

Abstract
Islet Ca2+-independent phospholipase A2 (iPLA2) is postulated to mediate insulin secretion by releasing arachidonic acid in response to insulin secretagogues. However, the significance of iPLA2 signaling in insulin secretion in vivo remains unexplored. Here we investigated the physiological role of iPLA2 in β-cell lines, isolated islets, and mice. We showed that small interfering RNA-specific silencing of iPLA2 expression in INS-1 cells significantly reduced insulin-secretory responses of INS-1 cells to glucose. Immunohistochemical analysis revealed that mouse islet cells expressed significantly higher levels of iPLA2 than pancreatic exocrine acinar cells. Bromoenol lactone (BEL), a selective inhibitor of iPLA2, inhibited glucose-stimulated insulin secretion from isolated mouse islets; this inhibition was overcome by exogenous arachidonic acid. We also showed that iv BEL administration to mice resulted in sustained hyperglycemia and reduced insulin levels during glucose tolerance tests. Clamp experiments demonstrated that the impaired glucose tolerance was due to insufficient insulin secretion rather than decreased insulin sensitivity. Short-term administration of BEL to mice had no effect on fasting glucose levels and caused no apparent pathological changes of islets in pancreas sections. These results unambiguously demonstrate that iPLA2 signaling plays an important role in glucose-stimulated insulin secretion under physiological conditions.
Phospholipase A2 (PLA2) is a diverse group of enzymes that catalyze the hydrolysis of the sn-2 fatty acyl bond of phospholipids to liberate free fatty acids and lysophospholipids (1). Growing evidence suggests that hydrolysis of arachidonic acid (AA) from the membrane phospholipids of β-cells by activation of intracellular PLA2 is involved in glucose-stimulated insulin secretion (2–5). Certain receptor-mediated insulin secretagogues, such as glucose-dependent insulinotropic polypeptide, cholecystokinin-8, and muscarinic agonist stimulate the release of AA by activation of iPLA2 (6–8). Both Ca2+-dependent and -independent PLA2 (cPLA2 and iPLA2, respectively) are reportedly involved in AA release and insulin secretion induced by insulin secretagogues (9–25). cPLA2, which requires micromolar Ca2+ for its enzymatic activity, was cloned from rat islet cells (26) and has been shown to play an important role in the maintenance of insulin stores (25, 27) and in the control of the rate of exocytosis in β-cells (28).
It is noteworthy that the majority of AA released in response to glucose stimulation is Ca2+ influx independent: AA accumulation occurs in Ca2+-free medium or in the presence of Ca2+-channel blockers (20, 29, 30). These findings suggest that glucose-induced AA release occurs before Ca2+ influx and, therefore, before insulin secretion. Indeed, AA has been found to stimulate a rise of cytosolic Ca2+ by multiple mechanisms (29, 31–33). Interestingly, like glucose-stimulated insulin secretion, glucose-stimulated AA release requires the transport and metabolization of glucose within β-cells, and ATP, thought to be a signal generated by glucose metabolism (34), can stimulate AA release in the absence of Ca2+ (20). An ATP-stimulated, Ca2+-independent PLA2 activity has also been identified in isolated islet and insulinoma cell lines (21). iPLA2 is an attractive candidate for mediating the Ca2+-independent, glucose metabolism-activated hydrolysis of AA from islet membrane phospholipids because bromoenol lactone (BEL), which selectively inhibits iPLA2 (35–37) while having no effect on β-cell metabolism (22), suppresses glucose-stimulated AA release, the rise of intracellular Ca2+, and insulin secretion (21, 22, 24, 38–40).
We have cloned an iPLA2 from rat and human pancreatic islets and confirmed that its catalytic activity is Ca2+ independent, stimulated by ATP, and inhibited by BEL (41, 42). We further examined the role of iPLA2 in mediating glucose-stimulated AA release and insulin secretion by overexpressing cloned iPLA2 cDNA in insulinoma INS-1 cells. Compared with controls, these cells exhibited increased AA release and insulin secretion in response to glucose stimulation; these qualities were further enhanced by elevation of cAMP and inhibited by BEL (24, 43).
Although experimental evidence from INS-1 cells with elevated iPLA2 expression supports the role of iPLA2 in glucose-stimulated insulin secretion, the physiological importance of iPLA2 in regulation of in vivo insulin levels remains unexplored. In this study, we further examined the function of iPLA2 in insulin secretion in INS-1 cells, isolated islets, and mice by using small interfering RNA (siRNA) silencing and BEL administration to block the expression or inhibit the activity of iPLA2. The results presented here demonstrate that inhibition of iPLA2 results in insufficient insulin secretion and hyperglycemia in normal mice.
RESULTS
Silencing of iPLA2 Expression by siRNA in INS-1 Cells Suppresses Glucose-Stimulated Insulin Secretion
It has previously been shown that iPLA2-overexpressing INS-1 cells secrete higher levels of insulin than parental INS-1 cells in response to glucose stimulation (24, 43, 44). To further verify the role of iPLA2 in insulin secretion, we applied RNA interference technology (45) to specifically silence iPLA2 expression in INS-1 cells. iPLA2-specific siRNA was constructed and evaluated as described in Materials and Methods. The psiRNA-iPLA2 construct that showed the highest degree of inhibition was used in all subsequent studies. As shown in Fig. 1B, about 50% of cells in the transfected plate, corresponding to the transfection efficiency of psiRNA-iPLA2, lost any detectable expression of iPLA2. In contrast, mock-transfected cells retained uniform iPLA2 expression (Fig. 1A). Western blot analysis confirmed that iPLA2 expression in siRNA-transfected cells was dramatically reduced compared with mock-transfected cells or to cells transfected with glyceraldehyde-3-phosphate dehydrogenase (GAPDH)-specific siRNA (Fig. 1C). The fact that some iPLA2 was still detectable in the Western blot analysis is likely due to the incomplete transfection of the cells with psiRNA-iPLA2 rather than to incomplete silencing of iPLA2 by the siRNA. For each subsequent batch of experiments, we confirmed iPLA2 expression silencing by measuring its activity before measuring the insulin-secretory response of the transfected cells. As shown in Fig. 1D, the glucose response was dramatically inhibited in siRNA-treated cells compared with control and mock-transfected cells. These data, along with our previous results showing a significant increase in insulin secretion when iPLA2 is overexpressed in INS-1 cells (24), demonstrate that iPLA2 signaling is important for glucose-stimulated insulin secretion.
Fig. 1. Specific Inhibition of iPLA2 Expression in INS-1 Cells by siRNA.
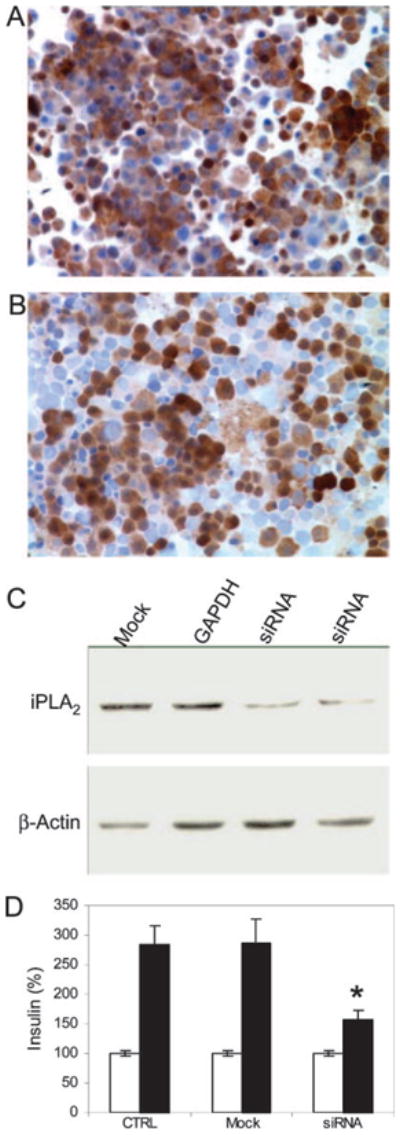
INS-1 cells transfected with mock (A) or siRNA (B) were spun on the slides and stained for iPLA2 (brown) and nuclei (light blue) (original magnification, ×100). About 50% of the cells expressed iPLA2, corresponding to the transfection efficiency of this method in INS-1 cells. C, Western blot analysis of siRNA inhibition of iPLA2 expression. INS-1 cells were transfected with either mock, GAPDH-specific siRNA, or iPLA2-specific siRNA (independent duplicates) constructs, respectively. Protein bands corresponding to iPLA2 and β-actin are indicated. D, Comparison of insulin secretion by INS-1 cells that were untransfected (CTRL), mock-transfected (Mock), or transfected with psiRNA-iPLA2 (siRNA). Insulin secretion (ng/ml) in the presence of 3 mM glucose (open bars) and 20 mM glucose (solid bars) is indicated. siRNA silencing of iPLA2 expression in INS-1 cells (n = 12) resulted in decreased levels of glucose-stimulated insulin secretion compared with untransfected (n = 18) or mock-transfected (n = 12) cells. Mean ± SEM is given. Data were compared with a two-tailed unpaired t test. *, P < 0.01.
