protects against oxygen and glucose deprivation (OGD)-induced cell death in neurons and human neuroblastoma cells (Filiano et al., 2008). The impact of TG2 in modulating stroke damage has not yet been investigated.
TG2 is a multifunctional protein. In addition to its transamidating activity, TG2 has GTPase, kinase, and protein disulphide isomerase activities, as well as scaffolding/linker properties (Fesus and Piacentini, 2002; Hasegawa et al., 2003; Milakovic et al., 2004; Mishra and Murphy, 2004). The ability of TG2 to interact with numerous proteins may be due to its ability to take on different structural conformations (Liu et al., 2002; Pinkas et al., 2007) and/or its subcellular localization (Gundemir and Johnson, 2009; Milakovic et al., 2004). A primary role of TG2 is modulating transcription by interacting with specific regulatory proteins; especially those affecting cell survival after insults (Ahn et al., 2008; Filiano et al., 2008; Lee et al., 2004).
TG2 belongs to a family of 9 transglutaminases, of which 8 have catalytic activity (Lorand and Graham, 2003). The transamidating reaction catalyzed by transglutaminases can yield protein crosslinks or incorporation of polyamines into proteins. Transglutaminases can also catalyze the deamidation of proteins (Lorand and Graham, 2003). While earlier studies focused on TG2’s transamidating activity, it is now clear that this activity is tightly controlled and predominately inactive (Begg et al., 2006; Siegel et al., 2008) and is not required to protect against OGD-induced cell death (Filiano et al., 2008) or death induced by other stressors (Milakovic et al., 2004).
TG2 is the primary transglutaminase in the brain (Bailey et al., 2004; Kim et al., 1999) where it is predominately expressed in neurons (Kim et al., 1999; Lesort et al., 1999; Perry and Haynes, 1993; Perry et al., 1995). It is largely excluded from the nucleus but translocates there in response to specific stressors (Campisi et al., 2003; Dardik and Inbal, 2006; Filiano et al., 2008; Lesort et al., 1998). Although the mechanisms and outcome of this translocation have not been elucidated, it puts TG2 in proximity to interact with components of transcriptional machinery.
In this study, we show that exogenous expression of human TG2 (hTG2) in mouse neurons results in significantly smaller infarct volumes after middle cerebral artery (MCA) ligation. Within hours post ligation, TG2 translocates to the nucleus of neurons. Further, nuclear localization of TG2 was observed in the area of stroke in human post-mortem tissue. TG2 binds HIF1β in vivo and attenuates the expression of HIF-dependent genes that are upregulated after MCA occlusion. These findings and our previous report (Filiano et al., 2008) indicate that TG2 protects against stroke damage by modulating hypoxia-mediated transcriptional events, possibly by attenuating HIF activation of pro-cell death genes.
Materials and Methods
Latex perfusions
Latex perfusions were modified from a previously reported study (Maeda et al., 1998). C57BL/6 mice were housed in microisolators and all animal studies were performed in accordance with ARP and UCAR-approved protocols. Mice were anesthetized with Nembutol (90mg/kg i.p.; Ovation Pharmaceuticals, Inc.) and given a lethal dose of papaverine (i.v. 50mg/kg; Sigma) in neutral buffered saline. An incision was made to expose the abdominal thoracic aorta. The descending aorta was cleaned of all fat and connective tissue under a dissecting microscope. Using a small 22G catheter (Terumo), the aorta was cannulated and tied in with 5-0 monofilament sutures (Ethicon). Warm latex (Chicago Latex Products; no. 563) was colored with Coomassie Brillant blue and maintained at 37°C. After cannulation, 2mls of pre-warmed latex was slowly injected and the mouse was immediately chilled on ice until the latex settled. Mice were decapitated and brains removed and post fixed overnight in 4% paraformaldehyde (PFA). Brains were placed in a mount to maintain appropriate perspectives and photographs were taken through a low power dissection microscope with a camera. Angles between MCAs and anterior cerebral arteries were measured using Image J software (NIH). It should be noted that these flat field images could not take into account the effect of the curvature of the brain.
Magnetic resonance imaging
Magnetic resonance imaging (MRI) investigation was performed using a horizontal Bruker Biospect 94/20 9.4T MR scanner (Billerica, MA, USA) equipped with a BGA12 gradient insert (max strength = 400 mT/m, Ø= 120 mm) employing a 72 mm resonator for radio-wave transmitting and an active-decoupled 20 mm surface coil for signal receiving. The mice were anesthetized by introduction of 5% isoflurane in O2 gas and placed in an animal bed equipped with circulating warm water to maintain body temperature at 37±0.5 °C. Anesthesia was maintained by continually delivering the mixed gas at 1.5-2% via a nose cone during MRI scans. T2-weighted MR images were acquired using spin echo sequence (RARE) with the following acquisition parameters: TR/TE = 5000/60 ms, 20 continuous slices with 0.5mm thickness which covered the whole stroke area, and FOV 25×25mm and matrix size of 256×256 to yield 0.098×0.098 mm in-plane resolution. The total acquisition time was approximately 11 minutes. Stroke infarct volumes were calculated from image stacks using Image J software.
3D MR angiographic imaging was performed using a 3D FLASH sequence without any contrast agent and the following imaging parameters: TR = 12 msec, TE = 2 msec, flip angle = 20°, NA (number of average) = 4, field of view (FOV) = 12×16×12mm, matrix size = 128 × 128 × 96, and acquisition time = 10 minutes.
Middle cerebral artery ligations
All mice used were 10-12 week old males that averaged approximately 26g in weight. The hTG2 mice were all hemizygous and have been described previously (Tucholski et al., 2006). No differences in weight were observed between wild type and transgenic hTG2 mice (data not shown). Mice were anesthetized by oxygen gas passed through an isoflurane vaporizer set to deliver 1.5%-2% isoflurane. The surgical area was shaved and prepared with alternating betadine scrub and ethanol. A small 5 mm vertical skin incision was cut between the left eye and ear to expose the skull. The temporal muscle was retracted and a small hole carefully removed with a 19 gauge needle at the junction of the zygomatic arch and squamosal bone. Using a dissecting microscope, further craniectomy with small bone rongeurs exposed the middle cerebral artery which was cauterized just distal of the lenticulostriate arteries. The incision was closed with sterile nylon monofilament sutures. Buprenorphine was used at 0.05mg/kg, im or sc prior to surgery, and then every 12 hours for two more doses. Body temperature was maintained at 37 °C with a heating pad during surgery (approx. 30 minutes) and recovery. For visualization of hypoxic tissue, hypoxyprobe (60mg/kg; Natural Pharmacia International) was injected i.p. just prior to anesthesia. Twenty four hours after surgery, brains were removed then processed for immunocytochemistry as described below and tissue stained with MAb1 Hypoxyprobe-1 (1:50; Natural Pharmacia International).
