

Abstract
Retinitis pigmentosa (RP) is the most common form of hereditary retinal degeneration, with a worldwide prevalence of 1 in 4000. Over 30 genes and loci have been implicated in nonsyndromic autosomal-recessive (ar) RP. Genome-wide homozygosity mapping was conducted in two sibships from an extended consanguineous Muslim Arab Israeli family segregating ar severe early-onset RP. A shared homozygous region on chromosome 17q25.3 was identified in both sibships, with an overlap of 4.7 Mb. One of the genes located in this interval is PDE6G, encoding for the inhibitory γ subunit of rod photoreceptor cyclic GMP-phosphodiesterase. Mutations in the genes encoding for the catalytic subunits of this holoenzyme, PDE6A and PDE6B, cause arRP. Sequencing of all coding exons, including exon-intron boundaries, revealed a homozygous single base change (c.187+1G>T) located in the conserved intron 3 donor splice site of PDE6G. This mutation cosegregated with the disease in the extended family. We used an in vitro splicing assay to demonstrate that this mutation leads to incorrect splicing. Affected individuals had markedly constricted visual fields. Both scotopic and photopic electroretinograms were severely reduced or completely extinct. Funduscopy showed typical bone spicule-type pigment deposits spread mainly at the midperiphery, as well as pallor of the optic disk. Macular involvement was indicated by the lack of foveal reflex and typical cystoid macular edema, proved by optical coherence tomography. These findings demonstrate the positive role of the γ subunit in maintaining phosphodiesterase activity and confirm the contribution of PDE6G to the etiology of RP in humans.
Main Text
Retinitis pigmentosa (RP, MIM 268000) is the most common form of hereditary retinal degeneration. The prevalence of RP in various western countries, including Israel, is approximately 1 in 4000.1,2 RP reflects a heterogeneous group of retinal dystrophies characterized by night blindness followed by visual field loss and often resulting in severe visual impairment. Ophthalmologic findings include characteristic pigmentation of the midperipheral retina, attenuation of retinal arterioles, and pale appearance of the optic disk.3 The disease is very heterogeneous, both clinically and genetically, and can be inherited as an autosomal-recessive (ar), autosomal-dominant (ad), or X-linked trait.1 A digenic pattern of inheritance has also been described.4 Over 40 genes and loci have been implicated in nonsyndromic RP, of which over 30 are associated with an ar mode of inheritance (Retnet: Retinal Information Network). However, it is estimated that the genes underlying 50% of RP cases are still unknown.
In an effort to identify previously unrecognized retinal degeneration genes, we ascertained two nuclear Israeli consanguineous families of Muslim Arab origin that belong to the same extended family (family TB14, Figures 1A and 1D). In each family, the parents are first cousins. In each of the families, there are two individuals affected with severe early-onset RP (Figure 1A). The study was approved by the institutional review board at Ha'Emek Medical Center and by the National Helsinki Committee for Genetic Research in Humans. Informed consent was obtained from all participants or their parents.
Figure 1.
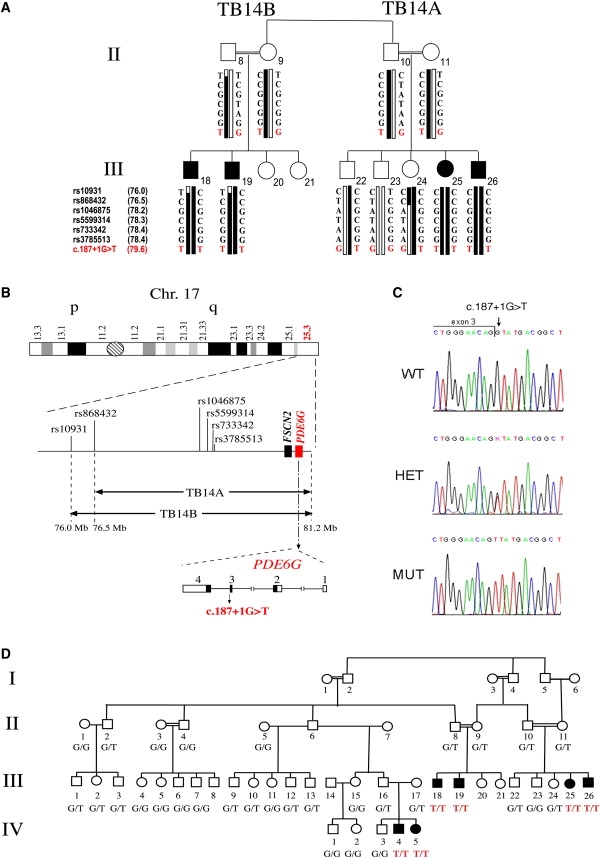
Genetic Analysis in Family TB14
(A) Shown are the two sibships segregating the c.187+1G>T mutation of PDE6G. SNP analysis performed on each of the sibships demonstrates cosegregation of a 17q25.3-linked haplotype with arRP. Double lines indicate consanguineous unions. Filled symbols represent affected individuals, whereas clear symbols represent unaffected individuals. Mutation-bearing haplotypes are marked by black bars. The position of the SNPs, based on the Human Genome Browser working draft hg18, is indicated between parentheses.
