

1. INTRODUCTION
The metabolic chart is not flat since all metabolites in various pathways are not equally represented in time and space within an organism. For the most part, we do not know the sources or flux rates of metabolites. Patients with metabolic defects are sometimes treated by dietary manipulations [1,2,3], because they have hypomorphic alleles, allowing for direct or indirect stimulation of an injured pathway. Here, we focus on a few Congenital Disorders of Glycosylation (CDG) and related diseases that impair sugar chain (glycan) synthesis with specific reference to the expectations, and surprises when monosaccharide supplements are given to patients or animals with inherited glycosylation deficiencies (table 1). These examples provide insights and cautions for studies covering the four-dimensional metabolic chart.
Table 1.
Disease names, genes, and enzymatic defects, and typical symptoms.
| Disease Name | Gene | Enzymatic Defect | Symptoms |
|---|---|---|---|
| CDG-Ia (PMM2-CDG) | PMM2 | Man-6-P→Man-l-P | psychomotor retardation, hypotonia, esotropia, lipodystrophy, cerebellar hypoplasia, storke-like episodes, seizures |
| CDG-Ib (MPI-CDG) | MPI | Man-6-P↔Fru-6-P | hepatic fibrosis, protein-losing enteropathy, coagulopathy, hypoglycemia |
| CDG-IIc (SLC35C – CDG) | SLC35C1 | GDP-Fuc(c)→GDP-Fuc(g) | recurrent infections, persistent neutrophilia, psychomotor retardation, microcephaly, hypotonia |
| HIBM/DMRV | GNE | UDP-GIcNAc→ManNAc-6-P | adult onset with progressive distal and proximal muscle weakness; spares quadriceps |
2. BIOSYNTHETIC PATHWAYS AND GLYCANS
2.1 Sources of Precursors
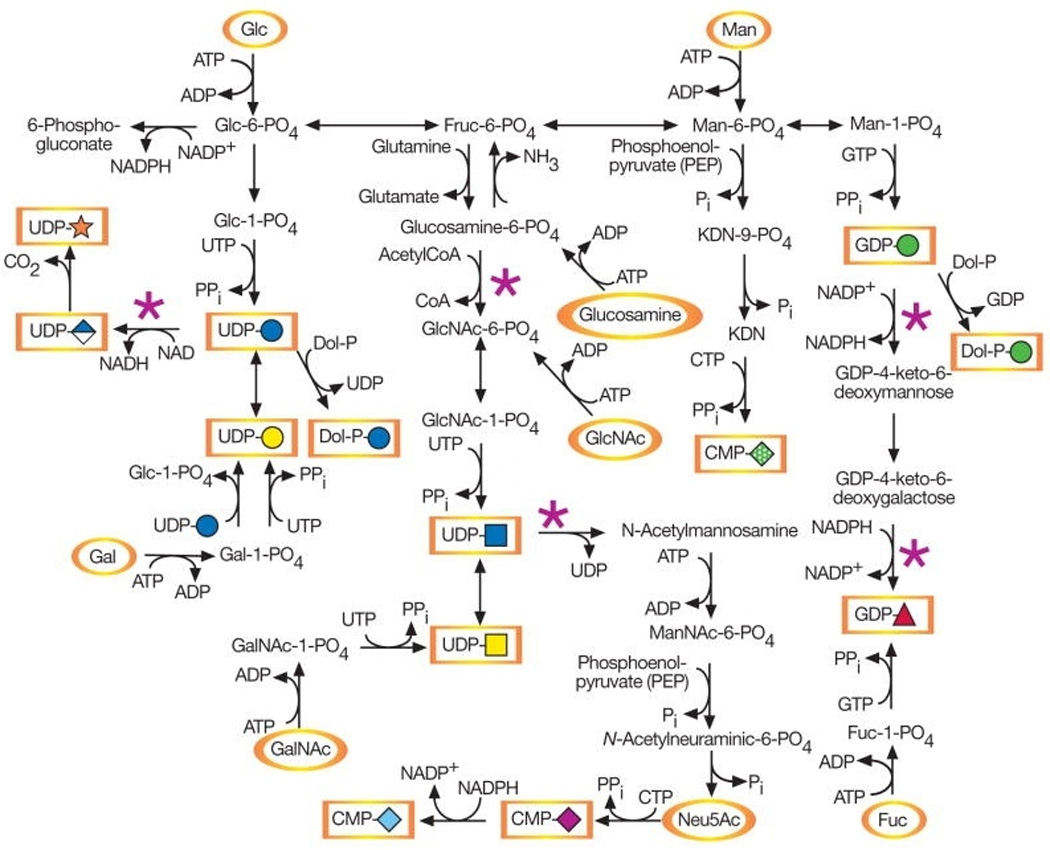
De novo and salvage metabolic pathways for monosaccharides are shown in Figure 1 [4]. Monosaccharides can be derived from the diet, or salvaged (reutilized) from degraded endogenous glycans.
Figure 1.
 Glc Glc |
 Man Man |
 Gal Gal |
 GlcNAc GlcNAc |
 GalNAc GalNAc |
 Sia Sia |
 Fuc Fuc |
 Xyl Xyl |
 GlcA GlcA |
Animal cell glycans made within the ER/Golgi contain nine precursor sugars: glucose, mannose, galactose, N-acetylglucosamine, N-acetylgalactosamine, sialic acid(s), fucose, xylose and glucuronic acid. All can be derived exclusively from glucose. Some occur in the diet or are indirectly salvaged from glycans degraded within lysosomes [4]. The relative contributions of de novo, salvage and dietary pathways are are quite variable but mostly unknown [5,6]. How the preferences translate into physiology/pathology probably depends on the relative biosynthetic loads of different tissues. For instance, during embryonic development chondrocytes carry a heavy glycosylation burden [7], and in postnatal growth, hepatocytes make most plasma glycoproteins.
2.2 Glycosylation Pathways
The N-glycan precursor is assembled on a carrier lipid (dolichol-P-P) into a 1sugar unit containing 9 mannose, 3 glucose and 2 N-acetylglucosamine residues which is transferred to Asn within a Asn-X-Thr/Ser sequence of newly synthesized proteins in the ER. The protein-bound chains are trimmed, processed and extended within the Golgi. Membrane-associated proteins in the ER, Golgi, lysosome and on the cell surface contain N-glycans as do extracellular matrix and secreted proteins. O-GalNAc linked glycans are usually found in large clusters on secreted and cell surface mucins typically produced by epithelial cells. Some of the O-GalNAc containing proteins also function as ligands for lectins such as the selectins, which are important for trafficking of leukocytes [8]. Glycophospholipid anchors and glycosphingolipids, often occur in lipid rafts [9]. Proteoglycans are made on a limited number of core proteins. They begin with xylose linked to Ser and are extended with glycosaminoglycan chains, e.g., heparan sulfate, chondroitin- and dermatan-sulfate, which influence morphogen and growth factor distribution [10]. Quantitatively smaller, more recently appreciated important glycosylation pathways include O-Fucose (on Notch receptors and ligands) [11], O-glucose (Notch receptors/igands) [12], and O-mannose (primarily on α-dystroglycan at neuromuscular junctions) [13,14]. The occurrence of monosaccharides in these pathways is shown in Table 2.
Table 2. Monosaccharides and Glycosylation Pathways in Animals.