Immunohistochemistry
Human brain sections from stroke cases were obtained from the Neuropathology core at the University of Rochester Medical Center. All studies on human tissue were reviewed by the IRB and qualified for exemption. Tissue for these studies was removed from unaffected brain areas, as well as from the infarcted area 11-27 hours post-mortem. The tissue was fixed for 10-14 hours in neutral buffered formalin; paraffin embedded, and cut into 4 μm sections. Paraffin was removed with Histoclear solution (Electron Microscopy Science) and the tissue rehydrated with isopropanol followed by boiling in citrate buffer for antigen retrieval. Slides were blocked in phosphate buffered saline (PBS) with 1% BSA and 0.2% non-fat dry milk for 1 hour before an overnight incubation in primary antibody. The dilution of antibodies was as followed: NSE (1:50, Abcam ab53025 rabbit polyclonal) and TG 4C1 (1:250, Iowa Developmental Studies Hybridoma Bank mouse monoclonal). Sections were subsequently incubated with fluorescent conjugated antibodies (1:1000; Invitrogen) and then for 5 minutes with DAPI (1μM) in PBS to stain nuclei. To eliminate autofluorescence of lipofuschin granules, slides were incubated with 0.3% Sudan Black in 70% ethanol for 5 minutes after incubation with the secondary antibody. Remnant Sudan Black was removed by washing slides in 70% ethanol followed by a PBS wash. Using Immunomount (Thermo Shandon), coverglass was mounted on the slides and immunofluorescence was viewed using an Axiovert inverted microscope at 40X with attached Axiocam-XMR camera (Zeiss).
For immunohistochemical analyses of the mouse brains, mice were injected with a lethal dose of Nembutol (100mg/kg i.p.) and subjected to trans-cardial perfusion with PBS followed with 4% PFA. Brains were removed and submersed in 4% PFA overnight. Brains were paraffin embedded and 7 μm sections were cut then processed and stained as above (TG2 Ab-1 1:500, Thermo Fisher Scientific). Nuclear quantification of hTG2 was performed with Image J software. Autothresholding of hTG2 and DAPI images were converted to binary, and overlapping pixels were counted and expressed as pixels per cell in the field. Microvasculature was analyzed with a modified, previously described technique (Hohenstein et al., 2005). Briefly, Deparaffinized slides were washed with PBS and incubated with fluorescein labeled Lycopersicon Esculentum (Tomato) Lectin (250μg/ml in PBS) for 1 hour at 37°C. Slides were then washed, mounted, and imaged as described above.
Co-immunoprecipitation
Mice were injected with a lethal dose of Nembutol 5 hours, 1 day, and 5 days post MCA ligations and brains were collected. Brains were quickly dissected on ice and cerebellum and olfactory bulbs were removed. Parallel cuts were made 2mm from the midline for collection of “at risk” (ipsilateral) and contra-lateral cortical tissue. Tissue was homogenized in IP Lysis Buffer [10 mM Tris pH 7.5, 10 mM NaCl, 3 mM MgCl2, 1 mM EGTA and 0.05% (v/v) NP-40] supplemented with the following protease inhibitors: 2 mM phenylmethylsulfonyl fluoride (PMSF), 20 μg/ml leupeptin, 20 μg/ml pepstatin and 10 μg/ml aprotinin. Lysates were sonicated and spun at 16000 g for 15 minutes. Supernatants of the soluble protein were collected and protein concentrations were determined by the bicinchoninic acid assay (Pierce). For immunoprecipitation, Dynabeads® M-280 magnetic beads that were coupled with sheep anti-mouse IgG antibodies (Invitrogen) were washed three times in Wash Buffer [PBS with 0.1% (w/v) BSA] and then incubated overnight with 2 μg of anti-HIF1β (Novus) antibody at 4°C with constant agitation. Beads were washed three times with Wash Buffer and incubated with 200 μg of brain lysate for immunoprecipitation of HIF1β for 3 hours at 4 °C with constant agitation. Beads were washed three times in Wash Buffer and bound protein was collected into 2X SDS Stop Buffer by boiling as described below. For immunoblot analysis, protein was diluted into 2X SDS Stop Buffer, boiled and electrophoresed on 8% SDS-polyacrylamide gels. Electrophoresed protein was then transferred to nitrocellulose membranes and nonspecific sites were blocked by incubating membranes in Blocking Solution [5% (w/v) skim milk powder in Tris-buffered saline containing Tween 20 (TBS-T) [20 mM Tris pH 7.5, 137 mM NaCl and 0.05% (v/v) Tween 20]] for 1 hour at room temperature. Membranes were washed with TBS-T and incubated with primary antibodies for hTG2 (1:5000; TG100) or HIF1β (1:2000; Novus) in Blocking Solution overnight at 4 °C. Membranes were then washed with TBS-T and incubated with a goat anti-mouse IgG (H+L) secondary antibody conjugated with horseradish peroxidase (HRP) (Bio-Rad) (1:2500) in Blocking Solution for 1 hour at 23 °C. Membranes were washed with TBS-T and developed by chemiluminescence following 1 minute incubation in Developing Buffer [48.3 mM Tris pH 8.6, 681 μM luminol, 32.3 μM para-coumeric acid and 0.0045% (v/v) H2O2]. Immunoblots that were reprobed were first incubated for 15 minutes in Stripping Buffer [62.5 mM Tris pH 6.8, 100 mM 2-mercaptoethanol and 2% (w/v) SDS] at 55 °C. Membranes were washed with TBS-T and then reprobed as described above starting at the blocking step.
RNA collection and quantitative real-time PCR (Q-PCR)
RNA was collected by adding 10X Trizol reagent (Invitrogen,) to tissue in cold PBS and homogenized. After isopropanol precipitation, RNA was treated with DNase I (Invitrogen) to eliminate any contamination of genomic DNA. RNA concentrations were measured and cDNAs were created by using SuperScript III Reverse Transcriptase and Random Primers (Invitrogen). cDNAs were diluted 1:10 and 2μl per well in a 96 well plate format were then used for Q-PCR with TG2 (Mm_00436980), Vegfa (Mm_01281449), Noxa (Mm_00451763), Bnip3 (Mm01275601), and PAI-1 (Mm_00435860) pre-made primer set and data was normalized to actin (Mm_02619580; Applied Biosystems). Amplification was performed using an ABI Prism 7300 Sequence Detection System (Applied Biosystems) at 50°C for 2 minutes, 95°C for 10 minutes, followed by 40 cycles of a 15-sec step at 95°C and a 1-minute step at 60°C. Data was analyzed using the ΔΔCt method as previously described (User Bulletin No. 2, Applied Biosystems).
Statistical analysis
Data from infarct studies are represented as volumes ± SD from 7 WT mice and 10 hTG2 mice. Statistical comparisons were done using a two-tailed unpaired t-test. The data from MCA angles and microvasculature analysis are represented as average from 4 and 3 (respectively) animals ± SD and also compared using a two-tailed unpaired t-test. Nuclear quatification of TG2 was analyze using a one-way analysis of variance (ANOVA) followed by Bonferroni posttest at each time point. For gene expression studies, statistical comparisons were conducted between wild type and TG2 expressing mice with MCA ligations using a two-way (ANOVA) followed by Bonferroni posttest at each time point. Significance was determined if p<0.05.
