

Abstract
By using a combination of array comparative genomic hybridization and a candidate gene approach, we identified nuclear factor I/X (NFIX) deletions or nonsense mutation in three sporadic cases of a Sotos-like overgrowth syndrome with advanced bone age, macrocephaly, developmental delay, scoliosis, and unusual facies. Unlike the aforementioned human syndrome, Nfix-deficient mice are unable to gain weight and die in the first 3 postnatal weeks, while they also present with a spinal deformation and decreased bone mineralization. These features prompted us to consider NFIX as a candidate gene for Marshall-Smith syndrome (MSS), a severe malformation syndrome characterized by failure to thrive, respiratory insufficiency, accelerated osseous maturation, kyphoscoliosis, osteopenia, and unusual facies. Distinct frameshift and splice NFIX mutations that escaped nonsense-mediated mRNA decay (NMD) were identified in nine MSS subjects. NFIX belongs to the Nuclear factor one (NFI) family of transcription factors, but its specific function is presently unknown. We demonstrate that NFIX is normally expressed prenatally during human brain development and skeletogenesis. These findings demonstrate that allelic NFIX mutations trigger distinct phenotypes, depending specifically on their impact on NMD.
Introduction
Overgrowth syndromes are defined on a height more than +2 standard deviations (SD) often present at birth and persistent in postnatal life. They tend to share several characteristics including macrocephaly, mental retardation, facial dysmorphic features, advanced bone age, hemihyperplasia, vascular malformations, and neoplasia but are distinct by the specific combination of these features.
The molecular causes of several overgrowth conditions such as Beckwith-Wiedemann syndrome (BWS [MIM 130650], Sotos syndrome (SS [MIM 117550]), Weaver syndrome (WS [MIM 277590]), or Simpson-Golabi-Behmel syndrome (SGBS [MIM 312870]) have been elucidated in recent years. The etiology of BWS is complex, involving a myriad of alterations to imprinted gene function in the chromosome 11p15 region.1 SS and some WS cases are caused by haploinsufficiency of the Nuclear receptor Set Domain containing protein 1 gene (MIM 606681), NSD1,2 which encodes a developmental regulatory protein exerting function(s) essential for early postimplantation development. NSD1 SET domain possesses intrinsic histone methyltransferase activity with specificity for Lys36 of histone H3 (H3-K36) and Lys20 of histone H4 (H4-K20). Finally, SGBS3 is due to mutations in GPC3 (MIM 300037, Glypican 3), which encodes an extracellular proteoglycan interacting with the insulin-like growth factor IGF2 and acting as a negative regulator of cell proliferation. Interestingly, several chromosomal duplications and deletions have been identified in patients with overgrowth, suggesting that some still unclassified overgrowth syndromes may be caused by subtle genomic imbalanced rearrangements. Tall stature is observed in patients with dup(4)(p16.3), probably due to dosage effect of the fibroblast growth factor receptor gene 3 (FGFR3 [MIM 134934]).4 Trisomy of 15q26-qter is frequently associated with tall stature and mental retardation caused by duplication of the IGF1R gene (MIM 147370).5 Finally, del(22)(q13) has also been associated with overgrowth and macrocephaly.6
Despite these recent advances, the disease-causing mechanisms remain unknown in 20%–40% of overgrowth cases. We recently reported a 1 Mb array-based comparative genomic hybridization (array-CGH) analysis of 93 patients with syndromic overgrowth. Our results identified nonrecurrent pathogenic chromosomal imbalances in 5% of cases.7 However, a large majority of patients remained without a known molecular etiology. We speculated that we failed to detect small cryptic rearrangement with the BAC-array used. This prompted us to evaluate unexplained cases with overgrowth via a higher resolution (Agilent 244K oligonucleotide) array-CGH. We present here the results of this analysis leading to the identification of NFIX (MIM 164005) deletions and point mutations in two distinct human disorders, a syndrome that shows some resemblance to SS and Marshall-Smith syndrome (MSS [MIM 602535]).
Subjects and Methods
Patients
For the 244K array-CGH, 18 patients and their parents were selected. Inclusion criteria were (1) unrelated parents; (2) developmental delay; (3) height >95th percentile and/or occipito-fronto circumference >95th percentile; and (4) at least two of the following features: (i) advanced bone age, (ii) dysmorphic craniofacial features, and (iii) congenital malformations.
For the NFIX mutations screening, 76 patients with unexplained syndromic overgrowth were first screened. The second screen tested 9 MSS patients fulfilling the diagnostic criteria for MS syndrome.8
Array-CGH Analysis
Blood samples from patients and unaffected relatives were obtained with the appropriate written informed consent, in accordance with the French ethical standards regarding human subjects, and genomic DNA was isolated from blood leucocytes with the Nucleon kit (Amersham) according to the manufacturer's instructions. Sample labeling for array-CGH was performed as described previously9 with 300 ng of input DNA. Array-CGH was performed on Agilent 244K oligonucleotide arrays (Agilent, Santa Clara, CA), according to the manufacturer's protocols.