Monosaccharides in Animal Glycans.
| Glc
|
Man
|
Gal
|
GlcNAc
|
GalNAc
|
Sia
|
Fuc
|
Xyl
|
GlcA
|
|
|---|---|---|---|---|---|---|---|---|---|
| Major Pathways | |||||||||
| N-GlcNAc | + | ++ | ++ | ++ | + | ++ | ++ | + | + |
| O-GalNAc | − | − | ++ | + | ++ | ++ | + | − | + |
| O-Xyl | |||||||||
| HS | − | − | + | +++ | − | − | − | + | +++ |
| CS/DS | − | − | + | − | +++ | − | − | + | +++ |
| GSL | ++ | − | ++ | + | + | ++ | + | − | + |
| GPI | − | +++ | + | +(GlcN) | − | + | − | − | − |
| Minor pathways | |||||||||
| O-Man | − | ++ | ++ | ++ | ++ | + | − | + | |
| O-Glc | ++ | − | − | − | − | − | + | − | |
| O-Fuc | − | − | + | ++ | − | + | ++ | − | − |
|
Glc |
Man |
Gal |
|
GlcNAc |
GalNAc |
Sia |
|
Fuc |
Xyl |
GlcA |
3. GLYCOSYLATION DISORDERS
An estimated 2–3% of the genome encodes proteins used for biosynthesis and/or recognition of glycans. In just over a decade, >40 different human genetic disorders were discovered that affect one or more glycosylation pathways [15]. Below we focus on several disorders where the patients’ pathology results from a limited supply of biosynthetic precursors.
3.1 CDG-Ia
3.1.1 Metabolic Steps
CDG-Ia and Ib involve the earliest steps of mannose metabolism. Mannose-P can be made from fructose-6-P using phosphomannose isomerase (MPI) or from mannose imported directly from an extracellular source (diet or turnover) via a hexose transporter. It is then converted into Man-1-P using phosphomannomutase 2 (PMM2, Man-6-P→ Man-1-P) which then leads to GDP-Man.
3.1.2 Patients
This disorder is caused by deficiency in PMM2 (<25% normal), which limits the amount of mannose flux into the glycosylation pathway resulting in sub-optimal occupancy of N-linked sites [16]. It is unknown whether other glycosylation pathways are also affected.
CDG-Ia patients are usually born at a normal weight, but are often symptomatic at birth and therefore show evidence of developmental abnormalities [17]. The babies often have an unusual distribution of fat above their buttocks along with inverted nipples. Symptoms which appear early after birth include stunted growth, crossed eyes, hypotonia (floppiness at the trunk) and highly variable developmental delay. They usually fail to develop a normal-sized, functional cerebellum. It is unknown whether this is due to disrupted migration of cells moving into the cerebellum, impaired growth, stress-related cell death or a combination of these. Children have abnormal skeletons, seizures, coagulopathy, and indications of liver damage [18,19,20,21]. The high demands placed on the chondrocytes that create the environment for proper bone ossification and on hepatocytes, which synthesize plasma proteins including clotting factors may help to explain some of these symptoms. Some children develop cardiomyopathy, pericardial effusions and ischemia. Mortality is about 20% in the first 5 years and is often due to severe infections.
3.1.3. Therapy
There is no biochemical or molecular therapy for this disorder. Impaired glycosylation seen in CDG-Ia patient cells improves when mannose is added to the culture medium. However, mannose supplements for patients are ineffective, probably because any additional Man-6-P formed by hexokinase would likely be catabolized by the fully active MPI. Mannose might be more effectively used if the degradation of pathway of Man-6-P via MPI were partially blocked [16]. Another idea is that the deficient substrate Man-1-P could be supplied as a cell-permeable pro-drug that would subsequently be converted into the parent compound for incorporation into glycans [16]. To date neither proposal has produced positive results. Direct enzymatic assays show that some patients have up to 25% residual enzymatic activity. Perhaps stabilizing mutated protein or modestly increasing the flux though the pathway could improve some symptoms in patients, especially those with high residual activity.
3.1.4. Animal Models
CDG-Ia is the most common of these rare glycosylation disorders with over 800 documented cases [22]. Ablation of the Pmm2 mouse gene is lethal within two days of fertilization [23]. Ablation of the first step in the N-glycosylation pathway also shows very early lethality [24]. True mouse models of CDG-Ia may require construction of hypomorphic alleles in mice based on homology with the patients’ mutations. Such constructs and studies are ongoing [25].
Recent studies in C.elegans used a sub-phenotypic dose of the N-glycosylation inhibitor tunicamycin along with RNAi technology to identify genes that caused a synthetic or enhanced deleterious phenotype. Of the 500 genes identified were a monosaccharide transporter, a subunit of the oligosaccharyl transferase complex, and several sugar transferases including 4 used for synthesis of the lipid precursor glycan. Nearly 200 candidate genes had unknown functions. Heterozygous mutations in some of these may help explain the wide phenotypic variability seen in CDG patients. [26].
3.2 CDG-Ib
3.2.1 Metabolic Steps
See 3.1.1
3.2.2 Patients
CDG-Ib is caused by mutations in MPI encoding phosphomannose isomerase (MPI, Fruc-6-P↔Man-6-P) which leads to under-occupied glycosylation sites similar to CDG-Ia. However, there are no obvious developmental abnormalities in utero. Ductal plate malformation in the liver during development contributes to later complications such as liver fibrosis [27]. Patients are born normally, but may suffer from hypoglycemia, failure to thrive (growth), coagulopathy, and intestinal abnormalities including protein-losing enteropthy (loss of plasma proteins via paracellular leakage in the small intestine) especially when patients are exposed to infection or stress. They have no dysmorphic features and have normal intelligence, neurologic and psychomotor development. This is surprising, since this defect occurs in the metabolic step immediately prior to PMM2, where patients have a much more severe phenotype. A number of CDG-Ib patients died prior to a correct diagnosis of impaired glycosylation [28,29,30].
3.2.3 Therapy
Daily dietary supplements of mannose reversed nearly all of the symptoms of this disorder, except for the liver fibrosis or bile duct abnormality [31]. Within a few weeks, abnormal glycosylation, coagulopathy, and enteric protein loss resolved [32]. A two-year-old child with CDG-Ib and short stature [33] became normal within a few months of starting mannose therapy. Patients usually have 2–15% residual MPI activity. Normally, human plasma mannose concentration is 1% that of glucose or ~55uM. Drinking 0.15gm/kg mannose 4 times per day raises the concentration to about 150–200uM with a turnover T1/2 of about 4 hr [34]. Mannose normally provides only about 2–4% of the mannose found in N-glycans in most tissues and cell types. Fibroblasts are an exception because they derive much more from mannose [35]. By far the great majority (96–98%) of mannose is derived from glucose (Sharma and Freeze, unpublished studies). Surprisingly, very small increases in mannose concentration is sufficient to rescue impaired glycosylation in the patients. Under normal conditions, MPI also “burns” Man-6-P by dispatching it into glycolysis as Fructose-6-P. No studies have been done to determine exactly the contributions of mannose and glucose to the glycosylation pathways in humans. It is likely to vary in different tissues depending on the ratio of PMM2: MPI since both compete for the same substrate, Man-6-P. Cell and animal models using stable isotopes and mass spectrometric analysis are likely to be informative [36].