Results
Infarct volumes are smaller in transgenic mice over-expressing human TG2 in neurons
To assess the molecular events that occur subsequent to an ischemic insult, we used the permanent MCA ligation model in C57BL/6 mice as described by Tamura and colleagues (Tamura et al., 1981). This results in consistent infarct volumes in the frontal and parietal cortex. The role of TG2 in ischemic stroke was evaluated in hemizygous transgenic mice overexpressing hTG2 under a prion promoter (Tucholski et al., 2006), and wild type littermate controls. The approximate location of the MCA ligation is indicated by the black bar in the image of the brain with latex perfused arteries shown in Figure 1A. Demonstration of disrupted blood flow after the permanent occlusion can be observed in the MR angiograph (bottom panel, white arrow; Fig. 1B). The top panel in Figure 1B is the same mouse 24 hours prior to ligation. Hypoxic infarct zones are distinct and confined to the frontal/parietal cortex with low variability between surgeries. Hypoxic tissue was identified by immunohistochemical staining of Hypoxyprobe-1 adducts formed in low oxygen (Fig. 1C). After MCA ligations, mice were allowed to recover for 24 hours then sequential (every 500 μm) T2 weighted MR images were acquired encompassing the entire infarct. An individual image is shown in Figure 1C. The yellow box corresponds to the area used for identification of hypoxic tissue (above). Upon volumetric analysis of infarct volumes, mice overexpressing hTG2 had infarct sizes of 21.7 mm3± 6.07 compared to 32.9 mm3± 5.84 (p < 0.005) in wild type controls (Fig. 1D). T2 weighted signal declines over time (3 and 5 days), however hTG2 mice always presented with smaller infarcts than littermate controls (data not shown).
Fig. 1.

Expression of hTG2 in mouse neurons reduces stroke volume after permanent MCA ligation. Permanent MCA ligation was performed on C57BL/6 mice at 10-12 weeks of age. A. Mouse brain macrovasculature is visualized using blue latex. The approximate location of MCA cauterization is represented by the black bar. B. Magnetic Resonance Angiograms of C57BL/6 pre (top) and 24 hours post MCA ligation (bottom). The position of the ligation of the MCA is indicated by white arrow. The absence of signal is due to an interruption of blood flow. C. Permanent ligation of the MCA results in a hypoxic/ischemic infarct that is restricted to the mouse cortex. Hypoxic area is visualized with an antibody that recognizes the chemical adducts formed in low oxygen with Hypoxyprobe, which is injected i.p. 30 minutes prior to ligation (left). The yellow box in the MRI scan represents area of histology. Infarcted tissue is recognized by increased T2 weighted MRI signal 24 hours post surgery on representative sections taken every 500μM throughout the mouse cortex (right). D. Calculated infarct volumes in hTG2 mice (21.70 mm3± 6.07; n=7) are 33% smaller than wild-type littermates (32.90 mm3± 5.84; n=10), p < 0.005.
hTG2 mice show no differences in brain vasculature
Although hTG2 expression is driven by the mouse prion promoter and almost exclusively expressed in neurons (Tucholski et al., 2006), it is important to determine if hTG2 expression has an effect on brain vasculature which may contribute to the reduction of stroke volumes. Evaluation of the vasculature is also important because TG2 can modulate HIF signaling (Filiano et al., 2008), and HIF signaling plays a pivotal role in angiogenesis and vascular development (Fong, 2008). Cerebral artery branches were visualized by perfusion of blue latex into the thoracic descending aorta (Fig. 2). No structural differences in macrovasculature were observed in the brains of mice overexpressing hTG2 compared to wild type mice. Angles between the MCA and anterior cerebral arteries were similar in wild type mice (86.5° ± 5.42) and hTG2 mice (85.1° ± 2.67; p=0.823; n=4 (Fig. 2). In both cohorts of mice, the posterior communicating artery was poorly developed or absent, but no differences between lines were observed. These data were expected, as the expression of hTG2 is driven by the prion promoter, and therefore predominantly neuronal (Tucholski et al., 2006).
Fig. 2.

Brain macrovasculature is not affected by neuronal expression of TG2. The primary vasculature of the brain of wild type (WT) and mice expressing hTG2 in neurons was labeled with blue latex. Angles between the MCA and horizontal (A1) segment of anterior cerebral artery were calculated using Image J. No differences in MCA branching angles from the Circle of Willis of WT and hTG2 mice were observed when calculated. p=0.8233; n=4.
The microvasculature was also analyzed by labeling 5 brain sections per mouse corresponding to approximately +1.95, +0.74, −0.94, −2.12, −3.80mm from bregma from hTG2 and wild type mice with fluorescein-conjugated lectin (Fig. 3). The average pixel densities per slice were calculated then averaged for hTG2 expressing (83.6 ±4.04) and wild type mice (80.9 ±4.41). No significant differences were observed (p=0.672; n=3; Fig. 3).
Fig. 3.

hTG2 overexpression does not affect mouse brain microvasculature. Microvasculature of the mouse brain was visualized by staining with Fluorescein-conjugated lectin. Representative photomicrographs are shown, wild type (WT) (top left) and hTG2 (top right). Microvasculature from 5 sections per brain was quantified and represented as pixels/brain section. No difference in staining was observed between WT and hTG2. p=0.6715; n=3 mice per group.
hTG2 translocates to the nucleus early after MCA ligations
Based on our recent findings that in response to OGD, TG2 translocates to the nucleus in a neuroblastoma cell line and suppresses HIF signaling (Filiano et al., 2008), we next investigated TG2 localization in the mouse post MCA ligation. Brains from hTG2 mice were collected 2 and 5 hours post surgery and subsequently prepared for immunohistochemical analysis. In the contra-lateral cortex (not shown) and sham-operated mice (Fig. 4A), hTG2 was exclusively detected in the cytoplasm of virtually all neurons (the sham surgery shown is at 5 hours post surgery). Two hours post MCA ligation, neuronal hTG2 staining was slightly elevated (Fig. 4B). Five hours after surgery this staining pattern was even more pronounced as TG2 increasingly localized to the nucleus (Fig. 4C). After 24 hours, although there was significant tissue loss in the infarcted area, virtually all remaining neurons presented with TG2 localized exclusively to the nucleus (data not shown). Quantification of images shows the nuclear localization of hTG2 to the nucleus increases over time suggesting hTG2 translocates to the nucleus in response to ischemia (Fig. 4D). Immunohistochemical identification of endogenous mouse TG2, unfortunately, could not be performed due to the fact that all available antibodies recognize a non-specific neuronal epitope in mouse brain (e.g., see (Bailey et al., 2004)).
Fig. 4.

