

Abstract
Heteroplasmy, the existence of multiple mtDNA types within an individual, has been previously detected by using mostly indirect methods and focusing largely on just the hypervariable segments of the control region. Next-generation sequencing technologies should enable studies of heteroplasmy across the entire mtDNA genome at much higher resolution, because many independent reads are generated for each position. However, the higher error rate associated with these technologies must be taken into consideration to avoid false detection of heteroplasmy. We used simulations and phiX174 sequence data to design criteria for accurate detection of heteroplasmy with the Illumina Genome Analyzer platform, and we used artificial mixtures and replicate data to test and refine the criteria. We then applied these criteria to mtDNA sequence reads for 131 individuals from five Eurasian populations that had been generated via a parallel tagged approach. We identified 37 heteroplasmies at 10% frequency or higher at 34 sites in 32 individuals. The mutational spectrum does not differ between heteroplasmic mutations and polymorphisms in the same individuals, but the relative mutation rate at heteroplasmic mutations is significantly higher than that estimated for all mutable sites in the human mtDNA genome. Moreover, there is also a significant excess of nonsynonymous mutations observed among heteroplasmies, compared to polymorphism data from the same individuals. Both mutation-drift and negative selection influence the fate of heteroplasmies to determine the polymorphism spectrum in humans. With appropriate criteria for avoiding false positives due to sequencing errors, next-generation technologies can provide novel insights into genome-wide aspects of mtDNA heteroplasmy.
Introduction
The mtDNA genome remains one of the most widely studied DNA segments in humans. It is particularly useful for studying population and evolutionary genetics because of its abundance in human cells, its uniparental, nonrecombining mode of inheritance, and its high mutation rate compared to that of the nuclear genome.1 Although each individual is typically characterized by a single mtDNA type, in fact each individual is a population of mtDNA genomes, and the presence of multiple mtDNA types within an individual is termed heteroplasmy.
Although little noted at the time, the first report of heteroplasmy in humans was in 1983, involving a study of a noncoding region of human mtDNA from 11 human placentas.2 Heteroplasmy has been investigated most often in correlation with mitochondrial disease, aging, and cancer.3–6 To date, more than 400 mtDNA mutations have been associated with human disease, and most were observed in heteroplasmic states, with pathogenic mutations coexisting with normal mitochondrial genomes.7 This suggests that the heteroplasmic level is of particular interest, as the disease phenotype becomes evident only when the percentage of mutant molecules exceeds a critical threshold value. Although this value differs for different mutations and in different tissues, it is usually in the range of 70%∼90%.8,9
Originally, heteroplasmy was believed to be quite rare in healthy individuals,10,11 but subsequent studies found many non-disease-related heteroplasmies.12–15 Moreover, heteroplasmy has started to play an important role in some forensic investigations.16,17 Thus, heteroplasmy can also be a useful genetic marker. Regarding heteroplasmy as the intermediate stage between the generation of mutations and the fixation of mutations in the individual or cell, it represents polymorphisms within the populations of mitochondrial genomes in one cell or tissue. Thus, it can be a potential resource for studying the mutational pattern, possible role of natural selection, and even the existence of recombination in mtDNA.18 For example, de novo mtDNA mutations in cancer tissues preferentially locate at the same positions as ancient variants in the human phylogeny, indicating similar selective constraints.19 Understanding the basis, extent, and forces influencing the occurrence and subsequent fate of heteroplasmic mtDNA mutations is one of the principal challenges facing scientists and clinicians in the field of mitochondrial genetics.
A variety of techniques have been employed for heteroplasmy detection, including Sanger capillary sequencing,13 high-performance liquid chromatography (HPLC),20 pyrosequencing,21,22 SnaPshot,23 high-resolution melt (HRM) profiling,24 a temporal temperature gradient gel electrophoresis (TTGE) strategy,25 the Invader assay,26 an amplification refractory mutation system,27 and surveyor nuclease.28 However, all of these methods have disadvantages, including the following: for some methods, the candidate heteroplasmic position needs to be defined first; the method may not allow determination of the actual heteroplasmic position; the level of heteroplasmy cannot be quantified accurately; and/or the method is too labor intensive to be applicable to large numbers of samples. Moreover, the efficiency of detection can vary substantially from laboratory to laboratory, even when the same method is applied, as a result of different instruments, chemistries, or standards for calling heteroplasmy.29,30 In addition, most of the previous studies have restricted their examination to the control region or the hypervariable segments thereof; very few studies have analyzed heteroplasmies in the coding region,31 even though these are likely to be of more importance for disease association. Therefore, more accurate and efficient methods are needed for the examination of heteroplasmy across the entire mtDNA genome.
Next-generation sequencing technologies can, in principle, provide the needed data, because millions of DNA reads can be produced in a single run at low cost.32 This technology has been widely used in the detection of SNPs on a genome-wide scale,33–37 and in one such study,34 pooled DNA from 66 individuals was used to successfully detect SNPs. More interestingly, this study also found that the allele frequency inferred from the reads had a significant correlation with that inferred from genotyping. Such DNA pools resemble the mtDNA heteroplasmy scenario and therefore suggest that this approach should be useful for heteroplasmy detection.
However, the relatively high sequence error associated with next-generation sequencing technologies can produce false positives (i.e., sequencing errors may be falsely considered to be heteroplasmies). Moreover, problems could arise if PCR was involved during preparation of the sequencing library, because PCR may alter the proportion of different alleles and amplification error may also result in false positives. In this study, we used simulations, phiX174 control sequence data, artificial mixtures, and replicates to design and evaluate criteria for accurate detection of heteroplasmic positions with the Illumina GA platform. We then applied these criteria to a data set of complete mtDNA genome sequence reads for 131 individuals from five Eurasian populations that had been generated via a parallel tagged approach and sequenced with the Illumina GA platform. The results of this large-scale investigation provide insights into genome-wide patterns of mtDNA heteroplasmy.
Material and Methods
Data
The mtDNA sequence reads used here were generated in a previous study (unpublished data). In brief, the entire mtDNA genome was amplified from 147 individuals from five populations (Georgia, Armenia, Azerbaijan, Iran, and Turkey) in two overlapping products of about 9.7 and 7.3 Kb and sequenced on the Illumina GAII platform (GAII; San Diego, CA, USA) via a multiplex sequencing protocol for sequencing libraries;38,39 details are described elsewhere (unpublished data). Out of 147 samples, the reads from 131 met the criteria for accurate detection of heteroplasmy, developed below. Of these, 97 were sequenced once with single-end 36 bp reads to an average coverage of 65×, 17 were sequenced twice with single-end 36 bp reads (because of insufficient coverage from the first lane) to an average of 78×, and 17 were sequenced once with single-end 76 bp reads to an average coverage of 211×. In addition, four samples originally sequenced once with 36 bp reads were resequenced (with the use of new PCR products amplified from the original DNA templates) with single-end 76 bp reads for assessment of reproducibility.
Assembly Strategy
The reads were assembled with the software MIA,40 the revised Cambridge reference sequence (rCRS) used as the reference.41 MIA is optimized for circular genome assembling and performs gapped assembling iteratively: after a consensus is called, the reads are realigned to the consensus and a new consensus is called; this process iterates until it converges on a single consensus sequence. For the 36 bp reads, maximally, three mismatches or two mismatches plus one gap were allowed to successfully map the read, whereas for the 76bp reads, five mismatches or four mismatches plus one gap were allowed. In addition, any read with more than two bases having a Phred-like quality score (QS) lower than 15 was removed (for 76 bp length reads, the threshold was five bases with QS < 10), and duplicate reads (reads mapping to the same position with same orientation on the reference) were removed, keeping the one with the highest QS.
Artificially Mixed Samples
The template DNAs from two individuals differing at 25 positions in their complete mtDNA genome sequences were mixed in different proportions (1:1, 1:3, and 1:9). DNA concentrations were measured by a NanoDrop ND-1000 spectrometer (NanoDrop Technologies, Wilmington, DE, USA), diluted and mixed in the desired proportions, and then used for long-rang PCR amplification and Illumina GAII sequencing with 76 bp reads. These artificially mixed samples were used for examination of the correlation between the heteroplasmy level (mixture proportion) and minor allele frequency estimated from the reads.