3.2.4 Animal models
Ablation of the mouse Mpi causes embryonic lethality at ~ 11.5d. The embryos showed growth retardation and placental hyperplasia. More than 90% failed to from yolk sac vasculature and one-third failed chorioallantoic fusion. Moreover, the embryos showed abnormalities in heart formation [37]. Glycosylation of the embryo appears to be sufficient based on normal lectin (concanavalin A) staining. Mouse sera contains ~100uM mannose, and adding mannose to the drinking water increases plasma mannose levels by up to 10-fold [38]. Attempts to rescue the Mpi null embryos by providing the dams with mannose had a decidedly negative effect: Mpi null embryos died even earlier and were resorbed [37] due to the accumulation of very high (mM) concentrations of Man-6-P, since it cannot be catabolized. Increased Man-6-P inhibits several glycolytic enzymes and depletes ATP, presumably by engaging in a futile cycle of phosphorylation and dephosphorylation. Other metabolic consequences may also occur as the embryos attempt to cope with the toxic levels of Man-6-P. Since Man-6-P is produced only within the cell, Man-6-P toxicity is cell autonomous. Temporal or tissue specific Mpi knockout constructs are unlikely to be useful since they would probably cause cell death wherever excision occurs.
As with lethal Pmm2 knockout, another option is to knock in a patient-like mutation to create a hypomorphic allele in the mouse. A recent abstract suggests that this approach may be successful since it yields viable embryos [39]. Whether they will model CDG-Ib is not yet clear, but this system should have the advantage of quantitatively measuring the contributions of glucose and mannose to various glycosylation pathways with and without mannose supplements.
No other animal models of CDG-Ib or MPI-deficiency have been reported. Zebrafish morphants should also be amenable to study [40].
3.3 CDG-IIc (Leukocyte Adhesion Deficiency, Type II)
3.3.1 Metabolic Steps
GDP-Fucose can be derived from two sources; 1.) GDP-Man via a two-step reaction in the cytoplasm or 2.) Fucose→Fucose-P→ GDP-Fucose, which must be transported into the ER or Golgi using GDP-Fucose transporters.
3.3.2 Patients
CDG IIc, aka, Leukocyte Adhesion Deficiency (LAD), type II is an autosomal recessive disorder first discovered in 1992 [41]. Patients had mental retardation, facial dysmorphia, recurrent infections and rare Bombay blood group, which is caused by the absence of the fucosylated H-antigen. The patients also had leukocytosis (elevated circulating leukocytes), immunodeficiency and lacked fucosylated glycans on the leukocyte cell surface due to a defective GDP-fucose transporter, SLC35C1 [42,43]. Fucosylated selectin ligands are required for leukocyte rolling on endothelial cells via their binding to E-, P- and L-selectins prior to firm adhesion and diapadesis.
3.3.3 Therapy
One CDG-IIc patient homozygous for SLC35C1 mutation received daily oral fucose supplements. Within two days, the leukocytosis resolved, recurrent infections ceased, and he gained significant weight. His psychomotor condition also improved, most likely as a result of a more stable medical status. Fucosylated selectin ligands were re-expressed within 40 days of starting therapy facilitating binding of neutrophils to endothelium [44]. Exogenous fucose also improved fucosylation of IgM, but surprisingly there was no expression of α-1,2-fucosylated H antigen even 10 years after beginning therapy. This indicates that the utilization of exogenous fucose was cell type specific and restricted to certain glycans. Presumably fucose was taken up by the recipient cells, phosphorylated to fucose-1-P, which then increased the endogenous pool of GDP-Fucose. Other CDG-IIc patients homozygous for another mutation did not benefit from fucose therapy [45], showing that elevated GDP-fucose levels cannot correct all patients.
3.3.4. Mouse Model of CDG-IIc
Slc35c1 knockout mice were generated [46] and about one-third mice died within first week. Slc35c1 deficient mice had 5.7% normal GDP-fucose transport activity. They showed leukocytosis, granulocytes that failed to adhere to E- and P-selectins and impaired leukocyte rolling in endothelial venules similar to CDG IIc patients. Another study showed defective leukocyte rolling to cremaster venules and lymphocyte homing to lymph nodes, but normal lymphocyte homing to the spleen, which probably is able to provide some adaptive immunity in Slc35c−/− mice [47]. Fucose supplements partially normalized fucosylation in Slc35c−/− fibroblasts thus suggesting an alternative transport mechanism.
3.3.5 Related Mouse Models
Hypofucosylation in other genetically engineered mice helps us to understand the mechanism of fucose rescue. The de novo pathway of GDP-Fucose synthesis begins with GDP-Man and requires two enzymes: GDP-mannose-4,6 dehydratase and GDP-4-keto-6-deoxymannose3,5-epimerase-4-reductase (FX). Breeding heterozygous Fx+/− mice very rarely produced Fx−/− progeny; most died in utero and before weaning [48]. Providing dietary fucose to the dams was unable to rescue these embryos to term, but maternally derived (salvaged) fucose did provide some fucosylated proteins. Two rare Fx−/− male survivors were crossed with Fx+/− females, which gave increased survival of Fx−/− animals from Fx−/− X Fx+/− crosses and Fx−/− intercrosses. A variable contribution of the mixed genetic background increased Fx−/− survival potential. Fx−/− mice were smaller, infertile and suffered from diarrhea and leukocytosis. However, fucose supplementation reversed these symptoms and restored fertility. It also led to a very rapid normal expression of P-selectin ligand, similar to CDG-IIc patients. How did such rapid improvement occur?
A likely explanation is that the loss of fucosylation not only prevents leukocyte diapadesis, but also induces myeloproliferation by signaling through the Notch pathway [49]. Multiple EGF modules on Notch receptors are modified by protein O-fucosyltransferase 1 (Pofut1). These are further elongated by the Fringe family of GlcNAc-glycosyltransferases [50]. Notch ligands normally suppress myeloid differentiation of progenitor cells and enhance expression of Notch target genes, but fucosylation-deficient myeloid progenitors are not sensitive to suppression by these ligands. In this mouse model there was also a secretory epithelial cell hyperplasia [51] and impaired lung fibrosis due to impaired myofibroblast differentiation during inflammation [52]. Providing fucose to these Fx−/− mice reversed the abnormal phenoptypes.
Another knockout mouse lacking a specific α1,6-fucosyl transferase (Fut8) for N-glycans shows growth retardation and loss of progeny during postnatal development [53]. Lack of this specific fucosylation also led to down-regulation of the EGF receptor, TGF-β receptor and integrins, which regulate several important events in growth and development. Impaired receptor signaling in CDG-IIc patients might explain growth and developmental retardation.
3.3.6. Models in other organisms
Gfr is the Drosophila ortholog of human GDP-fucose transporter. Gfr null mutants showed a wing phenotype typical of reduced Notch signaling [54], which could be rescued by wild type human GDP-fucose transporter, but not by human gene with the R147C mutation. The animal models and CDG-IIc patients underscore the importance of fucosylation in growth and development. Correcting the abnormal phenotypes with fucose in both patients and models helped us to understand (i) the underlying mechanisms leading to CDG-IIc pathology, (ii) the large influence of genetic background on survival (iii) distinction of contributions from salvage pathway vs. de novo pathways.