TG2 translocates to the nucleus following permanent MCA ligation. After sham surgeries, or 2 or 5 hours post permanent MCA ligation, the localization of hTG2 was visualized by immunohistochemistry. A. In sham surgeries (n=8), hTG2 is completely excluded from the nucleus (DAPI). This can be seen when the images are merged (A, far right). B. At 2 hours post MCA ligation (n=8), hTG2 colocalizes with numerous neuronal nuclei stained with DAPI. C. hTG2 translocation into the nucleus is observed in almost all neuron in the region of the infarct at 5 hours post ligation (n=6). D. Images were quantified and data is represented as TG2 and DAPI co-localizing pixels per cell ± SEM. *p<0.05
TG2 is localized to the nucleus in human stroke
Given the fact that hTG2 translocates to the nucleus in a murine model of stroke and that there is increasing evidence that TG2 plays a role in regulating transcription processes, we examined the neuronal distribution of TG2 in human ischemic tissue. For these studies post-mortem human brain sections obtained, both from the region of ischemia and from unaffected regions, were immunostained for TG2. As expected, TG2 in unaffected brain tissue showed primarily cytoplasmic staining with a distribution pattern similar to neuronal specific enolase (NSE; Fig. 5). In contrast, in the peri-infarct zone, neuronal TG2 was predominantly nuclear and co-localized with DAPI staining (Fig. 5), with the distribution of NSE being the same as in the unaffected areas. The images shown in Figure 5 are representative of the TG2 staining pattern observed in 4 out of the 5 cases analyzed (data not shown). It should be noted that TG2 is also in endothelial cells (resulting in staining of the vasculature), as well as in other non-neuronal cells in the brain which accounts for the TG2 staining pattern as previously described (Wilhelmus et al., 2009).
Fig. 5.

TG2 localizes to the nucleus of neurons after human stroke. Post mortem tissue from a human stroke case was immunostained for neuronal specific enolase (NSE) and TG2. In unaffected tissue, TG2 staining colocalized completely with NSE (top, yellow asterisks), suggesting that TG2 is expressed throughout neurons in human brain. In stroke tissue, TG2 colocalized with DAPI, yet NSE remained express in the entire neuronal body (middle, yellow asterisks). Higher magnifications of representative neurons are outlined in yellow and shown at the bottom. For clarity purposes, only TG2 (red) and DAPI (blue) are in included in the merged images.
Endogenous TG2 is upregulated after MCA ligation
WT mouse brain mRNA was isolated from the cortex containing the MCA ligation as well as contra-lateral cortex and analyzed by Q-PCR for TG2 transcripts (Fig. 6). All data are internally normalized to actin and represented as fold change relative to a sham control. Analysis of data shows that endogenous TG2 increases after MCA ligation (p<0.05) and with time (p<0.05). At 2 hours post ligation, TG2 transcripts were slightly decreased relative to contra-lateral cortex. Starting at 5 hours, TG2 mRNA level increased and peaked at 24 hours post surgery (*p<0.01).
Fig. 6.

Endogenous TG2 is upregulated after permanent MCA ligation. Mouse brain mRNA was collected 2 hours to 5 days post MCA ligation in WT mice. Ipsilateral and contralateral cortex was dissected and mRNA was isolated. Total mRNA was reverse transcribed and cDNA for TG2 was analyzed with quantitative real-time PCR. A. TG2 mRNA was normalized to actin and expressed relative to sham control, which is defined as 1. *p<0.05; B. data are show normalized to actin and expressed relative to contralateral cortex (n=4-7).
TG2 coimmunoprecipitates with HIF1β in mouse brain
We previously identified HIF1β as a binding protein for TG2 in human neuroblastoma SH-SY5Y cells, and a decrease in HIF signaling in response to hypoxia as measured using a synthetic reporter (Filiano et al., 2008). To determine whether TG2 bound HIF1β in vivo, a co-immunoprecipitation experiment was performed with hTG2 mouse brain lysate. Immunoprecipitation of HIF1β revealed that that hTG2 co-immunoprecipitates with HIF1β in lysates from sham surgery and after MCA ligation (Fig. 7). No differences in binding were observed in the two conditions.
Fig. 7.
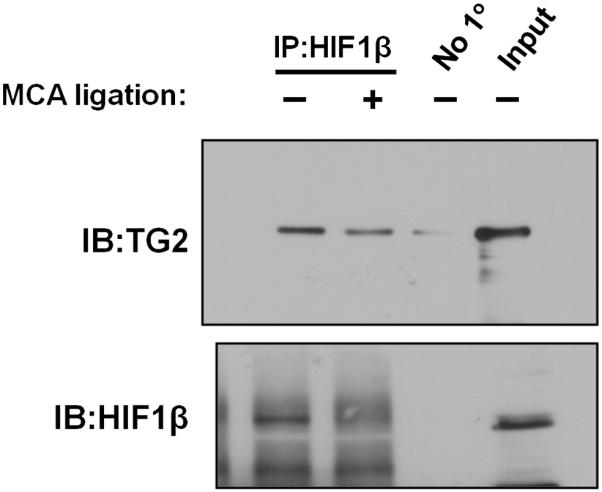
TG2 co-immunoprecipitates with HIF1β in mouse brain. Protein samples of mouse brain cortical hemispheres, with and without MCA ligation from 2 independent experiments were immunoprecipitated for HIF1β and immunoblotted for both HIF1β and TG2. The amount of TG2 that pulled out with HIF1β correlated with the amount of HIF1β initially immunoprecipitated. A control immunoprecipitation where the immunoprecipitating antibody was omitted is shown in lane 3.
hTG2 differentially regulates the expression of HIF responsive genes
We previously demonstrated that in a cell model increased expression of TG2 attenuated hypoxia-induced expression of Bnip3. To extend these studies, we analyzed expression of several HIF responsive genes post MCA ligation in mice overexpressing hTG2 and wild type mice. Gene expression was assessed 2, 5, 24, and 120 hours after ligation (Fig. 8). Using a two-way ANOVA to evaluate the effects of time and overexpression of hTG2 on gene expression post MCA ligation, we analyzed 4 known HIF controlled transcripts. 3 out of 4 HIF controlled genes analyzed were up regulated after MCA ligation in wild type mice. There was a significant upregulation in Vegf gene expression with time post-MCA ligation (p<0.005). Vegf was upregulated at 2 hours after MCA ligation and decreased back to baseline levels at 120 hours. When comparing mice that overexpress hTG2 to littermates, there was significantly less upregulation of Vegf mRNA (p<0.05). Noxa, a pro-apoptotic HIF dependent gene (Kim et al., 2004), was upregulated and peaked at 5 hours post MCA ligation in wild type mice. This upregulation was significantly inhibited in hTG2 mice (two-way ANOVA (p<0.05) using a post-hoc test (*p<0.05)). Immunoblotting revealed that Noxa protein levels were also lower in hTG2 mice compared to wild type mice 24 hours after stroke, although these differences did not reach significance (data not shown). This is not surprising given that Noxa is unstable and is rapidly turned over (Hagenbuchner et al, 2010). PAI-1 was activated in wild type mice and there was a slight yet not significant attenuation in mice overexpressing hTG2 (p=0.14). Bnip3 mRNA levels were not elevated in either cohort.