Mutation Screening of NFIX
Specific primers for PCR amplification of exons and splicing junctions of NFIX were designed based on its sequence data from the GenBank database (primer sequences are available in Table S1 available online). PCR products were purified with Exo-SAP (Amersham) and directly sequenced on an automated sequencer (ABI 3130xl, Applied Biosystems) with the Dye Terminator method.
Expression Analyses of NFIX by RT-PCR
Selected RNAs from the Total Human RNA Master Panel II (Clontech) were subjected to reverse transcription with the SuperScript first-strand synthesis system (Invitrogen). Specific primers for PCR amplification NFIX and Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) mRNA were designed based on their sequence data from the GenBank database.
In Situ Hybridization
Normal human embryos and fetal tissues were obtained after elective pregnancy termination in agreement with French legislation (law no. 2004-800), National Ethics Committee recommendations (report no. 1 of May 22, 1984), and the Necker Hospital ethics committee. Embryonic stages were established according to Carnegie staging (CS) classification. Adjacent sections of human embryos and fetuses were treated with antisense or with control sense riboprobes. Riboprobe labeling, tissue fixation, and hybridization were carried out according to standard protocols.10
Results
Array-CGH Analysis
Only de novo imbalances were further analyzed and the Database of Genomic Variants was used to exclude CNVs previously observed in control individuals. An abnormal pattern in two patients (patients A and B) was suggestive of a de novo 19p13.1 monosomy (Figure 1A). FISH analyses on metaphases and nuclei from blood lymphocytes via fosmid clones G248P84002A10 and G248P87616F9 for patient A and G248P87556E1 and G248P80518H5 for patient B confirmed array-CGH findings. The deletions involved a single common gene, nuclear factor I X-type, NFIX, which was therefore regarded as a strong candidate to cause the overgrowth in these patients (Figure 1A).
Figure 1.
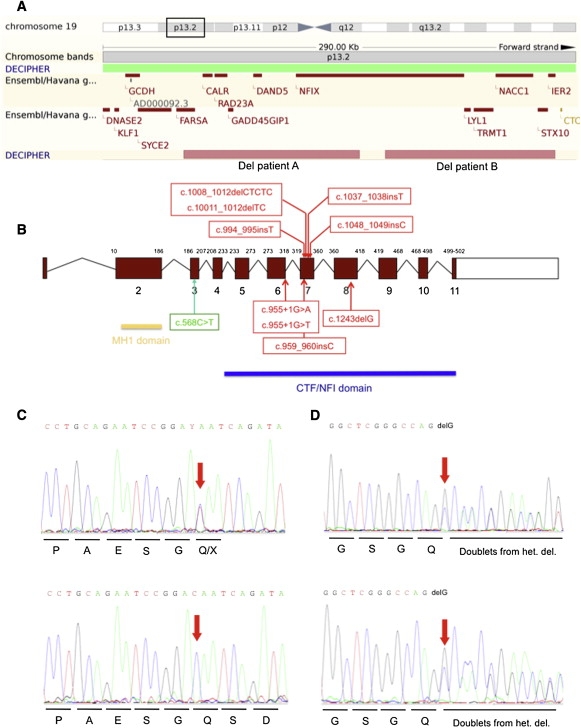
Genetic Analyses of Patients with Syndromic Overgrowth
(A) The figure shows the Ensembl display of the 19p13.2p13.3 region. Position of nonoverlapping NFIX deletions detected in patients A and B are represented by squares. For patient A, we found the proximal and distal breakpoints at 12 898 751 and 13 006 539 bp, respectively. For patient B, we found the proximal and distal breakpoints at 13 022 358 bp and 13 126 508 bp, respectively.
(B) The diagram shows the different mutations identified in NFIX genomic DNA. Open boxes represent untranslated regions and filled boxes represent coding regions. Mutation in green corresponds to the Sotos-like case while those in red indicate mutations found in MSS cases.
(C) Representative sequence traces of exon 3 in genomic DNA (top) or cDNA (bottom) of patient C (c.568C>T).
(D) Representative sequence traces of exon 8 sequences from genomic DNA (top) or cDNA (bottom) of patient F (c.1243 delG).
Mutation Screening of NFIX
A total of 76 patients with unexplained syndromic overgrowth were screened for NFIX mutations. The NFIX transcription unit encompasses 11 exons and encodes 6 isoforms that differ by alternative splicing of exon 7 and 9 and the use of alternative transcription initiation sites. A heterozygote de novo c.568C>T transition within exon 3 predicting a p.190Q>X nonsense mutation and causing premature translation termination codon (PTC) was identified in one patient (patient C diagnosed as “Sotos-like syndrome” based on slightly different facial features, Figures 1B and 2). This variant was not observed in 300 control chromosomes. Patients A–C presented a very similar phenotype consisting of postnatal overgrowth, macrocephaly, advanced bone age, long narrow face, high forehead, slender habitus, scoliosis, unusual behavior characterized especially by anxiety, and mental retardation (Figure 2, Table 1).
