

Abstract
Perrault syndrome is a recessive disorder characterized by ovarian dysgenesis in females, sensorineural deafness in both males and females, and in some patients, neurological manifestations. No genes for Perrault syndrome have heretofore been identified. A small family of mixed European ancestry includes two sisters with well-characterized Perrault syndrome. Whole-exome sequencing of genomic DNA from one of these sisters revealed exactly one gene with two rare functional variants: HSD17B4, which encodes 17β-hydroxysteroid dehydrogenase type 4 (HSD17B4), also known as D-bifunctional protein (DBP). HSD17B4/DBP is a multifunctional peroxisomal enzyme involved in fatty acid β-oxidation and steroid metabolism. Both sisters are compound heterozygotes for HSD17B4 c.650A>G (p.Y217C) (maternal allele) and HSB17B4 c.1704T>A (p.Y568X) (paternal allele). The missense mutation is predicted by structural analysis to destabilize the HSD17B4 dehydrogenase domain. The nonsense mutation leads to very low levels of HSD17B4 transcript. Expression of mutant HSD17B4 protein in a compound heterozygote was severely reduced. Mutations in HSD17B4 are known to cause DBP deficiency, an autosomal-recessive disorder of peroxisomal fatty acid β-oxidation that is generally fatal within the first two years of life. No females with DBP deficiency surviving past puberty have been reported, and ovarian dysgenesis has not previously been associated with this illness. Six other families with Perrault syndrome have wild-type sequences of HSD17B4. These results indicate that Perrault syndrome and DBP deficiency overlap clinically; that Perrault syndrome is genetically heterogeneous; that DBP deficiency may be underdiagnosed; and that whole-exome sequencing can reveal critical genes in small, nonconsanguineous families.
Main Text
Perrault syndrome (PS [MIM 233400]) is a sex-influenced disorder characterized by sensorineural deafness in both males and females and ovarian dysgenesis in females.1 Some patients also have neurological manifestations, including mild mental retardation and cerebellar and peripheral nervous system involvement.2,3 Clinical heterogeneity of the illness has prompted a possible classification of Perrault syndrome to type I, static and without neurological disease, and type II, with progressive neurological disease.
We evaluated an American family of mixed European ancestry with two daughters whose clinical manifestations have been thoroughly described.4,5 In the evaluation by Fiumara et al.,5 these patients are MK and LK. In addition to sensorineural deafness and ovarian dysgenesis, both sisters exhibited short stature, mild mental retardation, and progressive sensory and motor peripheral neuropathy. The older sister also had prominent cerebellar involvement, with dysarthria, intention tremor, and ataxia and MRI studies revealing “moderately severe” atrophy of the cerebellar hemispheres and vermis.
Because the family is small and not consanguineous, neither linkage analysis nor homozygosity mapping would have been informative in identifying the responsible gene. Instead, we applied whole-exome sequencing to identify the gene responsible for Perrault syndrome in this family. A paired-end library was prepared from genomic DNA of individual 4-01 (Figure 1A) and hybridized to biotinylated cRNA oligonucleotide baits from the SureSelect Human All Exon Kit (Agilent Technologies).6 The exome-enriched library was sequenced with paired-end 76 bp reads on two lanes of an Illumina Genome Analyzer IIx with the use of Illumina pipeline v1.6. We used MAQ v0.7.17 to align the sequence reads to the February 2009 human reference sequence of the UCSC Genome Browser (GRCh37/hg19). A total of 5.73 Gb of sequence (376,973,000 pairs of reads) aligned to the exome target. The proportion of the entire targeted exome covered by > 10 reads of Q30 or greater quality was 93.1%. DNA variants were filtered against dbSNP131 and Phase 3 of the 1000 Genomes project (March 2010 data release), then classified by predicted function to include all missense, nonsense, frameshift, or splice-site alleles. Given that dbSNP includes both disease-associated and benign alleles, we included in the analysis known SNPs identified by dbSNP as clinically associated. Because the pedigree structure of Perrault syndrome in the family is consistent with autosomal or X-linked recessive inheritance, the X chromosome was included in all analyses. The number of rare variants of each class identified in the proband is indicated in Table 1. We surveyed all genes harboring the 207 rare nonsense, missense, frameshift, or splice variants for any genes with two such alleles in this patient.