(B) Shown is chromosome 17 with the linkage intervals and the corresponding SNPs. FSCN2 and PDE6G reside within the region that is shared by all affected individuals. Also shown is the genomic structure of PDE6G. Coding exons are represented by black boxes. Noncoding exons are represented by white boxes. The location of the c.187+1G>T mutation is indicated.
(C) Sequence chromatograms for the c.187+1G>T mutation of PDE6G in a noncarrier individual (WT), an individual heterozygous for the mutation (het), and an affected individual homozygous for the mutant allele (mut). The exon-intron boundary is marked.
(D) A pedigree of the extended family TB14. Affected individuals are marked in black. Genotypes at the c.187+1 position are indicated.
To identify the mutated gene, we performed genome-wide homozygosity mapping with the Infinium Human Linkage 12 Genotyping Bead Chip (Illumina), which is capable of genotyping 6090 highly informative SNPs with an average genetic distance of 0.58 cM across the human genome. The only homozygous region shared among all four affected individuals was a 5.2 Mb interval at the telomeric end of chromosome 17 (17q25.3), between SNP rs10931 and the telomere (Figure 1B). SNP analysis in both families, including nonaffected siblings and parents, confirmed that the region segregates with arRP (Figure 1A). A recombination event in individual III-24 reduced the interval to 4.7 Mb between SNP rs868432 and the telomere (Figures 1A and 1B). This interval includes 203 genes. One of these genes is FSCN2 (fascin homolog 2, GenBank accession number NM_001077182), which is associated with adRP (MIM 607921, MIM 607643).5 Sequence analysis of the five coding exons of FSCN2 was performed with the Big Dye Terminator Cycle Sequencing Kit (PE Applied Biosystems). No mutation was detected in affected individuals.
Another gene located within the homozygous interval is PDE6G (GenBank accession number NM_002602, MIM 180073), which encodes for the inhibitory subunit of rod photoreceptor cyclic GMP-phosphodiesterase (cGMP-PDE), one of the key enzymes of the visual phototransduction cascade in the vertebrate retina. The holoenzyme is a heterotetrameric complex consisting of two large catalytic subunits, α (88 kDa) and β (84 kDa), and two identical inhibitory subunits, γ (11 kDa).6 Mutations in the genes encoding for the catalytic subunits of this holoenzyme, PDE6A and PDE6B, cause arRP in humans7,8 and retinal degeneration in animal models (MIM 180071, MIM 180072).9–12 Moreover, retinal degeneration that resembles human RP was identified in mice lacking the gene encoding for the γ inhibitory subunit Pde6g.13 The human PDE6G gene has been previously considered as a candidate for RP.14,15 However, pathogenic mutations of this gene have not been reported to date.
PDE6G harbors four exons. Sequence analysis of the three coding exons (exons 2–4), including exon-intron boundaries, was performed in an affected individual (individual III-26, Figure 1A). Primer sequences are listed in Table S1 available online. We identified a homozygous single base change, a G-to-T transversion located in the conserved intron 3 donor splice site (c.187+1G>T) (Figure 1C). This base change cosegregated with RP in the two sibships (Figure 1A). Analysis of 25 additional members of the extended family, including two additional affected individuals, confirmed cosegregation of the mutation with the disease. All unaffected individuals were either heterozygotes or carried two wild-type (WT) copies. Only the six affected individuals were homozygotes for the identified sequence change (Figure 1D). According to the splice-site consensus sequence in mammals, a G is located at position +1 of the donor site.16 To predict the effect of c.187+1G>T on splicing, we performed in silico analysis of the sequence with two different web-based tools (Splice Site Prediction by Neural Network and MaxEntScan). Both algorithms predicted that the c.187+1G>T change leads to elimination of intron 3 donor site.