3.4 Hereditary Inclusion Body Myopathy (HIBM)/Distal Myopathy with Rimmed Vacuoles (DMRV)
3.4.1. Metabolic Steps
Sialic acids (parent precursor N-acetylneuraminic acid) is efficiently salvaged from degraded glycans in the lysosome [55,56]. A small portion of dietary sialic acid is also incorporated into glycans [57,58]. Sialic acid is synthesized de novo from UDP-GlcNAc using the bifunctional enzyme UDP-GlcNAc epimerase/kinase (GNE) converts the substrate to N-acetyl mannosa-mine-6-phosphate (ManNAc-6-P), which then condenses with pyruvate to form N-acetylneuraminic acid and activated to CMP-Sialic acid by CTP. GlcNAc released from salvaged glycans also contributes to sialic acids by direct conversion to GlcNAc-6-P and epimerization to GlcNAc-1-P. The relaxed substrate specificity allows this enzyme to use ManNAc as well.
3.4.2 Patients
Hereditary Inclusion Body Myopathy (HIBM)/Distal Myopathy with Rimmed Vacuoles (DMRV) is an autosomal recessive, adult onset myopathy that does not affect children [59,60]. It begins in the distal leg muscles and progresses to the proximal muscles while sparing the quadriceps, and usually leads to wheelchair-restricted mobility. It is caused by mutations in GNE, which encodes N-acetyl glucosamine epimerase-kinase [61] that are rather mild with only 30–60% reduction in enzymatic activity [62]. Mutations can occur in either of the two catalytic domains with mutations in one domain affecting the activity of the other [63]. Cells from patients have sialic acid contents that overlap with controls, and depending on the study, some patients may show hyposialylation of α-dystroglycan and neural cell adhesion molecule (NCAM) [64].
The onset and rate of progression varies widely, even in patients with the same mutation and very similar genetic background. The observed penetrance is 99%, so there may be compensating or protective mechanisms in place [62]. Although the geographic distribution is broad, the highest incidence is in the Persian Jewish population where the carrier frequency of a founder mutation (M712T) is approximately 1:20 leading to a prevalence of 1:1600 [62]. Japan has a significant population (perhaps 400) of DMRV patients. The disease is also called Nonaka myopathy [65,66].
Several studies suggest that the production of sialylated glycans in the muscle is reduced, although this is still unsettled question. The enzyme is localized in the Golgi and in nucleus where CMP-Sialic acid synthase is found, leading some to speculate that it may also have a regulatory function [67].
The identification of GNE mutations in this disorder was surprising since mutations in an allosteric site of the protein that binds CMP-Sia to down regulate its activity, also produce another rare disorder called sialuria [68]. Patients with this condition accumulate large amounts of sialic acid in their cells and urine because they cannot decrease the rate of endogenous synthesis when salvaged or dietary sialic acid is sufficient. This implies that sialic acid flux is variable and needs to be is closely regulated. Unlike sialuria patients, HIBM2/DMRV patients do not accumulate sialic acid.
3.4.3 Therapy
There is no currently approved therapy for HIBM2/DMRV, although it is reasonable to suggest that sialic acid or ManNAc supplements might be beneficial. One pilot clinical study provided 4 patients with a one-month course of intravenous immune globulin that contains 8 µmoles of sialic acid/g. All patients showed a significant, measurable increase in localized muscle strength, but no improvement occurred based on immunohistochemical staining or immunoblotting of muscle biopsies for α-dystroglycan [69]. Presumably the transient improvement involved the endocytic uptake and lysosomal degradation of the infused sialylated protein, release of sialic acid within the lysosome, and its transport into the cytoplasm to bypass the block in the impaired muscle. Alternatively, the effects may have been indirect through an unknown mechanism. Uptake of exogenous free sialic acid occurs via non-clatherin mediated endocytosis [70] while ManNAc likely enters cells by diffusion directly into the cytosol. No clinical trials on ManNAc efficacy have been done since, ManNAc does not naturally occur in the diet unlike fucose and mannose and would require additional animal safety and toxicity studies after clearing pending regulatory approval. Since the pathway for ManNAc entry into cell is different from the uptake of a sialylated glycoprotein this would be expected to affect the tissue distribution and the efficiency of utilization of these different substrates.
3.4.4 Mouse models
GNE activity is high in liver, salivary glands and intestine, but very low in other organs and muscle [71]. Knockout of Gne in mice is lethal at 8.5 d in embryogenesis [72]. Proliferation of embryonic stem cells from these mice is directly correlated with GNE-expression and the cellular sialic acid concentration [73], which may explain embryonic lethality.
Two research groups made knockin mutations in mice. The first was the common Persian Jewish mutation (M712T) in a C57BL/6J background [74]. Residual enzymatic activity in homozygous mice was about 20% of control. Homozygotes were born, but died of kidney failure within the first 3 days. They showed glomerular hematuria, proteinuria, and podocytopathy. This resulted from an abnormal glomerular basement membrane and podocyte foot processes, with reduced sialylation of the major podocyte sialoprotein, podocalyxin. When ManNAc (1g/kg/day) was provided to the dams at mating and onward, 43% of the mice survived and they showed reduced pathology with improved morphology and function of the foot processes and glomerular basement membrane. Sialylation of podocalyxin improved with ManNAc treatment. The surviving mice showed no signs of muscular pathology. Providing ManNAc increased the enzymatic activity of Gne in both wild-type and homozygous mutant mice, possibly due to substrate stabilization.
Why this mutant strain showed kidney-specific pathology is unknown. Clearly, rescue with ManNAc suggests correction of a sialic acid deficiency. In that case it must have required maternal delivery of ManNAc to the embryo and utilization of the precursor for sialic acid synthesis.
The second group introduced a different mutation (D176V), which is prevalent in the Japanese population [75,76]. They bred Gne−/+ mice with transgenic mice carrying multiple copies of mutated human gene to produce a Gne null C57Bl/6 mice that carried five copies of the mutated human gene in the a Gne-null background. The mice were born normally but grew poorly and had a significantly higher mortality T1/2~50 weeks. Sialic acid content in proteins in plasma, liver and spleen were dramatically reduced by 60–90% and only by ~20–30%. in muscle and heart, They had poor muscle performance by 30 weeks of age and β-amyloid deposits accumulated in the myofibrils by 32 weeks. By 42 weeks they developed a muscular pathology resembling the human patients with accumulation of rimmed vacuole inclusions.
Most intriguing was their therapeutic intervention. Muscle atrophy and weakness are completely prevented by providing sialic acid, ManNAc, or sialyllactose in the drinking water of 5–6 week old mice. Even the lowest dose (20mg/kg body weight) of any of these sialic acid metabolites was equally effective in preventing the onset of disease. Sugar administration showed quantitative improvement in muscle performance, muscle sialic acid content, and reduced number of rimmed vacuoles and amyloid inclusions. It prevented atrophy of skeletal muscles and enhanced the proportion of muscle fibers with normal diameter and decreased the number of atrophic fibers. These results showed that sialic acid metabolism is indeed one of the key factors in the pathomechanism of the disorder. The doses are quite modest and most surprising for sialylactose, which is only 45% sialic acid. These preventive studies did not assess therapeutic reversal of established myopathy.