Pancreatic Islets Express High Levels of iPLA2
To assess the role of iPLA2 in insulin secretion in vivo, we first used immunohistochemical analysis to examine iPLA2 expression patterns in mouse pancreas, a gland with both endocrine (islet of Langerhans) and exocrine (acinar cells) functions. We removed the pancreas from C57BL/6J mice and stained three consecutive islet-containing sections for insulin, glucagon, and iPLA2, respectively (Fig. 2). Consistent with other reports, insulin, expressed by islet β-cells, was detectable in the core of the islet (Fig. 2A), and glucagon, expressed by islet α-cells, was found on the periphery (Fig. 2B). Interestingly, iPLA2 is expressed predominantly in islet cells costained by insulin but is barely detected in the exocrine acinar cells (Fig. 2C), suggesting that iPLA2 is more important in the endocrine tissue of the pancreas than in the exocrine cells. Consistent with these differences in expression levels, iPLA2 activity was higher in islets than in pancreatic acinar cells (Fig. 2D). Pretreatment of isolated islets with BEL (10 μM) reduced iPLA2 activity to close to the level of acinar cells.
Fig. 2. Immunohistochemical Analysis of iPLA2 Expression in Mouse Pancreas.
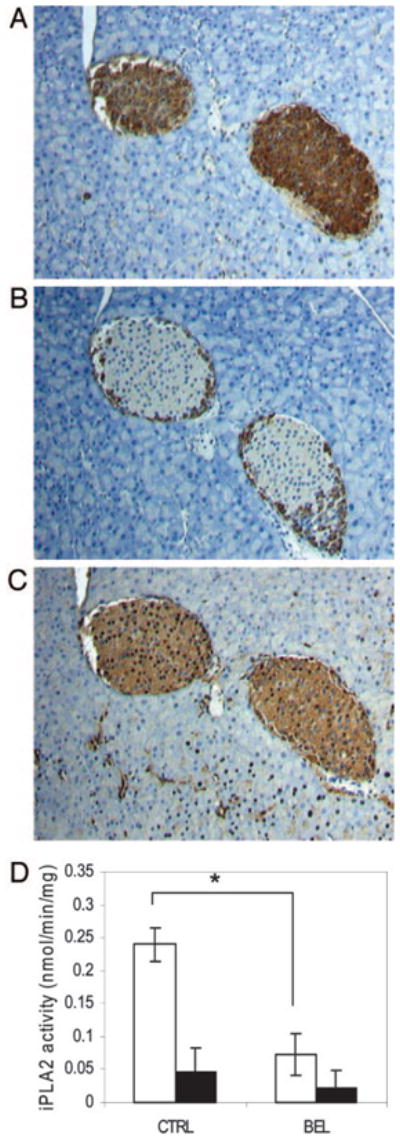
Three consecutive 4-μm-thick sections of pancreas from C57BL/6J mice were stained for insulin localized to the pancreatic β-cells of islet (A), glucagon localized to the pancreatic α-cells (B), and iPLA2 (1:500 dilutions of antibodies, T-14) (C). Slides were then counterstained with hematoxylin to identify cell nuclei (blue), (original magnification, ×100). D, Isolated islet (open bars) and nonislet cells (solid bars) were pretreated with either vehicle (CTRL) (n = 9) or 10 μM BEL (n = 8) for 30 min at 37 C, and iPLA2 activity was assessed and statistic analyses were conducted as described in Materials and Methods. *, P < 0.01.
BEL Treatment of Isolated Islets Inhibits Glucose-Stimulated Insulin Secretion and AA Reversed the BEL-Inhibitory Effect
We next examined whether inhibition of this islet-associated iPLA2 activity by BEL also suppresses glucose-stimulated insulin secretion in isolated mouse islets. Freshly isolated, randomly grouped islets from normal mice were used to assess glucose-stimulated insulin secretion by static incubation in the presence or absence of 10 μM BEL. As shown in Fig. 3, the islets secreted a large amount of insulin in response to 20 mM glucose in the absence of BEL. However, this secretory response was dramatically reduced in islets pretreated with BEL, consistent with previous reports on isolated rat islets (46).
Fig. 3. Effects of BEL or AA on Glucose-Stimulated Insulin Secretion in Isolated Islets.
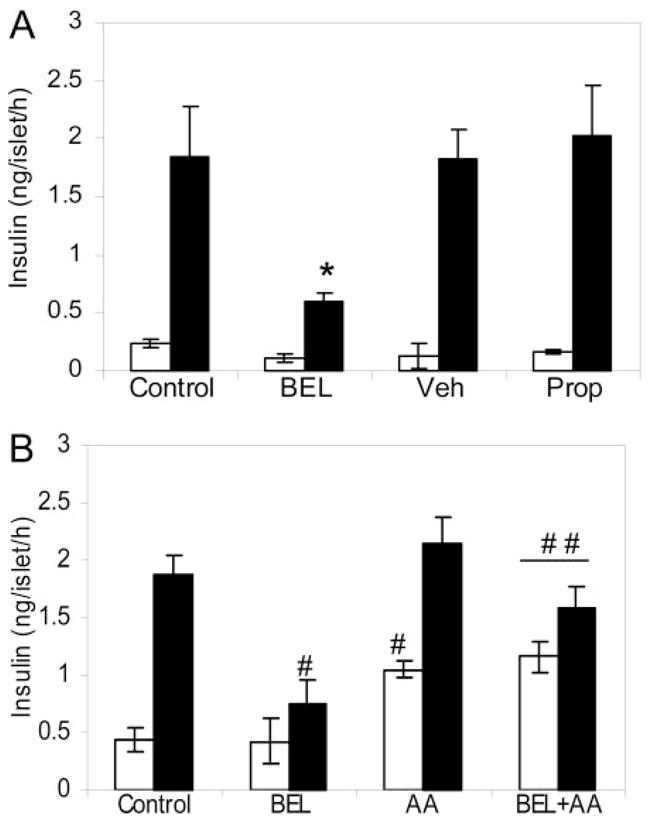
A, Batches of five islets in 0.5 ml of KRB buffer containing 0.1% BSA with designated substances were grouped into control (n = 6), BEL-treated (10 μM, n = 6), vehicle-treated (Veh) (n = 4), and propranolol-treated groups (Prop) (250 μM, n = 3) as indicated. Insulin secretion was determined by static incubations in the presence of 3 mM (open bars) and 20 mM (solid bars) glucose. *, P < 0.01. B, Insulin secretion was determined in the presence of 3 mM (open bars) or 20 mM (solid bars) glucose under the following conditions: control, islets treated with vehicle alone; BEL, islets pretreated with BEL (10 μM); AA, islets incubated with AA (100 μM); BEL+AA, islets pretreated with BEL and incubated with AA (100 μM). Mean ± SEM is indicated. Data were analyzed with a two-tailed unpaired t test. #, Compared with controls; ##, compared with the BEL-treated groups, P < 0.01.
BEL has recently been reported to inhibit magnesium-dependent cytosolic phosphatidate phosphohydrolase (PAP-1) in mouse P388D1 macrophages and in human amnionic WISH cells (47, 48). In isolated rat islets, 10 μM BEL inhibits about 23% of total PAP-1 activity (49). To differentiate the effect of BEL on iPLA2 from its effect on PAP-1, we treated the islets with propranolol, a well-known PAP-1-specific inhibitor (50–53). In accordance with other studies (52, 53), 250 μM propranolol completely inhibited PAP-1 activity in the islets. However, in contrast to BEL, propranolol administration caused glucose-stimulated insulin secretion in the islets to increase slightly (Fig. 3A). Taken together with our previous results showing that propranolol does not affect glucose-stimulated AA release (46) and insulin secretion (24), our findings indicate that the suppression of insulin secretion by BEL is not attributable to inhibition of PAP-1.
Inhibition of iPLA2 by BEL and suppression of AA release is thought to lead to the inhibition of glucose-stimulated insulin secretion (21, 22, 24, 38–40). We further examined whether exogenous AA can reverse the inhibitory effect of BEL on glucose-stimulated insulin secretion. Consistent with previous reports (19, 23), addition of exogenous AA significantly increased insulin secretion only in mouse islets incubated at substimulatory, but not stimulatory, glucose concentrations (Fig. 3B). Importantly, exogenous AA also reversed the inhibitory effect of BEL on glucose-stimulated insulin secretion. These data, along with our previous results showing that overexpressing iPLA2 in INS-1 cells significantly increases glucose-stimulated AA release (24), suggest that iPLA2 serves as part of a glucose-sensing pathway that rapidly generates AA to stimulate insulin exocytosis.