Figure 1.
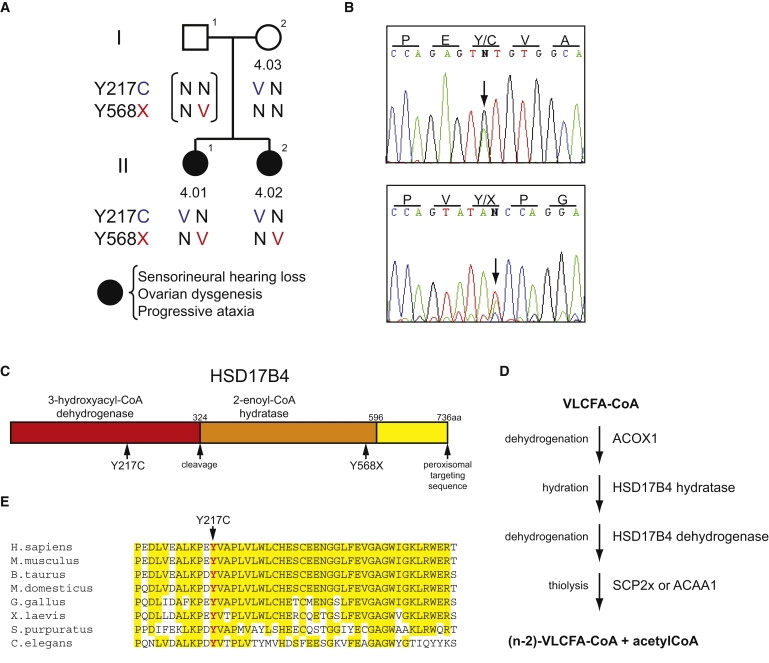
Coumpound Heterozygosity for HSD17B4 Mutations in Family 4
(A) Pedigree of Perrault syndrome family 4.
(B) Validation by Sanger sequencing of HSD17B4 mutations identified in individual 4-01 by whole-exome sequencing. Genomic DNA was sequenced after amplification with primers flanking exon 9 (5′- TGGAAAGGTGTGTCTGATAC-3′, 5′- CAAATGTAGAACTAAGTAAAAAC-3′) and exon 20 (5′- TTGAAAGACAAAGAATTGGC-3′, 5′-TGAAAACACCAGACAAGCTG-3′). Arrows indicate heterozygosity for chr5:118,824,914A>G, HSD17B4 c.650A>G (p.Y217C), in the upper panel, and for chr5:118,862,851T>A, HSB17B4 c.1704T>A (p.Y568X), in the lower panel.
(C) Schematic of the HSD17B4 protein.
(D) Diagram of the four steps of peroxisomal β-oxidation (left) and the enzymes by which they are catalyzed (right), using oxidation of very long chain fatty acids (VLCFA) as an example.
(E) Protein sequence alignment of HSD17B4 orthologs, showing the region surrounding the mutated Y217 (indicated in red). The project was approved by the Human Subjects Division of the University of Washington, and informed consent was obtained from all of the subjects.
Table 1.
Variants Revealed by Whole-Exome Sequencing in a Proband with Perrault Syndrome
| SBP | Indel | Total | |
|---|---|---|---|
| Number of variants with ≥ 15% of total reads | 790 | 356 | 1146 |
| Number of nonsense, missense, frameshift, or splice variants | 191 | 16 | 207 |
| Number of genes with ≥ 2 nonsense, missense, frameshift, or splice variants | 1 |
SBP indicates single base pair substitutions; Indel indicates small insertions or deletions.