Fig. 8.

TG2 attenuates the upregulation of HIF dependent genes after MCA ligations. Mice were subjected to MCA ligation and sacrificed 2 hours to 5 days post surgery. Data were normalized to β-actin and expressed relative to contralateral cortex. Expression of HIF dependent genes from WT and hTG2 mice are expressed as mean ± SEM from at least three mice per group. Two-way ANOVAs show significant effects of hTG2 on Vegf and Noxa expression (p<0.05) and an effect of time on Vegf expression (p<0.005) post MCA ligation. Asterisks denote a significant difference between WT and hTG2 mice at 5 hours (Bonferroni posttest, p < 0.05).
Discussion
In this study we demonstrate for the first time that increased neuronal expression of TG2 in the mouse brain protects against stroke damage. In response to stroke, TG2 mRNA was upregulated in WT mice. Nuclear translocation of hTG2 takes place within hours of MCA occlusion in the mouse permanent MCA ligation model, and this nuclear localization of TG2 was also observed in human stroke cases. TG2 represses HIF dependent genes, including Noxa, that are upregulated after MCA ligation and possibly maintains HIF activation at beneficial levels. These findings suggest that TG2 can affect signaling processes that result in neuronal protection against ischemic insult. We postulate that translocation of TG2 from the cytoplasm to the nucleus during in vivo ischemic insult to the brain may be essential to its protective role through regulation of transcriptional processes.
TG2 is predominately cytosolic but can translocate to the nucleus under certain conditions. By fractionating human neuroblastoma SH-SY5Y cells, it was found that ~7% of TG2 is localized to the nucleus under resting conditions (Lesort et al., 1998). However, increasing intracellular calcium concentrations led to a distinct nuclear translocation. TG2 is also upregulated and present in nuclei of astrocyte cultures exposed to glutamate (Campisi et al., 2003). The direct mechanisms of TG2 nuclear translocation are currently unknown. It is suggested that the interaction of TG2 with the nuclear transport protein, importin α-3 may be involved (Peng et al., 1999). Additionally, TG2 forms a complex with nucleoporin p62 (Singh et al., 1995). TG2 may also be shuttled into the nucleus by direct interactions with other proteins. We have previously shown that TG2 bind HIF1β and they co-immunoprecipitate in mouse brain. This interaction with HIF1β (Filiano et al., 2008), as well as other transcription factors such as c-Jun (Ahn et al., 2008), may also facilitate the nuclear translocation of TG2 in hypoxia/ischemia.
Evaluation of the role of TG2 in ischemia was performed using the permanent MCA occlusion model in mice overexpressing hTG2 in neurons. The permanent occlusion of the mouse MCA results in a distinct zone of ischemia in the cortex that is ideal for investigating the initial molecular mechanisms of TG2 in ischemia. Twenty-four hours after occlusion, infarcts in transgenic mice overexpressing hTG2 in neurons were 33% less in volume than wild type control mice when analyzed using T2 weighted MRI. Nuclear translocation of hTG2 was also observed as early as 2 hours post insult. This translocation was increasingly evident at 5 hours and all remaining neuronal cells contained nuclear hTG2 24 hours post stroke (data not shown). It would be extremely beneficial to investigate endogenous mouse TG2 in ischemia, but unfortunately all TG2 antibodies, to our knowledge, bind a non specific epitope in mouse neurons and cannot be used for immunohistochemistry (Bailey et al., 2004).
Even though hTG2 was expressed under the mouse prion promoter, which leads to predominately neuronal expression (Tucholski et al., 2006), it was important to rule out the possibility that hTG2 was attenuating stroke damage by altering the brain vasculature. Using latex perfusions and microvasculature staining, we revealed no discernible differences in the brain vasculature of hTG2 mice when compared with wild type mice. We conclude that decreased infarct volumes were not due to smaller MCA vascular beds but intracellular protective mechanisms.
Immunohistochemistry of TG2 after stroke in human post-mortem tissue parallels the mouse pathology. In human brain, TG2 is predominately excluded from the nucleus in neurons. However, when sections of post-mortem ischemic tissue were analyzed, TG2 was located in neuronal nuclei. This is intriguing given the fact that the ischemic insult occurred at least several days or more prior to tissue collection. It is possible that TG2 shuttles in the nucleus post stroke to limit infarct progression by regulating nuclear signaling events in ischemia.
There is an emerging appreciation that TG2 functions to modulate a number of transcriptional pathways. TG2 can form polymers of inhibitor of nuclear factor (NF)-κB (IκB) leading to increased NF-κB activation. The effect of NF-κB signaling in ischemia remains controversial but activation of NF-κB has shown to be neuronal protective in MCA occlusions (Li et al., 2008; Valerio et al., 2009). TG2 immunoprecipitates with c-Jun and can interfere with its interaction with c-fos and decrease c-Jun binding to AP-1 binding sites. This leads to down regulation of matrix metalloproteinase-9 (MMP-9) (Ahn et al., 2008). MMP-9 degrades the basal lamina in cerebral ischemia (Rosenberg et al., 1992). Although this is an attractive mechanism, we see no differences in Evan’s Blue extravasation in transgenic hTG2 mice compared to wildtype mice post stroke (data not shown). Increased cAMP levels upon adenylate cyclase activation leading to an augmented CREB signaling response was observed in SH-SY5Y cells that overexpress TG2 (Tucholski and Johnson, 2003). This could lead to neuronal protection and increasing levels of BDNF production in response to cerebral ischemia (Blanquet et al., 2006; Sonmez et al., 2007). TG2 can also modulate retinoblastoma protein (pRb) to protect cells against apoptosis (Boehm et al., 2002). Interestingly, a nuclear targeted, transamidating inactive TG2 mutant binds pRB and protects cells against intracellular calcium overloads (Milakovic et al., 2004). Additionally, ATP levels in hearts from TG2 -/- mice were 40% lower than wild type control (Szondy et al., 2006). The results of these last two studies may indicate that TG2 functions regulate mitochondrial genes needed to maintain ATP levels in the penumbra. In fact, cells overexpressing TG2 have higher mitochondria membrane potentials (Piacentini et al., 2002) that may lead to greater amounts of ATP generated. It is clear that TG2 can affect transcriptional responses in many paradigms.