BEL Administration to Mice Results in Impaired Glucose Tolerance and Reduced Insulin Levels
Because inhibition of iPLA2 activity by BEL effectively inhibited glucose-stimulated insulin secretion in mouse islets, we were interested in determining the physiological effect of BEL injection, and subsequent iPLA2 inhibition, in mice. Forty minutes before glucose tolerance testing, BEL (3 μg/g body wt) was injected into the tail vein of mice as described in Materials and Methods. As shown in Fig. 4A, fasting glucose levels in the BEL-treated mice were not significantly different from those in the vehicle-treated group. However, blood glucose levels in the BEL-treated mice were significantly higher than in the control group after ip glucose injection, and marked hyperglycemia persisted longer in the BEL-treated mice than in control mice. We did not observe any apparent difference between the sexes, and the data shown here were gathered from both male and female animals. As with the isolated islets, we also treated mice with propranolol to distinguish between iPLA2 and PAP-1 inhibition. Glucose levels in the propranolol-treated group exhibited no apparent difference from those of the control group (Fig. 4A), excluding the possibility that the hyperglycemic state observed in BEL-treated mice was attributable to PAP-1 inhibition.
Fig. 4. Glucose Tolerance Tests in Mice with or without BEL Administration.
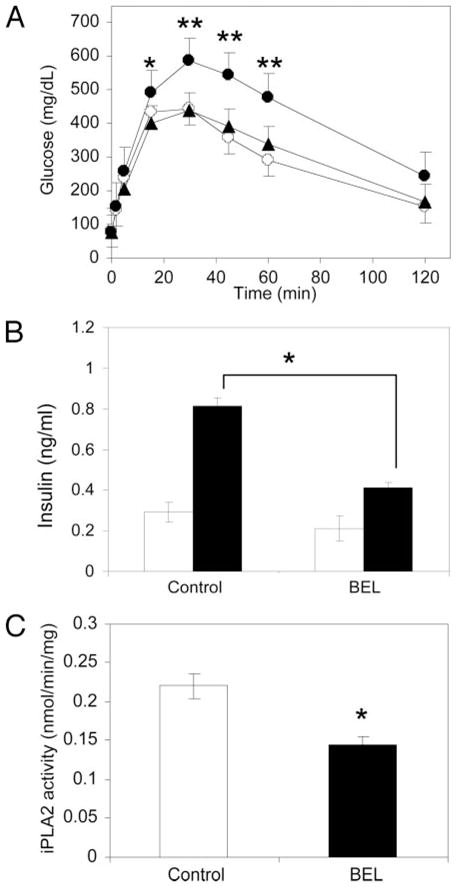
A, Glucose tolerance test. Age- and weight-matched mice were randomly grouped into BEL-treated (solid circles, n = 19), propranolol-treated (solid triangles, n = 8), and control groups (open circles, n = 23), and glucose tolerance tests were performed after ip injection of glucose (*, P < 0.05; **, P < 0.01). B, Plasma insulin levels in the control (n = 4) and BEL-treated (n = 5) mice before (fasting levels, open bars) and 10 min after ip glucose challenge (solid bars). C, The iPLA2 activity of islets isolated from vehicle (control) (n = 9) or BEL-treated mice (n = 11) after whole isolation procedure (P = 0.039). Mean ± SEM is indicated. Statistical analysis was conducted as described in Materials and Methods.
We also tested insulin levels in blood samples drawn from control and BEL-treated groups 10 min after glucose challenge. As shown in Fig. 4B, the insulin levels of the BEL-treated mice before glucose challenge were similar to those in the control group. After glucose challenge, however, insulin levels were significantly lower. These results demonstrate that BEL administration caused insufficient insulin secretion in the mice, which may contribute to impaired glucose tolerance. To confirm that BEL is capable of inhibiting iPLA2 in vivo, we isolated islets from the mice immediately after administration of BEL or vehicle and measured their iPLA2 activity. As shown in Fig. 4C, iPLA2 activity was significantly lower in islets isolated from the BEL-treated group than from the vehicle-treated group, even after the 4 h required for complete islet isolation. These data indicate that BEL administration to mice can inhibit iPLA2 activity in islets throughout the glucose tolerance testing period.
BEL Administration to Mice Does Not Affect Fasting Glucose Levels or Cause Pathological Changes to Pancreatic Islets
Next we examined whether BEL administration to mice affects fasting glucose levels or causes pathological changes to pancreatic islets. As shown in Fig. 5A, there was no apparent difference in fasting glucose levels between control and BEL-treated groups after 5 successive days of BEL administration. Additionally, as shown in Fig. 5B, immunohistochemical analysis of pancreatic islets revealed no morphological changes in islets from BEL-treated mice as compared with control mice, and expression of insulin, glucagon, and iPLA2 in the two groups was similar. Insulin content and iPLA2 activity were also not significantly different between control and BEL-treated mice (data not shown). These results indicate the architecture and protein expression profiles of pancreatic islet β-cells were not affected by short-term administration of BEL to mice.
Fig. 5. Immunohistochemical Examination of Pancreases from Mice with and without BEL Administration.
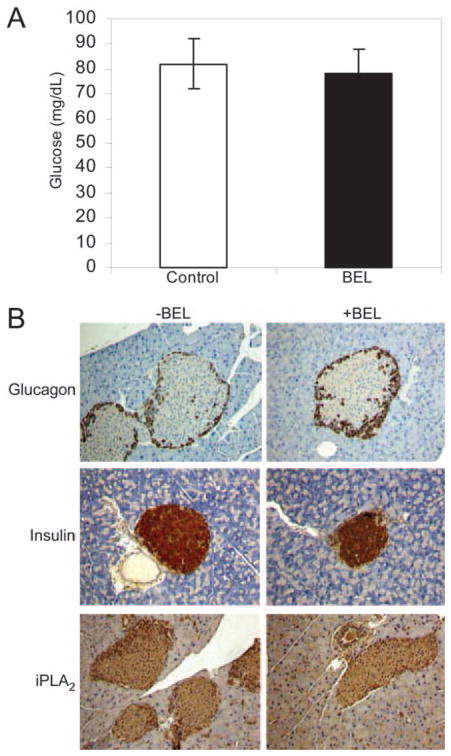
BEL (3 μg/g) or vehicle was injected into the tail vein of mice once a day for 5 d. A, Fasting glucose levels with either vehicle (open bar) or BEL (solid bar) were determined. Mean ± SEM is indicated. Data were compared with a two-tailed unpaired t test. B, The pancreatic sections were prepared and stained for insulin, glucagon, and iPLA2 (1:250 dilution of antibodies T-14). Slides were then counterstained with hematoxylin to identify cell nuclei (original magnification, ×100).
Inhibition of iPLA2 by BEL Has Little Effect on Insulin Sensitivity of Mice
The impaired glucose tolerance we observed in the BEL-treated group could be due either to reduced insulin sensitivity or insufficient insulin secretion. To resolve this question, we measured glucose disposal in the mice using a euglycemic hyperinsuline-mic clamp (54). Age- and weight-matched catheterized mice were iv injected with either vehicle or BEL (3 μg/g body wt), and blood glucose levels were clamped at 100 mg/dl in both groups (Fig. 6A). The insulin levels between groups during the clamp were not significantly different. As shown in Fig. 6B, the glucose infusion rate in the BEL-treated group is similar to that of the controls during clamps. We averaged the glucose infusion rate during the last 60 min to determine the glucose disposal rate as described previously (55). There was no significant difference in glucose disposal rates between the two groups (Fig. 6C). This finding indicates that insulin sensitivity under hyperinsulinemic conditions was not affected by BEL treatment.
Fig. 6. Effect of BEL Inhibition of iPLA2 on the Glucose Disposal Rate Analyzed by Hyperinsulinemic-Euglycemic Clamp Experiments.