Figure 2.
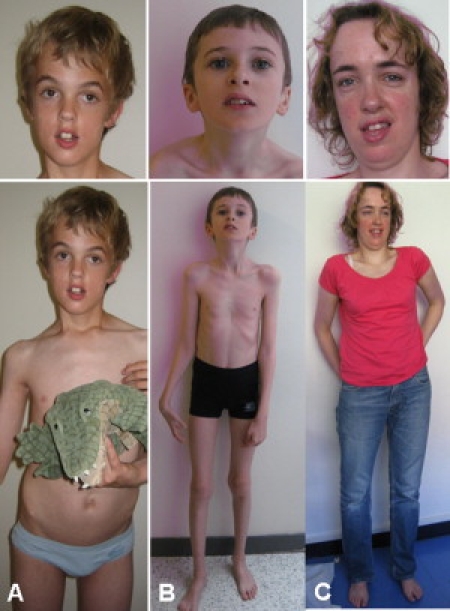
Front Views of the Three Overgrowth Syndrome Patients
Note the high forehead, long narrow face (patients A and C), and slender habitus.
Table 1.
Clinical Details of Patients with 19p13.1 Monosomy and c.568C>T NFI-X Mutation
| Patient A | Patient B | Patient C | |
|---|---|---|---|
| NFIX deletion/mutation | del 19p13.3 | del 19p13.3 | c.568C>T |
| Age at the last examination (yr) | 14 | 10 | 27 |
| Sex | M | M | F |
| Age mother/father (yr) | 31/33 | 25/30 | 31/31 |
| Ethnic origin | French | French | French |
| Prenatal growth | |||
| Birth weight (g) (centile) | 4500 (>95) | 3110 (10–50) | 3600 (50–90) |
| Birth height (cm) (centile) | 53 (95) | 49 (50) | 52 (95) |
| OFC (cm) (centile) | 38 (>95) | 33.5 (10) | 37.5 (>95) |
| Feeding problems in neonatal period | + | + | |
| Postnatal growth | |||
| Height > P98 | + | + | + |
| OFC > P98 | + | + | + |
| Height-weight ratio < P25 | + | + | |
| Developmental characteristics | |||
| Mental retardation | + | + | + |
| Speech delay | + | + | + |
| Autistic traits | + | + | + |
| Behavioral anomalies | + | + | + |
| Motor retardation | + | ||
| Hypotonia | + | + | |
| Brain MRI | |||
| Ventricular dilatation/hypoplasia corpus callosum | + | ||
| Ventricular dilatation | + | ||
| Craniofacial features | |||
| Long/narrow face | + | + | + |
| High forehead | + | + | + |
| Down-slanting palpebral fissures | + | + | |
| Small mouth | + | + | |
| Everted lower lip | + | + | |
| Prognathia | + | ||
| Eyes | |||
| Hypermotropia | + | + | |
| Strabismus | + | + | |
| Nystagmus | + | ||
| Astigmatism | + | ||
| Musculo-skeletal abnormalities | |||
| Pectus excavatum | + | + | |
| Scoliosis | + | + | |
| Advanced bone age | + | + | + |
| Abdominal wall hypotonia | + | + | |
| Coxa valga | + | + | |
| Hand/foot abnormalities | |||
| Long fingers | + | + | |
| Other abnormalities | |||
| Malformed nails | + | ||
| Premature eruption of teeth | + | ||
| Generalized livedo | + | ||
Two Nfix-deficient mice with marked differences in phenotypes have been described.11,12 They have distinct phenotypes but in one model, Nfix−/− newborn mice fail to thrive by P5, have a progressive kyphosis, delayed endochondral ossification, and reduction of trabecular bone formation, and usually die between days P21 and P28.11 These features were reminiscent of Marshall-Smith syndrome,8 which prompted us to consider NFIX as a candidate gene for MSS. Therefore, we screened nine MSS patients (Figure 3 and Table 2) for NFIX mutations and found seven independent heterozygous frameshift mutations (c.1011_1012 delTCfsX84, c.1037_1038insTfsX76, c.1008_1012 delCTCTCfsX83, c.1048_1049insCfsX70, c.1243 delGfsX48, c.994_995insTfsX88, c.959_960insCfsX10). In addition, two mutations were identified within the donor-splice site of exon 6 (c.955+1G>A, c.955+1G>T) (Figure 1B). All mutations occurred de novo and were not found in a panel of 300 control chromosomes.
Figure 3.