HIF has been shown to be the major transcriptional regulator in hypoxia. Activation of HIF results in the up regulation of genes needed for adaptation and cell survival (Guo et al., 2001; Sharp and Bernaudin, 2004; Tang et al., 2006). In addition to this function, recent data has shown that HIF also regulates a subset of proapoptotic genes including Bnip3, NIX, Noxa, and procaspase-3 (Baranova et al., 2007; Van Hoecke et al., 2007) and may have injurious effects upon activation. Neuronal cultures that express a dominant negative form of HIF1α, the oxygen dependent subunit of HIF, are protected against delayed hypoxic cell death (Halterman et al., 1999). HIF1α co-localizes with activated caspase-3 in ischemic infarcts and HIF1α binds to the procaspase-3 promoter following photothrombic cortical occlusion (Van Hoecke et al., 2007). Bilateral occlusion of the common carotid arteries in a conditional HIF1α knock out under a CaMKII-α-Cre neuronal specific promoter results in less cell neuronal death in the hippocampus compared to wild type mice (Helton et al., 2005). However, in another study, the same transgenic mice show an increase in cell death and reduced overall survival after transient focal cerebral ischemia (Baranova et al., 2007). In this study, a biphasic upregulation of HIF dependent genes is described. Q-PCR of specific HIF genes reveals an upregulation of proapoptotic genes 5 hours after transient MCA occlusion, and an additional upregulation of prosurvival genes at day 5 (Baranova et al., 2007). Inactivation of HIF1α using 2-methoxyestradiol or shRNA decreased infarct sizes in neonates exposed to transient unilateral carotid artery ligation (Chen et al., 2009; Chen et al., 2008). These reports suggest that there is more to HIF signaling than just activation under certain oxygen concentrations. HIF signaling needs to be differentially regulated in different tissues to control the severity of the response. In fact, differences in neuronal cell survival can be observed by differentially knocking out HIF1α in neurons or astrocytes in co-cultures (Vangeison et al., 2008). The complexity of HIF signaling is apparent in light of these studies and may not be resolved by complete inhibition of the HIF signaling pathway.
These major discrepancies in the role of HIF in ischemia are possibly due to the strength and duration of HIF activation, as well as the differential activation of HIF isoforms, such as HIF-2α. The transactivation domains of HIF 1 and 2α contribute to the differential specificity of each isoform’s target genes (Hu et al., 2007). This regulation at the transcriptional complex relies on specific co-activators (i.e. CBP/p300) that result in differential activation of specific HIF dependent genes (Kasper et al., 2005; Kasper and Brindle, 2006). TG2 attenuates HIF signaling in the human renal cell carcinoma cell line, 786-0 (Filiano et al., 2008). These cells have a mutation in VHL and rely primarily on a constitutively active HIF2α (Maxwell et al., 1999; Warnecke et al., 2004) and hence it is likely that TG2 attenuates signal of both orthologs. Although, HIF1α and HIF2α can bind identical hypoxic response elements therefore activating synthetic luciferase reports, they regulate distinct subsets of genes via their transactivation domains (Hu et al., 2007). Further studies may reveal that TG2 modulates the HIF orthologs distinctively. CBP/p300 are thought to be mandatory coactivators for HIF, yet many HIF genes are not affected by mutations of CBP/p300 (Kasper and Brindle, 2006). We have previously shown that overexpression of TG2 in a neuroblastoma, SH-SY5Y, cell line and in rat primary cortical neurons protects against oxygen and glucose deprivation (Filiano et al., 2008). TG2 binds to HIF1β in numerous assays and functions to attenuate HIF signaling in hypoxia. It is currently unknown how TG2 functions to decrease HIF activation without interrupting the formation of the HIF heterodimer, inhibiting nuclear translocation, or obstructing HIF from binding to the hypoxic response element. We hypothesize that TG2 may modulate the interactions between HIF and transcriptional activators. Furthermore, TG2 reduces the transcription of numerous proapoptotic genes in hypoxia. The mechanism by which TG2 attenuates HIF signaling has yet to be elucidated but is crucial to understanding how TG2 protects neurons against ischemia. Nonetheless, in the initial stages of ischemia TG2 translocates to the nucleus and remains there, most likely interacting with components of transcriptional machinery. This translocation is observed in humans and mice upon ischemic injury to the brain. All of these data strongly suggest that hTG2 regulates hypoxic signaling pathways in neurons and contributes to protection following cerebral ischemia.
Acknowledgements
The authors would like to thank Dr. Mahlon Johnson and The University of Rochester, Neuropathology core for supplying human stroke tissue and Linda Johnstone for paraffin embedding and slicing of mouse brains for immunohistochemistry. The surgical technique was mastered in the lab of Dr. W.D. Hill at the Medical College of Georgia. All MRI was performed with the assistance of Dr. Huadong Zeng at the University of Alabama at Birmingham. The authors would also like to acknowledge the labs of Drs. David Rempe and Bill Bowers, for the use of the ABI Thermocycler and dissection camera. This research was supported by NIH AG012396, NS051279, F31 NS064700 and AHA 30815697D.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aarts MM, Tymianski M. Molecular mechanisms underlying specificity of excitotoxic signaling in neurons. Curr Mol Med. 2004;4:137–47. doi: 10.2174/1566524043479202. [DOI] [PubMed] [Google Scholar]
- Ahn JS, et al. Tissue transglutaminase-induced down-regulation of matrix metalloproteinase-9. Biochem Biophys Res Commun. 2008;376:743–7. doi: 10.1016/j.bbrc.2008.09.048. [DOI] [PubMed] [Google Scholar]
- Aminova LR, et al. Prosurvival and prodeath effects of hypoxia-inducible factor-1alpha stabilization in a murine hippocampal cell line. J Biol Chem. 2005;280:3996–4003. doi: 10.1074/jbc.M409223200. [DOI] [PubMed] [Google Scholar]
- Bailey CD, et al. Validity of mouse models for the study of tissue transglutaminase in neurodegenerative diseases. Mol Cell Neurosci. 2004;25:493–503. doi: 10.1016/j.mcn.2003.11.016. [DOI] [PubMed] [Google Scholar]