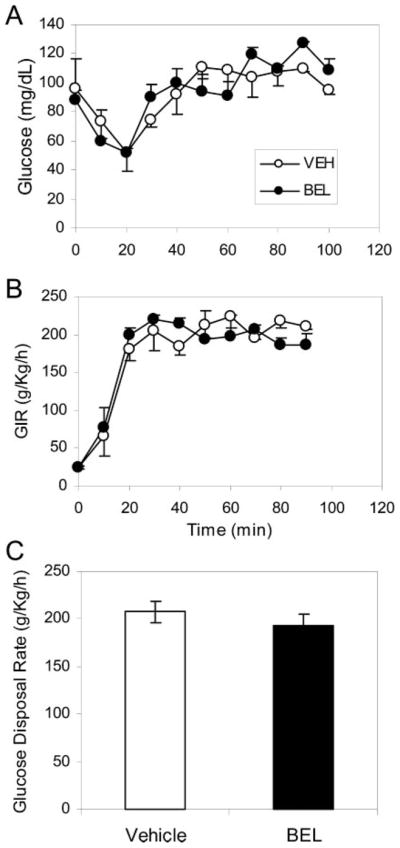
Age- and weight-matched C57BL/6J mice were randomly grouped to receive either BEL or vehicle treatment, and insulin (21 mU/kg/min) was prime-continuously infused throughout the 90-min experiment. A, Blood glucose was clamped at euglycemic levels (~100 mg/dl) with iv infusion of 10% glucose solution. B, The glucose infusion profiles from vehicle (open circles) and BEL-treated groups (solid circles) during clamps. C, The glucose disposal rates from the control group (solid bars, n = 5) and the BEL-treated group (filled bars, n = 5) were calculated as described in Materials and Methods. Mean ± SEM is indicated. Data were compared with a two-tailed unpaired t test. P = 0.97.
Inhibition of iPLA2 by BEL Administration Leads to Insufficient Insulin Secretion
Because BEL treatment does not significantly affect the glucose disposal rate in mice, we used hyperglycemic clamp experiments (56) to evaluate β-cell insulin secretion. Blood glucose levels were clamped at 3 times the normal range in both control and BEL-treated mice (Fig. 7A), and the glucose infusion rate was adjusted according to ongoing measurements of blood glucose levels (Fig. 7B). Although we began the experiment with the same infusion rate for both groups, glucose levels rose faster in the BEL-treated group than in the controls, and the mice in the control group required larger infusions of glucose within the first 30 min to elevate blood glucose to designated levels (Fig. 7B). As the experiment continued, the BEL-treated mice required less glucose to maintain the hyperglycemic state than did the controls. We then assessed insulin levels in blood samples from animals from both groups that had reached a steady-hyperglycemic state and found that average insulin levels in the BEL-treated group were significantly lower than in controls (Fig. 7C). In contrast, no differences were observed under basal conditions, consistent with the results of the glucose tolerance test (Fig. 4B). When these data are taken together, we can conclude that the hyperglycemia observed in the BEL-treated mice (Fig. 4A) results from insufficient insulin secretion rather than from decreased insulin sensitivity.
Fig. 7. Effect of BEL Inhibition of iPLA2 on Insulin Secretion Analyzed by Hyperglycemic Clamp Experiments.
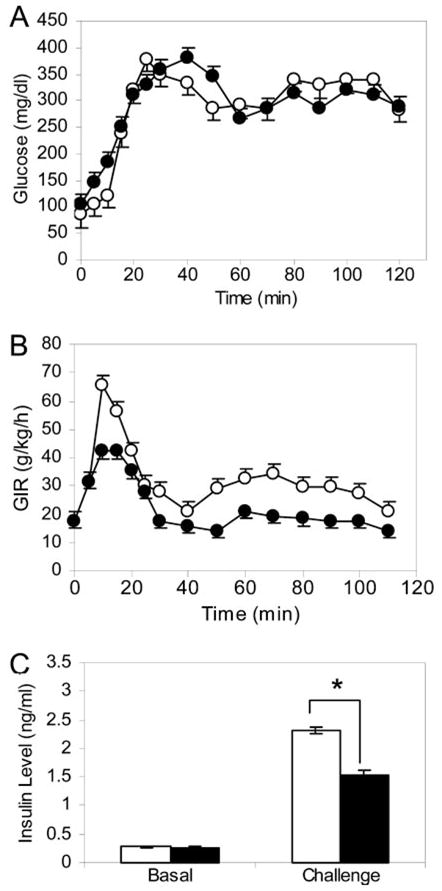
Age- and weight-matched C57BL/6J mice were randomly grouped into BEL-treated (solid circles, n = 5) and control groups (open circles, n = 5). A continuous hyperglycemic state was achieved by adjusting glucose infusion rates. A, Blood glucose level was clamped at approximately 300 mg/dl after a 30-min interval to reach hyperglycemia status. B, Glucose infusion rate (GIR) shown as an average of each group at each time point. C, The average plasma insulin levels (at 30, 60, 90, 120 min) in the BEL-treated (solid bars) and control (open bars) mice. Mean ± SEM is indicated. Data were compared with a two-tailed unpaired t test. *, P < 0.05.
DISCUSSION
It has long been proposed that glucose-stimulated hydrolysis of AA from islet membrane phospholipids by activation of PLA2 plays a signaling role in glucose-stimulated insulin secretion (4, 19, 57–59). However, a lack of in vivo studies has made it difficult to determine whether this hypothesis accurately represents the physiological process of insulin secretion. In the present study, we investigated the physiological role of iPLA2 in glucose-stimulated insulin secretion in β-cell lines, isolated islets, and C57BL/6J mice. The following results provide the first experimental evidence that iPLA2 signaling is important for sufficient insulin secretion in response to high glucose in vivo: 1) siRNA that specifically silences iPLA2 expression in cultured INS-1 cells significantly suppresses glucose-stimulated insulin secretion in those cells; 2) pancreatic islet β-cells express substantially more iPLA2 than do pancreatic exocrine acinar cells; 3) inhibition of iPLA2 catalytic activity by BEL in isolated mouse islets dramatically suppresses glucose-stimulated insulin secretion, and this inhibitory effect is reversed by exogenous AA; 4) inhibition of iPLA2 by administering BEL to mice causes sustained hyperglycemia and reduces insulin levels; and 5) BEL treatment of mice causes insufficient insulin secretion rather than alteration of insulin sensitivity.
If iPLA2 signaling is indeed physiologically important for insulin secretion, as we demonstrated in mice, the following questions should be considered: 1) Do islet cells contain a large pool of AA in their membrane phospholipids? Previous studies showed that AA was the most abundant fatty acid measured in the phospholipids of rodent and human islets and constituted more than 30% of the total esterified fatty acyl mass of islet glycerolipids (22, 38). It is also apparent that abundant AA in islet phospholipids is essential for insulin secretion by β-cells: reduced insulin secretion by late-passage INS-1 cells is associated with the loss of phospholipid-associated AA, and AA supplementation partially restores responses to insulin secretagogues in these cells (60). 2) Is the amount of unesterified AA released after glucose stimulation sufficient to stimulate insulin secretion under physiological conditions? It has been shown that intracellular concentrations of free AA can achieve levels of 100 μM or more during glucose stimulation (29, 30), well above the levels required to stimulate insulin secretion in in vitro studies. In these studies, 50 μM or greater of exogenous AA stimulates significant insulin secretion from intact or permeabilized islets even at substimulatory concentrations of glucose (19, 27, 57–59). The pattern of AA-induced insulin stimulation is also similar to the biphasic response to glucose (58). 3) Are islet β-cells capable of releasing AA quickly enough to stimulate insulin secretion? Exposure of islets to glucose causes a rapid release of AA (4), suggesting β-cells have an immediate response system. Because adjusting the amount of key enzyme through synthesis, degradation, and protein transport is too time consuming, most cells instead turn to ways of regulating the activity of enzymes already on hand when rapid responses to changing physiological conditions are necessary. We have shown here that islet cells express a substantial amount of iPLA2 (Fig. 2). Activation of this existing large pool upon glucose stimulation may contribute to the rapid release of AA. Consistent with this hypothesis, glucose-stimulated AA release in isolated islets was fully inhibited by BEL but not propranolol (46), iPLA2-overexpressing INS-1 cells release more AA than do parental INS-1 cells in response to stimulatory glucose concentrations, and this response can be inhibited by BEL (24).
Structural features of iPLA2 also support a physiologically important role in mediating glucose-stimulated AA release. iPLA2 contains an ATP-binding domain that is highly homologous to that of protein kinases (5, 61). The catalytic activity of islet iPLA2 is stimulated by ATP in vitro (41, 42), and iPLA2 protein binds to an ATP-sepharose column (62). The fact that glucose stimulates more AA release in iPLA2-overexpressing INS-1 cells than in parental INS-1 cells (5, 24) indicates that glucose can activate iPLA2, probably through its metabolite ATP. In addition, glucose stimulates iPLA2 translocation from the cytosol to the perinucleus, where it can access its membrane-phospholipid substrates. This translocation is further enhanced by elevation of cellular cAMP levels by 3-isobutyl-1-methylxanthine or forskolin, which is also accompanied by increased insulin secretion by INS-1 cells (24, 44). Taken together, we conclude that iPLA2 plays a key role in glucose-stimulated AA release and that the iPLA2/AA signaling pathway represents a distinct, physiologically relevant mechanism to mediate glucose-stimulated insulin secretion in vivo.