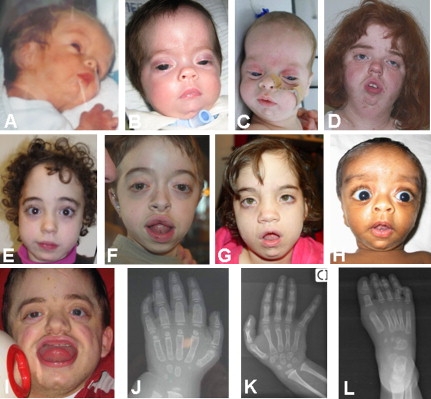
Pictures and Hand and Foot X-Rays of Patients with Marshall-Smith Syndrome
Facial features of the nine patients with Marshall-Smith syndrome (A–I) and hand X-rays at 1 year (J) and 4 years (K) of age and foot X-rays at 1 year of age (L). Note the high forehead, proptosis, underdeveloped midface, the advanced carpal and tarsal ossification, and the abnormal wide, bullet-shaped phalanges.
Table 2.
Clinical Details of the Nine Patients with Marshall-Smith Syndrome
|
Patient |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
8 |
9 |
|---|---|---|---|---|---|---|---|---|---|
| NFIX mutation | c.955+1G>A | c.955+1G>T | c.1011_1012 delTC | c.1037_1038 insT | c.1008_1012 delCTCTC | c.1048_1049 insC | c.1243 delG | c.994_995 insT | c.959_960 insC |
| Epidemiology | |||||||||
| Gender | M | F | M | M | F | F | M | F | M |
| Age at last examination | 3 weeksa | 6 months | 26 months | 3 years | 6 years | 7 years | 7 years | 16 years | 21 years |
| Ethnic origin | Belgium | Croatia | India | Netherlands | Brazil | Portugal | France | UK | UK |
| Growth | |||||||||
| Birth weight (g) | 2600 | 3400 | 2950 | 2975 | 1590 | 3230 | 2800 | 3860 | |
| Present weight (kg) | 2965 | 5 | 9.1 | 15 | 21 | 35 | 41 | ||
| Present height (cm) | 55 | 68 | 82 | 100 | 103 | 120 | 131 | ||
| Development | |||||||||
| Degree of delay | n.a. | Sev | Sev | Mod | Mod-Sev | Mod | Mod | Sev | Sev |
| First words (mths) | n.a. | n.a. | - | - | 60 | 36 | None | None | |
| Neurology | |||||||||
| High tone | - | - | - | - | - | - | N/A | N/A | |
| Brain MRI | dilated ventricles | pachygyria, hypoplasia callosal body | CTscan: N | hypoplasia callosal body | N/A | N/A | N/A | N/A | |
| Craniofacial features | |||||||||
| High forehead | + | + | + | + | + | + | + | + | + |
| Proptosis | + | + | + | + | + | + | + | + | + |
| Underdeveloped midface | + | + | + | + | + | - | + | + | + |
| Short nose | + | + | + | + | - | - | + | + | + |
| Prominent premaxilla | + | + | + | + | + | + | + | + | + |
| Everted lips | - | - | - | + | + | - | + | + | + |
| Gum hypertrophy | - | + | + | + | - | + | - | - | |
| Retrognathia | + | + | + | + | + | + | + | + | - |
| Eyes | |||||||||
| Myopia | - | - | - | −14 | −5/-8 | −12/-9 | - | - | - |
| Blue sclerae | + | + | + | + | + | - | + | + | + |
| Respiratory | |||||||||
| Choanal stenosis | - | + | - | - | - | + | - | - | |
| Respiratory problems | + | + | - | + | + | - | + | - | + |
| Musculo-Skeletal | |||||||||
| Abnormal bone maturation | + | + | + | + | + | + | + | + | + |
| Bone fractures | - | - | - | 1 | - | - | 4 | - | - |
| Scoliosis | - | - | - | - | + | - | + | + | + |
| Kyphosis | - | - | - | - | + | + | + | + | + |
| Umbilical hernia | - | + | - | - | - | + | - | - | |
| Other | |||||||||
| Cardiac defect | - | - | - | - | VSD | - | VSD | - | |
| Hearing loss | + | + | - | + | - | + | N/A | N/A | |
| Hypertrichosis | - | + | + | + | + | + | + | + | |
| Miscellaneous | IgA deficiency; vesico-ureteral reflux; long fingers | abnormal pinnae | hypospadias; glaucoma | long fingers | pyloric stenosis | Wilms tumor aged 4 | |||
Abbreviations: M, male; F, female; n.a., not applicable; N/A, not available; sev, severe; mod, moderate; VSD, ventricular septal defect.
Deceased.
RT-PCR Analyses of NFIX
To study the consequences of mutations causing either overgrowth or MSS on RNA level, we extracted, reverse transcribed, and sequenced RNA from skin fibroblasts of the overgrowth patient (patient C) and two MSS patients with a NFIX mutation. In the overgrowth patient, a single wild-type allele was observed (Figure 1C). In contrast, the MSS patients showed expression of both normal and mutated alleles (Figure 1D; Figures S1A and S1B). Similarly, RT-PCR analysis of RNA extracted of skin fibroblasts from the MSS patient carrying the c.955+1G>A mutation (disrupting proper mRNA splicing and causing partial inclusion of intron 6) detected both wild-type and mutated alleles (Figure S1C).
