

Abstract
Brachydactyly mental retardation syndrome (BDMR) is associated with a deletion involving chromosome 2q37. BDMR presents with a range of features, including intellectual disabilities, developmental delays, behavioral abnormalities, sleep disturbance, craniofacial and skeletal abnormalities (including brachydactyly type E), and autism spectrum disorder. To date, only large deletions of 2q37 have been reported, making delineation of a critical region and subsequent identification of candidate genes difficult. We present clinical and molecular analysis of six individuals with overlapping deletions involving 2q37.3 that refine the critical region, reducing the candidate genes from >20 to a single gene, histone deacetylase 4 (HDAC4). Driven by the distinct hand and foot anomalies and similar cognitive features, we identified other cases with clinical findings consistent with BDMR but without a 2q37 deletion, and sequencing of HDAC4 identified de novo mutations, including one intragenic deletion probably disrupting normal splicing and one intragenic insertion that results in a frameshift and premature stop codon. HDAC4 is a histone deacetylase that regulates genes important in bone, muscle, neurological, and cardiac development. Reportedly, Hdac4−/− mice have severe bone malformations resulting from premature ossification of developing bones. Data presented here show that deletion or mutation of HDAC4 results in reduced expression of RAI1, which causes Smith-Magenis syndrome when haploinsufficient, providing a link to the overlapping findings in these disorders. Considering the known molecular function of HDAC4 and the mouse knockout phenotype, taken together with deletion or mutation of HDAC4 in multiple subjects with BDMR, we conclude that haploinsufficiency of HDAC4 results in brachydactyly mental retardation syndrome.
Introduction
Chromosome deletions involving the 2q37 region result in brachydactyly mental retardation syndrome (BDMR [MIM 600430]), also known as Albright hereditary osteodystrophy-like syndrome (AHO-like). BDMR is a complex disorder that presents with a spectrum of clinical features, including developmental delay, obesity, autism spectrum disorder, and craniofacial and skeletal abnormalities, including brachydactyly type E in approximately 50% of cases.1,2 Cardiac defects are reported in ∼20% of cases,1 and behavioral problems and neurological anomalies are common.3–5 The deletion 2q37 syndrome phenotype consists of a variety of findings that overlap with other syndromes, including Smith-Magenis (SMS [MIM 182290]), Prader-Willi (PWS [MIM 176270]), Angelman (AS [MIM 105830]), and fragile-X (FRAX [MIM 300624]) syndromes.
Approximately 100 individuals have been reported with a deletion involving the chromosome 2q37 region.2 Genotype:phenotype correlations have been attempted in the past but have failed to identify a single gene that may contribute to the core findings observed in BDMR. Refining the critical region for deletion 2q37 syndrome has proven difficult because of poor genotype:phenotype correlation and a paucity of interstitial deletions that would help define minimal overlapping candidate regions. To date, the critical region for this syndrome has not been clearly defined, and a variety of phenotypes have been mapped to the 2q37.1→2qter region.2 Variable expressivity and reduced penetrance for most major features complicate genotype:phenotype correlations in this syndrome and further impair diagnosis.
In a study of 52 individuals referred for a phenotype consistent with Smith-Magenis syndrome (SMS) for whom no 17p11.2 deletion or RAI1 (MIM 607642) mutation could be found, we identified by whole-genome array comparative genomic hybridization (aCGH) several copy number variants responsible for the SMS-like phenotypes observed in these individuals.6 Interestingly, we identified two cases that carried overlapping deletions of the 2q37 region.6 After a literature review, we discovered that the phenotypic overlap between these two distinct syndromes was striking. Utilizing this unique aCGH data set and additional cases, we refined the critical region for BDMR, reducing the likely candidates to a single gene, HDAC4 (MIM 605314). Given the apparent phenotypic overlap between SMS and BDMR, we then investigated more closely the specific phenotypes in our cohorts with Smith-Magenis syndrome-like and/or AHO-like features and identified two subjects who lacked a 2q37 deletion but had striking phenotypic overlap with known 2q37 deletion cases. Sequencing of the coding region of HDAC4 in each of these cases revealed de novo mutations. Studies show that HDAC4 (histone deacetylase 4) is critical for proper skeletogenesis and chondrogenesis,7 as well as neuronal survival.8 Taking into consideration the overlapping deletion data and de novo mutations presented here, we assert that haploinsufficiency of HDAC4 results in the brachydactyly mental retardation syndrome.
Material and Methods
Subject Ascertainment
Subjects were not formally recruited. Samples and medical records were ascertained after informed consent was obtained by referral based on suspicion of Smith-Magenis syndrome or AHO-like features. Subjects were referred to the S.H.E. laboratory for molecular evaluation of SMS at Michigan State University or Virginia Commonwealth University or to the M.A.A. laboratory at the Cleveland Clinic for AHO-like phenotype. Samples and medical records were collected in accordance with Institutional Review Board-approved protocols from the appropriate institution. Peripheral blood was collected, and DNA and metaphase chromosomes were prepared according to standard methods. Phenotypic information was collected from medical records, geneticist reports, and patient photos.
Genomic DNA Sequence Analysis
Total DNA was isolated from cultured peripheral white blood cells with the QIAamp DNA Minikit (QIAGEN, Hilden, Germany). PCR was performed with intronic primers from the HDAC4 gene generated with Primer3 (v. 0.4.0), which flank both the 5′ and 3′ regions of each HDAC4 exon (Table S1 available online). PCR products were treated with ExoSAP-IT (USB, Cleveland, OH) to digest PCR primers. Sequencing and analysis of electropherograms were done as previously described.9 PCR products were sequenced and analyzed on both the forward and reverse DNA strands. Sequencing primers are provided in Tables S1 and S2.