MATERIALS AND METHODS
Materials
Tissue culture medium RPMI 1640 and fetal bovine serum (FBS) were purchased from Mediatech, Inc. (Herndon, VA). CMRL-1066, Hanks’ balanced salt solution, and penicillin/streptomycin were from Invitrogen (San Diego, CA). BSA, propranolol, Ficoll, and glucose solution were obtained from Sigma-Aldrich Co. (St. Louis, MO). BEL was from Cayman Chemical Co. (Ann Arbor, MI). Ultrasensitive insulin ELISA kit was from Crystal Chem, Inc. (Chicago, IL). Liberase RI was from Roche Molecular Biochemicals (Indianapolis, IN). FuGENE 6 transfection reagent was obtained from Roche. Radioimmunoprecipitation assay (RIPA) lysis buffer was from United States Biological (Swampscott, MA), and enhanced chemiluminescence Western blotting detection reagents were from Amersham Pharmacia Biotech (Piscataway, NJ). Human insulin was from Eli Lilly & Co. (Indianapolis, IN).
siRNA Construction and Expression in INS-1 Cells
pSilencer 2.0-U6 and pSilencer 3.0-H1 Vectors (Ambion, Inc., Austin, TX) were used to express iPLA2-specific siRNAs in INS-1 cells. pSilencer 2.0-U6 contains the human U6 promoter that has been used extensively to express siRNAs and ribozymes in mammalian cells, and pSilencer 3.0-H1 contains the H1 RNA promoter (H1 RNA is a component of ribonuclease P). The siRNA expression vectors were constructed according to the manufacturer’s instructions. Four pairs of siRNAs against rat iPLA2 mRNA were synthesized, annealed, and subcloned into both pSilencer 2.0-U6 and pSilencer 3.0-H1 Vectors. After verification and purification, all constructs were transfected into INS-1 cells to evaluate the inhibition of iPLA2 expression.
INS-1 cells were cultured in RPMI 1640 medium containing 10% FBS, 10 mM HEPES buffer, 2 mM glutamine, 1 mM sodium pyruvate, 50 mM β-mercaptoethanol, and 0.1% (wt/vol) of penicillin in a 5% CO2 in air atmosphere at 37 C. When the cells were approximately 70% confluent, they were transfected with FuGENE 6 transfection reagent according to the instruction manual. The cells were harvested and lysed, 3 d after transfection, in RIPA lysis buffer containing protease inhibitors. The supernatant from each transfectant was analyzed by the Western blot to determine whether iPLA2 expression was inhibited by the siRNA. The pSilencer 3.0-H1 construct corresponding to the sequence 5′-CTTGGCATTCGGGAGTGTT-3′ in the encoding region of iPLA2 mRNA showed the most inhibition in INS-1 cells. This vector was designated as psiRNA-iPLA2 and used to express siRNA in INS-1 cells in all of the experiments. GAPDH-specific siRNA was used as a negative control.
Western Blot Analysis
Cells were collected and lysed in RIPA buffer containing protease inhibitors. Aliquots containing 60 μg of protein were resolved on 8% SDS-PAGE. After electrophoresis, proteins were blotted onto nitrocellulose membranes, and iPLA2 was probed with goat antihuman iPLA2 polyclonal IgG (T-14, Santa Cruz Biotechnology, Inc., Santa Cruz, CA), followed by incubation with antigoat IgG conjugated with horseradish peroxidase. iPLA2 proteins were detected by enhanced chemiluminescence Western blotting detection reagents.
Immunohistochemistry
Isolated mouse pancreases were fixed overnight in 10% formalin at 4 C. Sections (4-μm) were cut using ThermoShandon Finesse Microtome (Thermo Shandon, Inc., Pittsburgh, PA). Slides were incubated in 4% H2O2 to block the endogenous peroxidase, and then Powerblock universal blocking reagent (10X, BioGenex Laboratories, Inc., San Ramon CA) was used to eliminate the background. Insulin, glucagon, and iPLA2 were stained with the rabbit antiinsulin antibodies (Santa Cruz Biotechnology), rabbit antiglucagon antibodies (DAKO Corp., Carpinteria, CA), or goat anti-iPLA2 antibodies (Santa Cruz Biotechnology), respectively. Biotinylated antirabbit IgG (BioGenex) or biotinylated rabbit antigoat IgG (Vector Laboratories, Inc., Burlingame, CA) were used as second antibody as appropriate, followed by a peroxidase-conjugated streptavidin (BioGenex) and DAKO liquid 3,3′-diaminobenzidine substrate-chromogen reagent (DAKO). Slides were then counterstained with hematoxylin to localize nuclei of cells. Finally, the slides were mounted with Cytoseal xylene-based mounting medium (Stephens Scientific, Kalamazoo, MI) for visualization. For immunocytochemical analysis, INS-1 cells were spun on a Silane-prep slide (Sigma Diagnostics, St. Louis, MO) using ThermoShandon Cytospin-4 (Thermo Shandon, Inc., Pittsburgh, PA) and stained with anti-iPLA2 antibodies. Immunoreactivity was detected as described above.
iPLA2 Activity Assay
The activity of iPLA2 was determined using a modified kit originally designed for cPLA2 (cPLA2 assay kit, Cayman Chemical Co.) as described previously (63, 64). Briefly, after specific treatments, tissues from mice or cultured cells are collected and homogenized in buffer (50 mM HEPES, pH 7.4; 1 mM EDTA) followed by centrifugation at 14,000 × g for 20 min at 4 C. The supernatant was removed, and the concentration of proteins was determined. iPLA2 activity was assayed by incubating the samples with arachidonoyl thio-PC (phosphorylcholine) for 1 h at 25 C in a Ca2+-free buffer (4 mM EGTA; 160 mM HEPES, pH 7.4; 300 mM NaCl; 8 mM Triton X-100; 60% glycerol; 2 mg/ml BSA). The reaction was stopped by addition of 5,5′-dithio-bis (nitrobenzoic acid) for 5 min, and the absorbance was determined at 414 nm using a μQuant microplate reader (Bio-Tek Instruments, Inc., Winooski, VT). The specific activity of iPLA2 was calculated and expressed in nanomoles/min/mg of total proteins. The background basal lipase activity, which was determined by inhibiting all specific iPLA2 activity in control samples with BEL, was subtracted from all readings as described elsewhere (63, 64).
Determination of Insulin Secretion by Static Incubation
For determination of insulin secretion by islets, isolated mouse islets were cultured overnight in RPMI-1640 medium containing 11 mM glucose, 2 mM L-glutamine, 10% FBS, 100 U/ml penicillin, and 100 μg/ml streptomycin. The following day, islets were transferred to CMRL-1066 medium containing 5.5 mM glucose and preincubated for 120 min at 37 C. The islets were then distributed in batches of five islets per well in 0.5 ml of Krebs-Ringer bicarbonate buffer (KRB) with or without 10 μM BEL for 30 min, and subsequently transferred under dissecting microscope to medium containing 3 mM glucose or 20 mM glucose or 100 μM AA incubating for 60 min. The medium was then removed from each well, and insulin levels were measured with an ultrasensitive insulin ELISA kit. Insulin secretion was also measured in INS-1 cells with and without transfected psiRNA-iPLA2. Cells in six-well plates were washed in KRB buffer and incubated in the KRB buffer containing 3 mM glucose for 60 min. The medium was then removed and replaced with KRB buffer containing either 3 mM or 20 mM glucose at 37 C for 60 min. The KRB buffer in each well was collected for insulin measurement.
Pancreatic Islet Isolation and Culture
Islets were isolated from C57BL/6J mice by liberase digestion, discontinuous Ficoll gradient separation, and hand pickup, a modification of the original method of Lacy and Kostianovsky (65). Complete islet isolation from mice takes about 4–5 h. Islets were cultured in an atmosphere of 95% air, 5% CO2 in CMRL-1066 medium containing 5.5 mM glucose, 2 mM L-glutamine, 10% (vol/vol) heat-inactivated FBS, 100 U/ml penicillin, and 100 μg/ml streptomycin.
Glucose Tolerance Test
Mice (8- to 12-wk old) were fasted overnight (16–18 h) and randomly assigned to receive BEL, vehicle (ethanol), or propranolol via tail vein injection. BEL (10 mM) was freshly prepared in 100% ethanol just before the injection. Glucose tolerance tests (ip) were carried out 40 min after injection. Blood was drawn from the tail vein before, and 5, 10, 15, 30, 45, 60, and 120 min after glucose administration (3 mg/g body wt) to the peritoneal cavity. Blood glucose levels were measured by a Glucometer Elite (Bayer Corp., Elkhart, IN) and Glucose Analyzer (Analox GM7 Analyser, Anolox Instruments USA, Inc. Lunenberg, MA). We determined the optimal amount of BEL by comparing the effect of 1, 3, and 5 μg/g body wt. Although glucose tolerance was affected by every level of BEL, 3 and 5 μg/g gave more consistent results. We used 3 μg BEL/g body wt in all experiments: approximately the same concentration (10 μM = 3.17 μg/ml) that inhibits iPLA2 activity and insulin secretion in islets (21, 22, 38–40). The amount of propranolol (5 μg/g body wt) was optimized based on the daily dosage (160 mg/83 kg body wt) used in humans (66).