Simulation Framework
To evaluate the impact of sequencing error on the detection of heteroplasmy, we performed simulations for varying levels of sequencing error, heteroplasmy level, and sequencing depth. For each simulation, one biallelic heteroplasmic position was assigned to the genome randomly, with the heteroplasmy level (minor allele frequency) set to be 5%, 10%, 20%, 30%, or 40%. Reads with a specific length (36bp, 76bp) were generated randomly from the rCRS to reach an average coverage of 36× or 76×. The sequencing error in the simulation was evenly distributed along the reads and along the mitochondrial genome, with the error rate set to be 1%, 0.5%, 0.3%, or 0.1%. Because Illumina GA uses the same laser to excite two pairs of nucleotides (A/C and G/T), the pairs produce similar emission spectra and are thus poorly distinguished by optical filters, which results in a higher proportion of errors involving A/C and G/T.39,42 On the other hand, heteroplasmy is highly biased toward transitions (A/G, C/T), as with mtDNA substitutions.13,14 We therefore specified that all sequencing errors would be A/C or G/T changes, whereas all heteroplasmy would involve A/G or C/T changes, so that we could readily distinguish sequencing errors from heteroplasmy in our simulations. For each setting, the simulation was repeated 100 times.
PhiX174 Data for Permutation Test
In each Illumina GA sequencing run, the phiX174 bacteriophage genome is sequenced on a separate lane to very high coverage (about 60,000×) and then used as a control for sequencing-error correction for that run. Theoretically, there should be no heteroplasmy in the phiX174 genome, given that it is from a single strain, although in fact Illumina technical support reports five SNPs in phiX174 genome sequences. Therefore, we applied our criteria for detecting heteroplasmy to the phiX174 data, to estimate the false-positive error rate associated with various criteria. To simulate different sequencing depths, we retrieved subsets of the reads from the phiX174 control, and for each depth (15-, 30-, 60-, 90-, 120-, 150-, 180-, 210-, 600-, and 1200-fold average coverage), we repeated the simulations 100 times.
MtDNA Polymorphism Data
Polymorphic positions were retrieved from Mitomap,7 mtDB,43 4775 whole mitochondrial genomes from GenBank,44 and 700 whole mitochondrial genomes from our laboratory (unpublished data). The relative mutation rate (RMR) for each polymorphic position was retrieved from Soares et al.45 Disease-associated positions were retrieved also from Mitomap7 (April 27, 2010 version); when defining a heteroplasmy as disease-associated, both the nucleotide position and the specific mutation were considered.
Haplogroup-defining positions were retrieved from Phylotree,46 and an in-house script was used to collect all the haplogroup-defining positions and the corresponding mutation type, which were then used to identify heteroplasmies occurring at such haplogroup-defining positions.
Validation of Heteroplasmies
To independently verify the heteroplasmic positions inferred from high-throughput sequencing, we performed single-base extension assays for nine heteroplasmic sites. Primers were designed to amplify shorter products around these sites for both control and heteroplasmic individuals with the use of original DNA as template (Table S1, available online). All single-base extension assays were carried out with the ABI Prism SnaPshot Kit (Applied Biosystems), with amplicons detected via capillary electrophoresis on an ABI Prism 3100 Genetic Analyzer according to the manufacturer's instructions.
Results
Sequencing Overview
A total of 45 million reads were generated for the 131 samples and four replicates, and 94% of the reads could be mapped to the rCRS. For the 36 bp reads, the average coverage was 67 ± 7 (mean ± standard deviation); meanwhile, 96.8% of the positions had coverage greater than 36× and at least ten reads for each strand. For the 76 bp reads, the average coverage was 211 ± 21, and 98.6% positions had coverage greater than 36× and at least ten reads for each strand.
Simulations
We used simulations to explore the effects of different sequencing error rates, coverage, and threshold of the minor allele frequency to define heteroplasmy, on the false-positive error (i.e., calling a sequencing error a heteroplasmy) and false-negative error (i.e., failing to call a true heteroplasmy) rates. Results are shown in Figure 1; 36× coverage is close to the lowest coverage that we have in our samples, whereas 76× coverage is close to the average coverage in our samples. The results illustrate the tradeoff that occurs between the false-positive and false-negative rates. When the heteroplasmy threshold is set relatively high (more than 10%), there are no false positives, even with a high sequencing error rate, but the false-negative rate is quite high, indicating that many true heteroplasmies will be missed. When the threshold is set relatively low (10% or less), the false-negative rate becomes very low, but now the false-positive rate becomes unacceptably high. Acceptable false-positive and false-negative rates can apparently be achieved only if the sequencing error rate can be reduced to 0.1% (Figure 1) or if the average coverage is increased.
Figure 1.
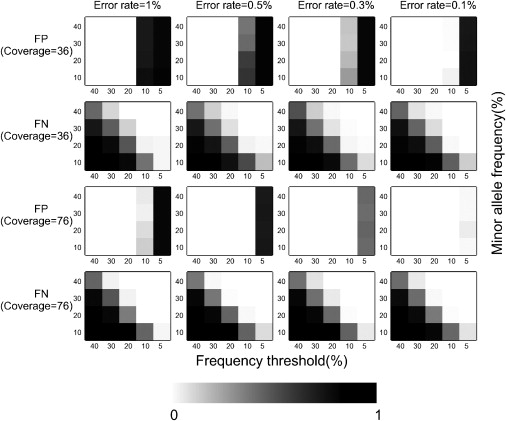
False-Negative Error Rate and False-Positive Error Rate in Detecting Heteroplasmy Inferred from Simulation
False-negative error rate and false-positive error rate calculated under different error rates (1%, 0.5%, 0.3%, 0.1%), coverage (36×, 76×), heteroplasmy levels (minor allele frequencies of 10%, 20%, 30%, 40%), and frequency thresholds used to define heteroplasmy (40%, 30%, 20%, 10%). For each setting, the simulation was repeated 100 times. FP denotes the false-positive error rate, FN denotes the false-negative error rate.
PhiX174 Analysis and Development of Criteria
The phiX174 genome is routinely sequenced to very high coverage in a separate lane in each Illumina GAII run, in order to provide baseline error rates for the base-calling software. Because in theory the phiX174 data should come from a single pure strain, there should not be any polymorphism, although five SNPs have been reported (Illumina technical support). Thus, any “heteroplasmic” position after assembly is presumably caused by sequencing error. We therefore investigated other ways to reduce false positives by applying various criteria to the phiX174 reads. Reads were randomly retrieved from the control lane to simulate a sequencing depth of 64-fold, and this procedure was repeated 100 times for each analysis. The number of heterozygous positions (defined as positions with at least one read with an alternative base) after assembly is shown in Table 1. Sequencing error is indeed an important issue, because in the absence of any QS filtering, 11% (583 out of 5386) of the positions were affected, and some of them can even reach a minor allele frequency between 20%∼30%. QS filtering can improve the situation, but even a fairly stringent QS (QS ≥ 23) leaves many heterozygous positions (Table 1). However, double-strand validation (i.e., requiring at least one read from each strand) considerably reduces the impact of sequencing error; with no quality filter, double-strand validation reduces the average number of heterozygous positions from 583 to 18 (Table 2). And with a combination of a stringent quality filter and double-strand validation, only two heterozygous positions are detected on average. Note that this better performance of double-strand validation was not caused by any bias in the number of reads in one direction, because all positions have a coverage greater than 40 and at least ten reads in each direction. Instead, sequence errors tend to be context specific, and hence the majority of sequence errors are found on only one strand.
Table 1.
Number of Heterozygous Positions Detected in the PhiX174 Genome under Different Criteria
| No Quality Filter | QS ≥ 20 | QS ≥ 23 | |
|---|---|---|---|
| Validated by one strand | 582.9 ± 40.9a | 192.8 ± 14.0 | 126.3 ± 24.6 |
| Validated by two strands | 17.9 ± 4.7 | 3.0 ± 1.4 | 1.9 ± 1.0 |
Standard deviation based on 100 resamplings of the data.
Table 2.
Number of Heteroplasmies Detected with PhiX174 Permutation Data
|
Frequency Threshold |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
|
One Direction |
Two Directions |
|||||||||
| Detection Rate | 0.05 | 0.1 | 0.2 | 0.3 | 0.4 | 0.05 | 0.1 | 0.2 | 0.3 | 0.4 |
| > 50% | 3 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| 25%–50% | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 10%–24% | 2 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 5%–9% | 2 | 2 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 |
| < 5% | 8 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
To investigate the performance of double-strand validation with various frequency thresholds for calling heterozygous positions, we carried out additional simulations using an average coverage of 66, to mimic our real data. Table 2 compares the results of single- versus double-strand validation for various thresholds (for other sequencing depths, see Table S2), requiring QS ≥ 20 at the position in question (and QS ≥ 15 for the 5 bp flanking sequence on each side). As expected, a lower frequency threshold resulted in a higher false-positive rate, but only one false positive was consistently detected with double-strand validation and a frequency threshold of 10% or more. This position (1301) is not included in the five SNPs previously reported, but it has been found to be polymorphic by another laboratory (T. Skelly, personal communication), so it is likely to be a novel SNP in the phiX174 genome.