SMS245 Allele Cloning
PCR products from SMS245 coding exon 4 were excised from 2% agarose gel and DNA isolated with QiaQuick kit (QIAGEN, Hilden, Germany). PCR products were then cloned into a StrataClone PCR Cloning vector via standard TA cloning techniques and protocol provided by the manufacturer (Agilent Technologies, Santa Clara, CA). Plasmid DNA was isolated with Fermentus GeneJET plasmid mini kit (Burlington, Ontario, Canada) according to standard manufacturer's instructions. Sequences were confirmed with vector primers and standard Sanger sequencing techniques.
Multiplex Ligation-Dependent Probe Amplification and SNP Array Analysis
MLPA was performed as previously described10 with five custom-designed probe sets that were approximately equally spaced across the genomic extent of HDAC4 (Table S3). Breakpoints outside of the HDAC4 gene were defined with Illumina CNV370 single nucleotide polymorphism arrays and Beadstudio software to identify copy number changes.
Quantitative Real-Time PCR
RNA was isolated from cultured patient and control lymphoblastoid cell lines or lymphocytes collected from blood via Trizol (Invitrogen, Carlsbad, CA) according to standard protocols. All cell lines were cultured for the same period of time to the same cell density, and all samples were processed in the same manner. Quantitative reverse transcriptase real-time PCR was performed as previously described11 via Taqman probes (Applied Biosystems, Foster City, CA) for GAPDH (MIM 138400, internal control), HDAC4, or RAI1. Control RNAs for real-time PCR assays came from “like” samples, i.e., whole blood or cultured cells, as described, and at least two different control samples were analyzed for each expression assay to ensure expression data were consistent.
Results
As listed and described in Table 1, common features of BDMR include mild facial dysmorphism, congenital heart defects, distinct brachydactyly type E (BDE), intellectual disabilities, developmental delay, seizures, autism spectrum disorder, and obesity.1,2 By utilizing recent studies of SMS-like6 and known deletion 2q37 subjects,1 we were able to make genotype:phenotype correlations allowing refinement of the critical region for BDMR to ∼chr2: 239,639,900-240,938,547 within the 2q37.3 band, including the HDAC4 gene (Figure 1).
Table 1.
Clinical Features of Cases with Deletions or Mutations Involving 2q37.3
| Phenotype | SMS117HDAC4 Mutation (c.2399_2400insC) | SMS245HDAC4 Mutation (c.490+56_121 del65) | SMS272del(2)(q37.3) Including HDAC46 | SMS320del(2)(q37) Including HDAC46 | 122del(2)(q37) Including HDAC41 | 10780del(2)(q37) Including HDAC43 | 2282 del(2)(q37.3) Not Including HDAC4 (current study) |
|---|---|---|---|---|---|---|---|
| Sex | F | F | F | F | F | F | F |
| Age at evaluation | 16 and 25 years | 10 and 16 years | 15 and 17 years | 3 years | 6 years | 12 years | 7 years |
| Developmental delay | + | + | + | + | + | + | + |
| Motor delay | + | + | - | + | + | + | + |
| Language impairment | + | + | + | + | + | + | + |
| Behavioral problems | self-injurious behavior; pulling out of fingernails | self-injurious behavior; skin picking; aggression | aggressive behavior; hand wringing; eye squint | self-injurious behavior; head banging; skin picking; hand biting; hyperactivity | receptive language and social communication disorders | N/A | autistic behavior; repetitive behaviors |
| Stereotypies | N/A | + | self-hug, tics | self-hug | N/A | N/A | N/A |
| Sleeping difficulties | + | + | - | + | N/A | N/A | N/A |
| Decreased sensitivity to pain | + | + | + | + | N/A | N/A | N/A |
| Hearing loss | + | - | N/A | - | - | - | - |
| Short stature | +/− | + | - | - | + | + | - |
| Seizures | essential tremor | - | - | - | - | + | + |
| Feeding difficulties | + | - | - | - | - | N/A | - |
| Obesity/overweight | + | ++ | + | + | - | - | + |
| Craniofacial manifestations | |||||||
| Broad face | + | + | + | + | + | + | + |
| Upslanting eyes | + | + | + | + | - | - | N/A |
| Brachycephaly | + | - | + | + | N/A | N/A | N/A |
| Midface hypoplasia | + | - | + | + | N/A | + | + |
| Broad, upturned nose | + | broad, not upturned | + | + | N/A | + | - |
| Skeletal abnormalities | |||||||
| Brachydactyly, type E | + | +, mild | + | N/A | + | + | - |
| Proximally placed 4th toe, shortened 4th metatarsal | + | N/A | + | - | + | + | - |
| Other | subvalvar aortic stenosis; mitral stenosis; pacemaker; hypothyroidism; hirsuitism; spina bifida occulta; very friendly | small chin; narrow palpebral fissures; constipation; hypermobility; bitemporal narrowing; dental anomalies | sinus arrhythmia; myopia; very friendly | 2-3 toe syndactyly | craniosynostosis | hypothyroidism; precocious puberty | |
Figure 1.
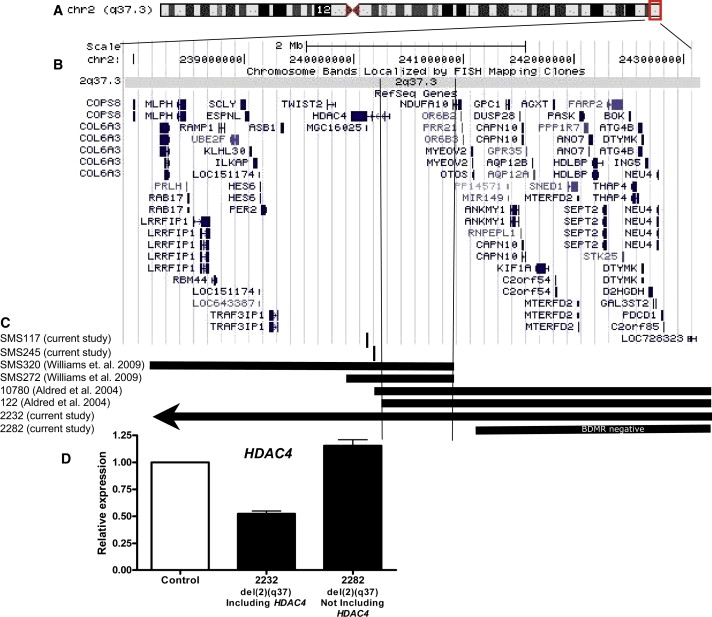
Delineation of the BDMR Critical Region
(A) Ideogram of chromosome 2 band q37.3.