Clamp Studies
At 4–5 months of age, male age-matched C57BL/6J mice (25–32 g) were anesthetized with a Ketamine/xylazine mixture, and an indwelling catheter was inserted into the right jugular vein as previously described (54). The venous catheter was used for the multiple infusions. Experiments were performed in awake, unrestrained, chronically catheterized mice. The catheterized mice were grouped into BEL-treated and vehicle-treated groups.
Insulin sensitivity was assessed using the euglycemic hyperinsulinemic clamp technique (67) with modification (54). Briefly, food was removed for 5–6 h before the catheterized mice were iv injected with either BEL (3 μg/g body wt) or vehicle. A prime-continuous insulin infusion (21 mU/kg/min) was administered during the 90-min experiment. Blood glucose was clamped at 100 mg/dl ± 10 mg/dl by adjusting the rate of 10% glucose infusion according to blood glucose measurements performed every 10 min using a Glucometer Elite (Bayer Corp.). The glucose infusion rate (mg/kg/min) was calculated as the mean value for each 10-min interval during the last 60 min of the clamp. Under the euglycemic hyperinsulinemic state, and the amount of glucose infused is considered equivalent to whole body glucose disposal rate. Glucose disposal rate during the last 60 min was expressed as mean ± SEM mg/kg/min.
A 120-min hyperglycemic clamp was used to evaluate insulin secretion in β-cells. A continuous infusion of 10% glucose was used to maintain plasma glucose concentrations at 3 times the normal range (~300 mg/dl) as described previously (56). BEL (10 mM solution in 100% ethanol; infusion rate at 1 μl/min) was continuously administered during the experiment to inhibit any newly synthesized iPLA2. During the first 30 min, the glucose infusion rate was adjusted at 5-min intervals to elevate glucose concentrations to approximately 300 mg/dl. After this time, glucose levels were maintained around this level by adjusting the infusion rate at 10-min intervals. Insulin concentrations were measured in plasma samples obtained before and 30, 60, 90, and 120 min after the experiment had begun. Insulin levels were measured using an ultrasensitive insulin ELISA kit (Crystal Chem, Inc.). We averaged the insulin levels from the last four time points to determine the hyperglycemic challenge state as described (55).
Animal Care
C57/BL6J mice (The Jackson Laboratory, Bar Harbor, ME) were housed under a standard 12-h light, 12-h dark cycle with standard mouse chow and water ad libitum. Age-matched mice were used to perform each experiment. All procedures involving animals were approved by the Institutional Animal Care and Usage Committee (IACUC) of the Mount Sinai School of Medicine.
Statistical Analysis
Results were expressed as mean ± SEM. The statistical significance of differences was analyzed with a two-tailed unpaired t test, where P < 0.05 was considered significant.
Acknowledgments
We thank Drs. Charles Mobbs, H. Henry Dong, and X. Sherry Chi for critical discussion of the manuscript.
This work was supported by grants from the National Institutes of Health/National Institutes of Diabetes and Digestive and Kidney Diseases NIDDK (RO1DK063076) and the Juvenile Diabetes Research Foundation, International JDRF (1-2002-646).
Abbreviations
- AA
Arachidonic acid
- BEL
bromoenol lactone
- cPLA2
Ca2+-dependent PLA2
- FBS
fetal bovine serum
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- iPLA2
Ca2+-independent PLA2
- KRB
Krebs-Ringer bicarbonate
- PAP-1
phosphatidate phosphohydrolase
- PLA2
phospholipase A2
- RIPA
radioimmunoprecipitation assay
- siRNA
small interfering RNA
References
- 1.Six DA, Dennis EA. The expanding superfamily of phospholipase A2 enzymes: classification and characterization. Biochim Biophys Acta. 2000;1488:1–19. doi: 10.1016/s1388-1981(00)00105-0. [DOI] [PubMed] [Google Scholar]
- 2.Metz SA. Pancreatic islet phospholipase A2: differential, Ca2+-dependent effects of lysophospholipids, arachidonic acid, and its lipoxygenase-derived metabolites on insulin release. Adv Prostaglandin Thromboxane Leukot Res. 1987;17B:668–676. [PubMed] [Google Scholar]
- 3.Jones PM, Persaud SJ. Arachidonic acid as a second messenger in glucose-induced insulin secretion from pancreatic β-cells. J Endocrinol. 1993;137:7–14. doi: 10.1677/joe.0.1370007. [DOI] [PubMed] [Google Scholar]
- 4.Turk J, Gross RW, Ramanadham S. Amplification of insulin secretion by lipid messengers. Diabetes. 1993;42:367–374. doi: 10.2337/diab.42.3.367. [DOI] [PubMed] [Google Scholar]
- 5.Ma Z, Turk J. The molecular biology of the group VIA Ca2+-independent phospholipase A2. Prog Nucleic Acid Res Mol Biol. 2001;67:1–33. doi: 10.1016/s0079-6603(01)67023-5. [DOI] [PubMed] [Google Scholar]
- 6.Simonsson E, Karlsson S, Ahren B. Ca2+-independent phospholipase A2 contributes to the insulino-tropic action of cholecystokinin-8 in rat islets: dissociation from the mechanism of carbachol. Diabetes. 1998;47:1436–1443. doi: 10.2337/diabetes.47.9.1436. [DOI] [PubMed] [Google Scholar]
- 7.Ehses JA, Lee SS, Pederson RA, McIntosh CH. A new pathway for glucose-dependent insulinotropic polypeptide (GIP) receptor signaling: evidence for the involvement of phospholipase A2 in GIP-stimulated insulin secretion. J Biol Chem. 2001;276:23667–23673. doi: 10.1074/jbc.M103023200. [DOI] [PubMed] [Google Scholar]
- 8.Konrad RJ, Jolly YC, Major C, Wolf BA. Carbachol stimulation of phospholipase A2 and insulin secretion in pancreatic islets. Biochem J. 1992;287:283–290. doi: 10.1042/bj2870283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Laychock SG. Phospholipase A2 activity in pancreatic islets is calcium-dependent and stimulated by glucose. Cell Calcium. 1982;3:43–54. doi: 10.1016/0143-4160(82)90036-7. [DOI] [PubMed] [Google Scholar]
- 10.Kato R, Yamamoto S, Nakadate T, Nakaki T. Possible involvement of phospholipase A2 activation and lipoxygenase product(s) in the mechanism of insulin secretion. Adv Prostaglandin Thromboxane Leukot Res. 1983;12:265–270. [PubMed] [Google Scholar]
- 11.Metz SA. Is phospholipase A2 a “glucose sensor” responsible for the phasic pattern of insulin release? Prostaglandins. 1984;27:147–158. doi: 10.1016/0090-6980(84)90228-4. [DOI] [PubMed] [Google Scholar]
- 12.Dunlop ME, Larkins RG. Activity of endogenous phospholipase C and phospholipase A2 in glucose stimulated pancreatic islets. Biochem Biophys Res Commun. 1984;120:820–827. doi: 10.1016/s0006-291x(84)80180-1. [DOI] [PubMed] [Google Scholar]
- 13.Dunlop M, Clark S. Glucose-induced phosphoryla-tion and activation of a high molecular weight cytosolic phospholipase A2 in neonatal rat pancreatic islets. Int J Biochem Cell Biol. 1995;27:1191–1199. doi: 10.1016/1357-2725(95)00093-5. [DOI] [PubMed] [Google Scholar]
- 14.Gagliardino JJ, Borelli MI, de Gagliardino EE, Garcia ME. Role of phospholipase and calmodulin inhibitors on insulin, arachidonic acid and prostaglandin E2 release. Diabetes Res Clin Pract. 1985;1:327–333. doi: 10.1016/s0168-8227(86)80045-6. [DOI] [PubMed] [Google Scholar]
- 15.Fujimoto WY, Metz SA. Phasic effects of glucose, phospholipase A2, and lysophospholipids on insulin secretion. Endocrinology. 1987;120:1750–1757. doi: 10.1210/endo-120-5-1750. [DOI] [PubMed] [Google Scholar]