On the basis of both the simulations and the phiX174 analyses, we decided on the following criteria for calling a heteroplasmic position: QS ≥ 20 at the position in question, QS ≥ 15 at the 5 bp flanking either side, a minor allele frequency of at least 10%, and the minor allele observed in at least one read in each direction. These criteria should result in no false positives, and we should have 100% power to detect heteroplasmies present at a frequency of 20% or more and 50% power to detect heteroplasmies at a frequency of 10%.
Artificially Mixed Samples and Improvement of the Criteria
To test the sensitivity and specificity of these criteria, we applied them to three artificially mixed samples consisting of DNA derived from two individuals in different ratios (1:9, 1:3, 1:1) whose mtDNA genome sequences differ at 25 positions. All 25 positions were observed to be heteroplasmic, with inferred frequencies closely corresponding to those expected (Figure 2), except for six positions for the 1:9 mixture, which fell close to, but below, the 10% threshold. The Pearson correlation coefficient between the heteroplasmic level estimated from sequencing and the actual proportion is 0.944 (p < 0.001), in good agreement with a previous study of artificially mixed samples analyzed by high-throughput sequencing.47 Four potential false positives were observed: position 7030AC (the first letter denotes the majority nucleotide and the second letter the minority nucleotide), with a minor allele frequency of 12% in the sample with a 1:1 ratio; position 13604GA, with minor allele frequencies of 10% and 12% in the 1:3 and 1:9 mixtures, respectively; and the A homopolymer region beginning at 12418, where 11% of the reads have 7A and 89% have 8A in the 1:9 mixture. However, as shown below, position 13604GA is actually inferred to be heteroplasmic, with a minor allele frequency of 11%, in the sample (Ir43) that was used as the major component in the mixed samples. Moreover, the A homopolymer region beginning at 12418 was heteroplasmic in both of the samples, with a minor allele (7A) frequency of 12% and 10%, respectively. Therefore, only position 7030 qualifies as a true false positive. A more stringent requirement of QS ≥ 23 at the heteroplasmic position (rather than QS ≥ 20) would eliminate this false positive, but it would also remove an unacceptably high number of other reads for the true heteroplasmies. However, requiring two distinct reads from each strand would also eliminate this false positive while retaining all of the other inferred heteroplasmies. Therefore, we adjusted the criteria for heteroplasmy to require that the minor allele was observed in at least two independent reads from each strand.
Figure 2.
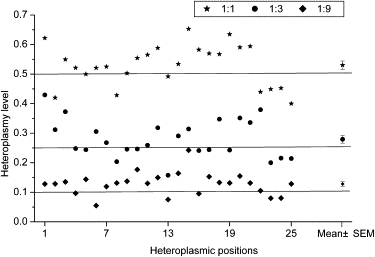
Heteroplasmy Level Estimated from Sequencing Reads for the Expected Heteroplasmic Positions in the Artificially Mixed Samples
Horizontal lines indicate the expected heteroplasmy levels. The rightmost column gives the mean and standard error of the mean value of all heteroplasmic positions under each mixture ratio.
Frequency Variation between Replicates
Four samples were sequenced twice independently, allowing us to assess how inferred minor allele frequencies varied between replicates. The average frequency variation between replicate pairs was 0.0057 ± 0.011, and there were 42 positions having a minor allele frequency difference greater than 0.1 (Figure 3). More stringent QS filtering (QS ≥ 23 for the position in question and QS ≥ 20 for the 5 bp flanking sequence on each side) reduced but did not eliminate the discrepancies; 19 positions still had a minor allele frequency difference greater than 0.1. Moreover, low coverage does not appear to play a significant role, because only five positions have a coverage lower than 10 in one of the replicates.
Figure 3.
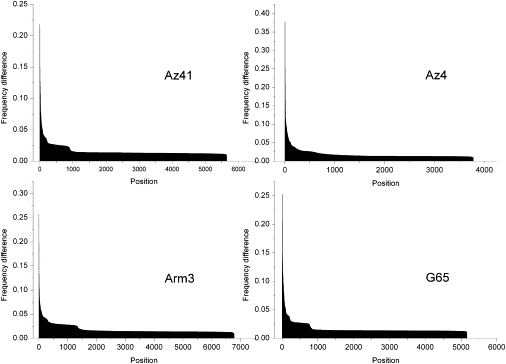
Distribution of Frequency Differences between Replicates
Only positions with a frequency difference greater than 0.01 are shown.
However, for 21 of the 42 positions with a minor allele frequency difference bigger than 0.1, the nucleotides involved were A and C (Table S3). Moreover, for many of these positions, the minor allele is observed on only one strand, indicating that the discrepancy between replicates is due to an increase in the minor allele on one strand only, consistent with sequencing errors. This same pattern was observed for another four positions in which A and T were involved (Table S3). The one exception to this pattern is position 385 for Az41, for which A and G are observed on both strands in both replicates. The frequency of the minor (G) allele was 20% versus 8.7% in the replicates; however, the difference was not significant (p = 0.059, Fisher's exact test). It is likely that this is a real heteroplasmic position and that the frequency difference between replicates is due to stochastic effects. All of the remaining 16 positions were located in the homopolymer region, for which inaccurate alignment can explain the large minor allele frequency difference.
Genome-wide Characterization of Heteroplasmy
Application of the criteria developed above to the full data set of 131 individuals resulted in the detection of 37 point heteroplasmies among 32 individuals (Table 3), or 24% of the individuals studied. The average coverage for the heteroplasmic positions is 79 (range: 28–170), and for only four positions (3014GT, 3492AC [twice], 10208CT) was the minor allele observed in fewer than six reads. Five individuals possessed two heteroplasmies each; overall, the number of heteroplasmies per individual did not differ significantly from the expectation when 37 heteroplasmies are randomly assigned to 131 individuals (p > 0.05). There were also no significant differences in how heteroplasmies were distributed among populations or among haplogroups. However, three heteroplasmies (146CT, 3492AC, 16223TC) occurred at the same position in two different individuals who belonged to different populations and had different mtDNA haplogroups; thus, these are likely to represent independent heteroplasmies. This is significantly more than expected by chance if heteroplasmies occur randomly across the mtDNA genome (p < 0.001).
Table 3.
Point Heteroplasmies Detected in 131 Individuals
| Position | Individual | Coverage | Major Allele | Frequency | Minor Allele | Frequency | Gene Annotationa |
|---|---|---|---|---|---|---|---|
| 64 | Ir28 | 82 | T | 0.87 | C | 0.13 | CR |
| 146 | Ir17 | 62 | T | 0.90 | C | 0.10 | CR |
| 146 | G65 | 186 | T | 0.80 | C | 0.20 | CR |
| 150b | Arm17 | 69 | C | 0.88 | T | 0.12 | CR |
| 152 | Ir11 | 71 | T | 0.89 | C | 0.11 | CR |
| 195b | G67 | 67 | T | 0.90 | C | 0.10 | CR |
| 203 | Az5 | 67 | A | 0.66 | G | 0.34 | CR |
| 204 | Arm17 | 75 | T | 0.89 | C | 0.11 | CR |
| 1552 | Ir54 | 69 | G | 0.87 | A | 0.13 | 12S |
| 3014 | Az10 | 44 | G | 0.86 | T | 0.14 | 16S |
| 3492 | Arm25 | 60 | A | 0.65 | C | 0.35 | NS(ND1; Lys>Asn) |
| 3492 | G20 | 63 | A | 0.78 | C | 0.22 | NS(ND1; Lys>Asn) |
| 3532 | Az14 | 73 | A | 0.82 | G | 0.18 | NS(ND1; Thr>Ala) |
| 4991 | Az4 | 173 | G | 0.65 | A | 0.35 | S(ND2) |
| 5460b | Az7 | 136 | A | 0.68 | G | 0.32 | NS(ND2; Thr>Ala) |
| 7754 | T186 | 73 | A | 0.89 | G | 0.11 | NS(COX2; Asn>Asp) |
| 8152 | Az46 | 80 | G | 0.88 | A | 0.13 | S(COX2) |
| 8551 | Ir30 | 67 | C | 0.54 | T | 0.46 | S/NS(ATP6/ATP8) |
| 8743 | Ir29 | 75 | G | 0.68 | A | 0.32 | NS(ATP6; Val>Met) |
| 10208 | Ir10 | 57 | C | 0.89 | T | 0.11 | S(ND3) |
| 10427 | Az39 | 68 | G | 0.85 | A | 0.15 | tRNA-ARG |
| 11253b | G1 | 68 | C | 0.76 | T | 0.24 | NS(ND4; Thr>Ile) |
| 11692 | G38 | 72 | A | 0.76 | C | 0.24 | S(ND4) |
| 11809 | G82 | 72 | T | 0.78 | C | 0.22 | S(ND4) |
| 12654 | Arm37 | 69 | A | 0.86 | G | 0.14 | S(ND5) |
| 13368 | Arm19 | 196 | G | 0.54 | A | 0.45 | S(ND5) |
| 13604 | Ir43 | 68 | G | 0.90 | A | 0.10 | NS(ND5; Ser>Asn) |
| 14527 | G25 | 66 | A | 0.89 | G | 0.11 | S(ND6) |
| 14561 | T9 | 63 | A | 0.78 | G | 0.22 | NS(ND6; Asp>Gly) |
| 14770 | Az14 | 77 | T | 0.55 | C | 0.45 | S(CYTB) |
| 15046 | T186 | 62 | G | 0.74 | A | 0.26 | S(CYTB) |
| 15314 | Arm20 | 206 | G | 0.84 | A | 0.14 | NS(CYTB; Ala>Thr) |
| 16093 | Az49 | 147 | C | 0.91 | T | 0.08 | CR |
| 16217 | T30 | 65 | C | 0.91 | T | 0.09 | CR |
| 16223 | G73 | 178 | T | 0.89 | C | 0.11 | CR |
| 16223 | Arm20 | 190 | T | 0.89 | C | 0.11 | CR |
| 16362 | G67 | 76 | T | 0.87 | C | 0.13 | CR |
The abbreviations used for gene annotation: CR: control region; 12S: 12S rRNA; 16S: 16S rRNA; S: synonymous; NS: non-synonymous. Gene name is displayed if it happens in the protein-coding genes, and amino acid change is displayed for non-synonymous mutation.