In Situ Hybridization
To gain insight into the function of NFIX gene product and establish correlations with the clinical manifestations of the patients, we studied the expression of NFIX in various human embryos and fetal and adult tissues. RT-PCR analysis demonstrated an ubiquitous expression of NFIX in the human cell types and tissues tested, including chondrocytes and osteoblasts (Figure S2). In situ hybridization in normal human embryos at CS17 (gestational day 42 [d42]) showed a nearly ubiquitous expression of NFIX, with prominent expression in the central nervous system and the peripheral nervous system (Figures 4A–4E). In fetal brain (22 WG), NFIX was expressed in the cerebral cortex, in the hippocampus, and faintly in the thalamus. Finally, in the skeleton, NFIX expression was first noticed at CS17 in the mandibular arch, the cartilage primordium of the humerus, the scapula, and the vertebrae. In the limb, NFIX expression was first observed at CS17 in the perichondrium. At 14 weeks of gestation (WG), NFIX was highly expressed in the proliferating zone of the digit (Figures 4F and 4G).
Figure 4.
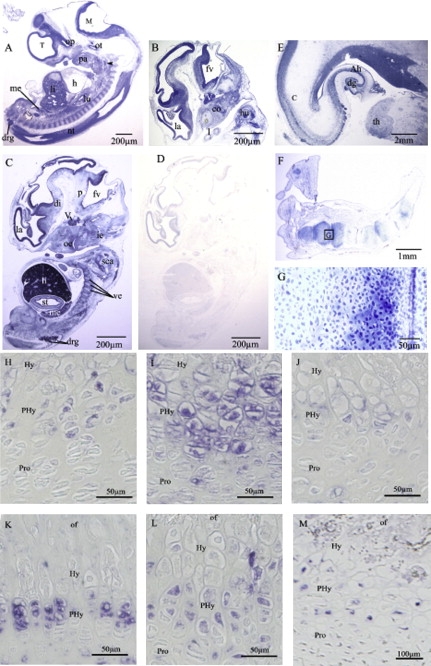
Pattern of NFIX Expression in Early Human Development and Fetal Epiphyseal Growth Plate
(A–G) Adjacent sections of human embryos and fetuses treated with antisense (A–C, E–G) or with control sense (D) riboprobes.
(A) Carnegie stage 17 (CS17) human embryo section.
(B–D) CS19 human embryo sections.
(E) Frontal section of a fetal (22WG) brain.
(F and G) Sagittal section of a fetal (14WG) hand.
The hybridization with control sense probe does not give any signal (D).
Note the expression in the neuroepithelium of the prosencephalon, the mesencephalon, the rhombencephalon, and the spinal cord as well as in the cranial and dorsal root ganglia.
Abbreviations: Ah, Ammon's horn; c, cerebral cortex; co, cochlea; dg, dentate gyrus; di, diencephalon; drg, dorsal root ganglia; fv, fourth ventricule; h, heart; hu, humerus; ie, inner ear; la, lateral ventricule; li, liver; lu, lung; M, mesencephalon; me, mesonephros; nt, neuroepithelium of the neural tube; oe, olfactory epithelium; op, optic stalk; ot, otic vesicle; p, pons; pa, pharingial arch; sca, scapula; st, stomach; T, Telencephalon; th, thalamus; V, trigeminal ganglia; ve, cartilage primordia of vertebrae.
(H–M) NFIX immunohistochemostry performed in 1-week-old mice (H), 2-week-old mice (I), 3-week-old mice (J), 4-week-old mice (K), 5-week-old mice (L), and 20 weeks of gestation human fetus (M). Note the restricted expression in the prehypertrophic chondrocytes and the expression in bone.
Abbreviations: of, ossification front, hy, hypertrophic zone; PHy, prehypertrophic zone; Pro, proliferative zone.
The study of distal femoral growth plates of 1- to 5-week-old mice and human fetus revealed strong NFIX expression in bone and in prehypertrophic chondrocytes (Figures 4H–4M).
Discussion
We report here NFIX deletions and nonsense mutation in three patients with an overgrowth syndrome that shows some resemblance to Sotos syndrome and which is characterized by postnatal overgrowth, macrocephaly, advanced bone age, long narrow face, high forehead, slender habitus, scoliosis, anxiety, and mental retardation.