- 16.Zawalich W, Zawalich K. Effect of exogenous phospholipase A2 on insulin secretion from perifused rat islets. Diabetes. 1985;34:471–476. doi: 10.2337/diab.34.5.471. [DOI] [PubMed] [Google Scholar]
- 17.Turk J, Wolf BA, Lefkowith JB, Stump WT, McDaniel ML. Glucose-induced phospholipid hydrolysis in isolated pancreatic islets: quantitative effects on the phospholipid content of arachidonate and other fatty acids. Biochim Biophys Acta. 1986;879:399–409. doi: 10.1016/0005-2760(86)90232-8. [DOI] [PubMed] [Google Scholar]
- 18.Konrad RJ, Jolly YC, Major C, Wolf BA. Inhibition of phospholipase A2 and insulin secretion in pancreatic islets. Biochim Biophys Acta. 1992;1135:215–220. doi: 10.1016/0167-4889(92)90139-3. [DOI] [PubMed] [Google Scholar]
- 19.Band AM, Jones PM, Howell SL. Arachidonic acid-induced insulin secretion from rat islets of Langerhans. J Mol Endocrinol. 1992;8:95–101. doi: 10.1677/jme.0.0080095. [DOI] [PubMed] [Google Scholar]
- 20.Turk J, Mueller M, Bohrer A, Ramanadham S. Arachidonic acid metabolism in isolated pancreatic islets. VI. Carbohydrate insulin secretagogues must be metabolized to induce eicosanoid release. Biochim Biophys Acta. 1992;1125:280–291. doi: 10.1016/0005-2760(92)90057-3. [DOI] [PubMed] [Google Scholar]
- 21.Gross RW, Ramanadham S, Kruszka KK, Han X, Turk J. Rat and human pancreatic islet cells contain a calcium ion independent phospholipase A2 activity selective for hydrolysis of arachidonate which is stimulated by adenosine triphosphate and is specifically localized to islet β-cells. Biochemistry. 1993;32:327–336. doi: 10.1021/bi00052a041. [DOI] [PubMed] [Google Scholar]
- 22.Ramanadham S, Gross RW, Han X, Turk J. Inhibition of arachidonate release by secretagogue-stimulated pancreatic islets suppresses both insulin secretion and the rise in β-cell cytosolic calcium ion concentration. Biochemistry. 1993;32:337–346. doi: 10.1021/bi00052a042. [DOI] [PubMed] [Google Scholar]
- 23.Loweth AC, Scarpello JH, Morgan NG. A specific inhibitor of cytosolic phospholipase A2 activity, AA-COCF3, inhibits glucose-induced insulin secretion from isolated rat islets. Biochem Biophys Res Commun. 1996;218:423–427. doi: 10.1006/bbrc.1996.0075. [DOI] [PubMed] [Google Scholar]
- 24.Ma Z, Ramanadham S, Wohltmann M, Bohrer A, Hsu FF, Turk J. Studies of insulin secretory responses and of arachidonic acid incorporation into phospholipids of stably transfected insulinoma cells that overexpress group VIA phospholipase A2 (iPLA2β) indicate a signaling rather than a housekeeping role for iPLA2β. J Biol Chem. 2001;276:13198–13208. doi: 10.1074/jbc.M010423200. [DOI] [PubMed] [Google Scholar]
- 25.Persaud SJ, Roderigo-Milne HM, Squires PE, Sugden D, Wheeler-Jones CP, Marsh PJ, Belin VD, Luther MJ, Jones PM. A key role for β-cell cytosolic phospholipase A2 in the maintenance of insulin stores but not in the initiation of insulin secretion. Diabetes. 2002;51:98–104. doi: 10.2337/diabetes.51.1.98. [DOI] [PubMed] [Google Scholar]
- 26.Ma Z, Ramanadham S, Hu Z, Turk J. Cloning and expression of a group IV cytosolic Ca2+-dependent phospholipase A2 from rat pancreatic islets. Comparison of the expressed activity with that of an islet group VI cytosolic Ca2+-independent phospholipase A2. Biochim Biophys Acta. 1998;1391:384–400. doi: 10.1016/s0005-2760(98)00027-7. [DOI] [PubMed] [Google Scholar]
- 27.Jones PM, Burns CJ, Belin VD, Roderigo-Milne HM, Persaud SJ. The role of cytosolic phospholipase A2 in insulin secretion. Diabetes. 2004;53(Suppl 1):S172–S178. doi: 10.2337/diabetes.53.2007.s172. [DOI] [PubMed] [Google Scholar]
- 28.Juhl K, Hoy M, Olsen HL, Bokvist K, Efanov AM, Hoffmann EK, Gromada J. cPLA2α-evoked formation of arachidonic acid and lysophospholipids is required for exocytosis in mouse pancreatic β-cells. Am J Physiol Endocrinol Metab. 2003;285:E73–E81. doi: 10.1152/ajpendo.00086.2003. [DOI] [PubMed] [Google Scholar]
- 29.Wolf BA, Turk J, Sherman WR, McDaniel ML. Intra-cellular Ca2+ mobilization by arachidonic acid. Comparison with myoinositol 1,4,5-trisphosphate in isolated pancreatic islets. J Biol Chem. 1986;261:3501–3511. [PubMed] [Google Scholar]
- 30.Wolf BA, Pasquale SM, Turk J. Free fatty acid accumulation in secretagogue-stimulated pancreatic islets and effects of arachidonate on depolarization-induced insulin secretion. Biochemistry. 1991;30:6372–6379. doi: 10.1021/bi00240a004. [DOI] [PubMed] [Google Scholar]
- 31.Ramanadham S, Gross R, Turk J. Arachidonic acid induces an increase in the cytosolic calcium concentration in single pancreatic islet β cells. Biochem Biophys Res Commun. 1992;184:647–653. doi: 10.1016/0006-291x(92)90638-2. [DOI] [PubMed] [Google Scholar]
- 32.Ramanadham S, Turk J. ω-Conotoxin inhibits glucose- and arachidonic acid-induced rises in intracellular [Ca2+] in rat pancreatic islet β-cells. Cell Calcium. 1994;15:259–264. doi: 10.1016/0143-4160(94)90065-5. [DOI] [PubMed] [Google Scholar]
- 33.Shuttleworth TJ. Arachidonic acid activates the noncapacitative entry of Ca2+ during [Ca2+]i oscillations. J Biol Chem. 1996;271:21720–21725. doi: 10.1074/jbc.271.36.21720. [DOI] [PubMed] [Google Scholar]
- 34.Meglasson MD, Matschinsky FM. Pancreatic islet glucose metabolism and regulation of insulin secretion. Diabetes Metab Rev. 1986;2:163–214. doi: 10.1002/dmr.5610020301. [DOI] [PubMed] [Google Scholar]
- 35.Hazen SL, Zupan LA, Weiss RH, Getman DP, Gross RW. Suicide inhibition of canine myocardial cytosolic calcium-independent phospholipase A2. Mechanism-based discrimination between calcium-dependent and -independent phospholipases A2. J Biol Chem. 1991;266:7227–7232. [PubMed] [Google Scholar]
- 36.Ackermann EJ, Conde-Frieboes K, Dennis EA. Inhibition of macrophage Ca2+-independent phospholipase A2 by bromoenol lactone and trifluoromethyl ketones. J Biol Chem. 1995;270:445–450. doi: 10.1074/jbc.270.1.445. [DOI] [PubMed] [Google Scholar]
- 37.Balsinde J, Balboa MA, Insel PA, Dennis EA. Regulation and inhibition of phospholipase A2. Annu Rev Pharmacol Toxicol. 1999;39:175–189. doi: 10.1146/annurev.pharmtox.39.1.175. [DOI] [PubMed] [Google Scholar]
- 38.Ramanadham S, Bohrer A, Gross RW, Turk J. Mass spectrometric characterization of arachidonate-containing plasmalogens in human pancreatic islets and in rat islet β-cells and subcellular membranes. Biochemistry. 1993;32:13499–13509. doi: 10.1021/bi00212a015. [DOI] [PubMed] [Google Scholar]
- 39.Ramanadham S, Bohrer A, Mueller M, Jett P, Gross RW, Turk J. Mass spectrometric identification and quantitation of arachidonate-containing phospholipids in pancreatic islets: prominence of plasmenylethanolamine molecular species. Biochemistry. 1993;32:5339–5351. doi: 10.1021/bi00071a009. [DOI] [PubMed] [Google Scholar]
- 40.Ramanadham S, Wolf MJ, Jett PA, Gross RW, Turk J. Characterization of an ATP-stimulatable Ca2+-independent phospholipase A2 from clonal insulin-secreting HIT cells and rat pancreatic islets: a possible molecular component of the β-cell fuel sensor. Biochemistry. 1994;33:7442–7452. doi: 10.1021/bi00189a052. [DOI] [PubMed] [Google Scholar]