Disease-associated positions reported by Mitomap.7
We also detected three indel heteroplasmies (outside homopolymer regions) among three individuals (Table 4). One of these was located in a tRNA gene, whereas the other two were in the control region. In addition, we observed another 112 indels in the following homopolymer and STR regions (with the variation observed indicated in parentheses): 66∼71 (5C-6C), 303∼309 (6C-9C), 514∼523 (5CA-6CA), 12418∼12425 (7A-8A), and 16184∼16193 (9C-12C). Even if we only use the reads that span these homopolymer and STR regions, we still observed these length variations in at least two distinct reads per strand. However, slippage of the DNA polymerase during replication can also result in length variation, which is frequently observed in the homopolymer and STR region.48,49 Therefore, the indel heteroplasmic regions would need additional validation, ideally by a method that does not require PCR amplification, and for this reason we do not include these regions in our analysis of heteroplasmy.
Table 4.
Indel Heteroplasmies Detected in 131 Individuals
| Position | Individual | Coverage | Allele 1 | Frequency | Allele 2 | Frequency | Gene Annotation |
|---|---|---|---|---|---|---|---|
| 57 | Ir28 | 82 | C | 0.84 | - | 0.16 | CR |
| 15940 | Arm2 | 208 | - | 0.85 | T | 0.15 | tRNA-THR |
| 248 | Ir10 | 68 | A | 0.84 | - | 0.16 | CR |
Validation of Heteroplasmies
We selected nine heteroplasmic sites for independent validation via single-base extension assays. For each site, the predicted heteroplasmy state was observed in the corresponding individual (Figure S1), whereas control individuals lacking the heteroplasmy displayed only single peaks in the assays (data not shown). These results indicate that the heteroplasmies inferred by analysis of high-throughput sequencing reads are indeed true heteroplasmies, because they are validated by another method.
Patterns of Heteroplasmy Variation
Thirteen of the 34 heteroplasmic positions are located in the control region, which is more than expected by chance if heteroplasmic positions are distributed randomly across the mtDNA genome (p < 0.001, chi-square test). The ratio of transitions to transversions at heteroplasmies is 8.25, which is not significantly different (p = 0.616, Fisher's exact test) from the ratio of 8.09 for polymorphic positions in the same individuals. Moreover, the mutational spectrum for heteroplasmies does not differ significantly from that for polymorphisms in the same populations, or from the mutational spectrum for polymorphisms reported in the Mitomap database (Figure 4).
Figure 4.
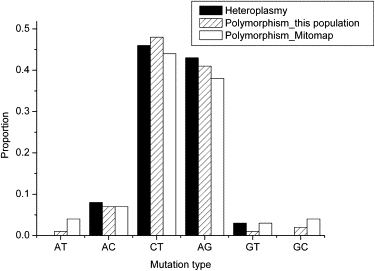
Mutation Spectrum for the Heteroplasmy and Polymorphism Data
For investigation of the correlation between the incidence of heteroplasmy and mutation rates, the RMR for each heteroplasmic position was obtained from a previous study.45 We classified the heteroplasmic positions into three categories: entire genome, control region, and coding region; the comparisons of RMR between heteroplasmic positions with all positions and all mutable positions in each category are shown in Table 5. Heteroplasmic positions have higher mutation rates in all categories, significantly so except when compared to all mutable positions in the coding region. Overall, heteroplasmies are occurring preferentially at positions with high mutation rates. This is particularly evident in the control region, in which six of the ten positions with the highest mutation rates (Soares et al.45) were found to exhibit heteroplasmy in our study.
Table 5.
Relative Mutation Rate at Heteroplasmic Positions
| Heteroplasmic Positions | All Positions | All Mutable Positions | |
|---|---|---|---|
| Whole genome | 24.95 | 0.645a | 2.72a |
| Control region | 64.08 | 3.74a | 7.01a |
| Coding region | 4.2 | 0.469a | 2.05 |
Significantly different from that of heteroplasmic positions at the p < 0.001 level in the Mann-Whitney test.
We detected 21 heteroplasmies at 20 positions in the protein-coding genes (3492AC is shared by two individuals). Ten of these are nonsynonymous mutations, ten are synonymous mutations, and one is located in the overlap region involving the ATP6 (MIM 516060) and ATP8 (MIM 516070). There is an excess of nonsynonymous mutations at the heteroplasmic positions when compared to polymorphism data from the same individuals (164 nonsynonymous and 385 synonymous mutations; p = 0.051, Fisher's exact test) or to a previous study50 (413 nonsynonymous and 1037 synonymous mutations; p = 0.045, Fisher's exact test).
Five of the ten nonsynonymous heteroplasmies are located at positions with an assigned RMR,45 and one of them is located at the position with the highest RMR among 824 nonsynonymous mutations (5460AG, RMR = 36). Two other nonsynonymous heteroplasmies also have high RMR (15314GA, RMR = 12; 11253CT, RMR = 5), whereas the other two (7754AG, 8743GA) both have RMR = 1. The heteroplasmic nonsynonymous sites tend to have higher mutation rates than previously reported nonsynonymous mutations in polymorphism data45 (p = 0.068, Mann-Whitney test). The other nonsynonymous heteroplasmies—3492AC (which occurred twice), 3532AG, 14561AG, and 13604AG—have never been observed as polymorphisms (based on sequences retrieved from Mitomap,7 mtDB,43 and GenBank, as well as 700 unpublished mtDNA genome sequences from our laboratory). The overall ratio of nonsynonymous heteroplasmies at sites without previously reported mutations, 5/10, is significantly higher than that observed in polymorphism data from the same individuals, 23/164 (p = 0.01, Fisher's exact test). There is thus a dichotomous tendency for nonsynonymous heteroplasmies to occur either at rapidly evolving sites or at sites that have not been observed as polymorphisms. Eight of the ten synonymous mutations have an assigned RMR;45 the average RMR for these eight positions is 3.0 ± 2.6, which is higher than the overall RMR of 2.0 for synonymous mutations, but not significantly so (p = 0.187, Mann-Whitney test). The other two synonymous mutations have not been previously observed as polymorphisms and hence do not have an assigned RMR.
Finally, two heteroplasmic positions (3014GT, 1552AG) are located in the stems of rRNA genes,51,52 with the major alleles identical to the rCRS, and were not observed to be polymorphic in the database. One heteroplasmic position (10427AG) is located in the connection of two stems of the tRNA-ARG (MIM 590005),53 and this position has an RMR of 2.0.
Disease-Associated Heteroplasmies
Four of the 34 (11.8%) heteroplasmic positions are reported as disease-associated in Mitomap7 (Table 3), which is more than expected by chance (2.5% of all mtDNA positions are reported to be disease-associated in Mitomap;7 p = 0.010, permutation test). However, all of these positions have also been reported previously as polymorphisms; moreover, there is also an excess number of polymorphisms at disease-associated sites in the mtDNA genome sequences from the same individuals (6.1%; p < 0.001, permutation test).