NFIX belongs to the Nuclear factor one (NFI) family of transcription factors. Members of this family act as homo- and heterodimers and bind with high affinity to the palindromic consensus sequence TTGGC(N5)GCCAA.13 In vertebrates, the Nfi gene family consists of four closely related genes (Nfia-c and Nfix).14,15 They encode proteins with a conserved N-terminal DNA-binding and dimerization domain16 and a C-terminal transactivation/repression domain. Two Nfix-deficient mice with marked differences in phenotypes have been described.11,12 They have in common brain anomalies (enlarged ventricles, corpus callosum agenesis, or expanded cortex or distortion of the hippocampus) but have distinct phenotypes from those observed in overgrowth patients. In one model, Nfix−/− newborn mice are indistinguishable from wild-type and heterozygous littermates, but fail to thrive by P5 and usually die between days P21 and P28.11 They also present with progressive kyphosis, delayed endochondral ossification, and reduction of trabecular bone formation. These features resemble Marshall-Smith syndrome.8 MSS was first described in 1971 and was originally considered an overgrowth condition based on the accelerated skeletal maturation. Main features include high forehead, proptosis, blue sclerae, underdeveloped midface, short nose, and retrognathia. The outcome is characterized by mental retardation, failure to thrive, life-threatening respiratory difficulties, and skeletal anomalies including osteopenia, kyphoscoliosis, fractures, and hyperextensibility.8 The identification of NFIX mutations in the nine MSS patients further demonstrates the usefulness of mouse model in selecting candidate genes for human disorders.
The present results address the question of how allelic NFIX mutations can cause two different phenotypes. Either triggering or escaping NMD may explain this observation. Indeed, although nonsense and frameshift mutations resulting in PTC are expected to be natural targets of the NMD RNA surveillance system, several studies have shown that escaping NMD may lead the translated protein to convey potent dominant-negative or gain-of-function effects.17–20 Whereas the overgrowth phenotype is due to NFIX haploinsufficiency, the presence of both normal and mutated alleles in MSS supports that mutated RNAs escape NMD surveillance. We therefore suggest that NFIX mutations causing MSS generate mutant proteins able to exert a dominant-negative effect over the wild-type allele and result in a more severe phenotype closely resembling the Nfix−/− mouse phenotype.
Previous reports have demonstrated that NMD can modify phenotypes conveyed by allelic truncating mutations. SOX 10 (MIM 602229) dominant-negative mutations are responsible for a severe phenotype of peripheral demyelinating neuropathy, central dysmyelinating leukodystrophy, Waardenburg syndrome, and Hirschsprung disease (PCWH syndrome [MIM 609136]) whereas nonsense mutations activating NMD are responsible for the less severe phenotype combining Waardenburg and Hirschsprung only.21 Similarly, either triggering or escaping NMD is responsible for the various phenotypes caused by mutations within the myelin protein zero (MIM 159440) or elastin genes (MIM 130160).22–25 In mammalian cells, an exon-exon junction complex of proteins deposited 20–24 nucleotides upstream of exon-exon junctions during pre-mRNA splicing is considered to be a primary determinant of NMD.26,27 Consequently, translation termination at a nonsense codon located more than 50–55 nucleotides upstream of an exon-exon junction generally triggers NMD. Most of the NFIX mutations causing MSS that caused PTC were located more than 50–55 nucleotides upstream of an exon-exon junction. Why these mutations escaped NMD remains hitherto unexplained. Finally, one cannot exclude variable consequences of the mutations on the six different isoforms encoded by NFIX.
The NFI proteins have been shown to be involved in replication, signal transduction, and transcription.28 The NFI family has also been largely studied in respect to brain development and loss of function of NFI members in mice is associated with corpus callosum agenesis, a feature observed in overgrowth patients from this study. Our observation of a prominent expression in the nervous system in human embryo and of a specific expression in fetal brain is also in agreement with the findings of mental retardation in overgrowth and MSS patients.
Finally, the presence of a specific expression in prehypertrophic chondrocytes and bone is also in correlation with the clinical manifestations observed in the two human phenotypes, including advanced bone age in overgrowth patients and advanced carpal ossification, kyphosis, and osteopenia in MSS. Interestingly, the restricted expression to the prehypertrophic chondrocytes is quite similar to the parathyroid-related peptide receptor (PTHR1 [MIM 168468]) pattern of expression.29 These findings suggest a key role of NFIX in chondrocyte differentiation and bone formation and its specific involvement in the endochondral ossification process. The overgrowth in patients with dominant NFIX mutations supports a dysregulation of the switch between proliferation and differentiation stages and also suggests that NFIX could act as a negative regulator of the endochondral ossification process. The decreased bone density and spontaneous fractures in MSS patients could be directly linked to the role of NFIX in bone formation.
In conclusion, the present study demonstrates that NFIX haploinsufficiency is responsible for a novel clinically recognizable overgrowth syndrome condition and shows that NFIX is a disease-causing gene of Marshall-Smith syndrome. We provide further evidence that nonsense-mediated mRNA decay modulates the clinical consequences of the mutations. Finally, our findings support a key role of NFIX in human brain development and skeletogenesis.
Supplemental Data
Supplemental Data include two figures and one table and can be found with this article online at http://www.cell.com/AJHG/.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Ensembl Genome Browser, http://www.ensembl.org/
National Center for Biotechnology Information, http://www.ncbi.nlm.nih.gov/
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
Acknowledgments
We are grateful to the patients and their family and to the French association “L'éveil” for their participation in the study. This study was supported by the Centre National de la Recherche Scientifique, the GIS-Maladies Rares, the Agence de BioMédecine, and the international Marshall-Smith Support group. The authors declare no competing financial interests.