- Baranova O, et al. Neuron-specific inactivation of the hypoxia inducible factor 1 α increases brain injury in a mouse model of transient focal cerebral ischemia. J Neurosci. 2007;27:6320–32. doi: 10.1523/JNEUROSCI.0449-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begg GE, et al. Mechanism of allosteric regulation of transglutaminase 2 by GTP. Proc Natl Acad Sci U S A. 2006;103:19683–8. doi: 10.1073/pnas.0609283103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanquet PR, et al. Identification of a biphasic signaling pathway involved in ischemic resistance of the hippocampal dentate gyrus. Exp Neurol. 2006;202:357–72. doi: 10.1016/j.expneurol.2006.06.016. [DOI] [PubMed] [Google Scholar]
- Boehm JE, et al. Tissue transglutaminase protects against apoptosis by modifying the tumor suppressor protein p110 Rb. J Biol Chem. 2002;277:20127–30. doi: 10.1074/jbc.C200147200. [DOI] [PubMed] [Google Scholar]
- Campisi A, et al. Glutamate-induced increases in transglutaminase activity in primary cultures of astroglial cells. Brain Res. 2003;978:24–30. doi: 10.1016/s0006-8993(03)02725-2. [DOI] [PubMed] [Google Scholar]
- Chen C, et al. Early inhibition of HIF-1α with small interfering RNA reduces ischemic-reperfused brain injury in rats. Neurobiol Dis. 2009;33:509–17. doi: 10.1016/j.nbd.2008.12.010. [DOI] [PubMed] [Google Scholar]
- Chen W, et al. HIF-1α inhibition ameliorates neonatal brain injury in a rat pup hypoxic-ischemic model. Neurobiol Dis. 2008;31:433–41. doi: 10.1016/j.nbd.2008.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dardik R, Inbal A. Complex formation between tissue transglutaminase II (tTG) and vascular endothelial growth factor receptor 2 (VEGFR-2): proposed mechanism for modulation of endothelial cell response to VEGF. Exp Cell Res. 2006;312:2973–82. doi: 10.1016/j.yexcr.2006.05.019. [DOI] [PubMed] [Google Scholar]
- Dayan F, et al. The oxygen sensor factor-inhibiting hypoxia-inducible factor-1 controls expression of distinct genes through the bifunctional transcriptional character of hypoxia-inducible factor-1α. Cancer Res. 2006;66:3688–98. doi: 10.1158/0008-5472.CAN-05-4564. [DOI] [PubMed] [Google Scholar]
- Fesus L, Piacentini M. Transglutaminase 2: an enigmatic enzyme with diverse functions. Trends Biochem Sci. 2002;27:534–9. doi: 10.1016/s0968-0004(02)02182-5. [DOI] [PubMed] [Google Scholar]
- Filiano AJ, et al. Transglutaminase 2 protects against ischemic insult, interacts with HIF1β, and attenuates HIF1 signaling. Faseb J. 2008;22:2662–75. doi: 10.1096/fj.07-097709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fong GH. Mechanisms of adaptive angiogenesis to tissue hypoxia. Angiogenesis. 2008;11:121–40. doi: 10.1007/s10456-008-9107-3. [DOI] [PubMed] [Google Scholar]
- Gundemir S, Johnson GV. Intracellular localization and conformational state of transglutaminase 2: implications for cell death. PLoS ONE. 2009;4:e6123. doi: 10.1371/journal.pone.0006123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo K, et al. Hypoxia induces the expression of the pro-apoptotic gene BNIP3. Cell Death Differ. 2001;8:367–76. doi: 10.1038/sj.cdd.4400810. [DOI] [PubMed] [Google Scholar]
- Hagenbuchner J, et al. The anti-apoptotic protein BCL2L1/Bcl-xL is neutralized by pro-apoptotic PMAIP1/Noxa in neuroblastoma, thereby determining bortezomib sensitivity independent of prosurvival MCL1 expression. J Biol Chem. 2010;285:6904–12. doi: 10.1074/jbc.M109.038331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halterman MW, et al. Hypoxia-inducible factor-1α mediates hypoxia-induced delayed neuronal death that involves p53. J Neurosci. 1999;19:6818–24. doi: 10.1523/JNEUROSCI.19-16-06818.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa G, et al. A novel function of tissue-type transglutaminase: protein disulphide isomerase. Biochem J. 2003;373:793–803. doi: 10.1042/BJ20021084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helton R, et al. Brain-specific knock-out of hypoxia-inducible factor-1α reduces rather than increases hypoxic-ischemic damage. J Neurosci. 2005;25:4099–107. doi: 10.1523/JNEUROSCI.4555-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohenstein B, et al. Vasodilator-stimulated phosphoprotein-deficient mice demonstrate increased platelet activation but improved renal endothelial preservation and regeneration in passive nephrotoxic nephritis. J Am Soc Nephrol. 2005;16:986–96. doi: 10.1681/ASN.2004070591. [DOI] [PubMed] [Google Scholar]
- Hu CJ, et al. The N-terminal transactivation domain confers target gene specificity of hypoxia-inducible factors HIF-1α and HIF-2α. Mol Biol Cell. 2007;18:4528–42. doi: 10.1091/mbc.E06-05-0419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ientile R, et al. Transglutaminase activity and transglutaminase mRNA transcripts in gerbil brain ischemia. Neurosci Lett. 2004;363:173–7. doi: 10.1016/j.neulet.2004.04.003. [DOI] [PubMed] [Google Scholar]
- Jang GY, et al. Transglutaminase 2 suppresses apoptosis by modulating caspase 3 and NF-κB activity in hypoxic tumor cells. Oncogene. 2009;29:356–67. doi: 10.1038/onc.2009.342. [DOI] [PubMed] [Google Scholar]
- Kasper LH, et al. Two transactivation mechanisms cooperate for the bulk of HIF-1-responsive gene expression. Embo J. 2005;24:3846–58. doi: 10.1038/sj.emboj.7600846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasper LH, Brindle PK. Mammalian gene expression program resiliency: the roles of multiple coactivator mechanisms in hypoxia-responsive transcription. Cell Cycle. 2006;5:142–6. doi: 10.4161/cc.5.2.2353. [DOI] [PubMed] [Google Scholar]
- Kim JY, et al. BH3-only protein Noxa is a mediator of hypoxic cell death induced by hypoxia-inducible factor 1alpha. J Exp Med. 2004;199:113–24. doi: 10.1084/jem.20030613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SY, et al. Differential expression of multiple transglutaminases in human brain. Increased expression and cross-linking by transglutaminases 1 and 2 in Alzheimer’s disease. J Biol Chem. 1999;274:30715–21. doi: 10.1074/jbc.274.43.30715. [DOI] [PubMed] [Google Scholar]
- Lee MJ, et al. Identification of the hypoxia-inducible factor 1 alpha-responsive HGTD-P gene as a mediator in the mitochondrial apoptotic pathway. Mol Cell Biol. 2004;24:3918–27. doi: 10.1128/MCB.24.9.3918-3927.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesort M, et al. Distinct nuclear localization and activity of tissue transglutaminase. J Biol Chem. 1998;273:11991–4. doi: 10.1074/jbc.273.20.11991. [DOI] [PubMed] [Google Scholar]
- Lesort M, et al. Tissue transglutaminase is increased in Huntington’s disease brain. J Neurochem. 1999;73:2018–27. [PubMed] [Google Scholar]
- Li J, et al. Cell death and proliferation in NF-κB p50 knockout mouse after cerebral ischemia. Brain Res. 2008;1230:281–9. doi: 10.1016/j.brainres.2008.06.130. [DOI] [PubMed] [Google Scholar]