Previous studies on the metabolism of sialic acid and sialyl lactose in rats showed that both could be absorbed from the intestine, and 60–90% of the sialic acid is excreted into the urine within 6 hr [11]. Intravenous injection gave similar results. Only a small amount of radiolabeled material remained in the tissue. Most of it was degraded and not salvaged. These few animal studies suggest that the majority of these molecules go to “waste”.
Whether sialic acid, ManNAc or sialylactose will be effective therapies for patients is an open question. Expense is one consideration. Reagent grade sialic acid, ManNAc and sialyl lactose have not been tested for safety, which is required it they are intended to treat a specific disorder.
One of the most abundant sources of sialylactose is human milk [77], specifically the whey, but it is also abundant in milk from elephants and spiny anteaters [78,79]. The potential availability of these untested sugars for a population of patients aware of their 99% probable prognosis is a powerful motivation to look past issues of safety and dosage [62]. Again, this exposes our limited basic knowledge of the non-flat metabolic chart and the need to understand contributions of de novo, dietary, and salvage pathways.
4. PERSPECTIVES and CONCLUSIONS
These examples of human glycosylation disorders illustrate several points.
Small changes in glycan precursor flux cause significant but unpredictable changes in glycosylation in selected cells and tissues.
Hypomorphic alleles in different genetic backgrounds can influence the ability of sugars to rescue pathological phenotypes.
The relative contributions of de novo, salvage and dietary sources of monosaccharides are mostly unknown beyond a few experiments in cultured cells.
The fate of monosaccharides released from turnover of endogenous glycans and dietary sources needs to be investigated since it may have therapeutic impact for patients.
Additional models of impaired glycosylation are needed to assess the precursor contributions to major and minor pathways. Tractable experimental systems such as zebrafish and state-of-the-art metabolomic approaches may be useful to track sugar precursor origin and utilization.
Better fundamental understanding of the 4 dimensional metabolic chart may help create new therapeutic opportunities.
The public’s positive attitude toward nutritional/dietary alterations as potential “natural” treatments provide both reason and opportunity to investigate basic science underlying sugar metabolism.
Many human glycosylation disorders were discovered in the last decade. Most likely, new ones that affect glycosylation will be discovered the near future using state-of-the-art genetic mapping and high throughput sequencing technologies. Both forward and reverse genetic approaches in model systems will certainly encounter glycosylation insufficiencies or stressful conditions that unmask them. In these conditions, it will be important to consider sugar dietary modifications to ameliorate the phenotype as shown in the examples here and in settings where providing GlcNAc in the drinking water modulates signaling pathways and branching of N-glycans [80]
Acknowledgements
This work was supported by R01 DK55615, The Rocket Fund, and by Challenge Grant 1RC1HD0641501. HHF is a Sanford Professor.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Berry GT. Metabolic profiling. Nestle Nutr Workshop Ser Pediatr Program. 2008;62:55–75. doi: 10.1159/000146249. discussion 75–80. [DOI] [PubMed] [Google Scholar]
- 2.Brockmann K. The expanding phenotype of GLUT1-deficiency syndrome. Brain Dev. 2009;31:545–552. doi: 10.1016/j.braindev.2009.02.008. [DOI] [PubMed] [Google Scholar]
- 3.Skvorak KJ. Animal models of maple syrup urine disease. J Inherit Metab Dis. 2009;32:229–246. doi: 10.1007/s10545-009-1086-z. [DOI] [PubMed] [Google Scholar]
- 4.Freeze HH, Elbein AD. In: Essentials of Glycobiology. 2nd ed. Varki A, Cummings RD, Esko JD, Freeze HH, Stanley P, Bertozzi CR, Hart GW, Etzler ME, editors. New York: Cold Spring Harbor Laboratory Pr; 2009. pp. 47–61. [PubMed] [Google Scholar]
- 5.Diaz S, Varki A. Metabolic radiolabeling of animal cell glycoconjugates. In: Cooligan JE, Dunn BM, Ploegh HL, Speicher DW, Wingfield PT, editors. Curr Protoc Protein Sci. Unit 12.2. Ch 12. New Jersey: John Wiley & Sons; 2009. pp. 1–55. [Google Scholar]
- 6.Mobasheri A, Bondy CA, Moley K, Mendes AF, Rosa SC, Richardson SM, Hoyland JA, Barrett-Jolley R, Shakibaei M. Facilitative glucose transporters in articular chondrocytes. Expression, distribution and functional regulation of GLUT isoforms by hypoxia, hypoxia mimetics, growth factors and pro-inflammatory cytokines. Adv Anat Embryol Cell Biol. 2008;200:1–84. [PubMed] [Google Scholar]
- 7.Onyekwelu I, Goldring MB, Hidaka C. Chondrogenesis, joint formation, and articular cartilage regeneration. J Cell Biochem. 2009;107:383–392. doi: 10.1002/jcb.22149. [DOI] [PubMed] [Google Scholar]
- 8.Sperandio M, Gleissner CA, Ley K. Glycosylation in immune cell trafficking. Immunol Rev. 2009;230:97–113. doi: 10.1111/j.1600-065X.2009.00795.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schnaar RL, Suzuki A, Stanley P. In: Essentials of Glycobiology. 2nd ed. Varki A, Cummings RD, Esko JD, Freeze HH, Stanley P, Bertozzi CR, Hart GW, Etzler ME, editors. New York: Cold Spring Harbor Laboratory Pr; 2009. pp. 129–141. [PubMed] [Google Scholar]
- 10.Esko JD, Kimata K, Lindahl U. In: Essentials of Glycobiology. 2nd ed. Varki A, Cummings RD, Esko JD, Freeze HH, Stanley P, Bertozzi CR, Hart GW, Etzler ME, editors. New York: Cold Spring Harbor Laboratory Pr; 2009. pp. 229–246. [PubMed] [Google Scholar]
- 11.Freeze HH, Haltiwanger RS. In: Essentials of Glycobiology. 2nd ed. Varki A, Cummings RD, Esko JD, Freeze HH, Stanley P, Bertozzi CR, Hart GW, Etzler ME, editors. New York: Cold Spring Harbor Laboratory Pr; 2009. pp. 163–173. [PubMed] [Google Scholar]