Contributor Information
Laurence Colleaux, Email: laurence.colleaux@inserm.fr.
Valérie Cormier-Daire, Email: valerie.cormier-daire@inserm.fr.
References
- 1.Weksberg R., Smith A.C., Squire J., Sadowski P. Beckwith-Wiedemann syndrome demonstrates a role for epigenetic control of normal development. Hum. Mol. Genet. 2003;12(Spec No 1):R61–R68. doi: 10.1093/hmg/ddg067. [DOI] [PubMed] [Google Scholar]
- 2.Visser R., Matsumoto N. Genetics of Sotos syndrome. Curr. Opin. Pediatr. 2003;15:598–606. doi: 10.1097/00008480-200312000-00010. [DOI] [PubMed] [Google Scholar]
- 3.Neri G., Gurrieri F., Zanni G., Lin A. Clinical and molecular aspects of the Simpson-Golabi-Behmel syndrome. Am. J. Med. Genet. 1998;79:279–283. doi: 10.1002/(sici)1096-8628(19981002)79:4<279::aid-ajmg9>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 4.Partington M.W., Fagan K., Soubjaki V., Turner G. Translocations involving 4p16.3 in three families: Deletion causing the Pitt-Rogers-Danks syndrome and duplication resulting in a new overgrowth syndrome. J. Med. Genet. 1997;34:719–728. doi: 10.1136/jmg.34.9.719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Faivre L., Gosset P., Cormier-Daire V., Odent S., Amiel J., Giurgea I., Nassogne M.C., Pasquier L., Munnich A., Romana S. Overgrowth and trisomy 15q26.1-qter including the IGF1 receptor gene: report of two families and review of the literature. Eur. J. Hum. Genet. 2002;10:699–706. doi: 10.1038/sj.ejhg.5200879. [DOI] [PubMed] [Google Scholar]
- 6.Phelan M.C., Rogers R.C., Saul R.A., Stapleton G.A., Sweet K., McDermid H., Shaw S.R., Claytor J., Willis J., Kelly D.P. 22q13 deletion syndrome. Am. J. Med. Genet. 2001;101:91–99. doi: 10.1002/1096-8628(20010615)101:2<91::aid-ajmg1340>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- 7.Malan V., Chevallier S., Soler G., Coubes C., Lacombe D., Pasquier L., Soulier J., Morichon-Delvallez N., Turleau C., Munnich A. Array-based comparative genomic hybridization identifies a high frequency of copy number variations in patients with syndromic overgrowth. Eur. J. Hum. Genet. 2010;18:227–232. doi: 10.1038/ejhg.2009.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Adam M.P., Hennekam R.C., Keppen L.D., Bull M.J., Clericuzio C.L., Burke L.W., Ormond K.E., Hoyme E.H. Marshall-Smith syndrome: Natural history and evidence of an osteochondrodysplasia with connective tissue abnormalities. Am. J. Med. Genet. A. 2005;137:117–124. doi: 10.1002/ajmg.a.30580. [DOI] [PubMed] [Google Scholar]
- 9.Fiegler H., Geigl J.B., Langer S., Rigler D., Porter K., Unger K., Carter N.P., Speicher M.R. High resolution array-CGH analysis of single cells. Nucleic Acids Res. 2007;35:e15. doi: 10.1093/nar/gkl1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Delous M., Baala L., Salomon R., Laclef C., Vierkotten J., Tory K., Golzio C., Lacoste T., Besse L., Ozilou C. The ciliary gene RPGRIP1L is mutated in cerebello-oculo-renal syndrome (Joubert syndrome type B) and Meckel syndrome. Nat. Genet. 2007;39:875–881. doi: 10.1038/ng2039. [DOI] [PubMed] [Google Scholar]
- 11.Driller K., Pagenstecher A., Uhl M., Omran H., Berlis A., Grunder A., Sippel A.E. Nuclear factor I X deficiency causes brain malformation and severe skeletal defects. Mol. Cell. Biol. 2007;27:3855–3867. doi: 10.1128/MCB.02293-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Campbell C.E., Piper M., Plachez C., Yeh Y.T., Baizer J.S., Osinski J.M., Litwack E.D., Richards L.J., Gronostajski R.M. The transcription factor Nfix is essential for normal brain development. BMC Dev. Biol. 2008;8:52. doi: 10.1186/1471-213X-8-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Roulet E., Bucher P., Schneider R., Wingender E., Dusserre Y., Werner T., Mermod N. Experimental analysis and computer prediction of CTF/NFI transcription factor DNA binding sites. J. Mol. Biol. 2000;297:833–848. doi: 10.1006/jmbi.2000.3614. [DOI] [PubMed] [Google Scholar]