Because of the limited expression pattern of PDE6G, we could not evaluate the effect of c.187+1G>T on splicing in patient-derived RNA. Alternatively, we used an in vitro splicing assay approach. For this purpose, we created a minigene construct. This construct harbors PDE6G exons 2, 3, and 4, flanked by 82–263 bp of intronic sequences, downstream of a cytomegalovirus (CMV) promoter (Figure 2A). Two different constructs were created: a construct harboring the WT allele (c.187+1G) and a construct harboring the mutant allele (c.187+1T). Each construct was transfected into the Y79 retinoblastoma-derived cell line, followed by RNA extraction and RT-PCR analysis. To analyze the results, we sequenced the splicing products derived from each construct. RNA derived from the WT construct yielded a main correctly spliced product of 433 bp, which included exons 2, 3, and 4. RNA derived from the mutant construct yielded a main aberrantly spliced product of 461 bp. In this product, a cryptic donor splice site, located 28 bp downstream of the mutant splice site within intron 3, was used. Both WT and mutant constructs also yielded an additional minor product of 392 bp, in which exon 3 was skipped (Figure 2).
Figure 2.
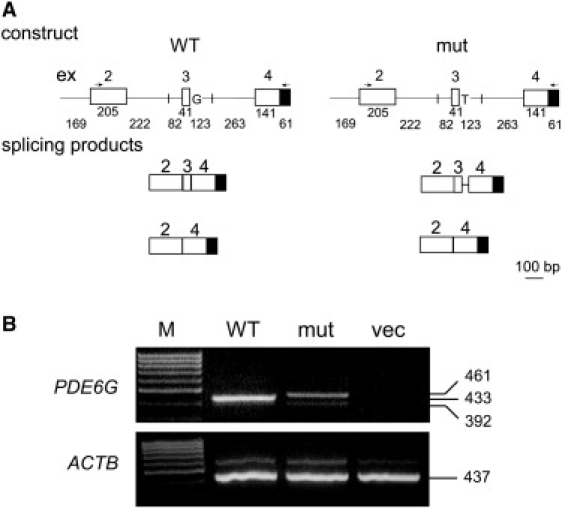
Minigene Constructs and Products Obtained in the In Vitro Splicing Assay
(A) Shown is a schematic representation of the constructs, which include PDE6G exons 2 to 4 (represented by white boxes) flanked by 82–263 bp of intronic sequences (represented by straight lines). Vector-derived sequences are represented by a black box. Constructs harbor either the WT or the c.187+1G>T mutant allele at the donor splice site of intron 3. The locations of primers used for RT-PCR analysis are indicated by arrows (a forward primer located in exon 2 and a vector-derived reverse primer). Also shown are the obtained splicing products. Sizes of exonic and intronic fragments are indicated below them.
(B) WT and mutant (mut) constructs were transfected into Y79 cells, followed by RNA extraction and RT-PCR analysis. No PCR products were obtained from cells transfected with the empty pCMV-script vector (vec). β-actin (ACTB) served as an internal control for RNA quality and quantity. M denotes size marker.
To date, two PDE6G splice variants have been reported: variant 1 (GenBank accession number NM_002602) includes exons 1–4 and encodes for the full-length protein of 87 amino acids, whereas variant 2 (GenBank accession number NR_026872) is noncoding as a result of the skipping of exon 2. To test whether alternative splicing of exon 3 occurs in vivo, we performed RT-PCR analysis on human retina RNA with primers derived from exons 2 and 4. Only one product was obtained, which included exon 3 (data not shown). In addition, BLAST searches against RNA and EST databases revealed no indication for the existence of PDE6G transcripts lacking exon 3. We therefore concluded that exon 3 skipping may be an artifact of the in vitro splicing assay.
The in vitro splicing assay we performed demonstrates that an intron 3 donor splice site harboring the c.187+1G>T change is not efficiently recognized by the human splicing machinery. Although the exact effect of this splicing mutation on PDE6G transcripts in human retina is not known, the expected outcome is incorrect splicing, leading to an abnormal protein product. The length of WT PDE6G protein is 87 amino acids. The use of a cryptic intron 3 donor splice site is expected to yield a protein of 114 amino acids, in which the last 52 amino acids (at positions 63–114) are incorrect.