Only one gene, HSD17B4 (MIM 601860) on chromosome 5q23.1, fulfilled this criterion (Table 2). The patient's DNA included mutations at chr5:118,824,914A>G, corresponding to HSD17B4 c.650A>G (p.Y217C) in exon 9, and at chr5:118,862,851T>A, corresponding to HSB17B4 c.1704T>A (p.Y568X) in exon 20 (accession no. NM_000414). An intronless partial pseudogene of HSD17B4 is located on chr8q24.13. Because the pseudogene duplicates HSD17B4 exon 20, only 19% of total reads aligned to chr5:118,862,851 harbor the nonsense mutation. We encountered similar asymmetry in read number as the result of pseudogenization of CHEK28. In contrast, the pseudogene does not include HSD17B4 exon 9, and 45% of total reads aligned to chr5:118,824,914 harbor the missense mutation. PCR amplification and Sanger sequencing demonstrated that both the proband (4-01) and her sister (4-02) were compound heterozygotes for both mutations, with the nonsense allele inherited from their mother (4-03) and the father an obligate carrier of the missense mutation (Figures 1A and 1B). The PCR primers used for mutation validation were unique to the functional copy on chromosome 5 and did not detect the pseudogene. Primers for sequencing HSD17B4 are listed in Table S1, available online.
Table 2.
Number of Wild-Type and Variant Reads at HSD17B4
| Chr. | Coordinate | Variant | Wild-Type Reads | Variant Reads | Effect |
|---|---|---|---|---|---|
| 5 | 118,824,914 | A>G | 159 | 129 | Y217C |
| 5 | 118,862,851 | T>A | 198 | 47 | Y568stop |
In order to determine whether both mutant transcripts were expressed, we sequenced lymphoblast cDNA from patient 4-02 and from a control individual with wild-type HSD17B4 sequence. The HSB17B4 c.1704T>A (p.Y568X) transcript is expressed at very low levels relative to the HSD17B4 c.650A>G (p.Y217C) transcript (Figure 2A). This observation suggests that the HSB17B4 c.1704T>A (p.Y568X) transcript is subjected to nonsense-mediated decay whereas the HSD17B4 c.650A>G (p.Y217C) is more stably expressed.
Figure 2.
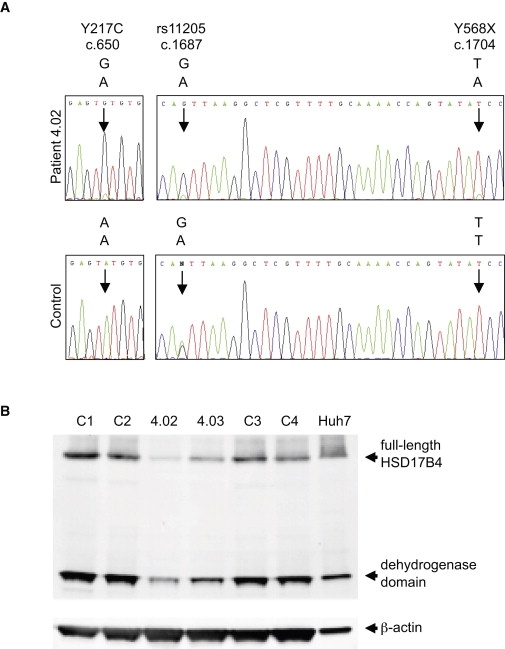
Expression of HSD17B4 Mutant Transcripts and Protein
(A) Expression of the two mutant transcripts of HSD17B4. Gene-specific cDNA for HSD17B4 was prepared from lymphoblast RNA of patient 4-02 and an unrelated control. cDNA was generated from total RNA with a primer located in exon 23 (5′- CCAGAACCACTTTTCAGGTCAATAGTCCAC-3′), amplified with primer pairs spanning exons 7–12 (5′-GGGTTCATTCCAAGTGACAC-3′, 5′-TCCTGATGTTGCTGTAGACG-3′) and exons 17–22, (5′- GCCATACCTAATAGACCTCC-3′, 5′- CAGCATTTACTTTCTTCACC-3′), and Sanger sequenced. The upper panels indicate sequence from patient 4-02, whose genomic DNA is heterozygous at nucleotide positions c.650, c.1687 (rs11205), and c.1704. The cDNA sequence harboring the GGT haplotype, which carries the missense allele G at c.650, the G allele at c.1687, and the wild-type allele T at c.1704 is more highly expressed than the sequence harboring the AAA haplotype, which carries the wild-type allele A at c.650, the A allele at c.1687, and the nonsense allele A at c.1704. The lower panels indicate sequence from an unrelated control, who is heterozygous at c.1687 and wild-type at c.650 and c.1704. The two alleles of the SNP at c.1687 appear equally expressed in the lymphoblast cDNA of the control.