(B) Schematic representation of 2q37.3 region and RefSeq Genes included.
(C) Horizontal bars indicate the regions of deletion in each of these key 2q37 deletion syndrome cases. The brachydactyly mental retardation syndrome critical region is indicated by the vertical bars. The SMS117 HDAC4 point mutation is indicated by the small vertical line. The SMS245 HDAC4 intronic deletion is indicated by the small vertical line.
(D) Quantitative real-time PCR of mRNA from lymphoblastoid cell lines from subject 2232 (del2q37.3 including HDAC4) and subject 2282 (del2q37.3 not including HDAC4) relative to control subject (no HDAC4 mutation or deletion). Assay performed twice in triplicate. Bars represent standard deviation from the mean. Data show ∼50% reduction in HDAC4 expression in 2232 and no change in 2282.
Refinement of the BDMR Critical Region
Because the deletions we identified map to a small overlapping region within the 2q37.3 band,1,6 we were able to narrow the critical region to ∼200 kb region including HDAC4 (Figure 1), with SMS272 critical to this analysis resulting from the very small deletion identified by aCGH.
SMS272 carries a small, 1.17 Mb, 2q37.3 deletion (Figure 1).6 She is an obese Native American with cognitive and developmental delays who was originally referred for features of Smith-Magenis syndrome (SMS).6 Skeletal features include brachydactyly type E (shortened 3rd, 4th, and 5th metacarpals with shortened and proximately paced 4th left toe), midface hypoplasia, and brachycephaly (Figure 2, Table 1). She also exhibits stereotypies and aggressive behavior but lacks two of the key features, sleep disturbance and self-abusive behavior, which are common in SMS. SMS320, who was also referred for an SMS-like phenotype,6 carries a larger deletion of 2q37.3 that spans 3.02 Mb (Figure 1) and was referred for developmental, speech, and motor delays, sleep disturbance, stereotypies, attention seeking, and self-injurious behaviors. Her craniofacial and skeletal phenotype consists of brachydactyly, brachycephaly, midface hypoplasia, tented upper lip, and a broad, square face (Table 1).
Figure 2.
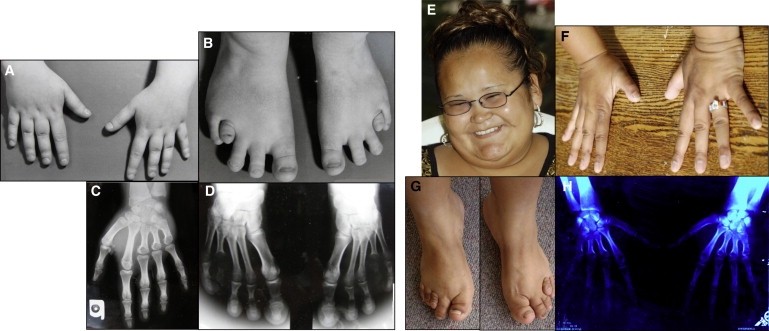
Skeletal Anomalies Observed in BDMR
(A) Hands of case SMS117.
(B) Feet of SMS117, showing shortened 4th metatarsal and wide spacing between toes.
(C) Radiograph of left hand of SMS117 showing shortened 3rd, 4th, and 5th metacarpals.
(D) Radiograph of SMS117 feet showing proximally placed and shortened 4th metatarsal and bilaterally widely spaced 1st, 2nd, and 3rd toes.
(E) Photo of SMS272 at 17 years of age.
(F) Hands of SMS272. Note shortened 3rd and 4th fingers.
(G) Feet of SMS272 with shortened 3rd toes and proximally placed and shortened 4th toes.
(H) Radiograph documenting shortened 3rd and 4th metacarpals in the hands of SMS272.
Two del(2)(q37.3) subjects were analyzed by MLPA to assess the specific location of the breakpoints observed (Table 1; Figure S1). As shown in Figure 1, the proximal breakpoints of the deletions are within the HDAC4 gene. The breakpoint identified in case 122 is in the second intron of HDAC4 (3.21 Mb deletion of 2q37), whereas the breakpoint for case 10780 is in intron 9 (3.2 Mb deletion of 2q37) (Figure 1; Figure S1). Both deletions should result in haploinsufficiency for HDAC4 because the promoter and initial coding exons are deleted in both cases (HDAC4 is transcribed on the minus strand of DNA). Subject 122 presented with developmental delay, mild facial dysmorphism, brachydactyly type E, and receptive language and social communication disorder. Case 10780 presented with developmental delay, short stature, facial dysmorphism, brachydactyly type E, and grand mal seizures (Table 1). Detailed clinical descriptions of both cases have been published previously1,3 (Table 1).
Further supporting the deletion of HDAC4 as responsible for the brachydactyly type E phenotype is the fact that subject 2282 has a 2.68 Mb terminal deletion with a breakpoint that is distal of HDAC4 (Figure 1C). This individual presented with minor craniofacial abnormalities, developmental delay, obesity, autistic behaviors, and a history of seizures but had no evidence of metacarpal or metatarsal shortening and no evidence of cardiac or other major organ defect (Table 1). The phenotype of subject 2282 indicates that other aspects of 2q37 deletion syndrome are complex and like many deletion/mutation syndromes, multiple genes may contribute to the full spectrum of the phenotype. An additional case, subject 2232, was diagnosed with a terminal 2q37 deletion approximately 7 Mb in size (Figure 1). Unfortunately, no clinical details were available and therefore this case is not included in Table 1. However, this terminal deletion of chromosomal band 2q37 (including HDAC4) results in a ∼50% reduction in HDAC4 expression (Figure 1D), whereas the deletion carried by case 2282 (not deleted for HDAC4) has normal HDAC4 expression, providing evidence for haploinsufficiency when HDAC4 is deleted.