To screen control DNA samples for c.187+1G>T, we used a restriction-based assay. No carriers of c.187+1G>T were identified among 256 Muslim Israeli control subjects, including 135 Muslim Arabs from Northern Israel, 70 Muslim Arabs from Central Israel, and 51 Bedouins. However, c.187+1G>T was found heterozygously in 7 of 84 random adults from the same village in which family TB14 resides (village E), thus indicating a carrier frequency of 8.3% (95% confidence interval, 3.6%–15.4%) in this village. c.187+1G>T is therefore a founder mutation of village E and is rare in the surrounding Muslim Arab population. Most of the Israeli Arab population has been living in small, relatively isolated localities, which were originally settled by a small number of founders. Most of the genetic diseases frequent among Israeli Arabs are due to founder effects.17 For example, we have recently identified a single founder mutation of PRCD that leads to a high frequency of RP in another Muslim Arab Israeli village, in which the carrier frequency is 10%.18 The identification of c.187+1G>T as a common RP-causing mutation in village E will allow for sensitive and cost-effective use of genetic testing for carrier screening and diagnostic purposes.
In an effort to identify additional PDE6G mutations, we sequenced the three coding exons of 119 unrelated Israeli patients with RP and Leber's Congenital Amaurosis (LCA), but no mutations were found. In addition, we analyzed whole-genome homozygosity mapping data of 90 unrelated RP and LCA patients from consanguineous families. Four of these patients had homozygous regions harboring PDE6G, but sequencing analysis revealed no mutations. These patients had multiple large homozygous regions, and we predict that their disease is caused by other genes. In total, we analyzed data from 209 RP and LCA patients, with negative results, indicating that PDE6G mutations are very rare and that their contribution to the overall prevalence of RP and/or LCA in the Israeli population is minor. Similar results were obtained by Hahn et al.,15 who screened the PDE6G gene by SSCP analysis in a total of 704 unrelated patients with various forms of hereditary retinal degeneration, including 471 patients with RP and 41 patients with LCA, and found no mutations.
Clinically, the affected individuals described here have severe early-onset RP. Both scotopic and photopic electroretinograms (ERGs) were markedly reduced or completely extinct by as early as 4 years of age. Visual evoked potentials (VEPs) were of normal waveforms but prolonged implicit time (data not shown). Funduscopic examination revealed relatively mild yet typical bone spicule-type pigment deposits in the periphery and midperiphery. In the younger patients (aged 10 and 12), blood vessels and optic disk were normal, whereas in their older siblings (aged 15 and 16), blood vessel attenuation and pallor of the optic disk were observed. Macular involvement in all patients was indicated by the lack of foveal reflex and cystoid macular edema documented by optical coherence tomography (OCT). All patients had markedly constricted visual fields, ranging from 10° to less than 5°, with tiny residual islands of central vision. Nevertheless, best-corrected visual acuity was relatively good (Table 1; Figure 3). These findings are similar to those reported in RP patients due to mutations in PDE6A and PDE6B.7,8,19
Table 1.
Clinical Characteristics of Individuals Homozygous for the PDE6G c.187+1G>T Mutation
| Patient Number, Sex | Age (yrs) | Eye | Refractive Error | Visual Acuitya | Visual Field |
Full-Field Electroretinogram |
||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
LA: Single Flashb |
LA: Flicker (30 Hz)c |
|||||||||||
| Amp (μV) | Latency (mS) | Amp (μV) | Latency (mS) | DA: Dim (μV)d | DA: Moderate (μV)e | DA: Bright (μV)f | ||||||
| III-25, F | 4 | OD | ND | ND | ND | 10 | 44 | 9 | 27 | b 0 | a 17, b 12 | a 27, b 45 |
| OS | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ||
| 12 | OD | −2.25/−0.75 × 170° | 6/12 | <5° | ND | ND | ND | ND | ND | ND | ND | |
| OS | −1.50/−1.00 × 40° | 6/12 | 10° | ND | ND | ND | ND | ND | ND | ND | ||
| III-26, M | 16 | OD | −3.00/−2.50 × 180° | 6/12 | 5° | ND | ND | ND | ND | ND | ND | ND |
| OS | −1.50/−2.75 × 180° | 6/12 | 5° | ND | ND | ND | ND | ND | ND | ND | ||
| III-18, M | 10 | OD | ND | 6/12+ | ND | 0 | − | 0 | − | b 0 | a 0, b 0 | a 0, b 0 |
| OS | ND | 6/12+ | ND | ND | ND | ND | ND | ND | ND | ND | ||
| 15 | OD | −1.00/−0.25 × 160° | 6/18 | 5° | NA | NA | NA | NA | NA | NA | NA | |
| OS | −0.25/0.00 | 6/12 | 10° horizontal, 5° vertical | NA | NA | NA | NA | NA | NA | NA | ||
| III-19, M | 7 | OD | ND | 6/12 | ND | 0 | − | 0 | − | b 0 | a 0, b 0 | a 0, b 0 |
| OS | ND | 6/12 | ND | ND | ND | ND | ND | ND | ND | ND | ||
| 10 | OD | +1.75/−1.25 × 180° | 6/18− | <5° | NA | NA | NA | NA | NA | NA | NA | |
| OS | +1.75/−2.00 × 170° | 6/18− | <5° | NA | NA | NA | NA | NA | NA | NA | ||
The following abbreviations are used: F, female; M, male; OD, right eye; OS, left eye; ND, not determined; LA, light adaptation (cone ERG); DA, dark adaptation (rod ERG); NA, not applicable.