(B) Lymphoblast cell lysates (50 μg) of patients (4-02 and 4-03) and controls (C1–C4) and Huh7 lysates (10 μg) were analyzed by immunoblotting using primary antibodies rabbit anti-HSD17B4 (Sigma) and mouse anti-β-actin (Sigma) and IRDye-conjugated secondary antibodies (LI-COR Biosciences). Full-length HSD17B4 (79 kD) and the processed dehydrogenase domain (35 kD) are indicated.
The protein encoded by HSD17B4, 17β-hydroxysteroid dehydrogenase type 4 (HSD17B4), also known as D-bifunctional protein (DBP) and multifunctional protein 2 (MFP-2), is a multifunctional peroxisomal enzyme involved in fatty acid β-oxidation and steroid metabolism.9,10 HSD17B4 contains three functional domains: an N-terminal 3-hydroxyacyl-CoA dehydrogenase domain, a central 2-enoyl-CoA hydratase domain, and a C-terminal sterol carrier protein 2-like domain (SCP-2L), the final three amino acids of which form the peroxisomal targeting signal11 (Figure 1C). The 79 kD full-length HSD17B4 protein is proteolytically cleaved after peroxisomal import to yield a 35 kD dehydrogenase unit and a 45 kD hydratase/SCP-2L unit, both of which are homodimeric.12,13,14,15 The hydratase and dehydrogenase domains catalyze sequential steps in fatty acid β-oxidation (Figure 1D). In addition, the dehydrogenase domain functions in steroid metabolism to convert the more active 17β-estradiol to the less active estrone.8 The sequence and multifunctional nature of HSD17B4 are highly conserved.8,16 In particular, the Y217 residue that is mutant in these patients is identical from humans to C. elegans and is located in a very highly conserved region of the protein (Figure 1E). HSD17B4 p.Y217C is predicted by the PolyPhen-2 algorithm to be “probably damaging” with a score of 0.991 and by the Xvar algorithm to have “medium functional impact” with a score of 1.615. Neither HSD17B4 c.650A>G (p.Y217C) nor HSB17B4 c.1704T>A (p.Y568X) was present in 1092 control individuals (2184 control chromosomes).
Mutations in HSD17B4 cause DBP deficiency (MIM 261515), an autosomal-recessive disorder of peroxisomal fatty acid β-oxidation that is generally fatal within the first two years of life. Patients with classical DBP deficiency present with hypotonia and seizures by one month of age. They have dysmorphic features and hearing and vision impairments, and they generally make no developmental progress.9 DBP deficiency results in the accumulation of β-oxidation substrates, including very long chain fatty acids (VLCFA), branched chain fatty acids, and bile acid intermediates that are all detectable in serum or cultured patient fibroblasts. In addition, the presence of DBP can be directly examined by immunoblotting, and DBP enzyme activity can be measured in fibroblast samples.9 DBP deficiency can be classified into three types, each based on which of the two DBP enzymatic activities are diminished. In type I, both hydratase and dehydrogenase activities are deficient, type II is a specific hydratase deficiency, and type III is a specific dehydrogenase deficiency.9 Different classes of HSD17B4 mutations are associated with each type of DBP deficiency. All DBP deficiency patients described to date are homozygous or compound heterozygous for mutations in HSD17B4. Type I deficiency is generally associated with nonsense mutations, frameshift mutations, or in-frame deletions of 20 or more residues in the dehydrogenase domain. In patients with type I deficiency, HSD17B4 protein is almost always undetectable in fibroblasts. Type II deficiency is associated with missense mutations or in-frame deletions in the hydratase domain, and type III deficiency is associated with missense mutations or single amino acid deletions in the dehydrogenase domain.17