These molecular and clinical data refined the BDMR critical region to a single gene, HDAC4, and supported mutation analysis of HDAC4 as the logical next step toward identification of the causative gene that, when deleted or mutated, results in BDMR. Further support for the conclusion that HDAC4 mutations cause BDMR is the Hdac4−/− mouse,7 which has a significant skeletal phenotype.7 Although important in electron transport and complex I function in mitochondria, NDUFA10 (MIM 603835) was eliminated as a probable cause for BDMR given that mitochondrial dysfunction does not appear to be a key finding in these patients.
HDAC4 Mutation Causes BDMR
Two cases, SMS117 and SMS245, were selected for mutation analysis of HDAC4 because of their phenotypic overlap with the deletion 2q37 cases discussed above (Figure 2, Table 1). All coding exons for HDAC4 were sequenced and assessed for mutation in SMS117 and SMS245 (PCR primers in Table S1). For SMS117, analysis of the nucleotide sequence for exon 19 revealed a de novo insertion of a single cytosine (c.2399_2400insC; Figure 3). Because this insertion lies within a run of five cytosines, the exact location of the insertion cannot be determined, but this does not impact the resulting location of the premature stop codon (p.Gly801TrpfsX77) (Figure 3). This insertion does not confer a reduction in HDAC4 mRNA expression (Figure 1D), suggesting that this mutation does not result in significant mRNA decay and probably produces a nonfunctional HDAC4 protein. SMS117 is a female French-Canadian born at 43 weeks to nonconsanguineous parents (Figure 3, Table 1). Birth was complicated by two induction attempts. Weight at birth was 2.85 kg (10%–25%) and length was 51 cm (25%–50%). The first year was marked by feeding difficulties, and subvalvular aortic stenosis was identified at 4 months. Psychomotor development was delayed, with walking at 18 months and speaking in sentences at 6 years of age. The clinical phenotype worsened with age. She received a mitral valve replacement and permanent pacemaker at 5 years. At 13.5 years, she was referred to genetics where she presented with dysmorphic features, including midface hypoplasia, broad face and nose, brachycephaly, frontal bossing, a down-turned lower lip, and upslanted eyes. Decreased deep tendon reflexes and a wide-based gate were observed, as was sensorineural hearing loss (>40 dB) for which she wears two hearing aids. She had a decreased sensitivity to pain, onychotillomania, hyperactivity, and a decreased attention span. Radiological studies also revealed cerebral atrophy and spina bifida occulta. In addition to the admission for heart surgery, she was hospitalized for hypersomnolence and precipitous loss of consciousness. During 44 hr of EEG video telemetry, a complete absence of REM sleep was noted. Sleep abnormalities began at 3 years of age with several arousals throughout the night, which lasted until the age of 8 years; however, at 25 years of age, she could sleep for up to 18 hr if not awakened. Physical examination at 25 years of age revealed obesity (96 kg, >95%; 157cm, 10%–25%) and confirmed the previously described physical and neurological findings. She had bilateral proximal placement of the 3rd, 4th, and 5th fingers, bilateral proximally placed 4th toes, and bilateral widely spaced 1st, 2nd, and 3rd toes. Brachydactyly type E was confirmed with shortened 3rd, 4th, and 5th metacarpals and bilaterally shortened 4th metatarsals on X-ray (Figure 2). Total hand length was 14 cm (<3%) and total foot length was 22.5 cm (10%–25%). There is a striking similarity between the morphology of the hands and feet of SMS117 (HDAC4 mutation) and SMS272 (2q37.3 deletion) (Figure 2), lending further support to the importance of HDAC4 in this syndrome. Family history was unremarkable; however, the patient's father died at age 50 years of a pulmonary embolism, and no other abnormalities were reported.
Figure 3.
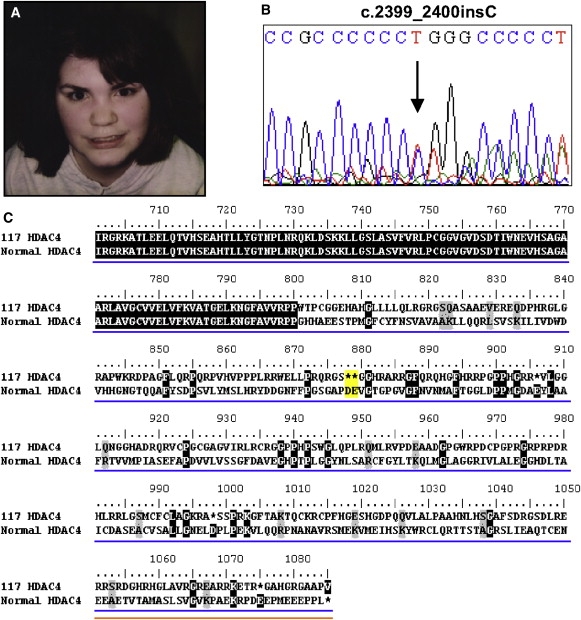
SMS117 HDAC4 Mutation and Impact on HDAC4 Protein
(A) SMS117 at 13 years.
(B) Electropherogram trace showing nucleotide insertion of a single cytosine at c.2399 in the HDAC4 gene.
(C) Partial amino acid alignment of the normal HDAC4 protein with the altered protein probably produced in SMS117 resulting from frameshift mutation. Premature stop codon highlighted in yellow. The histone deacetylase domain resides at amino acids 655–1084 (blue line) and the nuclear export signal is at amino acids 1051–1084 (orange line). The mutation disrupts the HDAC domain and removes the nuclear export signal.