It seems likely that disease-associated mutations with mild effects will be observed as polymorphisms in “normal” individuals more frequently than disease-associated mutations with strong effects. Overall, 22.4% of the disease-associated mutations in Mitomap are also present in normal populations and 77.4% are absent in normal populations. All of the four disease-associated heteroplasmic positions are observed as polymorphisms in normal populations, which is significantly more than expected (p = 0.003, permutation test). Moreover, the average RMR for these four disease-associated heteroplasmic positions is 51, which is significantly higher than the average RMR of 2.72 for all mutable positions (p < 0.001, Mann-Whitney test) and significantly higher than the average RMR of 8.8 for all disease-associated positions that also present as polymorphisms in normal populations (p = 0.007, Mann-Whitney test).
Discussion
We first discuss issues related to the criteria for detecting heteroplasmy, and we then discuss the implications of our results.
Reducing the Impact of Sequencing Errors
Although the high-throughput sequencing technologies have a higher per-base sequencing error rate compared with the traditional capillary sequencers, the new technologies still give highly accurate sequences because of the higher sequencing depth.39,42,54 However, this holds only for assembling a consensus sequence or calling SNPs, in which using a “majority rule” eliminates the impact of sequencing error and produces the desired result. Trying to detect heteroplasmy presents different issues, because the minor allele at the heteroplasmic position may be indistinguishable from sequencing error. QS filtering can help eliminate sequencing errors but is insufficient to completely solve the problem (Table 1).
The most obvious way to distinguish between sequencing errors and heteroplasmy is to invoke a threshold. Sequencing errors happen at a specific rate (10−2∼10−3, based on our unpublished data and other studies39), so frequency of an error observed at low coverage should decrease to this level with higher coverage, whereas the level of heteroplasmy should not change with the coverage. However, sequencing errors are reported to be position dependent and context dependent.36,39,42 In fact, we have observed several sequencing error “hot spots” (including 257AC, 3492AC, 3511AC, 4774TA, 5290AT, 9801GT, 10306AC, 10792AC, 11090AC), defined here as meeting the following criteria: (1) ≥ 10% of the individuals have the minor allele with frequency ≥ 10% at this position, but (2) the minor allele for ≥ 90% of the individuals cannot be validated by reads from both strands. As expected, most of the sequencing-error hotspots involve misidentification of A to C; thus, some errors can reach a high enough frequency to pass a simple frequency-threshold criterion and contribute to false positives.
However, given that heteroplasmy is not strand dependent and that it is highly unlikely that an error-prone context would exist for the same position on both strands, a useful filter is double-strand validation: heteroplasmies are called only if both of alleles can be detected by reads from both strands. Double-strand validation alone significantly reduced the number of heterozygous positions detected in the phiX174 sequence data (Table 1). However, even double-strand validation is not sufficient to eliminate all sequencing errors: one position (7030AC) in the artificially mixed samples passed this filter but is a false positive. This can occur because sequencing error also occurs in the other direction by the usual background error rate (e.g., with 40 reads in the non-error-prone direction and an error rate of 0.3%, there is an 11% chance of observing at least one error). We therefore implemented the requirement of observing both alleles in at least two independent reads from each direction to validate the heteroplasmy, and the number of reads required on both strands should be increased with a higher sequencing depth. Although this requirement appears to be sufficient to eliminate virtually all false positives due to sequence errors, it does increase the probability of false negatives due to failure to observe a true minor allele in at least two reads from each direction, especially when coverage is low.
Another aspect of Illumina GA sequencing errors is that they tend to be position dependent: typically, the error rate at the end of the read is approximately 2- to 6-fold higher than at the beginning. One potential way to reduce the false-positive errors that are caused by position-dependent error might therefore be to require that heteroplasmies are validated by the beginning or middle of reads. Indeed, this requirement significantly reduced the number of heterozygous positions in the phiX174 reads (see details in Table S4). However, our data also suggest that the error at the ends of the reads is equivalent to that in the middle of the reads under the QS filtering that we applied (QS ≥ 20 for the position in question and QS ≥ 15 for the 5 bp flanking sequence on each side), which may reflect the fact that QS is correlated with the position in the read. We therefore did not include the requirement of confirmation by the beginning or middle part of the reads in order to call a heteroplasmy.
Impact of Assembly Strategy on Calling of Heteroplasmies
In addition to sequencing errors, inaccurate assembly of the reads can create artificial heteroplasmies, because the minor allele could come from reads originating from another part of the mtDNA genome. To evaluate whether heteroplasmy is caused by improper alignment, we removed all reads with multiple best hits on the final consensus sequence (i.e., mapping quality equals 0 when assembling the reads by using Burrows Wheeler alignment as implemented in the software BWA55), and none of the inferred heteroplasmies disappeared. Therefore, incorrect assembly does not appear to be influencing our results.
Another question concerning the assembly is whether to use all reads or only unique reads (i.e., those with different start and/or endpoints) from high-throughput sequencing data.56–58 The concern is that duplicate reads may represent copies from the same molecule rather than independent reads. We examined this question in the artificially mixed samples and found that using unique reads versus all of the reads does not influence the detection and level of heteroplasmy (results not shown). However, when we resampled the reads from the 1:3 mixture to investigate the influence of sequencing depth, using duplicate reads resulted in higher false-positive and false-negative error rates than using unique reads (Table S5). Apparently, with low coverage even a small number of duplicate reads can have a large impact on the inferred minor allele frequency. Because a large fraction (∼27%) of the positions in our samples have a sequencing depth less than 150×, we used only the unique reads to infer heteroplasmies.
Impact of PCR on Calling of Heteroplasmies
PCR is another factor influencing the detection of heteroplasmy; biased amplification can alter the minor allele frequency, and sequence error introduced during PCR amplification may be falsely detected as heteroplasmies.59,60 However, several observations indicated that PCR has had little or no effect on our analyses: nine inferred heteroplasmy were genotyped by single-base extension assays, and all nine were validated; the inferred heteroplasmy level in artificially mixed samples was close to the mixture ratio; and sequencing of four samples in duplicate showed no frequency difference greater than 10% that could be validated by the double-strand criterion. It should be noted that all PCR products used in the above studies were independently amplified from the original DNA template. Therefore, possible artifacts from the PCR process do not appear to be influencing our inferences concerning heteroplasmy.
Other Factors that Influence the Detection of Heteroplasmy
Besides sequencing error, assembly error, and PCR artifacts, there are other processes that could potentially influence the identification of heteroplasmic positions by generating false positives. These include jumping PCR,61 inadvertent sequencing of nuclear mitochondrial pseudogenes (numts),48,62 contamination during the library preparation, and the “stochastic effect” produced by random sampling of sequencing reads. To what extent could these be responsible for the heteroplasmies that we detected in this study?
The first possibility is jumping PCR, which could result in the incorrect allocation of a read to an individual, because a parallel tagged approach was used in our study.61 However, after the indexing PCR of seven cycles, no additional PCR was performed on the pooled samples, so jumping PCR should not be a problem.
Numts are another potential source of contamination during mtDNA PCR amplification, because there are 46 paralogous nuclear DNA fragments that represent the entire mitochondrial genome.62 However, instead of short-range PCR or capture-based methods, which may be more severely influenced by numts, we utilized long-range PCR. No secondary amplification products were observed, and moreover, 94% of the reads could be mapped to the mtDNA genome, indicating that the reads are indeed originating from authentic mtDNA and not from numts.
Inadvertent sample mixtures or contamination could also produce the appearance of heteroplasmy (indeed, this was the basis for the artificially mixed samples). To verify that such contamination does not explain inferred heteroplasmies, we checked whether the content of the heteroplasmies in one individual could be used to define two different haplogroups in Phylotree.46 In fact, several individuals in the original study (unpublished data) did exhibit numerous putative heteroplasmic positions that could be explained by sample mixtures involving two different haplogroups; these individuals were excluded from further analysis. In the 131 individuals used in this study, sample mixtures involving any known haplogroups cannot explain the inferred heteroplasmies.
A final concern is the “stochastic effect,” i.e., how much variation can occur in sample replicates, and how this would influence the inference of heteroplasmy. In the four replicates, everything was repeated from the template DNA via the same methods; thus, any allele frequency variation for each position between these replicate pairs should be caused by the stochastic effect. Although the “stochastic effect” could result in frequency variation greater than 0.1, none could be validated by the double-strand criteria, indicating that stochastic effects are unlikely to produce false positives under our criteria for detecting heteroplasmy.