The genotype of the affected sisters in family 4 does not correspond exactly to any of these types. HSD17B4 p.Y568X is a mutation of the sort associated with type I deficiency. HSD17B4 p.Y217C is a missense mutation in the dehydrogenase domain, so generally associated with type III deficiency. Interestingly, one of the more mildly affected cases in the literature, who was alive at 7.5 years, is a compound heterozygote for type I and type II alleles.16 Differences in transcript expression of the two HSD17B4 mutant alleles in the patients in family 4 (Figure 2A) motivated us to examine expression of HSD17B4 protein in these patients by immunoblotting. Using an antibody directed against the dehydrogenase domain of HSD17B4, we compared the expression of HSD17B4 in lymphoblast lysate from affected individual 4-02 and her mother, 4-03, versus HSD17B4 expression in lymphoblast lysate from four unrelated, healthy individuals with wild-type HSD17B4 sequences and in the human liver cell line Huh7. In all control samples and in Huh7, full-length HSD17B4 protein and the 35 kD dehydrogenase domain are detected (Figure 2B). In the sample from the unaffected mother, 4-03, who is heterozygous for HSD17B4 p.Y568X, there is a noticeable reduction in the amounts of full-length 79 kD protein and processed 35 kD dehydrogenase domain, and there is no evidence of a truncated protein. The reduced level of the transcript encoding HSD17B4 p.Y568X (Figure 2A) suggests that nonsense-mediated decay of the mutant RNA is the primary explanation for the reduced protein level, but the stability of the truncated protein may also be reduced. In the sample from affected sister 4-02, who is compound heterozygous for HSD17B4 p.Y568X and HSD17B4 p.Y217C, the amounts of both full-length protein and the dehydrogenase domain are further reduced. These results are consistent with previous observations that nonsense and other truncating HSD17B4 mutations generally lead to a complete loss of protein,14,16,18 which would explain the dramatic decrease in the full-length and 35 kD proteins in individual 4-03. Some missense mutations also appear to lead to a decrease in the amount of protein detected,16,17 consistent with the further reduction in the amount of HSD17B4 in individual 4-02.
Because the affected sisters likely express only protein with the HSD17B4 p.Y217C mutation, we predict that they have type III DBP deficiency, with a defect in dehydrogenase activity. A crystal structure of the human dehydrogenase domain in complex with nicotinamide adenine dinucleotide (NAD [PDB 1GZ6]) allows us to examine the structural role of Y217 and to predict the effect of the HSD17B4 p.Y217C substitution on dehydrogenase function. The HSD17B4 dehydrogenase domain exists as a “domain-swapped” dimer, in which the C-terminal region of one polypeptide chain crosses over to complete the functional structure of the other polypeptide chain and vice versa (Figure 3). In particular, the C-terminal helix αH of chain B (dark blue) forms a lid over the NAD-binding site and interacts with two short helices in chain A (dark green) that surround the NAD-binding site and contribute catalytic residues. The aromatic side chain of Y217 points away from the NAD-binding site and is involved in a network of interactions predicted to stabilize the functional dimeric structure. Specifically, Y217 from chain A (yellow and red spheres) is in close contact to the side chain of R258 from chain B (gray and blue spheres) in a cation-π orbital interaction.19 R258 is also in a position to make a favorable charged interaction with the negatively charged side chain of D303 on chain B (gray and red spheres). D303 is at the extreme C-terminal end of helix αH, so the interaction network is likely responsible for holding this helix in place, thereby completing the NAD-binding site. R258 is part of the interaction surface for the coenzyme-A moiety of the substrate,20 so its proper placement in the structure may affect substrate binding as well. Substitution of the aromatic side chain of Y217 with a small, hydrophilic cysteine residue is predicted to destabilize the conformation of the dehydrogenase domain and thereby decrease specific activity of the enzyme. A known DBP type III deficiency mutation at the adjacent position V218,16,21 whose side chain points directly into the NAD-binding site, is predicted to disrupt NAD binding16 and is likely to be more severe than Y217C.
Figure 3.