Sequencing of the HDAC4 gene in SMS245 included all coding sequence and known splice sites and identified a de novo intron 5 deletion of 65 bp at Chr2:239,763,222-239,763,157 (c.490+56_121del65) (Figure 4), probably altering splicing of exons 5–6. These findings were confirmed by cloning, sequencing, and aligning both alleles of this region (Figure 4). This finding was not present in either parent, nor in the Database of Genomic Variants. SMS245 is a 16-year-old intellectually disabled female who is obese, weight 152.3 kg (+7 SD) and height 159.8 cm (5th centile). She was born preterm at 35 weeks gestation and experienced feeding difficulties during infancy and has chronic constipation. She has a small chin, large asymmetrically placed ears, bitemporal narrowing, and narrow palpebral fissures (Figure 4). Motor activities and speech were delayed with first steps taken at 2.5 years and first sentence at 7 years. Neurological and behavioral abnormalities include aggressive tantrum-like behavior, decreased sensitivity to pain, head banging, self-biting, skin picking resulting in scarring, stereotypies, food-seeking behaviors, and sleep disturbance with multiple nighttime awakenings and daily naps at age 16. Musculoskeletal features include hypotonia, hypermobility, pes planus, and type E brachydactyly with mildly shortened 3–5 metacarpals and metatarsals. Brachydactyly type E ranging from absent to severe has been reported in 2q37.3 deletion cases.
Figure 4.
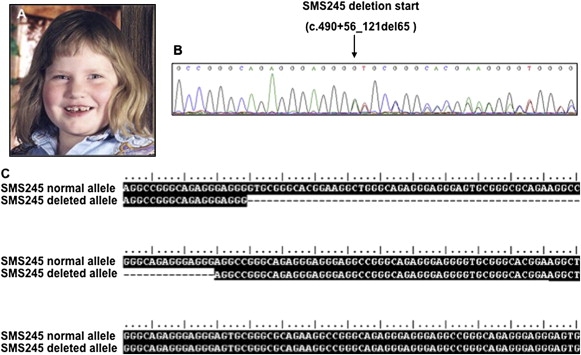
SMS245 HDAC4 Mutation
(A) SMS245, age 10.
(B) Electropherogram trace showing deletion start of SMS245 HDAC4 intron 5 at bp Chr2:239,763,222 (c.490+56_121del65).
(C) Alignment of SMS245 normal allele with SMS245 deleted allele. Note 65 bp intronic deletion. Performed with ClustalW accessory application in BioEdit sequence alignment editor.
Effect of HDAC4 Mutation or Deletion on Overlapping Pathways
Several cases presented here were initially referred for suspicion of Smith-Magenis syndrome (SMS320, SMS272, SMS117, and SMS245), which is caused by deletion or mutation of the retinoic acid induced 1 gene (RAI1). Based upon the significant phenotypic overlap between BDMR and SMS, we questioned whether HDAC4 and RAI1 might function in similar or overlapping molecular pathways which, when disrupted, lead to similar phenotypic effects, including sleep disturbances, behavioral problems, obesity, and craniofacial dysmorphism. As shown in Figure 5, RAI1 expression is reduced in SMS117, SMS245, and subject 2232 (del2q37.3), which indicates that RAI1 may function downstream of HDAC4 and is either directly or indirectly regulated by HDAC4. These data provide a molecular basis for the observed phenotypic similarities between these two disorders.
Figure 5.
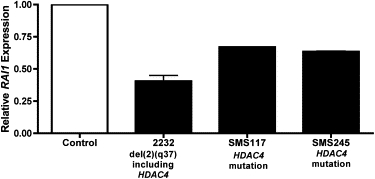
RAI1 Expression Is Reduced in HDAC4 Deletion and Mutation Cases
Quantitative real-time PCR of mRNA from lymphoblastoid cells from subject 2232 (del2q37.3) and SMS117 (HDAC4 mutation) or lymphocytes from whole blood (SMS245, HDAC4 mutation) relative to control subjects (no HDAC4 mutation or deletion), as described in Material and Methods. Assay performed twice in triplicate. Bars represent standard deviation from the mean. Control RNA for real-time PCR assays came from “like” samples, i.e., blood or cultured cells.
Discussion
We describe six individuals with del(2)(q37.3), five of which include or interrupt the HDAC4 gene, as well as two individuals who have mutations within the HDAC4 gene, pointing to HDAC4 as the primary causative gene for 2q37 deletion syndrome. Additional cases from the literature supporting this conclusion include patient 2 from Shrimpton et al. (2004)12 with brachydactyly type E, short stature, and cognitive delay and a recent report of a patient with brachydactyly type E, developmental delay, autism, and seizures (Felder et al., 2009).13 Although HDAC4 was not specifically examined in these two cases, the breakpoints mapped in close proximity to HDAC4, suggesting that this gene was probably disrupted. Expression of HDAC4 was also reduced to ∼50% in 2q37.3 deletion patient cells in the current study. Taken together, we conclude that haploinsufficiency of HDAC4 results in brachydactyly mental retardation syndrome.
HDAC4
HDAC4 is a class II HDAC, which acts as a corepressor for DNA-binding transcription factors. Acting in concert with HDAC9 and HDAC3, HDAC4 acts to inhibit a variety of transcription factors, including MEF2C (MIM 600662) and RUNX2 (MIM 600211), both of which are essential for proper skeletal development and serve to deacetylate histones. This deacetylation condenses chromatin, making it inaccessible to factors that drive transcription. Additionally, class II HDACs are antagonists of cardiac hypertrophy needed for proper cardiac development.14 Because of this molecular evidence, it is logical to conclude that deletion or mutation of HDAC4 would lead to the core phenotypes seen in BDMR. In addition, it is important to note that all HDAC4 deletion or mutation cases evaluated for RAI1 expression showed reduced mRNA expression (Figure 5). This is not surprising given the strong phenotypic overlap with BDMR and SMS. These data indicate that HDAC4 and RAI1 function in a common pathway, which, in turn, leads to common phenotypic effects when disrupted.