Best-corrected visual acuity.
Normal amplitude (amp) is 100–300 μV; normal latency is 26–30 mS.
Normal amplitude (amp) is 60–180 μV; normal latency is 25–30 mS.
Normal b wave is 130–330 μV.
Normal a wave is 150–320 μV; normal b wave is 290–500 μV.
Normal a wave is 320–520 μV; normal b wave is 410–610 μV.
Figure 3.
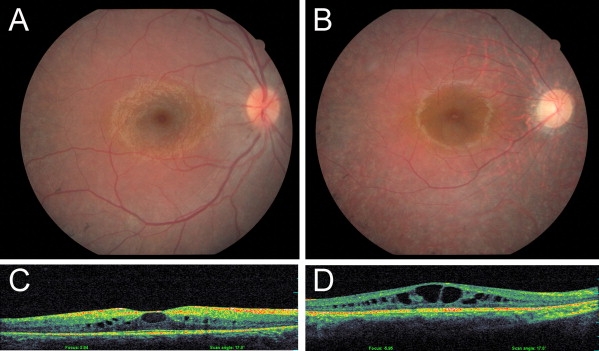
Fundus Photographs and Optical Coherence Tomography in Affected Individuals from Family TB14
(A and C) Fundus photograph (A) and OCT (C) of individual III-19 at the age of 10 years, demonstrating normal blood vessels and optic disk, peripheral bone spicule-type pigment deposits, absence of a foveal reflex, and mild cystoid macular edema.
(B and D) Fundus photograph (B) and OCT (D) of individual III-26 at the age of 16 years, demonstrating attenuation of retinal blood vessels, pale optic disk, tapetoretinal degeneration with unhealthy discoloration of the macula, absence of a foveal reflex, and severe cystoid macular edema.
As indicated before, cGMP-PDE is a key enzyme in the visual phototransduction cascade. Vision begins when a molecule of visual pigment, 11-cis-retinal, is photoisomerized by a photon of light, leading to the generation of metarhodopsin II (R∗). R∗ activates the G protein transducin, which in turn stimulates the activity of its effector enzyme, cGMP-PDE. This activation is achieved by binding of transducin to the Pγ subunit, thereby removing the inhibitory constraint from the Pα and Pβ catalytic subunits. The activated PDE lowers the intracellular concentration of cGMP, thereby closing cGMP-gated cation channels located on the rod plasma membrane and initiating a neural response to light.20
As can be expected, elimination of Pα and Pβ activity results in arRP in humans and retinal degeneration in mice.7–12 In contrast, the absence of Pγ might be expected to reduce rod cGMP levels by allowing the constitutively uninhibited activity of Pαβ. Consequently, the cGMP-gated cation channels would be permanently closed, eliminating the rod's response to light. Interestingly, a heterozygous missense mutation in Pβ was identified in a family segregating autosomal-dominant congenital stationary night blindness (CSNB), a condition in which rods have reduced light sensitivity but do not degenerate. This missense mutation was eventually found to impair the inhibitory interaction between Pβ and Pγ.4,21 Moreover, the p.W70A missense mutation in Pγ in mice led to rod desensitization and delayed response, a phenotype similar to CSNB in humans.22 Surprisingly, complete elimination of Pγ in mice resulted in a different phenotype that involved retinal degeneration and resembled RP in humans. Retinal cGMP levels in Pde6g null mice were increased, not decreased as may have been expected. The authors hypothesized that the high cGMP concentrations may keep cGMP-gated cation channels open continuously and lead to an excessive energy load on rod photoreceptors, resulting in degeneration.13 A transgenic allele of Pγ lacking the last 7 amino acids of its C terminus did not rescue the null phenotype.23 The conclusion was that the interaction of Pγ with Pαβ through its C terminus has a positive role for the proper activation of PDE and that all three subunits are essential for assembly of a stable, active holoenzyme.13,23 Further support for these conclusions came from biochemical and structural analyses. These indicated that Pγ domains in charge of transducin binding (T62-I87) and Pαβ inhibition (N74-I87) are located in the C terminus and are overlapping.24–26 The current hypothesis is that the interaction between activated transducin and Pγ triggers a conformational change and results in a rigid body movement of Pγ C terminus away from Pαβ, whereas Pγ central region could stay bound to Pαβ until the binding is weakened by lowered cGMP levels.27