- 12.Luther KB, Haltiwanger RS. Role of unusual O-glycans in intercellular signaling. Int J Biochem Cell Biol. 2009;41:1011–1024. doi: 10.1016/j.biocel.2008.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Martin PT. Congenital muscular dystrophies involving the O-mannose pathway. Curr Mol Med. 2007;7:417–425. doi: 10.2174/156652407780831601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Muntoni F, Torelli S, Brockington M. Muscular dystrophies due to glycosylation defects. Neurotherapeutics. 2008;5:627–632. doi: 10.1016/j.nurt.2008.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Freeze HH. Genetic defects in the human glycome. Nat Rev Genet. 2006;7:537–551. doi: 10.1038/nrg1894. [DOI] [PubMed] [Google Scholar]
- 16.Freeze HH. Towards a therapy for phosphomannomutase 2 deficiency, the defect in CDG-Ia patients. Biochim Biophys Acta. 2009;1792:835–840. doi: 10.1016/j.bbadis.2009.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jaeken J, Matthijs G. Congenital disorders of glycosylation: a rapidly expanding disease family. Annu Rev Genomics Hum Genet. 2007;8:261–278. doi: 10.1146/annurev.genom.8.080706.092327. [DOI] [PubMed] [Google Scholar]
- 18.Coman D, Irving M, Kannu P, Jaeken J, Savarirayan R. The skeletal manifestations of the congenital disorders of glycosylation. Clin Genet. 2008;73:507–515. doi: 10.1111/j.1399-0004.2008.01015.x. [DOI] [PubMed] [Google Scholar]
- 19.Grunewald S. The clinical spectrum of phosphomannomutase 2 deficiency (CDG-Ia) Biochim Biophys Acta. 2009;1792:827–834. doi: 10.1016/j.bbadis.2009.01.003. [DOI] [PubMed] [Google Scholar]
- 20.Eklund EA, Freeze HH. The congenital disorders of glycosylation: a multifaceted group of syndromes. NeuroRx. 2006;3:254–263. doi: 10.1016/j.nurx.2006.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Leroy JG. Congenital disorders of N-glycosylation including diseases associated with O- as well as N-glycosylation defects. Pediatr Res. 2006;60:643–656. doi: 10.1203/01.pdr.0000246802.57692.ea. [DOI] [PubMed] [Google Scholar]
- 22.Haeuptle MA, Hennet T. Congenital disorders of glycosylation: an update on defects affecting the biosynthesis of dolichol-linked oligosaccharides. Hum Mutat. 2009;30:1628–1641. doi: 10.1002/humu.21126. [DOI] [PubMed] [Google Scholar]
- 23.Thiel C, Lubke T, Matthijs G, von Figura K, Korner C. Targeted disruption of the mouse phosphomannomutase 2 gene causes early embryonic lethality. Mol Cell Biol. 2006;26:5615–5620. doi: 10.1128/MCB.02391-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Marek KW, Vijay IK, Marth JD. A recessive deletion in the GlcNAc-1-phophotransferase gene results in peri-implantation embryonic lethality. Glycobiology. 1999;9:1263–1271. doi: 10.1093/glycob/9.11.1263. [DOI] [PubMed] [Google Scholar]
- 25.Grunewald S. The clinical spectrum of phosphomannomutase 2 deficiency (CDG-Ia) Biochim Biophys Acta. 2009;1792:827–834. doi: 10.1016/j.bbadis.2009.01.003. [DOI] [PubMed] [Google Scholar]
- 26.Struwe WB, Hughes BL, Osborn DW, Boudreau ED, Shaw KM, Warren CE. Modeling a congenital disorder of glycosylation type I in C. elegans: a genome-wide RNAi screen for N-glycosylation-dependent loci. Glycobioloby. 2009;19:1554–1562. doi: 10.1093/glycob/cwp136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.de Lonlay P, Seta N. The clinical spectrum of phosphomannose isomerase deficiency, with an evaluation of mannose treatment for CDG-lb. Biochim Biophys Acta. 2009;1792:841–843. doi: 10.1016/j.bbadis.2008.11.012. [DOI] [PubMed] [Google Scholar]
- 28.Vuillaumier-Barrot S, Le Bizec C, de Lonlay P, Barnier A, Mitchell G, Pelletier V, Prevost C, Saudubray JM, Durand G, Seta N. Protein losing enteropathy-hepatic fibrosis syndrome in Saguenay-Lac St-Jean, Quebec is a congenital disorder of glycosylation type Ib. J Med Genet. 2002;39:849–851. doi: 10.1136/jmg.39.11.849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Westphal V, Kjaergaard S, Davis JA, Peterson SM, Skovby F, Freeze HH. Genetic and metabolic analysis of the first adult with congenital disorder of glycosylation type Ib: long-term outcome and effects of mannose supplementation. Mol Genet Metab. 2001;73:77–85. doi: 10.1006/mgme.2001.3161. [DOI] [PubMed] [Google Scholar]
- 30.Jaeken J, Matthijs G, Saudubray JM, Dionisi-Vici C, Bertini E, de Lonlay P, Henri H, Carchon H, Schollen E, Van Schaftingen E. Phosphomannose isomerase deficiency: a carbohydrate-deficient glycoprotein syndrome with hepatic-intestinal presentation. Am J Hum Genet. 1998;62:1535–1539. doi: 10.1086/301873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.de Lonlay P, Seta N. The clinical spectrum of phosphomannose isomerase deficiency, with an evaluation of mannose treatment for CDG-Ib. Biochim Biophys Acta. 2009;1792:841–843. doi: 10.1016/j.bbadis.2008.11.012. [DOI] [PubMed] [Google Scholar]
- 32.Niehues R, Hasilik M, Alton G, Korner C, Schiebe-Sukumar M, Koch HG, Zimmer KP, Wu R, Harms E, Reiter K, von Figura K, Freeze HH, Harms HK, Marquardt T. Carbohydrate-deficient glycoprotein syndrome type Ib. Phosphomannose isomerase deficiency and mannose therapy. J Clin Invest. 1998;101:1414–1420. doi: 10.1172/JCI2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Miller BS, Khosravi MJ, Patterson MC, Conover CA. IGF system in children with congenital disorders of glycosylation. Clin Endocrinol (Oxf) 2009;70:892–897. doi: 10.1111/j.1365-2265.2009.03531.x. [DOI] [PubMed] [Google Scholar]
- 34.Alton G, Kjaergaard S, Etchison JR, Skovby F, Freeze HH. Oral ingestion of mannose elevates blood mannose levels: a first step toward a potential therapy for carbohydrate-deficient glycoprotein syndrome type I. Biochem Mol Med. 1997;60:127–133. doi: 10.1006/bmme.1997.2574. [DOI] [PubMed] [Google Scholar]
- 35.Panneerselvam K, Etchison JR, Freeze HH. Human fibroblasts prefer mannose over glucose as a source of mannose for N-glycosylation. Evidence for the functional importance of transported mannose. J Biol Chem. 1997;272:23123–23129. doi: 10.1074/jbc.272.37.23123. [DOI] [PubMed] [Google Scholar]
- 36.Shimizu K. Metabolic flux analysis based on 13C-labeling experiments and integration of the information with gene and protein expression patterns. Adv Biochem Eng Biotechnol. 2004;91:1–49. doi: 10.1007/b94204. [DOI] [PubMed] [Google Scholar]