- 41.Ma Z, Ramanadham S, Kempe K, Chi XS, Ladenson J, Turk J. Pancreatic islets express a Ca2+-independent phospholipase A2 enzyme that contains a repeated structural motif homologous to the integral membrane protein binding domain of ankyrin. J Biol Chem. 1997;272:11118–11127. [PubMed] [Google Scholar]
- 42.Ma Z, Wang X, Nowatzke W, Ramanadham S, Turk J. Human pancreatic islets express mRNA species encoding two distinct catalytically active isoforms of group VI phospholipase A2 (iPLA2) that arise from an exon-skipping mechanism of alternative splicing of the transcript from the iPLA2 gene on chromosome 22q13.1. J Biol Chem. 1999;274:9607–9616. doi: 10.1074/jbc.274.14.9607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ma Z, Bohrer A, Wohltmann M, Ramanadham S, Hsu FF, Turk J. Studies of phospholipid metabolism, proliferation, and secretion of stably transfected insulinoma cells that overexpress group VIA phospholipase A2. Lipids. 2001;36:689–700. doi: 10.1007/s11745-001-0774-9. [DOI] [PubMed] [Google Scholar]
- 44.Ma Z, Zhang S, Turk J, Ramanadham S. Stimulation of insulin secretion and associated nuclear accumulation of iPLA2β in INS-1 insulinoma cells. Am J Physiol Endocrinol Metab. 2002;282:E820–E833. doi: 10.1152/ajpendo.00165.2001. [DOI] [PubMed] [Google Scholar]
- 45.Hannon GJ. RNA interference. Nature. 2002;418:244–251. doi: 10.1038/418244a. [DOI] [PubMed] [Google Scholar]
- 46.Ramanadham S, Hsu FF, Bohrer A, Ma Z, Turk J. Studies of the role of group VI phospholipase A2 in fatty acid incorporation, phospholipid remodeling, lysophosphatidylcholine generation, and secretagogue-induced arachidonic acid release in pancreatic islets and insulinoma cells. J Biol Chem. 1999;274:13915–13927. doi: 10.1074/jbc.274.20.13915. [DOI] [PubMed] [Google Scholar]
- 47.Balsinde J, Dennis EA. Bromoenol lactone inhibits magnesium-dependent phosphatidate phosphohydrolase and blocks triacylglycerol biosynthesis in mouse P388D1 macrophages. J Biol Chem. 1996;271:31937–31941. doi: 10.1074/jbc.271.50.31937. [DOI] [PubMed] [Google Scholar]
- 48.Balboa MA, Balsinde J, Dennis EA. Involvement of phosphatidate phosphohydrolase in arachidonic acid mobilization in human amnionic WISH cells. J Biol Chem. 1998;273:7684–7690. doi: 10.1074/jbc.273.13.7684. [DOI] [PubMed] [Google Scholar]
- 49.Hsu FF, Ma Z, Wohltmann M, Bohrer A, Nowatzke W, Ramanadham S, Turk J. Electrospray ionization/mass spectrometric analyses of human promonocytic U937 cell glycerolipids and evidence that differentiation is associated with membrane lipid composition changes that facilitate phospholipase A2 activation. J Biol Chem. 2000;275:16579–16589. doi: 10.1074/jbc.M908342199. [DOI] [PubMed] [Google Scholar]
- 50.Abdel-Latif AA, Smith JP. Studies on the effects of magnesium ion and propranolol on iris muscle phosphatidate phosphohydrolase. Can J Biochem Cell Biol. 1984;62:170–177. doi: 10.1139/o84-024. [DOI] [PubMed] [Google Scholar]
- 51.Pappu AS, Hauser G. Propranolol-induced inhibition of rat brain cytoplasmic phosphatidate phosphohydrolase. Neurochem Res. 1983;8:1565–1575. doi: 10.1007/BF00964158. [DOI] [PubMed] [Google Scholar]
- 52.Meier KE, Gause KC, Wisehart-Johnson AE, Gore AC, Finley EL, Jones LG, Bradshaw CD, McNair AF, Ella KM. Effects of propranolol on phosphatidate phosphohydrolase and mitogen-activated protein kinase activities in A7r5 vascular smooth muscle cells. Cell Signal. 1998;10:415–426. doi: 10.1016/s0898-6568(97)00140-x. [DOI] [PubMed] [Google Scholar]
- 53.Johnson CA, Balboa MA, Balsinde J, Dennis EA. Regulation of cyclooxygenase-2 expression by phosphatidate phosphohydrolase in human amnionic WISH cells. J Biol Chem. 1999;274:27689–27693. doi: 10.1074/jbc.274.39.27689. [DOI] [PubMed] [Google Scholar]
- 54.Rossetti L, Chen W, Hu M, Hawkins M, Barzilai N, Efrat S. Abnormal regulation of HGP by hyperglycemia in mice with a disrupted glucokinase allele. Am J Physiol Endocrinol Metab. 1997;273:E743–E750. doi: 10.1152/ajpendo.1997.273.4.E743. [DOI] [PubMed] [Google Scholar]
- 55.Niswender KD, Shiota M, Postic C, Cherrington AD, Magnuson MA. Effects of increased glucokinase gene copy number on glucose homeostasis and hepatic glucose metabolism. J Biol Chem. 1997;272:22570–22575. doi: 10.1074/jbc.272.36.22570. [DOI] [PubMed] [Google Scholar]
- 56.Niswender KD, Postic C, Jetton TL, Bennett BD, Piston DW, Efrat S, Magnuson MA. Cell-specific expression and regulation of a glucokinase gene locus transgene. J Biol Chem. 1997;272:22564–22569. doi: 10.1074/jbc.272.36.22564. [DOI] [PubMed] [Google Scholar]
- 57.Metz SA. Exogenous arachidonic acid promotes insulin release from intact or permeabilized rat islets by dual mechanisms. Putative activation of Ca2+ mobilization and protein kinase C. Diabetes. 1988;37:1453–1469. doi: 10.2337/diab.37.11.1453. [DOI] [PubMed] [Google Scholar]
- 58.Landt M, Easom RA, Colca JR, Wolf BA, Turk J, Mills LA, McDaniel ML. Parallel effects of arachidonic acid on insulin secretion, calmodulin-dependent protein kinase activity and protein kinase C activity in pancreatic islets. Cell Calcium. 1992;13:163–172. doi: 10.1016/0143-4160(92)90044-s. [DOI] [PubMed] [Google Scholar]
- 59.Band AM, Jones PM, Howell SL. The mechanism of arachidonic acid-induced insulin secretion from rat islets of Langerhans. Biochim Biophys Acta. 1993;1176:64–68. doi: 10.1016/0167-4889(93)90178-r. [DOI] [PubMed] [Google Scholar]
- 60.Ramanadham S, Hsu F, Zhang S, Bohrer A, Ma Z, Turk J. Electrospray ionization mass spectrometric analyses of phospholipids from INS-1 insulinoma cells: comparison to pancreatic islets and effects of fatty acid supplementation on phospholipid composition and insulin secretion. Biochim Biophys Acta. 2000;1484:251–266. doi: 10.1016/s1388-1981(00)00022-6. [DOI] [PubMed] [Google Scholar]
- 61.Hanks SK, Quinn AM, Hunter T. The protein kinase family: conserved features and deduced phylogeny of the catalytic domains. Science. 1988;241:42–52. doi: 10.1126/science.3291115. [DOI] [PubMed] [Google Scholar]
- 62.Wolf MJ, Gross RW. Expression, purification, and kinetic characterization of a recombinant 80-kDa intracellular calcium-independent phospholipase A2. J Biol Chem. 1996;271:30879–30885. doi: 10.1074/jbc.271.48.30879. [DOI] [PubMed] [Google Scholar]
- 63.Smani T, Zakharov SI, Csutora P, Leno E, Trepakova ES, Bolotina VM. A novel mechanism for the store-operated calcium influx pathway. Nat Cell Biol. 2004;6:113–120. doi: 10.1038/ncb1089. [DOI] [PubMed] [Google Scholar]
- 64.Smani T, Zakharov SI, Leno E, Csutora P, Trepakova ES, Bolotina VM. Ca2+-independent phospholipase A2 is a novel determinant of store-operated Ca2+ entry. J Biol Chem. 2003;278:11909–11915. doi: 10.1074/jbc.M210878200. [DOI] [PubMed] [Google Scholar]
- 65.Lacy PE, Kostianovsky M. Method for the isolation of intact islets of Langerhans from the rat pancreas. Diabetes. 1967;16:35–39. doi: 10.2337/diab.16.1.35. [DOI] [PubMed] [Google Scholar]
- 66.Ferrara LA, Capaldo B, Rivellese AA, Genovese S, Iovine C, Russo L, Mancini M. Effects of β-receptor blockade on carbohydrate metabolism. J Hypertens. 1985;3(Suppl 3):S199–S201. [PubMed] [Google Scholar]
- 67.DeFronzo RA, Tobin JD, Andres R. Glucose clamp technique: a method for quantifying insulin secretion and resistance. Am J Physiol Endocrinol Metab. 1979;237:E214–E223. doi: 10.1152/ajpendo.1979.237.3.E214. [DOI] [PubMed] [Google Scholar]