- Liu S, et al. Structural basis for the guanine nucleotide-binding activity of tissue transglutaminase and its regulation of transamidation activity. Proc Natl Acad Sci U S A. 2002;99:2743–7. doi: 10.1073/pnas.042454899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorand L, Graham RM. Transglutaminases: crosslinking enzymes with pleiotropic functions. Nat Rev Mol Cell Biol. 2003;4:140–56. doi: 10.1038/nrm1014. [DOI] [PubMed] [Google Scholar]
- Maeda K, et al. Differences in the cerebrovascular anatomy of C57black/6 and SV129 mice. Neuroreport. 1998;9:1317–9. doi: 10.1097/00001756-199805110-00012. [DOI] [PubMed] [Google Scholar]
- Maxwell PH, et al. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature. 1999;399:271–5. doi: 10.1038/20459. [DOI] [PubMed] [Google Scholar]
- Milakovic T, et al. Intracellular localization and activity state of tissue transglutaminase differentially impacts cell death. J Biol Chem. 2004;279:8715–22. doi: 10.1074/jbc.M308479200. [DOI] [PubMed] [Google Scholar]
- Mishra S, Murphy LJ. Tissue transglutaminase has intrinsic kinase activity: identification of transglutaminase 2 as an insulin-like growth factor-binding protein-3 kinase. J Biol Chem. 2004;279:23863–8. doi: 10.1074/jbc.M311919200. [DOI] [PubMed] [Google Scholar]
- Peng X, et al. Interaction of tissue transglutaminase with nuclear transport protein importin-α3. FEBS Lett. 1999;446:35–9. doi: 10.1016/s0014-5793(99)00018-6. [DOI] [PubMed] [Google Scholar]
- Perry MJ, Haynes LW. Localization and activity of transglutaminase, a retinoid-inducible protein, in developing rat spinal cord. Int J Dev Neurosci. 1993;11:325–37. doi: 10.1016/0736-5748(93)90004-w. [DOI] [PubMed] [Google Scholar]
- Perry MJ, et al. Transglutaminase C in cerebellar granule neurons: regulation and localization of substrate cross-linking. Neuroscience. 1995;65:1063–76. doi: 10.1016/0306-4522(94)00556-k. [DOI] [PubMed] [Google Scholar]
- Piacentini M, et al. Transglutaminase overexpression sensitizes neuronal cell lines to apoptosis by increasing mitochondrial membrane potential and cellular oxidative stress. J Neurochem. 2002;81:1061–72. doi: 10.1046/j.1471-4159.2002.00898.x. [DOI] [PubMed] [Google Scholar]
- Pinkas DM, et al. Transglutaminase 2 undergoes a large conformational change upon activation. PLoS Biol. 2007;5:e327. doi: 10.1371/journal.pbio.0050327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg GA, et al. TIMP-2 reduces proteolytic opening of blood-brain barrier by type IV collagenase. Brain Res. 1992;576:203–7. doi: 10.1016/0006-8993(92)90681-x. [DOI] [PubMed] [Google Scholar]
- Sharp FR, Bernaudin M. HIF1 and oxygen sensing in the brain. Nat Rev Neurosci. 2004;5:437–48. doi: 10.1038/nrn1408. [DOI] [PubMed] [Google Scholar]
- Sharp FR, et al. Multiple molecular penumbras after focal cerebral ischemia. J Cereb Blood Flow Metab. 2000;20:1011–32. doi: 10.1097/00004647-200007000-00001. [DOI] [PubMed] [Google Scholar]
- Siegel M, et al. Extracellular transglutaminase 2 is catalytically inactive, but is transiently activated upon tissue injury. PLoS ONE. 2008;3:e1861. doi: 10.1371/journal.pone.0001861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh US, et al. Identification and biochemical characterization of an 80 kilodalton GTP-binding/transglutaminase from rabbit liver nuclei. Biochemistry. 1995;34:15863–71. doi: 10.1021/bi00048a032. [DOI] [PubMed] [Google Scholar]
- Sonmez A, et al. Erythropoietin attenuates neuronal injury and potentiates the expression of pCREB in anterior horn after transient spinal cord ischemia in rats. Surg Neurol. 2007;68:297–303. doi: 10.1016/j.surneu.2006.11.045. discussion 303. [DOI] [PubMed] [Google Scholar]
- Szondy Z, et al. Tissue transglutaminase (TG2) protects cardiomyocytes against ischemia/reperfusion injury by regulating ATP synthesis. Cell Death Differ. 2006;13:1827–9. doi: 10.1038/sj.cdd.4401889. [DOI] [PubMed] [Google Scholar]
- Tamura A, et al. Focal cerebral ischaemia in the rat: 1. Description of technique and early neuropathological consequences following middle cerebral artery occlusion. J Cereb Blood Flow Metab. 1981;1:53–60. doi: 10.1038/jcbfm.1981.6. [DOI] [PubMed] [Google Scholar]
- Tang Y, et al. Effect of hypoxic preconditioning on brain genomic response before and following ischemia in the adult mouse: identification of potential neuroprotective candidates for stroke. Neurobiol Dis. 2006;21:18–28. doi: 10.1016/j.nbd.2005.06.002. [DOI] [PubMed] [Google Scholar]
- Tolentino PJ, et al. Increased expression of tissue-type transglutaminase following middle cerebral artery occlusion in rats. J Neurochem. 2004;89:1301–7. doi: 10.1111/j.1471-4159.2004.02436.x. [DOI] [PubMed] [Google Scholar]
- Tucholski J, Johnson GV. Tissue transglutaminase directly regulates adenylyl cyclase resulting in enhanced cAMP-response element-binding protein (CREB) activation. J Biol Chem. 2003;278:26838–43. doi: 10.1074/jbc.M303683200. [DOI] [PubMed] [Google Scholar]
- Tucholski J, et al. Tissue transglutaminase overexpression in the brain potentiates calcium-induced hippocampal damage. J Neurochem. 2006;97:582–94. doi: 10.1111/j.1471-4159.2006.03780.x. [DOI] [PubMed] [Google Scholar]
- Valerio A, et al. Leptin is induced in the ischemic cerebral cortex and exerts neuroprotection through NF-κB/c-Rel-dependent transcription. Stroke. 2009;40:610–7. doi: 10.1161/STROKEAHA.108.528588. [DOI] [PubMed] [Google Scholar]
- Van Hoecke M, et al. Evidence of HIF-1 functional binding activity to caspase-3 promoter after photothrombotic cerebral ischemia. Mol Cell Neurosci. 2007;34:40–7. doi: 10.1016/j.mcn.2006.09.009. [DOI] [PubMed] [Google Scholar]
- Vangeison G, et al. The good, the bad, and the cell type-specific roles of hypoxia inducible factor-1 alpha in neurons and astrocytes. J Neurosci. 2008;28:1988–93. doi: 10.1523/JNEUROSCI.5323-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang GL, et al. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci U S A. 1995;92:5510–4. doi: 10.1073/pnas.92.12.5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warnecke C, et al. Differentiating the functional role of hypoxia-inducible factor (HIF)-1α and HIF-2α (EPAS-1) by the use of RNA interference: erythropoietin is a HIF-2α target gene in Hep3B and Kelly cells. Faseb J. 2004;18:1462–4. doi: 10.1096/fj.04-1640fje. [DOI] [PubMed] [Google Scholar]
- Wilhelmus MM, et al. Transglutaminases and transglutaminase-catalyzed cross-links colocalize with the pathological lesions in Alzheimer’s disease brain. Brain Pathol. 2009;19:612–22. doi: 10.1111/j.1750-3639.2008.00197.x. [DOI] [PMC free article] [PubMed] [Google Scholar]