Impact of HDAC4 Mutation
The impact of the c.2399_2400insC found in SMS117 on the HDAC4 protein is significant. The premature stop codon interrupts the histone deacetylase domain, potentially limiting or preventing all histone deacetylase activity from this HDAC4 allele. This deacetylase activity is essential for protein function, as shown by the fact that HDAC4 has the ability to deacetylate all four core histones.15 If amino acids 1002–1058 or 803–846 of HDAC4 (the most conserved domain of HDAC4 across species) are deleted, interaction with HDAC3 (MIM 605166) and all histone deacetylase activity are eliminated.16 As shown in Figure 3, the insertion of a cytosine at codon 801 causes a frameshift that significantly reduces amino acid identity (5%) or similarity (5%) between the SMS117 mutant and wild-type alleles. Taken together, these data provide evidence that the mutant HDAC4 protein is completely void of histone deacetylase activity and can no longer bind with HDAC3. Further, when mutated from histidine to alanine, a charged polar to nonpolar change at p.802 and p.803, Fischle et al. (2002) showed that binding to NCOR1 (MIM 600849) was eliminated.16 NCOR1 is essential for HDAC3 recruitment and HDAC activity of the complex. The SMS117 mutation of HDAC4 shows a p.H802T and p.H803P change at these essential amino acids leading to charged polar to uncharged polar and charged polar to nonpolar changes, respectively, potentially having a great impact on the activity of this mutant protein (Figure 3). Alternatively, the premature protein truncation might lead to nonsense-mediated RNA decay and haploinsufficiency. SMS245 carries a 65 bp de novo deletion in intron 5, which could result in incorrect splicing of the HDAC4 mRNA (Figure 4). This splicing error probably leads to RNA decay and haploinsufficiency of HDAC4.
Hdac4−/− and Related Mouse Models
The Hdac4−/− mouse exhibits a variety of significant phenotypes and was shown to have early-onset chondrocyte hypertrophy and premature bone ossification.7 These mice present with severe bone malformations and are much smaller than wild-type littermates because of premature ossification of the developing bone. A reflection of the importance of HDAC4 in bone malformation in humans can be seen when studying the hands and feet of SMS117 and SMS272 (Figure 2). In addition, in the same manuscript by Vega et al. (2004), it was shown that Hdac4 also directly controls Runt-related transcription factor 2 (Runx2) activity. The Hdac4−/− mouse model directly mimics the phenotype seen with constitutive Runx2 expression in chondrocytes.17,18
MEF2C activity, which is necessary for proper chondrocyte hypertrophy and bone development, is also regulated by HDAC4.19,20 Mice deleted for the Mef2c gene have impaired chondrocyte hypertrophy, cartilage angiogenesis, ossification, and longitudinal bone growth, a reciprocal phenotype to that of the Hdac4−/− mouse,21 consistent with HDAC4 acting as a negative corepressor of MEF2C.
Clinical Implications
In the analysis of 20 2q37 deletion cases, Aldred et al. (2004) concluded that the region most probably associated with congenital heart defects in the BDMR syndrome included the HDAC4 gene.1 In this report, we support this finding with the fact that SMS117 has a significant cardiac defect. The role of HDAC4 in cardiac development is further supported by data that implicate MEF2C, which is regulated by HDAC4, in cardiac development.22 Additional studies and cases are required to confirm this hypothesis.
Based upon our findings reported here, we conclude that haploinsufficiency of the HDAC4 gene results in BDMR. Data support a significant role for HDAC4 in normal skeletal development, specifically in metacarpal and metatarsal growth and craniofacial development, and in neuronal function, with a prominent role in behavior. Autism or autism spectrum disorder (ASD) has been reported in many cases with 2q37.3 deletions and is consistent with the behavioral and developmental findings in the cases we report here.1,13,23,24 However, two of our older cases are reported to be “very friendly” but with poor communication skills, whereas the younger subjects have significant speech delay and poor communication with behavioral problems often prohibiting positive interactions. These findings suggest that reduced expression or function of HDAC4 is a contributor to the ASD phenotype observed in many cases with 2q37 deletion but that interpersonal skills may improve with age and with reduction of negative behaviors. Given that the genetic basis of autism is highly complex, it is not surprising that autistic behavior was also reported in case 2282 with a terminal 2q deletion that does not include HDAC4. This phenotypic outcome was also found in a published case with a much smaller subtelomeric deletion, suggesting that multiple genes at 2q37 may be involved in this phenotype.25 A more proximal locus, CENTG2 at 2q37 (now renamed AGAP1 [MIM 608651]), has also been suggested by two-point linkage analysis in autism families, although this was not supported by multipoint analysis.26
Additionally, a recent publication by Klopocki et al. (2010)27 has linked mutations in the parathyroid hormone related protein (PTHLH [MIM 168470]) to result in brachydactyly type E. Interestingly, Pthlh was identified as an inhibitor of Runx2 (regulated by Hdac4) in mice,28 and Pthlh−/− mice exhibit lethal short-limbed chondrodysplasia. This finding speaks to the probability of a common molecular pathway involved in the development of metacarpals and metatarsals in the hands and feet in humans.
Given the significant phenotypic overlap with Smith-Magenis syndrome and the fact that RAI1 expression is reduced in BDMR cases, BDMR and SMS should be considered together in the differential diagnosis. Although type E brachydactyly is unique to BDMR, it is only penetrant in 50%–60% of cases, so its absence does not remove the possibility of a positive diagnosis for BDMR. Also, other phenotypes within the BDMR syndrome show variable expressivity, especially heart defects and behavioral problems, and this phenotypic variability is commonly observed among complex behavioral syndromes. Given the range of phenotypic expression, a genetic “two-hit” model is very probably applicable to these complex neuropsychiatric disorders.29 The range of phenotypes and variable expressivity observed are probably dependent on deletion size and compounded by other CNVs in the genome, which is consistent with regard to BDMR. The diagnostic challenges and the elucidation of core phenotypes from syndrome to syndrome make it important to note all molecular findings and detail specific phenotypic findings in all cases. Gene expression data shown here support common molecular pathways among these complex neurodevelopmental disorders and lay the foundation for delineating the etiology behind the common phenotypes, which will allow for improved diagnosis and, ultimately, rational therapeutic design for pharmacological and behavioral interventions.