- 37.DeRossi C, Bode L, Eklund EA, Zhang F, Davis JA, Westphal V, Wang L, Borowsky AD, Freeze HH. Ablation of mouse phosphomannose isomerase (Mpi) causes mannose 6-phosphate accumulation, toxicity, and embryonic lethality. J Biol Chem. 2006;281:5916–5927. doi: 10.1074/jbc.M511982200. [DOI] [PubMed] [Google Scholar]
- 38.Davis JA, Freeze HH. Studies of mannose metabolism and effects of long-term mannose ingestion in the mouse. Biochim Biophys Acta. 2001;1528:116–126. doi: 10.1016/s0304-4165(01)00183-0. [DOI] [PubMed] [Google Scholar]
- 39.Sharma V, Srivastava A, Nayak J, Ng B, Bode L, Derossi C, Guess C, Ichikawa M, Krajewski S, Freeze HH. A Viable Mouse Model for Congenital Disorders of Glycosylation, CDG-Ib. Program and Abstracts for the 2009 Meeting of the Society for Glycobiology; 2009. p. 1330. [Google Scholar]
- 40.Knepik E. Zebrafish Model of CDG-Ij. Glycobiology. 2009;19:1292. [Google Scholar]
- 41.Etzioni A, Frydman M, Pollack S, Avidor I, Phillips ML, Paulson JC, Gershoni-Baruch R. Brief report: recurrent severe infections caused by a novel leukocyte adhesion deficiency. N Engl J Med. 1992;327:1789–1792. doi: 10.1056/NEJM199212173272505. [DOI] [PubMed] [Google Scholar]
- 42.Luhn K, Wild MK, Eckhardt M, Gerardy-Schahn R, Vestweber D. The gene defective in leukocyte adhesion deficiency II encodes a putative GDP-fucose transporter. Nat Genet. 2001;28:69–72. doi: 10.1038/ng0501-69. [DOI] [PubMed] [Google Scholar]
- 43.Lubke T, Marquardt T, Etzioni A, Hartmann E, von Figura K, Korner C. Complementation cloning identifies CDG-IIc, a new type of congenital disorders of glycosylation, as a GDP-fucose transporter deficiency. Nat Genet. 2001;28:73–76. doi: 10.1038/ng0501-73. [DOI] [PubMed] [Google Scholar]
- 44.Marquardt T, Lhun K, Srikrishna G, Freeze HH, Harms E, Vestweber D. Correction of leukocyte adhesion deficiency type II with oral fucose. Blood. 1999;94:3976–3985. [PubMed] [Google Scholar]
- 45.Etzioni A, Tonetti M. Fucose supplementation in leukocyte adhesion deficiency type II. Blood. 2000;95:3641–3643. [PubMed] [Google Scholar]
- 46.Hellbusch CC, Sperandio M, Frommhold D, Yakubenia S, Wild MK, Popovici D, Vestweber D, Grone HJ, von Figura K, Lubke T, Komer C. Golgi GDP-fucose transporter-deficient mice mimic congenital disorder of glycosylation llc/leukocyte adhesion deficiency II. J Biol Chem. 2007;282:10762–10772. doi: 10.1074/jbc.M700314200. [DOI] [PubMed] [Google Scholar]
- 47.Yakubenia S, Frommhold D, Scholch D, Hellbusch CC, Komer C, Petri B, Jones C, Ipe U, Bixel MG, Krempien R, Sperandio M, Wild MK. Leukocyte trafficking in a mouse model for leukocyte adhesion deficiency II/congenital disorder of glycosylation IIc. Blood. 2008;112:1472–1481. doi: 10.1182/blood-2008-01-132035. [DOI] [PubMed] [Google Scholar]
- 48.Smith PL, Myers JT, Rogers CE, Zhou L, Petrynaik B, Becker DJ, Homeister JW, Lowe JB. Conditional control of selectin ligand expression and global fucosylation events in mice with a targeted mutation at the FX locus. J Cell Biol. 2002;158:801–815. doi: 10.1083/jcb.200203125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhou L, Li LW, Yan Q, Petryniak B, Man Y, Su C, Shim J, Chervin S, Lowe JB. Notch-dependent control of myelopoiesis is regulated by fucosylation. Blood. 2008;112:308–319. doi: 10.1182/blood-2007-11-115204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Moloney DJ, Panin VM, Johnston SH, Chen J, Shao L, Wilson R, Wang Y, Stanley P, Irvine KD, Haltiwanger RS, Vogt TF. Fringe is a glycosyltransferase that modifies Notch. Nature. 2000;406:369–375. doi: 10.1038/35019000. [DOI] [PubMed] [Google Scholar]
- 51.Waterhouse C, Johnson S, Phillipson M, Zbytnuik L, Petri B, Kelly M, Lowe J, Kubes P. Secretory Cell Hyperplasia and Defects in Notch Activity in a Mouse Model of Leukocyte Adhesion Deficiency Type II. Gastroenterology. doi: 10.1053/j.gastro.2009.10.049. In press. [DOI] [PubMed] [Google Scholar]
- 52.Liu T, Hu B, Choi YY, Chung M, Ullenbruch M, Yu H, Lowe JB, Phan SH. Notch1 signaling in FIZZ1 induction of myofibroblast differentiation. Am J Pathol. 2009;174:1745–1755. doi: 10.2353/ajpath.2009.080618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang X, Inoue S, Gu J, Miyoshi E, Noda K, Li W, Misuno-Horikawa Y, Nakano M, Asahi M, Takahashi M, Uozumi N, Ihara S, Lee SH, Ikeda Y, Yamaguchi Y, Aze Y, Tomiyama Y, Fujii J, Suzuki K, Kondo A, Shapiro SD, Lopez-Otin C, Kuwaki T, Okabe M, Honke K, Taniguchi N. Dysregulation of TGF-beta1 receptor activation leads to abnormal lung development and emphysema-like phenotype in core fucose-deficient mice. Proc Natl Acad ScieU S A. 2005;102:15791–15796. doi: 10.1073/pnas.0507375102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ishikawa HO, Higashi S, Ayukawa T, Sasamura T, Kitagawa M, Harigaya K, Aoki K, Ishida N, Sanai Y, Matsuno K. Notch deficiency implicated in the pathogenesis of congenital disorder of glycosylation IIc. Proc Natl Acad Sci U S A. 2005;102:18532–18537. doi: 10.1073/pnas.0504115102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tettamanti G. Ganglioside/glycosphingolipid turnover: new concepts. Glycoconj J. 2004;20:301–317. doi: 10.1023/B:GLYC.0000033627.02765.cc. [DOI] [PubMed] [Google Scholar]
- 56.Tettamanti G, Bassi R, Viani P, Riboni L. Salvage pathways in glycosphingolipid metabolism. Biochimie. 2003;85:423–437. doi: 10.1016/s0300-9084(03)00047-6. [DOI] [PubMed] [Google Scholar]
- 57.Nohle U, Schauer R. Metabolism of Sialic Acids from Exogeneously Administered Sialyllactose and Mucin in Mouse and Rat. Physiol Chem. 1984;365:1457–1467. doi: 10.1515/bchm2.1984.365.2.1457. [DOI] [PubMed] [Google Scholar]
- 58.Nohle U, Schauer R. Uptake Metabolism and Excretion of Orally and Intravenously Administered, 14C- and 3H-Labeled N-Acetylneuraminic Acid Mixture in the Mouse and Rat. Physiol Chem. 1981;362:1495–1506. doi: 10.1515/bchm2.1981.362.2.1495. [DOI] [PubMed] [Google Scholar]
- 59.Argov Z, Mitrani-Rosenbaum S. Hereditary inclusion body myopathies. In: Karpati G, Hilton-jones D, Bushby K, Griggs R, editors. Disorders of Voluntary Muscle. 8th ed. Cambridge University Press; 2009. pp. 492–498. [Google Scholar]