Genome-wide Insights into Heteroplasmy
As discussed above, the heteroplasmies detected by our criteria are unlikely to be caused by sequencing errors or other artifacts. We now discuss the insights provided by this large-scale examination of mtDNA genome-wide heteroplasmy. We identified 37 point heteroplasmies and three indel heteroplasmies among 33 of 131 individuals (25%), which is higher than the range of 3.8%–6% reported in previous studies that used other methods.13,63 We attribute this higher rate to the increased sensitivity that high-throughput sequencing offers for detecting heteroplasmy and to the fact that we are analyzing heteroplasmy across the entire mtDNA genome, whereas previous studies focused on the control region.
The familiar transition bias in human mtDNA muations64 was observed, in that 89% of the point heteroplasmies are transitions. Thirteen point heteroplasmies (35%) were found in the control region, which is more than expected, and all of them are located in hypermutable positions (RMR ≥ 6). An association between heteroplasmy and hypermutable positions was found previously,65 and overall, these results suggest that mutation is the major driving force behind heteroplasmy and that a mutation-drift process can explain how heteroplasmies arise, drift to high frequencies within an individual, and eventually become “fixed” as polymorphisms among individuals.
However, in the protein-coding region, mutations are located in both hypermutable positions and hypomutable positions. Altogether, eight heteroplasmies are located in positions that have never been reported to mutate, which is more than expected by chance (p = 0.04). Moreover, there is a significant excess of heteroplasmies involving nonsynonymous changes in comparison to polymorphism data. It thus seems as if deleterious mutations arise as heteroplasmies and can reach appreciable frequencies (> 10%) within individuals but do not drift to fixation. Instead, purifying selection must be operating on some heteroplasmies to prevent their fixation within individuals. Although purifying selection on mtDNA has been inferred from other studies,66–68 no previous evidence of such selection involving heteroplasmy has been found,69 which may reflect the limited number of heteroplasmies previously studied in the coding region.
Significantly more heteroplasmies were detected in disease-associated positions than expected by chance, but all of them were also observed to be polymorphic in the same population or in our polymorphism data set and, moreover, are associated with high mutation rates. These are not characteristics expected to be associated with deleterious mutations and hence may reflect limitations on the accuracy of studies that attempt to elucidate disease-associated mtDNA mutations. Or, these may be associated with mild effects on the phenotype. Still, we found that even in comparison to the polymorphism profile, there is an excess number of heteroplasmies in disease-associated positions. It is thus possible that these disease-associated heteroplasmies may drift to high enough frequencies within an individual to result in the disease phenotype and/or that they will be removed by purifying selection.
Detecting Heteroplasmy by High-Throughput Sequencing Technology
We have developed a set of criteria to detect heteroplasmy in complete human mtDNA genomes from the reads generated by Illumina GAII technology. Recently, another study used the same sequencing technology to investigate mtDNA heteroplasmy.14 This study sequenced only one individual per lane, to an average coverage of ∼16,000, and hence used a correspondingly lower threshold for the minor allele frequency of 1.6% to call heteroplasmic positions. They detected 40 heteroplasmies above this threshold in ten healthy individuals;14 of these, seven heteroplasmies in four individuals had a minor allele frequency greater than the 10% threshold that we used. This is significantly more than the 37 point heteroplasmies that we detected among 131 individuals (p = 0.02, resampling test). Possible reasons for this difference include: a higher false-negative error rate in our study, which is due to lower sequencing depth; the different tissues used (blood and saliva in our study, colonic mucosae in their study14); and/or the relatively old age of individuals in their study.14
Although sequencing to a much higher depth obviously increases the power to detect heteroplasmies by lowering the minor allele frequency threshold, stringent quality control procedures are still necessary to determine a proper frequency threshold. In fact, we note that in the previous analysis of heteroplasmic variants among tissues from a single individual (Table 2 of He et al.14), five of 14 heteroplasmies (36%) involve AC/GT changes, which is the most common sequencing error on the Illumina GA platform. This is significantly more than the 9% frequency of AC/GT changes among heteroplasmies in our study (p < 0.05), and it therefore suggests that some of these are likely to reflect false positives due to sequencing errors.
The strategy used to detect heteroplasmic variants can therefore vary, depending on the goal of the study (and available funding). Sequencing one sample per lane (or even one sample on multiple lanes) will lower the threshold for detecting heteroplasmic variants but will increase the cost and therefore limit the number of samples that can be studied. Parallel tagged approaches, which were used here, result in lower coverage and hence require a higher minor allele frequency to call a heteroplasmy, but they are a more cost-efficient approach for analyzing heteroplasmy in a large number of samples. Indeed, we predict that the next few years will see a huge increase in complete mtDNA genome sequences generated to an average coverage of ∼50×–100× with next-generation sequencing platforms. These data will provide a rich resource for further investigation of heteroplasmy. Regardless of the strategy and average coverage obtained, our results indicate that accurate calling of heteroplasmic positions requires the analysis of control data and an appropriate statistical model to generate appropriate criteria. With such appropriate criteria for avoiding false positives due to sequencing errors, high-throughput sequencing platforms can provide a reliable genome-wide heteroplasmy map, which can provide further insights into mtDNA-related diseases and the evolution of mtDNA.
Supplemental Data
Supplemental Data include five tables and one figure can be found with this article online at http://www.cell.com/AJHG/.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Mapping Iterative Assembler (MIA), http://sourceforge.net/projects/mia-assembler/
Mitomap, http://www.mitomap.org
mtDB - Human Mitochondrial Genome Database, http://www.genpat.uu.se/mtDB/
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
Phylotree, http://www.phylotree.org
Acknowledgments
We thank all individuals who kindly donated a sample for the original study; the MPI-EVA sequencing group and M. Kircher for technical support; J. Li for producing Figure 1; and M. Whitten, R. E. Green, E. Gunnarsdóttir, and D. Hughes for helpful discussion. Funding was provided by the Max Planck Society.
Contributor Information
Mingkun Li, Email: mingkun_li@eva.mpg.de.
Mark Stoneking, Email: stoneking@eva.mpg.de.
References
- 1.Pakendorf B., Stoneking M. Mitochondrial DNA and human evolution. Annu. Rev. Genomics Hum. Genet. 2005;6:165–183. doi: 10.1146/annurev.genom.6.080604.162249. [DOI] [PubMed] [Google Scholar]
- 2.Greenberg B.D., Newbold J.E., Sugino A. Intraspecific nucleotide sequence variability surrounding the origin of replication in human mitochondrial DNA. Gene. 1983;21:33–49. doi: 10.1016/0378-1119(83)90145-2. [DOI] [PubMed] [Google Scholar]
- 3.Wallace D.C. Mitochondrial DNA sequence variation in human evolution and disease. Proc. Natl. Acad. Sci. USA. 1994;91:8739–8746. doi: 10.1073/pnas.91.19.8739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chinnery P.F., Samuels D.C., Elson J., Turnbull D.M. Accumulation of mitochondrial DNA mutations in ageing, cancer, and mitochondrial disease: is there a common mechanism? Lancet. 2002;360:1323–1325. doi: 10.1016/S0140-6736(02)11310-9. [DOI] [PubMed] [Google Scholar]