As such, we recommend molecular evaluation in cases with phenotypic findings consistent with BDMR and/or SMS for deletions involving chromosomes 2q37.3 and 17p11.2, respectively (karyotype, FISH, and/or aCGH). For those cases without 2q37.3 deletion and with type E brachydactyly, sequencing of HDAC4 is appropriate. For those subjects without a deletion of 17p11.2 and without type E brachydactyly, RAI1 sequence analysis should be undertaken, and if negative, followed by HDAC4 sequencing. We anticipate additional mutations in the HDAC4 gene will be identified, lending further support to this gene playing an essential role in proper cognitive, bone, and cardiac development in humans.
Supplemental Data
Supplemental Data include one figure and three tables and can be found with this article online at http://www.cell.com/AJHG/.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Database of Genomic Variants, http://projects.tcag.ca/variation/
Etoology, http://www.etoology.net/index.php/software/genetics/100-bioedit-709.html
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
Primer3, http://frodo.wi.mit.edu/primer3/
UCSC Genome Bioinformatics, http://www.genome.ucsc.edu, accessed Feb. 25, 2010, build GRCh37/hg19
Acknowledgments
We thank all of the patients and families involved with this study. We thank Emily Edelman for data collection efforts and Jackie Morris for sample and data collection efforts. This work was supported, in part, by a grant from the Fondation Jérôme Lejeune.
References
- 1.Aldred M.A., Sanford R.O., Thomas N.S., Barrow M.A., Wilson L.C., Brueton L.A., Bonaglia M.C., Hennekam R.C., Eng C., Dennis N.R. Molecular analysis of 20 patients with 2q37.3 monosomy: definition of minimum deletion intervals for key phenotypes. J. Med. Genet. 2004;41:433–439. doi: 10.1136/jmg.2003.017202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Falk R.E., Casas K.A. Chromosome 2q37 deletion: Clinical and molecular aspects. Am. J. Med. Genet. C. Semin. Med. Genet. 2007;145C:357–371. doi: 10.1002/ajmg.c.30153. [DOI] [PubMed] [Google Scholar]
- 3.Wilson L.C., Leverton K., Oude Luttikhuis M.E., Oley C.A., Flint J., Wolstenholme J., Duckett D.P., Barrow M.A., Leonard J.V., Read A.P. Brachydactyly and mental retardation: An Albright hereditary osteodystrophy-like syndrome localized to 2q37. Am. J. Hum. Genet. 1995;56:400–407. [PMC free article] [PubMed] [Google Scholar]
- 4.Phelan M.C., Rogers R.C., Clarkson K.B., Bowyer F.P., Levine M.A., Estabrooks L.L., Severson M.C., Dobyns W.B. Albright hereditary osteodystrophy and del(2) (q37.3) in four unrelated individuals. Am. J. Med. Genet. 1995;58:1–7. doi: 10.1002/ajmg.1320580102. [DOI] [PubMed] [Google Scholar]
- 5.Sebat J., Lakshmi B., Malhotra D., Troge J., Lese-Martin C., Walsh T., Yamrom B., Yoon S., Krasnitz A., Kendall J. Strong association of de novo copy number mutations with autism. Science. 2007;316:445–449. doi: 10.1126/science.1138659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Williams S.R., Girirajan S., Tegay D., Nowak N., Hatchwell E., Elsea S.H. Array comparative genomic hybridisation of 52 subjects with a Smith-Magenis-like phenotype: Identification of dosage sensitive loci also associated with schizophrenia, autism, and developmental delay. J. Med. Genet. 2009;47:223–229. doi: 10.1136/jmg.2009.068072. [DOI] [PubMed] [Google Scholar]
- 7.Vega R.B., Matsuda K., Oh J., Barbosa A.C., Yang X., Meadows E., McAnally J., Pomajzl C., Shelton J.M., Richardson J.A. Histone deacetylase 4 controls chondrocyte hypertrophy during skeletogenesis. Cell. 2004;119:555–566. doi: 10.1016/j.cell.2004.10.024. [DOI] [PubMed] [Google Scholar]
- 8.Chen B., Cepko C.L. HDAC4 regulates neuronal survival in normal and diseased retinas. Science. 2009;323:256–259. doi: 10.1126/science.1166226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Girirajan S., Elsas L.J., Devriendt K., Elsea S.H. RAI1 variations in Smith-Magenis syndrome patients without 17p11.2 deletions. J. Med. Genet. 2005;42:820–828. doi: 10.1136/jmg.2005.031211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Aldred M.A., Vijayakrishnan J., James V., Soubrier F., Gomez-Sanchez M.A., Martensson G., Galie N., Manes A., Corris P., Simonneau G. BMPR2 gene rearrangements account for a significant proportion of mutations in familial and idiopathic pulmonary arterial hypertension. Hum. Mutat. 2006;27:212–213. doi: 10.1002/humu.9398. [DOI] [PubMed] [Google Scholar]
- 11.Girirajan S., Truong H.T., Blanchard C.L., Elsea S.H. A functional network module for Smith-Magenis syndrome. Clin. Genet. 2009;75:364–374. doi: 10.1111/j.1399-0004.2008.01135.x. [DOI] [PubMed] [Google Scholar]
- 12.Shrimpton A.E., Braddock B.R., Thomson L.L., Stein C.K., Hoo J.J. Molecular delineation of deletions on 2q37.3 in three cases with an Albright hereditary osteodystrophy-like phenotype. Clin. Genet. 2004;66:537–544. doi: 10.1111/j.1399-0004.2004.00363.x. [DOI] [PubMed] [Google Scholar]