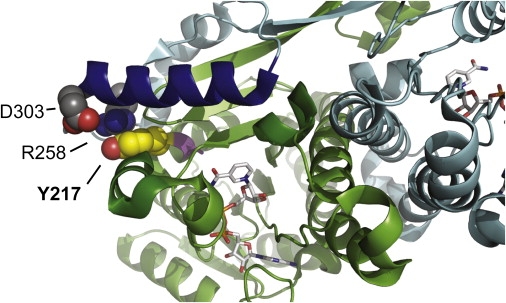
Structure of the HSD17B4 Dehydrogenase Domain
A ribbon representation of the HSD17B4 dehydrogenase domain in complex with NAD (PDB 1GZ6), showing the region surrounding the mutated residue Y217. The A and B chains of the homodimer are colored green and blue, respectively. NAD is represented as sticks within the binding site. As explained in the text, substitution of cysteine for tyrosine at position 217 is predicted to abrogate the cation-π orbital interaction of Y217 and R258 and thus the critical charged interaction of R258 with D303, leading to destabilization of the dehydrogenase domain and loss of specific activity of the enzyme.
Although DBP deficiency is commonly fatal in early childhood, a small number of type II and type III patients have been documented as surviving beyond 8 yrs of age.16,20,22,23 All of these cases were associated with missense mutations in the hydratase or dehydrogenase domain that, in most cases, were predicted to result in only minor structural effects and probable retention of residual enzyme activity. In many of these cases, there was little or no accumulation of β-oxidation substrates in serum, and DBP deficiency was detectable only by analysis of cultured fibroblasts.
It is likely that the sisters in our study are even more mildly affected than these previously documented cases. They were last examined at the ages of 27 and 16 and are therefore older than almost all documented cases, but notably, they share the critical features of older DBP-deficiency cases. An 8-yr-old DBP-deficient girl described by Soorani-Lunsing et al.21 had hearing loss and ataxia, peripheral polyneuropathy, and cerebellar atrophy. Her clinical course was characterized by a period of delayed development followed by progressive neurological deterioration, all similar to that of the older sister we studied. Serum VLCFA levels were normal in this girl,21 as in the older sister we report, who at age 23 had normal serum levels of VLCFA and phytanic acid.5 Hearing loss, polyneuropathy, cerebellar atrophy, and progressive loss of motor skills have also been observed in other long-term DBP-deficiency survivors, again with little or no abnormalities in VLCFA and other β-oxidation substrates.16,24 An important conclusion to gather from these observations is that mild DBP deficiency may be underdiagnosed if cases with normal VLCFA or phytanic acid levels are not tested for enzymatic HSD17B4/DBP activity.
Except for the milder course of their disease, the young women we report have ovarian dysgenesis as the major clinical feature distinguishing them from other DBP-deficiency cases, and the combination of deafness and ovarian dysgenesis led to the diagnosis of Perrault syndrome. Although progressive neurological symptoms have been described in Perrault syndrome, ovarian dysgenesis has not previously been associated with DBP deficiency. However, very few patients have survived to the age of puberty, and none of those documented patients have been female. Our results suggest that mutations in HSD17B4 leading to DBP deficiency that is mild enough to allow survival to the age of puberty are likely to cause ovarian dysgenesis in females in addition to the known neurological defects.
HSD17B4 mutations are the first identified cause of Perrault syndrome. Although HSD17B4 should be examined in other Perrault cases, especially those with neurological features, it is clear that Perrault syndrome is genetically heterogeneous. We have ruled out mutations in HSD17B4 as the cause of Perrault syndrome in six other families with hearing loss and ovarian dysgenesis, both with and without neurological symptoms.
