

Abstract
Palindrome-mediated genomic instability has been associated with chromosomal translocations, including the recurrent t(11;22)(q23;q11). We report a syndrome characterized by extremity anomalies, mild dysmorphia, and intellectual impairment caused by 3:1 meiotic segregation of a previously unrecognized recurrent palindrome-mediated rearrangement, the t(8;22)(q24.13;q11.21). There are at least ten prior reports of this translocation, and nearly identical PATRR8 and PATRR22 breakpoints were validated in several of these published cases. PCR analysis of sperm DNA from healthy males indicates that the t(8;22) arises de novo during gametogenesis in some, but not all, individuals. Furthermore, demonstration that de novo PATRR8-to-PATRR11 translocations occur in sperm suggests that palindrome-mediated translocation is a universal mechanism producing chromosomal rearrangements.
Introduction
Approximately one in 600 individuals is the carrier of a balanced constitutional translocation.1 Most balanced-translocation carriers are healthy and do not come to medical attention until they experience infertility or the birth of an abnormal child with an unbalanced form of the translocation.2 The majority of non-Robertsonian constitutional translocations are unique. Only two translocations, the t(11;22)(q23;q11) and the t(4;8)(pl6;p23), have been reported to recur de novo multiple times.3–5 Both are identified after meiotic malsegregation that results in abnormal offspring. The constitutional t(11;22) segregates 3:1 and has been described as the most common recurrent non-Robertsonian translocation in humans.6 Although t(11;22) balanced-translocation carriers are phenotypically normal, males are occasionally subject to infertility, whereas females may experience recurrent pregnancy loss. Carriers are often identified after the birth of unbalanced offspring with the supernumerary der(22)t(11;22) syndrome (Emanuel syndrome [MIM 609029]).7 Unlike the t(11;22), the t(4;8) undergoes 2:2 malsegregation. Patients who carry the der(4)t(4;8) have Wolf-Hirschhorn syndrome (MIM 194190), whereas patients who carry the der(8)t(4;8) have a different phenotype, which includes mental retardation.8
These two translocations are hypothesized to recur by distinct mechanisms. The t(4;8) is postulated to be mediated by homologous recombination between olfactory receptor gene clusters.9,10 Alternatively, the breakpoints of numerous unrelated t(11;22) cases have been consistently shown to be located within palindromic AT-rich repeats (PATRRs) on 11q23 and 22q11.11–14 This suggests that the t(11;22) is caused by the formation of hairpin/cruciform structures that lead to double-strand DNA breaks and chromosomal translocation. Interestingly, a t(17;22) has been identified in two unrelated cases. Similar to the t(11;22), both of the reported t(17;22)s have been shown to be mediated by the same PATRR on 22q11.2 and a PATRR in intron 31 of the NF1 gene.15 These translocations involving the PATRR at 22q11 suggested the possibility for other PATRR-mediated rearrangements in humans, given that palindrome-induced genetic instability has been clearly demonstrated in numerous model organisms, including bacteria, yeast, and mice.16–18
Indeed, the PATRR on chromosome 22 has been described as a hotspot for translocation breakpoints.19 Recent findings of PATRR-like or palindromic sequences at the translocation breakpoints of other chromosome 22 partner chromosomes (chromosomes 1p, 4q, and 8q) support the hypothesis that palindrome-mediated genomic rearrangement is a pathway for producing chromosomal translocations.15,20–23 The majority of the t(11;22) as well as the two t(17;22) breakpoints have been localized at the center of the PATRRs, suggesting that the center of the palindrome is susceptible to breakage as a prerequisite to chromosomal rearrangement. However, until now, the only translocations involving PATRRs identified more than once were the t(11;22) and the t(17;22).
In this manuscript, we describe multiple individuals with balanced and unbalanced forms of the t(8;22)(q24.13;q11.2). Most have been previously reported, and one is a new example seen at our institution. However, this translocation had not previously been recognized as a recurrent rearrangement with a consistent mechanism. Interestingly, the 8q breakpoint region is also involved in a t(3;8) which has been reported to segregate with renal cell carcinoma in two independent families.24,25 Similar to the t(11;22), the 8;22 translocation segregates 3:1 to produce unbalanced karyotypes, and all examples studied occur at nearly identical breakpoints in PATRRs on 8q and 22q. The reports of at least 12 cases of this rearrangement and the detection of this translocation in sperm from chromosomally normal males suggest that it is another recurrent palindrome-mediated non-Robertsonian translocation. These findings provide additional support for the role of palindromic sequences in genomic instability.