- 13.Felder B., Radlwimmer B., Benner A., Mincheva A., Todt G., Beyer K.S., Schuster C., Bolte S., Schmotzer G., Klauck S.M. FARP2, HDLBP and PASK are downregulated in a patient with autism and 2q37.3 deletion syndrome. Am. J. Med. Genet. 2009;149A:952–959. doi: 10.1002/ajmg.a.32779. [DOI] [PubMed] [Google Scholar]
- 14.Zhang C.L., McKinsey T.A., Chang S., Antos C.L., Hill J.A., Olson E.N. Class II histone deacetylases act as signal-responsive repressors of cardiac hypertrophy. Cell. 2002;110:479–488. doi: 10.1016/s0092-8674(02)00861-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Grozinger C.M., Hassig C.A., Schreiber S.L. Three proteins define a class of human histone deacetylases related to yeast Hda1p. Proc. Natl. Acad. Sci. USA. 1999;96:4868–4873. doi: 10.1073/pnas.96.9.4868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fischle W., Dequiedt F., Hendzel M.J., Guenther M.G., Lazar M.A., Voelter W., Verdin E. Enzymatic activity associated with class II HDACs is dependent on a multiprotein complex containing HDAC3 and SMRT/N-CoR. Mol. Cell. 2002;9:45–57. doi: 10.1016/s1097-2765(01)00429-4. [DOI] [PubMed] [Google Scholar]
- 17.Takeda S., Bonnamy J.P., Owen M.J., Ducy P., Karsenty G. Continuous expression of Cbfa1 in nonhypertrophic chondrocytes uncovers its ability to induce hypertrophic chondrocyte differentiation and partially rescues Cbfa1-deficient mice. Genes Dev. 2001;15:467–481. doi: 10.1101/gad.845101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ueta C., Iwamoto M., Kanatani N., Yoshida C., Liu Y., Enomoto-Iwamoto M., Ohmori T., Enomoto H., Nakata K., Takada K. Skeletal malformations caused by overexpression of Cbfa1 or its dominant negative form in chondrocytes. J. Cell Biol. 2001;153:87–100. doi: 10.1083/jcb.153.1.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Black B.L., Olson E.N. Transcriptional control of muscle development by myocyte enhancer factor-2 (MEF2) proteins. Annu. Rev. Cell Dev. Biol. 1998;14:167–196. doi: 10.1146/annurev.cellbio.14.1.167. [DOI] [PubMed] [Google Scholar]
- 20.McKinsey T.A., Zhang C.L., Olson E.N. MEF2: A calcium-dependent regulator of cell division, differentiation and death. Trends Biochem. Sci. 2002;27:40–47. doi: 10.1016/s0968-0004(01)02031-x. [DOI] [PubMed] [Google Scholar]
- 21.Arnold M.A., Kim Y., Czubryt M.P., Phan D., McAnally J., Qi X., Shelton J.M., Richardson J.A., Bassel-Duby R., Olson E.N. MEF2C transcription factor controls chondrocyte hypertrophy and bone development. Dev. Cell. 2007;12:377–389. doi: 10.1016/j.devcel.2007.02.004. [DOI] [PubMed] [Google Scholar]
- 22.Munoz J.P., Collao A., Chiong M., Maldonado C., Adasme T., Carrasco L., Ocaranza P., Bravo R., Gonzalez L., Diaz-Araya G. The transcription factor MEF2C mediates cardiomyocyte hypertrophy induced by IGF-1 signaling. Biochem. Biophys. Res. Commun. 2009;388:155–160. doi: 10.1016/j.bbrc.2009.07.147. [DOI] [PubMed] [Google Scholar]
- 23.Smith M., Escamilla J.R., Filipek P., Bocian M.E., Modahl C., Flodman P., Spence M.A. Molecular genetic delineation of 2q37.3 deletion in autism and osteodystrophy: Report of a case and of new markers for deletion screening by PCR. Cytogenet. Cell Genet. 2001;94:15–22. doi: 10.1159/000048775. [DOI] [PubMed] [Google Scholar]
- 24.Reddy K.S. Cytogenetic abnormalities and fragile-X syndrome in autism spectrum disorder. BMC Med. Genet. 2005;6:3. doi: 10.1186/1471-2350-6-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wolff D.J., Clifton K., Karr C., Charles J. Pilot assessment of the subtelomeric regions of children with autism: Detection of a 2q deletion. Genet. Med. 2002;4:10–14. doi: 10.1097/00125817-200201000-00002. [DOI] [PubMed] [Google Scholar]
- 26.Wassink T.H., Piven J., Vieland V.J., Jenkins L., Frantz R., Bartlett C.W., Goedken R., Childress D., Spence M.A., Smith M. Evaluation of the chromosome 2q37.3 gene CENTG2 as an autism susceptibility gene. Am. J. Med. Genet. B. Neuropsychiatr. Genet. 2005;136B:36–44. doi: 10.1002/ajmg.b.30180. [DOI] [PubMed] [Google Scholar]
- 27.Klopocki E., Hennig B.P., Dathe K., Koll R., de Ravel T., Baten E., Blom E., Gillerot Y., Weigel J.F., Kruger G. Deletion and point mutations of PTHLH cause brachydactyly type E. Am. J. Hum. Genet. 2010;86:434–439. doi: 10.1016/j.ajhg.2010.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Guo J., Chung U.I., Yang D., Karsenty G., Bringhurst F.R., Kronenberg H.M. PTH/PTHrP receptor delays chondrocyte hypertrophy via both Runx2-dependent and -independent pathways. Dev. Biol. 2006;292:116–128. doi: 10.1016/j.ydbio.2005.12.044. [DOI] [PubMed] [Google Scholar]
- 29.Girirajan S., Rosenfeld J.A., Cooper G.M., Antonacci F., Siswara P., Itsara A., Vives L., Walsh T., McCarthy S.E., Baker C. A recurrent 16p12.1 microdeletion supports a two-hit model for severe developmental delay. Nat. Genet. 2010;42:203–209. doi: 10.1038/ng.534. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.