Acknowledgments
This work was supported by National Institute of Deafness and other Communication Disorders of the NIH (R01DC005641) with a supplement from the American Recovery and Reinvestment Act.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes, http://www.1000genomes.org/page.php?page=home
Genome Reference Consortium, http://www.ncbi.nlm.nih.gov/projects/genome/assembly/grc/
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
PolyPhen-2, http://genetics.bwh.harvard.edu/pph2/
RSCB Protein Data Bank, http://www.pdb.org
UCSC Genome Browser, http://genome.ucsc.edu/
Xvar, http://Xvar.org
References
- 1.Perrault M., Klotz B., Housset E. Deux cas de syndrome de Turner avec surdi-mudité dans une même fratrie. [Two cases of Turner syndrome with deaf-mutism in two sisters] Bull. Mem. Soc. Med. Hop. Paris. 1951;67:79–84. [PubMed] [Google Scholar]
- 2.Nishi Y., Hamamoto K., Kajiyama M., Kawamura I. The Perrault syndrome: clinical report and review. Am. J. Med. Genet. 1988;31:623–629. doi: 10.1002/ajmg.1320310317. [DOI] [PubMed] [Google Scholar]
- 3.Gottschalk M.E., Coker S.B., Fox L.A. Neurologic anomalies of Perrault syndrome. Am. J. Med. Genet. 1996;65:274–276. doi: 10.1002/(SICI)1096-8628(19961111)65:4<274::AID-AJMG5>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 4.McCarthy D.J., Opitz J.M. Perrault syndrome in sisters. Am. J. Med. Genet. 1985;22:629–631. doi: 10.1002/ajmg.1320220324. [DOI] [PubMed] [Google Scholar]
- 5.Fiumara A., Sorge G., Toscano A., Parano E., Pavone L., Opitz J.M. Perrault syndrome: evidence for progressive nervous system involvement. Am. J. Med. Genet. A. 2004;128A:246–249. doi: 10.1002/ajmg.a.20616. [DOI] [PubMed] [Google Scholar]
- 6.Walsh T., Shahin H., Elkan-Miller T., Lee M.K., Thornton A.M., Roeb W., Abu Rayyan A., Loulus S., Avraham K.B., King M.-C., Kanaan M. Whole exome sequencing and homozygosity mapping identify mutation in the cell polarity protein GPSM2 as the cause of nonsyndromic hearing loss DFNB82. Am. J. Hum. Genet. 2010;87:90–94. doi: 10.1016/j.ajhg.2010.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li H., Ruan J., Durbin R. Mapping short DNA sequencing reads and calling variants using mapping quality scores. Genome Res. 2008;18:1851–1858. doi: 10.1101/gr.078212.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Walsh T., Lee M.K., Casadei S., Thornton A.M., Stray S.M., Pennil C., Nord A.S., Mandell J.B., Swisher E.M., King M.-C. Detection of inherited mutations for breast and ovarian cancer using genomic capture and massively parallel sequencing. Proc. Natl. Acad. Sci. USA. 2010;107:12629–12633. doi: 10.1073/pnas.1007983107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.de Launoit Y., Adamski J. Unique multifunctional HSD17B4 gene product: 17beta-hydroxysteroid dehydrogenase 4 and D-3-hydroxyacyl-coenzyme A dehydrogenase/hydratase involved in Zellweger syndrome. J. Mol. Endocrinol. 1999;22:227–240. doi: 10.1677/jme.0.0220227. [DOI] [PubMed] [Google Scholar]
- 10.Wanders R.J.A., Barth P.G., Heymans H.S.A. Single peroxisomal enzyme deficiencies. In: Scriver C.R., Beaudet A.L., Sly W.S., Valle D., editors. The Molecular and Metabolic Bases of Inherited Disease. McGraw-Hill; New York: 2001. pp. 3219–3256. [Google Scholar]
- 11.Leenders F., Husen B., Thole H.H., Adamski J. The sequence of porcine 80 kDa 17 beta-estradiol dehydrogenase reveals similarities to the short chain alcohol dehydrogenase family, to actin binding motifs and to sterol carrier protein 2. Mol. Cell. Endocrinol. 1994;104:127–131. doi: 10.1016/0303-7207(94)90114-7. [DOI] [PubMed] [Google Scholar]