Based on our in vitro splicing assay, the splicing product obtained from the c.187+1G>T mutant allele encodes for an abnormal Pγ protein in which the last 25 amino acids (D63-I87) are replaced by 52 irrelevant amino acids. The missing amino acids include the overlapping regions responsible for transducin binding and Pαβ inhibition. Based on the data presented above, this mutant protein will not enable the formation of an active PDE holoenzyme. This is in agreement with the observed phenotype, which resembles the phenotype of Pde6g null mice13 and involves severe early-onset retinal degeneration. Nevertheless, it is possible that different mutant alleles of PDE6G will be discovered in the future in patients with CSNB.
Unlike the other components of rod PDE, Pγ is expressed in nonocular tissues and was found to have important functions, including regulation of p42/p44 MAPK-dependent signaling in HEK293 cells,28 prevention of phosphorylation and activation of lung PDE5 by protein kinase A,29 and interaction with SH3-containing proteins.30 Based on these findings, it could be expected that elimination of Pγ would elicit extraocular phenotypes. However, such phenotypes were not detected in Pde6g null mice13 or in our human RP patients.
In summary, the crucial role played by cGMP-PDE in normal retinal function is well established. Mutations in the catalytic α and β subunits of this holoenzyme are a known cause of arRP. The data presented here demonstrate the positive role of the γ subunit in maintaining PDE activity and confirm the contribution of PDE6G to the etiology of RP in humans.
Acknowledgments
We are grateful to the patients and their relatives for their participation in this study. We thank Karl Skorecki and Dror Sharon for DNA samples and Liliana Mizrahi-Meissonnier for technical help. This work was supported by research grants from the Legacy Heritage Bio-Medical program of the Israel Science Foundation (to T.B.-Y.) and from the R.L. Kohns Eye Research Fund (to T.B.-Y. and S.A.S.).
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Basic Local Alignment Search Tool (BLAST), http://blast.ncbi.nlm.nih.gov/Blast.cgi
Berkeley Drosophila Genome Project: Splice Site Prediction by Neural Network, http://www.fruitfly.org/seq_tools/splice.html
GenBank, http://www.ncbi.nih.gov/Genbank/
MaxEntScan, http://genes.mit.edu/burgelab/maxent/Xmaxentscan_scoreseq.html
Retnet: Retinal Information Network, http://www.sph.uth.tmc.edu/Retnet/
UCSC Genome Browser, http://www.genome.ucsc.edu/
References
- 1.Rosner M., Hefetz L., Abraham F.A. The prevalence of retinitis pigmentosa and congenital stationary night blindness in Israel. Am. J. Ophthalmol. 1993;116:373–374. doi: 10.1016/s0002-9394(14)71358-3. [DOI] [PubMed] [Google Scholar]
- 2.Hamel C. Retinitis pigmentosa. Orphanet J. Rare Dis. 2006;1:40. doi: 10.1186/1750-1172-1-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hartong D.T., Berson E.L., Dryja T.P. Retinitis pigmentosa. Lancet. 2006;368:1795–1809. doi: 10.1016/S0140-6736(06)69740-7. [DOI] [PubMed] [Google Scholar]
- 4.Gal A., Orth U., Baehr W., Schwinger E., Rosenberg T. Heterozygous missense mutation in the rod cGMP phosphodiesterase beta-subunit gene in autosomal dominant stationary night blindness. Nat. Genet. 1994;7:64–68. doi: 10.1038/ng0594-64. [DOI] [PubMed] [Google Scholar]