- 5.Szibor M., Holtz J. Mitochondrial ageing. Basic Res. Cardiol. 2003;98:210–218. doi: 10.1007/s00395-003-0421-z. [DOI] [PubMed] [Google Scholar]
- 6.Wallace D.C. Mitochondrial genetics: a paradigm for aging and degenerative diseases? Science. 1992;256:628–632. doi: 10.1126/science.1533953. [DOI] [PubMed] [Google Scholar]
- 7.MITOMAP. A Human Mitochondrial Genome Database. http://www.mitomap.org, 2009. [DOI] [PMC free article] [PubMed]
- 8.Rossignol R., Faustin B., Rocher C., Malgat M., Mazat J.P., Letellier T. Mitochondrial threshold effects. Biochem. J. 2003;370:751–762. doi: 10.1042/BJ20021594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhu D.P., Economou E.P., Antonarakis S.E., Maumenee I.H. Mitochondrial DNA mutation and heteroplasmy in type I Leber hereditary optic neuropathy. Am. J. Med. Genet. 1992;42:173–179. doi: 10.1002/ajmg.1320420208. [DOI] [PubMed] [Google Scholar]
- 10.Monnat R.J., Loeb L.A. Nucleotide sequence preservation of human mitochondrial DNA. Proc. Natl. Acad. Sci. USA. 1985;82:2895–2899. doi: 10.1073/pnas.82.9.2895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Monnat R.J., Reay D.T. Nucleotide sequence identity of mitochondrial DNA from different human tissues. Gene. 1986;43:205–211. doi: 10.1016/0378-1119(86)90208-8. [DOI] [PubMed] [Google Scholar]
- 12.Comas D., Pääbo S., Bertranpetit J. Heteroplasmy in the control region of human mitochondrial DNA. Genome Res. 1995;5:89–90. doi: 10.1101/gr.5.1.89. [DOI] [PubMed] [Google Scholar]
- 13.Irwin J.A., Saunier J.L., Niederstätter H., Strouss K.M., Sturk K.A., Diegoli T.M., Brandstätter A., Parson W., Parsons T.J. Investigation of heteroplasmy in the human mitochondrial DNA control region: a synthesis of observations from more than 5000 global population samples. J. Mol. Evol. 2009;68:516–527. doi: 10.1007/s00239-009-9227-4. [DOI] [PubMed] [Google Scholar]
- 14.He Y., Wu J., Dressman D.C., Iacobuzio-Donahue C., Markowitz S.D., Velculescu V.E., Diaz L.A., Kinzler K.W., Vogelstein B., Papadopoulos N. Heteroplasmic mitochondrial DNA mutations in normal and tumour cells. Nature. 2010;464:610–614. doi: 10.1038/nature08802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jazin E.E., Cavelier L., Eriksson I., Oreland L., Gyllensten U. Human brain contains high levels of heteroplasmy in the noncoding regions of mitochondrial DNA. Proc. Natl. Acad. Sci. USA. 1996;93:12382–12387. doi: 10.1073/pnas.93.22.12382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Salas A., Carracedo A., Macaulay V., Richards M., Bandelt H.J. A practical guide to mitochondrial DNA error prevention in clinical, forensic, and population genetics. Biochem. Biophys. Res. Commun. 2005;335:891–899. doi: 10.1016/j.bbrc.2005.07.161. [DOI] [PubMed] [Google Scholar]
- 17.Salas A., Lareu M.V., Carracedo A. Heteroplasmy in mtDNA and the weight of evidence in forensic mtDNA analysis: a case report. Int. J. Legal Med. 2001;114:186–190. doi: 10.1007/s004140000164. [DOI] [PubMed] [Google Scholar]
- 18.Zsurka G., Kraytsberg Y., Kudina T., Kornblum C., Elger C.E., Khrapko K., Kunz W.S. Recombination of mitochondrial DNA in skeletal muscle of individuals with multiple mitochondrial DNA heteroplasmy. Nat. Genet. 2005;37:873–877. doi: 10.1038/ng1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhidkov I., Livneh E.A., Rubin E., Mishmar D. MtDNA mutation pattern in tumors and human evolution are shaped by similar selective constraints. Genome Res. 2009;19:576–580. doi: 10.1101/gr.086462.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Meierhofer D., Mayr J.A., Ebner S., Sperl W., Kofler B. Rapid screening of the entire mitochondrial DNA for low-level heteroplasmic mutations. Mitochondrion. 2005;5:282–296. doi: 10.1016/j.mito.2005.06.001. [DOI] [PubMed] [Google Scholar]
- 21.Ballana E., Govea N., de Cid R., Garcia C., Arribas C., Rosell J., Estivill X. Detection of unrecognized low-level mtDNA heteroplasmy may explain the variable phenotypic expressivity of apparently homoplasmic mtDNA mutations. Hum. Mutat. 2008;29:248–257. doi: 10.1002/humu.20639. [DOI] [PubMed] [Google Scholar]
- 22.White H.E., Durston V.J., Seller A., Fratter C., Harvey J.F., Cross N.C. Accurate detection and quantitation of heteroplasmic mitochondrial point mutations by pyrosequencing. Genet. Test. 2005;9:190–199. doi: 10.1089/gte.2005.9.190. [DOI] [PubMed] [Google Scholar]
- 23.Cassandrini D., Calevo M.G., Tessa A., Manfredi G., Fattori F., Meschini M.C., Carrozzo R., Tonoli E., Pedemonte M., Minetti C. A new method for analysis of mitochondrial DNA point mutations and assess levels of heteroplasmy. Biochem. Biophys. Res. Commun. 2006;342:387–393. doi: 10.1016/j.bbrc.2006.01.152. [DOI] [PubMed] [Google Scholar]
- 24.Dobrowolski S.F., Hendrickx A.T., van den Bosch B.J., Smeets H.J., Gray J., Miller T., Sears M. Identifying sequence variants in the human mitochondrial genome using high-resolution melt (HRM) profiling. Hum. Mutat. 2009;30:891–898. doi: 10.1002/humu.21003. [DOI] [PubMed] [Google Scholar]
- 25.Wong L.J., Chen T.J., Tan D.J. Detection of mitochondrial DNA mutations using temporal temperature gradient gel electrophoresis. Electrophoresis. 2004;25:2602–2610. doi: 10.1002/elps.200406016. [DOI] [PubMed] [Google Scholar]
- 26.Mashima Y., Nagano M., Funayama T., Zhang Q., Egashira T., Kudho J., Shimizu N., Oguchi Y. Rapid quantification of the heteroplasmy of mutant mitochondrial DNAs in Leber's hereditary optic neuropathy using the Invader technology. Clin. Biochem. 2004;37:268–276. doi: 10.1016/j.clinbiochem.2003.11.011. [DOI] [PubMed] [Google Scholar]
- 27.Bai R.K., Wong L.J.C. Detection and quantification of heteroplasmic mutant mitochondrial DNA by real-time amplification refractory mutation system quantitative PCR analysis: a single-step approach. Clin. Chem. 2004;50:996–1001. doi: 10.1373/clinchem.2004.031153. [DOI] [PubMed] [Google Scholar]
- 28.Bannwarth S., Procaccio V., Paquis-Flucklinger V. Surveyor Nuclease: a new strategy for a rapid identification of heteroplasmic mitochondrial DNA mutations in patients with respiratory chain defects. Hum. Mutat. 2005;25:575–582. doi: 10.1002/humu.20177. [DOI] [PubMed] [Google Scholar]
- 29.Hancock D.K., Tully L.A., Levin B.C. A Standard Reference Material to determine the sensitivity of techniques for detecting low-frequency mutations, SNPs, and heteroplasmies in mitochondrial DNA. Genomics. 2005;86:446–461. doi: 10.1016/j.ygeno.2005.06.006. [DOI] [PubMed] [Google Scholar]
- 30.Prieto L., Alonso A., Alves C., Crespillo M., Montesino M., Picornell A., Brehm A., Ramírez J.L., Whittle M.R., Anjos M.J. 2006 GEP-ISFG collaborative exercise on mtDNA: reflections about interpretation, artefacts, and DNA mixtures. Forensic Sci. Int.; Genet. 2008;2:126–133. doi: 10.1016/j.fsigen.2007.10.010. [DOI] [PubMed] [Google Scholar]
- 31.Melton T. Mitochondrial DNA heteroplasmy. Forensic Science Review. 2004;16:1–20. [PubMed] [Google Scholar]
- 32.Mardis E.R. The impact of next-generation sequencing technology on genetics. Trends Genet. 2008;24:133–141. doi: 10.1016/j.tig.2007.12.007. [DOI] [PubMed] [Google Scholar]
- 33.Hillier L.W., Marth G.T., Quinlan A.R., Dooling D., Fewell G., Barnett D., Fox P., Glasscock J.I., Hickenbotham M., Huang W. Whole-genome sequencing and variant discovery in C. elegans. Nat. Methods. 2008;5:183–188. doi: 10.1038/nmeth.1179. [DOI] [PubMed] [Google Scholar]
- 34.Van Tassell C.P., Smith T.P., Matukumalli L.K., Taylor J.F., Schnabel R.D., Lawley C.T., Haudenschild C.D., Moore S.S., Warren W.C., Sonstegard T.S. SNP discovery and allele frequency estimation by deep sequencing of reduced representation libraries. Nat. Methods. 2008;5:247–252. doi: 10.1038/nmeth.1185. [DOI] [PubMed] [Google Scholar]
- 35.Bansal V., Harismendy O., Tewhey R., Murray S.S., Schork N.J., Topol E.J., Frazer K.A. Accurate detection and genotyping of SNPs utilizing population sequencing data. Genome Res. 2010;20:537–545. doi: 10.1101/gr.100040.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Out A.A., van Minderhout I.J., Goeman J.J., Ariyurek Y., Ossowski S., Schneeberger K., Weigel D., van Galen M., Taschner P.E., Tops C.M. Deep sequencing to reveal new variants in pooled DNA samples. Hum. Mutat. 2009;30:1703–1712. doi: 10.1002/humu.21122. [DOI] [PubMed] [Google Scholar]