- 12.Malila L.H., Siivari K.M., Mäkelä M.J., Jalonen J.E., Latipää P.M., Kunau W.H., Hiltunen J.K. Enzymes converting D-3-hydroxyacyl-CoA to trans-2-enoyl-CoA. Microsomal and peroxisomal isoenzymes in rat liver. J. Biol. Chem. 1993;268:21578–21585. [PubMed] [Google Scholar]
- 13.Jiang L.L., Kobayashi A., Matsuura H., Fukushima H., Hashimoto T. Purification and properties of human D-3-hydroxyacyl-CoA dehydratase: medium-chain enoyl-CoA hydratase is D-3-hydroxyacyl-CoA dehydratase. J. Biochem. 1996;120:624–632. doi: 10.1093/oxfordjournals.jbchem.a021458. [DOI] [PubMed] [Google Scholar]
- 14.Jiang L.L., Miyazawa S., Souri M., Hashimoto T. Structure of D-3-hydroxyacyl-CoA dehydratase/D-3-hydroxyacyl-CoA dehydrogenase bifunctional protein. J. Biochem. 1997;121:364–369. doi: 10.1093/oxfordjournals.jbchem.a021596. [DOI] [PubMed] [Google Scholar]
- 15.van Grunsven E.G., van Berkel E., Mooijer P.A., Watkins P.A., Moser H.W., Suzuki Y., Jiang L.L., Hashimoto T., Hoefler G., Adamski J., Wanders R.J. Peroxisomal bifunctional protein deficiency revisited: resolution of its true enzymatic and molecular basis. Am. J. Hum. Genet. 1999;64:99–107. doi: 10.1086/302180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Breitling R., Marijanović Z., Perović D., Adamski J. Evolution of 17beta-HSD type 4, a multifunctional protein of beta-oxidation. Mol. Cell. Endocrinol. 2001;171:205–210. doi: 10.1016/s0303-7207(00)00415-9. [DOI] [PubMed] [Google Scholar]
- 17.Ferdinandusse S., Ylianttila M.S., Gloerich J., Koski M.K., Oostheim W., Waterham H.R., Hiltunen J.K., Wanders R.J.A., Glumoff T. Mutational spectrum of D-bifunctional protein deficiency and structure-based genotype-phenotype analysis. Am. J. Hum. Genet. 2006;78:112–124. doi: 10.1086/498880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.van Grunsven E.G., Mooijer P.A., Aubourg P., Wanders R.J. Enoyl-CoA hydratase deficiency: identification of a new type of D-bifunctional protein deficiency. Hum. Mol. Genet. 1999;8:1509–1516. doi: 10.1093/hmg/8.8.1509. [DOI] [PubMed] [Google Scholar]
- 19.Dougherty D.A. Cation-pi interactions in chemistry and biology: a new view of benzene, Phe, Tyr, and Trp. Science. 1996;271:163–168. doi: 10.1126/science.271.5246.163. [DOI] [PubMed] [Google Scholar]
- 20.Haapalainen A.M., Koski M.K., Qin Y.M., Hiltunen J.K., Glumoff T. Binary structure of the two-domain (3R)-hydroxyacyl-CoA dehydrogenase from rat peroxisomal multifunctional enzyme type 2 at 2.38 A resolution. Structure. 2003;11:87–97. doi: 10.1016/s0969-2126(02)00931-0. [DOI] [PubMed] [Google Scholar]
- 21.Paton B.C., Pollard A.N. Molecular changes in the D-bifunctional protein cDNA sequence in Australasian patients belonging to the bifunctional protein complementation group. Cell Biochem. Biophys. 2000;32:247–251. doi: 10.1385/cbb:32:1-3:247. [DOI] [PubMed] [Google Scholar]
- 22.Soorani-Lunsing R.J., van Spronsen F.J., Stolte-Dijkstra I., Wanders R.J., Ferdinandusse S., Waterham H.R., Poll-The B.T., Rake J.P. Normal very-long-chain fatty acids in peroxisomal D-bifunctional protein deficiency: a diagnostic pitfall. J. Inherit. Metab. Dis. 2005;28:1172–1174. doi: 10.1007/s10545-005-0149-z. [DOI] [PubMed] [Google Scholar]
- 23.Paton B.C., Sharp P.C., Crane D.I., Poulos A. Oxidation of pristanic acid in fibroblasts and its application to the diagnosis of peroxisomal beta-oxidation defects. J. Clin. Invest. 1996;97:681–688. doi: 10.1172/JCI118465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ferdinandusse S., Denis S., Mooyer P.A., Dekker C., Duran M., Soorani-Lunsing R.J., Boltshauser E., Macaya A., Gärtner J., Majoie C.B. Clinical and biochemical spectrum of D-bifunctional protein deficiency. Ann. Neurol. 2006;59:92–104. doi: 10.1002/ana.20702. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.