- 14.Rupp R.A., Kruse U., Multhaup G., Gobel U., Beyreuther K., Sippel A.E. Chicken NFI/TGGCA proteins are encoded by at least three independent genes: NFI-A, NFI-B and NFI-C with homologues in mammalian genomes. Nucleic Acids Res. 1990;18:2607–2616. doi: 10.1093/nar/18.9.2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kruse U., Qian F., Sippel A.E. Identification of a fourth nuclear factor I gene in chicken by cDNA cloning: NFI-X. Nucleic Acids Res. 1991;19:6641. doi: 10.1093/nar/19.23.6641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gounari F., De Francesco R., Schmitt J., van der Vliet P., Cortese R., Stunnenberg H. Amino-terminal domain of NF1 binds to DNA as a dimer and activates adenovirus DNA replication. EMBO J. 1990;9:559–566. doi: 10.1002/j.1460-2075.1990.tb08143.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mendell J.T., Dietz H.C. When the message goes awry: Disease-producing mutations that influence mRNA content and performance. Cell. 2001;107:411–414. doi: 10.1016/s0092-8674(01)00583-9. [DOI] [PubMed] [Google Scholar]
- 18.Holbrook J.A., Neu-Yilik G., Hentze M.W., Kulozik A.E. Nonsense-mediated decay approaches the clinic. Nat. Genet. 2004;36:801–808. doi: 10.1038/ng1403. [DOI] [PubMed] [Google Scholar]
- 19.Kuzmiak H.A., Maquat L.E. Applying nonsense-mediated mRNA decay research to the clinic: Progress and challenges. Trends Mol. Med. 2006;12:306–316. doi: 10.1016/j.molmed.2006.05.005. [DOI] [PubMed] [Google Scholar]
- 20.Chang Y.F., Imam J.S., Wilkinson M.F. The nonsense-mediated decay RNA surveillance pathway. Annu. Rev. Biochem. 2007;76:51–74. doi: 10.1146/annurev.biochem.76.050106.093909. [DOI] [PubMed] [Google Scholar]
- 21.Cossais F., Wahlbuhl M., Kriesch J., Wegner M. SOX10 structure-function analysis in the chicken neural tube reveals important insights into its role in human neurocristopathies. Hum. Mol. Genet. 2010;19:2409–2420. doi: 10.1093/hmg/ddq124. [DOI] [PubMed] [Google Scholar]
- 22.Warner L.E., Hilz M.J., Appel S.H., Killian J.M., Kolodry E.H., Karpati G., Carpenter S., Watters G.V., Wheeler C., Witt D. Clinical phenotypes of different MPZ (P0) mutations may include Charcot-Marie-Tooth type 1B, Dejerine-Sottas, and congenital hypomyelination. Neuron. 1996;17:451–460. doi: 10.1016/s0896-6273(00)80177-4. [DOI] [PubMed] [Google Scholar]
- 23.Inoue K., Khajavi M., Ohyama T., Hirabayashi S., Wilson J., Reggin J.D., Mancias P., Butler I.J., Wilkinson M.F., Wegner M. Molecular mechanism for distinct neurological phenotypes conveyed by allelic truncating mutations. Nat. Genet. 2004;36:361–369. doi: 10.1038/ng1322. [DOI] [PubMed] [Google Scholar]
- 24.Metcalfe K., Rucka A.K., Smoot L., Hofstadler G., Tuzler G., McKeown P., Siu V., Rauch A., Dean J., Dennis N. Elastin: Mutational spectrum in supravalvular aortic stenosis. Eur. J. Hum. Genet. 2000;8:955–963. doi: 10.1038/sj.ejhg.5200564. [DOI] [PubMed] [Google Scholar]
- 25.Micale L., Turturo M.G., Fusco C., Augello B., Jurado L.A., Izzi C., Digilio M.C., Milani D., Lapi E., Zelante L. Identification and characterization of seven novel mutations of elastin gene in a cohort of patients affected by supravalvular aortic stenosis. Eur. J. Hum. Genet. 2010;18:317–323. doi: 10.1038/ejhg.2009.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Le Hir H., Izaurralde E., Maquat L.E., Moore M.J. The spliceosome deposits multiple proteins 20-24 nucleotides upstream of mRNA exon-exon junctions. EMBO J. 2000;19:6860–6869. doi: 10.1093/emboj/19.24.6860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Le Hir H., Gatfield D., Izaurralde E., Moore M.J. The exon-exon junction complex provides a binding platform for factors involved in mRNA export and nonsense-mediated mRNA decay. EMBO J. 2001;20:4987–4997. doi: 10.1093/emboj/20.17.4987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gronostajski R.M. Roles of the NFI/CTF gene family in transcription and development. Gene. 2000;249:31–45. doi: 10.1016/s0378-1119(00)00140-2. [DOI] [PubMed] [Google Scholar]
- 29.Hopyan S., Gokgoz N., Poon R., Gensure R.C., Yu C., Cole W.G., Bell R.S., Juppner H., Andrulis I.L., Wunder J.S. A mutant PTH/PTHrP type I receptor in enchondromatosis. Nat. Genet. 2002;30:306–310. doi: 10.1038/ng844. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.