- 37.Shen Y., Wan Z., Coarfa C., Drabek R., Chen L., Ostrowski E.A., Liu Y., Weinstock G.M., Wheeler D.A., Gibbs R.A., Yu F. A SNP discovery method to assess variant allele probability from next-generation resequencing data. Genome Res. 2010;20:273–280. doi: 10.1101/gr.096388.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Meyer M., Kircher M. Illumina sequencing library preparation for highly multiplexed target capture and sequencing. Cold Spring Harb Protoc. 2010;2010:t5448. doi: 10.1101/pdb.prot5448. [DOI] [PubMed] [Google Scholar]
- 39.Kircher M., Stenzel U., Kelso J. Improved base calling for the Illumina Genome Analyzer using machine learning strategies. Genome Biol. 2009;10:R83. doi: 10.1186/gb-2009-10-8-r83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Briggs A.W., Good J.M., Green R.E., Krause J., Maricic T., Stenzel U., Lalueza-Fox C., Rudan P., Brajkovic D., Kucan Z. Targeted retrieval and analysis of five Neandertal mtDNA genomes. Science. 2009;325:318–321. doi: 10.1126/science.1174462. [DOI] [PubMed] [Google Scholar]
- 41.Andrews R.M., Kubacka I., Chinnery P.F., Lightowlers R.N., Turnbull D.M., Howell N. Reanalysis and revision of the Cambridge reference sequence for human mitochondrial DNA. Nat. Genet. 1999;23:147. doi: 10.1038/13779. [DOI] [PubMed] [Google Scholar]
- 42.Dohm J.C., Lottaz C., Borodina T., Himmelbauer H. Substantial biases in ultra-short read data sets from high-throughput DNA sequencing. Nucleic Acids Res. 2008;36:e105. doi: 10.1093/nar/gkn425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ingman M., Gyllensten U. mtDB: Human Mitochondrial Genome Database, a resource for population genetics and medical sciences. Nucleic Acids Res. 2006;34(Database issue, Database issue):D749–D751. doi: 10.1093/nar/gkj010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Benson D.A., Karsch-Mizrachi I., Lipman D.J., Ostell J., Sayers E.W. GenBank. Nucleic Acids Res. 2009;37(Database issue, Database issue):D26–D31. doi: 10.1093/nar/gkn723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Soares P., Ermini L., Thomson N., Mormina M., Rito T., Röhl A., Salas A., Oppenheimer S., Macaulay V., Richards M.B. Correcting for purifying selection: an improved human mitochondrial molecular clock. Am. J. Hum. Genet. 2009;84:740–759. doi: 10.1016/j.ajhg.2009.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.van Oven M., Kayser M. Updated comprehensive phylogenetic tree of global human mitochondrial DNA variation. Hum. Mutat. 2009;30:E386–E394. doi: 10.1002/humu.20921. [DOI] [PubMed] [Google Scholar]
- 47.Tang S., Huang T. Characterization of mitochondrial DNA heteroplasmy using a parallel sequencing system. Biotechniques. 2010;48:287–296. doi: 10.2144/000113389. [DOI] [PubMed] [Google Scholar]
- 48.Hirano M., Shtilbans A., Mayeux R., Davidson M.M., DiMauro S., Knowles J.A., Schon E.A. Apparent mtDNA heteroplasmy in Alzheimer's disease patients and in normals due to PCR amplification of nucleus-embedded mtDNA pseudogenes. Proc. Natl. Acad. Sci. USA. 1997;94:14894–14899. doi: 10.1073/pnas.94.26.14894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Seo S.B., Jang B.S., Zhang A., Yi J.A., Kim H.Y., Yoo S.H., Lee Y.S., Lee S.D. Alterations of length heteroplasmy in mitochondrial DNA under various amplification conditions. J. Forensic Sci. 2010;55:719–722. doi: 10.1111/j.1556-4029.2010.01321.x. [DOI] [PubMed] [Google Scholar]
- 50.Kivisild T., Shen P., Wall D.P., Do B., Sung R., Davis K., Passarino G., Underhill P.A., Scharfe C., Torroni A. The role of selection in the evolution of human mitochondrial genomes. Genetics. 2006;172:373–387. doi: 10.1534/genetics.105.043901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ballana E., Morales E., Rabionet R., Montserrat B., Ventayol M., Bravo O., Gasparini P., Estivill X. Mitochondrial 12S rRNA gene mutations affect RNA secondary structure and lead to variable penetrance in hearing impairment. Biochem. Biophys. Res. Commun. 2006;341:950–957. doi: 10.1016/j.bbrc.2006.01.049. [DOI] [PubMed] [Google Scholar]
- 52.Burk A., Douzery E.J.P., Springer M.S. The secondary structure of mammalian mitochondrial 16S rRNA molecules: refinements based on a comparative phylogenetic approach. J. Mamm. Evol. 2002;9:225–252. [Google Scholar]
- 53.Florentz C., Sohm B., Tryoen-Tóth P., Pütz J., Sissler M. Human mitochondrial tRNAs in health and disease. Cell. Mol. Life Sci. 2003;60:1356–1375. doi: 10.1007/s00018-003-2343-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Harismendy O., Ng P.C., Strausberg R.L., Wang X., Stockwell T.B., Beeson K.Y., Schork N.J., Murray S.S., Topol E.J., Levy S., Frazer K.A. Evaluation of next generation sequencing platforms for population targeted sequencing studies. Genome Biol. 2009;10:R32. doi: 10.1186/gb-2009-10-3-r32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gomez-Alvarez V., Teal T.K., Schmidt T.M. Systematic artifacts in metagenomes from complex microbial communities. ISME J. 2009;3:1314–1317. doi: 10.1038/ismej.2009.72. [DOI] [PubMed] [Google Scholar]
- 57.Ng S.B., Turner E.H., Robertson P.D., Flygare S.D., Bigham A.W., Lee C., Shaffer T., Wong M., Bhattacharjee A., Eichler E.E. Targeted capture and massively parallel sequencing of 12 human exomes. Nature. 2009;461:272–276. doi: 10.1038/nature08250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li R., Fan W., Tian G., Zhu H., He L., Cai J., Huang Q., Cai Q., Li B., Bai Y. The sequence and de novo assembly of the giant panda genome. Nature. 2010;463:311–317. doi: 10.1038/nature08696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Calloway C.D., Reynolds R.L., Herrin G.L., Anderson W.W. The frequency of heteroplasmy in the HVII region of mtDNA differs across tissue types and increases with age. Am. J. Hum. Genet. 2000;66:1384–1397. doi: 10.1086/302844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Grzybowski T., Malyarchuk B.A., Czarny J., Miścicka-Sliwka D., Kotzbach R. High levels of mitochondrial DNA heteroplasmy in single hair roots: reanalysis and revision. Electrophoresis. 2003;24:1159–1165. doi: 10.1002/elps.200390149. [DOI] [PubMed] [Google Scholar]
- 61.Liu S., Thaler D.S., Libchaber A. Signal and noise in bridging PCR. BMC Biotechnol. 2002;2:13. doi: 10.1186/1472-6750-2-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Parr R.L., Maki J., Reguly B., Dakubo G.D., Aguirre A., Wittock R., Robinson K., Jakupciak J.P., Thayer R.E. The pseudo-mitochondrial genome influences mistakes in heteroplasmy interpretation. BMC Genomics. 2006;7:185. doi: 10.1186/1471-2164-7-185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Santos C., Sierra B., Alvarez L., Ramos A., Fernández E., Nogués R., Aluja M.P. Frequency and pattern of heteroplasmy in the control region of human mitochondrial DNA. J. Mol. Evol. 2008;67:191–200. doi: 10.1007/s00239-008-9138-9. [DOI] [PubMed] [Google Scholar]
- 64.Brown G.G., Simpson M.V. Novel features of animal mtDNA evolution as shown by sequences of two rat cytochrome oxidase subunit II genes. Proc. Natl. Acad. Sci. USA. 1982;79:3246–3250. doi: 10.1073/pnas.79.10.3246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Stoneking M. Hypervariable sites in the mtDNA control region are mutational hotspots. Am. J. Hum. Genet. 2000;67:1029–1032. doi: 10.1086/303092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rand D.M. The units of selection on mitochondrial DNA. Annu. Rev. Ecol. Syst. 2001;32:415–448. [Google Scholar]
- 67.Elson J.L., Turnbull D.M., Howell N. Comparative genomics and the evolution of human mitochondrial DNA: assessing the effects of selection. Am. J. Hum. Genet. 2004;74:229–238. doi: 10.1086/381505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ruiz-Pesini E., Wallace D.C. Evidence for adaptive selection acting on the tRNA and rRNA genes of human mitochondrial DNA. Hum. Mutat. 2006;27:1072–1081. doi: 10.1002/humu.20378. [DOI] [PubMed] [Google Scholar]
- 69.Wonnapinij P., Chinnery P.F., Samuels D.C. The distribution of mitochondrial DNA heteroplasmy due to random genetic drift. Am. J. Hum. Genet. 2008;83:582–593. doi: 10.1016/j.ajhg.2008.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.