- 60.Nonaka I, Noguchi S, Nishino I. Distal myopathy with rimmed vacuoles and hereditary inclusion body myopathy. Curr Neurol Neurosci Rep. 2005;5:61–65. doi: 10.1007/s11910-005-0025-0. [DOI] [PubMed] [Google Scholar]
- 61.Eisenberg I, Avidan N, Potikha T, Hochner H, Chen M, Olender T, Barash M, Shemesh M, Sadeh M, Grabov-Nardini G, Shmilevich I, Friedmann A, Karpati G, Bradley WG, Baumbach L, Lancet D, Asher EB, Beckmann JS, Argov Z, Mitrani-Rosenbaum S. The UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase gene is mutated in recessive hereditary inclusion body myopathy. Nat Genet. 2001;29:83–87. doi: 10.1038/ng718. [DOI] [PubMed] [Google Scholar]
- 62.Argov Z Mitrani-Rosenbaum. The Hereditary Inclusion Body Myopathy Enigma and its Future Therapy. Neurotherapeutics: The Journal of the American Society for Experimental NeuroTherapeutics. 2008;Vol 5:633–637. doi: 10.1016/j.nurt.2008.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sparkes SE, Ciccone C, Lalor M, Orvisky E, Klootwijk R, Savelkoul PJ, Dalakas MC, Krasnewich DM, Gahl WA, Huizing M. Use of a cell-free system to determine UDP-N-acetylglucosamine 2-epimerase and N-acetylmannosamine kinase activities in human hereditary inclusion body myopathy. Glycobiology. 2005;15:1102–1110. doi: 10.1093/glycob/cwi100. [DOI] [PubMed] [Google Scholar]
- 64.Salama I, Hinderlich S, Shlomai Z, Eisenberg I, Krause S, Yarema K, Argov Z, Lochmuller H, Reutter W, Dabby R, Sadeh M, Ben-bassat H, Mitrani-Rosenbaum S. No overall hyposialylation in hereditary inclusion body myopathy myoblasts carrying the homozygous M712T GNC mutation. Biochem Biophys Res Commun. 2005;328:221–226. doi: 10.1016/j.bbrc.2004.12.157. [DOI] [PubMed] [Google Scholar]
- 65.Nishino I, Malicdan MC, Murayama K, Nonaka I, Hayashi YK, Noguchi S. Molecular pathomechansim of distal myopathy with rimmed vacuoles. Acta Myol. 2005;24:80–83. [PubMed] [Google Scholar]
- 66.Nonaka I, Noguchi S, Nishino I. Distal myopathy with rimmed vacuoles and hereditary inclusion body myopathy. Curr Neurol Neurosci Rep. 2005;5:61–65. doi: 10.1007/s11910-005-0025-0. [DOI] [PubMed] [Google Scholar]
- 67.Krause S, Hinderlich S, Amsili S, Horstkorte R, Wiendl H, Argov Z, Mitrani-Rosenbaum S, Lochmuller H. Localization of UDP-GlcNAc 2-epimerase/ManAc kinase (GNE) in the Golgi complex and the nucleus of mammalian cells. Exp Cell Res. 2005;304:365–379. doi: 10.1016/j.yexcr.2004.11.010. [DOI] [PubMed] [Google Scholar]
- 68.Leroy JG, Seppala R, Huizing M, Dacremont G, De Simpel H, Van Coster RN, Orvisky E, Krasnewich DM, Gahl WA. Dominant inheritance of sialuria, an inborn error of feedback inhibition. Am J Hum Genet. 2001;68:1419–1427. doi: 10.1086/320598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sparks S, Rakocevic G, Joe G, Manoli I, Shrader J, Harris-Love M, Sonies B, Ciccone C, Dorward H, Krasnewich D, Huizing M, Dalakas MC, Gahl WA. Intravenous immune globulin in hereditary inclusion body myopathy: a pilot study. BMC Neurol. 2007;7:3. doi: 10.1186/1471-2377-7-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bardor M, Nguyen DH, Diaz S, Varki A. Mechanism of uptake and incorporationof the non-human sialic acid N-glycolylneuraminic acid into human cells. J Biol Chem. 2005;280:4228–4237. doi: 10.1074/jbc.M412040200. [DOI] [PubMed] [Google Scholar]
- 71.Stasche R, Hinderlich S, Weise C, Effertz K, Lucka L, Moormann P, Reutter W. A bifunctional enzyme catalyzes the first two steps in N-acetylneuraminic acid biosythesis of rat liver. Molecular cloning and functional expression of UDP-N-acetyl-glucosamine 2-epimerase/N-acetylmannosamine kinase. J Biol Chem. 1997;272:24319–24324. doi: 10.1074/jbc.272.39.24319. [DOI] [PubMed] [Google Scholar]
- 72.Schwarzkopf M, Knobeloch KP, Rohde E, Hinderlich S, Wiechens N, Lucka L, Horak I, Reutter W, Horstkorte R. Sialylation is essential for early development in mice. Proc Natl Acad Sci U S A. 2002;99:5267–5270. doi: 10.1073/pnas.072066199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Weidemann W, Klukas C, Klein A, Simm A, Schreiber F, Horstkorte R. Lessons from GNE-deficient embryonic stem cells: sialic acid biosynthesis is involved in proliferation and gene expression. Glycobiology. 2010;20:107–117. doi: 10.1093/glycob/cwp153. [DOI] [PubMed] [Google Scholar]
- 74.Huizing M, Rakocevic G, Sparks SE, Mamali I, Shatunov A, Goldfarb L, Krasnewich D, Gahl WA, Dalakas MC. Hypoglycosylation of alpha-dystroglycan in patients with hereditary IBM due to GNE mutations. Mol Genet Metab. 2004;81:196–202. doi: 10.1016/j.ymgme.2003.11.012. [DOI] [PubMed] [Google Scholar]
- 75.Malicdan MC, Noguchi S, Nonaka I, Hayashi YK, Nishino I. A Gne knockout mouse expressing human GNE D176V mutation develops features similar to distal myopathy with rimmed vacuoles or hereditary inclusion body myopathy. Hum Mol Genet. 2007;16:2669–2682. doi: 10.1093/hmg/ddm220. [DOI] [PubMed] [Google Scholar]
- 76.Malicdan MC, Noguchi S, Hayashi YK, Nonaka I, Nishino I. Prophylactic treatment with sialic acid metabolites precludes the development of the mypathic phenotype in the DMRV-hIBM mouse model. Nat Med. 2009;15:690–695. doi: 10.1038/nm.1956. [DOI] [PubMed] [Google Scholar]
- 77.Bode L. Recent advances on structure, metabolism, and function of human milk oligosaccharides. J Nutr. 2006;136:2127–2130. doi: 10.1093/jn/136.8.2127. [DOI] [PubMed] [Google Scholar]
- 78.Osthoff G, Dickens L, Urashima T, Bonnet SL, Uemura Y, van der Westhuizen JH. Structural characterization of oligosaccharides in the milk of an African elephant (Loxodonta africana africana) Comp Biochem Physiol B biochem Mol Biol. 2008;150:74–84. doi: 10.1016/j.cbpb.2008.01.010. [DOI] [PubMed] [Google Scholar]
- 79.Kamerling JP, Dorland L, van Halbeek H, Vliegenthart JF, Messer M, Schauer R. Structural studies of 4-O-acetyl-alpha-N-acetylneuraminyl-(2 goes to 3)-lactose, the main oligosaccharide in echidna milk. Carbohydr Res. 1982;100:331–340. doi: 10.1016/s0008-6215(00)81046-0. [DOI] [PubMed] [Google Scholar]
- 80.Grigorian A, Lee SU, Tian W, Chen IJ, Gao G, Mendelsohn R, Dennis JW, Demetriou M. Control of T Cell-mediated autoimmunity by metabolite flux to N-glycan biosynthesis. J Biol Chem. 2007;282:20027–20035. doi: 10.1074/jbc.M701890200. [DOI] [PubMed] [Google Scholar]