- 5.Wada Y., Abe T., Takeshita T., Sato H., Yanashima K., Tamai M. Mutation of human retinal fascin gene (FSCN2) causes autosomal dominant retinitis pigmentosa. Invest. Ophthalmol. Vis. Sci. 2001;42:2395–2400. [PubMed] [Google Scholar]
- 6.Ionita M.A., Pittler S.J. Focus on molecules: Rod cGMP phosphodiesterase type 6. Exp. Eye Res. 2007;84:1–2. doi: 10.1016/j.exer.2005.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McLaughlin M.E., Sandberg M.A., Berson E.L., Dryja T.P. Recessive mutations in the gene encoding the beta-subunit of rod phosphodiesterase in patients with retinitis pigmentosa. Nat. Genet. 1993;4:130–134. doi: 10.1038/ng0693-130. [DOI] [PubMed] [Google Scholar]
- 8.Huang S.H., Pittler S.J., Huang X., Oliveira L., Berson E.L., Dryja T.P. Autosomal recessive retinitis pigmentosa caused by mutations in the alpha subunit of rod cGMP phosphodiesterase. Nat. Genet. 1995;11:468–471. doi: 10.1038/ng1295-468. [DOI] [PubMed] [Google Scholar]
- 9.Pittler S.J., Baehr W. Identification of a nonsense mutation in the rod photoreceptor cGMP phosphodiesterase beta-subunit gene of the rd mouse. Proc. Natl. Acad. Sci. USA. 1991;88:8322–8326. doi: 10.1073/pnas.88.19.8322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chang B., Hawes N.L., Pardue M.T., German A.M., Hurd R.E., Davisson M.T., Nusinowitz S., Rengarajan K., Boyd A.P., Sidney S.S. Two mouse retinal degenerations caused by missense mutations in the beta-subunit of rod cGMP phosphodiesterase gene. Vision Res. 2007;47:624–633. doi: 10.1016/j.visres.2006.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sakamoto K., McCluskey M., Wensel T.G., Naggert J.K., Nishina P.M. New mouse models for recessive retinitis pigmentosa caused by mutations in the Pde6a gene. Hum. Mol. Genet. 2009;18:178–192. doi: 10.1093/hmg/ddn327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tuntivanich N., Pittler S.J., Fischer A.J., Omar G., Kiupel M., Weber A., Yao S., Steibel J.P., Khan N.W., Petersen-Jones S.M. Characterization of a canine model of autosomal recessive retinitis pigmentosa due to a PDE6A mutation. Invest. Ophthalmol. Vis. Sci. 2009;50:801–813. doi: 10.1167/iovs.08-2562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tsang S.H., Gouras P., Yamashita C.K., Kjeldbye H., Fisher J., Farber D.B., Goff S.P. Retinal degeneration in mice lacking the gamma subunit of the rod cGMP phosphodiesterase. Science. 1996;272:1026–1029. doi: 10.1126/science.272.5264.1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cotran P.R., Bruns G.A., Berson E.L., Dryja T.P. Genetic analysis of patients with retinitis pigmentosa using a cloned cDNA probe for the human gamma subunit of cyclic GMP phosphodiesterase. Exp. Eye Res. 1991;53:557–564. doi: 10.1016/0014-4835(91)90213-x. [DOI] [PubMed] [Google Scholar]
- 15.Hahn L.B., Berson E.L., Dryja T.P. Evaluation of the gene encoding the gamma subunit of rod phosphodiesterase in retinitis pigmentosa. Invest. Ophthalmol. Vis. Sci. 1994;35:1077–1082. [PubMed] [Google Scholar]
- 16.Mount S.M. A catalogue of splice junction sequences. Nucleic Acids Res. 1982;10:459–472. doi: 10.1093/nar/10.2.459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zlotogora J. Molecular basis of autosomal recessive diseases among the Palestinian Arabs. Am. J. Med. Genet. 2002;109:176–182. doi: 10.1002/ajmg.10328. [DOI] [PubMed] [Google Scholar]
- 18.Nevet M.J., Shalev S.A., Zlotogora J., Mazzawi N., Ben-Yosef T. Identification of a prevalent founder mutation in an Israeli Muslim Arab village confirms the role of PRCD in the aetiology of retinitis pigmentosa in humans. J. Med. Genet. 2010 doi: 10.1136/jmg.2009.073619. Published online May 27, 2010. [DOI] [PubMed] [Google Scholar]
- 19.Tsang S.H., Tsui I., Chou C.L., Zernant J., Haamer E., Iranmanesh R., Tosi J., Allikmets R. A novel mutation and phenotypes in phosphodiesterase 6 deficiency. Am. J. Ophthalmol. 2008;146:780–788. doi: 10.1016/j.ajo.2008.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Arshavsky V.Y., Lamb T.D., Pugh E.N., Jr. G proteins and phototransduction. Annu. Rev. Physiol. 2002;64:153–187. doi: 10.1146/annurev.physiol.64.082701.102229. [DOI] [PubMed] [Google Scholar]
- 21.Muradov K.G., Granovsky A.E., Artemyev N.O. Mutation in rod PDE6 linked to congenital stationary night blindness impairs the enzyme inhibition by its gamma-subunit. Biochemistry. 2003;42:3305–3310. doi: 10.1021/bi027095x. [DOI] [PubMed] [Google Scholar]
- 22.Salchow D.J., Gouras P., Doi K., Goff S.P., Schwinger E., Tsang S.H. A point mutation (W70A) in the rod PDE-gamma gene desensitizing and delaying murine rod photoreceptors. Invest. Ophthalmol. Vis. Sci. 1999;40:3262–3267. [PMC free article] [PubMed] [Google Scholar]
- 23.Tsang S.H., Yamashita C.K., Lee W.H., Lin C.S., Goff S.P., Gouras P., Farber D.B. The positive role of the carboxyl terminus of the gamma subunit of retinal cGMP-phosphodiesterase in maintaining phosphodiesterase activity in vivo. Vision Res. 2002;42:439–445. doi: 10.1016/s0042-6989(01)00213-9. [DOI] [PubMed] [Google Scholar]
- 24.Slep K.C., Kercher M.A., He W., Cowan C.W., Wensel T.G., Sigler P.B. Structural determinants for regulation of phosphodiesterase by a G protein at 2.0 A. Nature. 2001;409:1071–1077. doi: 10.1038/35059138. [DOI] [PubMed] [Google Scholar]
- 25.Song J., Guo L.W., Muradov H., Artemyev N.O., Ruoho A.E., Markley J.L. Intrinsically disordered gamma-subunit of cGMP phosphodiesterase encodes functionally relevant transient secondary and tertiary structure. Proc. Natl. Acad. Sci. USA. 2008;105:1505–1510. doi: 10.1073/pnas.0709558105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Barren B., Gakhar L., Muradov H., Boyd K.K., Ramaswamy S., Artemyev N.O. Structural basis of phosphodiesterase 6 inhibition by the C-terminal region of the gamma-subunit. EMBO J. 2009;28:3613–3622. doi: 10.1038/emboj.2009.284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guo L.W., Hajipour A.R., Ruoho A.E. Complementary interactions of the rod PDE6 inhibitory subunit with the catalytic subunits and transducin. J. Biol. Chem. 2010;285:15209–15219. doi: 10.1074/jbc.M109.086116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wan K.F., Sambi B.S., Frame M., Tate R., Pyne N.J. The inhibitory gamma subunit of the type 6 retinal cyclic guanosine monophosphate phosphodiesterase is a novel intermediate regulating p42/p44 mitogen-activated protein kinase signaling in human embryonic kidney 293 cells. J. Biol. Chem. 2001;276:37802–37808. doi: 10.1074/jbc.M105087200. [DOI] [PubMed] [Google Scholar]
- 29.Tate R.J., Lochhead A., Brzeski H., Arshavsky V., Pyne N.J. The gamma-subunit of the rod photoreceptor cGMP-binding cGMP-specific PDE is expressed in mouse lung. Cell Biochem. Biophys. 1998;29:133–144. doi: 10.1007/BF02737832. [DOI] [PubMed] [Google Scholar]
- 30.Morin F., Vannier B., Houdart F., Regnacq M., Berges T., Voisin P. A proline-rich domain in the gamma subunit of phosphodiesterase 6 mediates interaction with SH3-containing proteins. Mol. Vis. 2003;9:449–459. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.