Material and Methods
Fluorescence In Situ Hybridization
Cytogenetic and fluorescence in situ hybridization (FISH) analyses were performed on metaphase spreads from peripheral-blood lymphocyte cultures as described in Gotter et al.,23 with the use of the bacterial artificial chromosome clone RP11–158k1. This research study was approved by the institutional review board of the Children's Hospital of Philadelphia, and written consent was obtained from all patients or their parents.
Sequence Analysis of t(8;22) Junction Sequences
Genomic DNA was extracted from peripheral blood or lymphoblast cell lines via previously described methods.23,26–28 Nested PCR was performed with the use of the primers described in Gotter et al.23 Primer sequences are available in Table S1, available online. In brief, der(8) products were amplified with the use of the following primers: primary amplification: 8q24-7F with 22.B2R; secondary amplification: 8q24-8F with 22.B4R; der(22) product amplification: primary amplification: 22.B1 and 8q24-7R, secondary amplification: 22.B3 and 8q24-8R. Samples were subjected to PCR purification (Qiaquick PCR Purification Kit, QIAGEN) and sequenced bidirectionally by capillary electrophoresis (ABI3730 Genetic Analyzer, Applied Biosytems). Sequences were analyzed with the Sequencher analysis program (Gene Codes). Kalign (Tool for Fast Multiple Protein and Nucleotide Sequence Alignment) was used to align the resulting sequences.
Genome-wide SNP Array
Genomic DNA samples were analyzed with the use of the Affymetrix SNP 6.0 Array platform according to the manufacturer's instructions (Affymetrix). Copy-number data were analyzed with the Partek Genomics Suite (Partek).
Determination of t(8;22) De Novo Frequency
Anonymous sperm samples were obtained from normal, healthy volunteers and tested for the der(8)t(8;22) and/or the der(22)t(8;22) PCR fragment. DNA was extracted from fresh or frozen semen samples with the Puregene DNA purification kit (QIAGEN) according to the manufacturer's instructions. PCR was performed as above, with the use of 24 100 ng aliquots of each sperm DNA sample and a 5′ fluorescently labeled reverse primer (6-carboxyfluorescein). Products were separated by agarose gel electrophoresis or capillary electrophoresis on an ABI 3730 DNA Analyzer (Applied Biosystems). Sizes were estimated against an internal standard (ROX 500, 6-Carboxyl-X-Rhodamine) with GeneMapper software (Applied Biosystems). The translocation frequency was calculated via the methods described in Kurahashi et al.29
PATRR8 Characterization
Genomic DNA was amplified with primers flanking the PATRR8 (primer sequences are available in Table S1). PCR was performed with Advantage2 Polymerase (Clontech) or the LA Taq DNA Polymerase system (Takara). Products were separated by agarose gel electrophoresis and excised (QIAquick Gel Extraction Kit, QIAGEN). The purified products were cloned into pCR2.1TOPO (Invitrogen), and transformed into SURE (Stop Unwanted Rearrangement Events) strain E.Coli (Stratagene). Plasmid DNA was extracted and was sequenced as above.
t(8;11)(q24.1,q23) and t(1;22)(p21.2,q11.2) Amplification in Sperm DNA
Der(8) and der(11)t(8;11) products were amplified with the use of the primers described in Gotter et al.23 and Kurahashi et al.13 PCR was performed with the LA Taq DNA Polymerase system (Takara), with the use of 250 ng aliquots of each sperm DNA sample. The resulting products were separated by gel electrophoresis and sequenced as above. A similar assay was performed for the detection of t(1;22) junction fragments with the use of the primers described in Gotter et al.22 and listed in Table S1.
Results
Identification of a t(8;22) with Nearly Identical PATRR Breakpoints
We previously reported a patient with +der(22)t(8;22)(q24.1;q11.2).23 The breakpoints at 8q24.1 and 22q11.2 occur in PATRRs. A second patient with a molecularly similar supernumerary der(22)t(8;22) was referred to our attention. Like the father of the first patient, the mother of this second patient carries what cytogenetically appeared to be a similar t(8;22)(q24.1;q11.2) as a balanced rearrangement. Comparison of the translocation breakpoints of both cases by FISH, PCR of the breakpoints, and genome-wide SNP array indicates that they are nearly identical (Figure 1 and Figure 2; cases 10 and 11).
Figure 1.
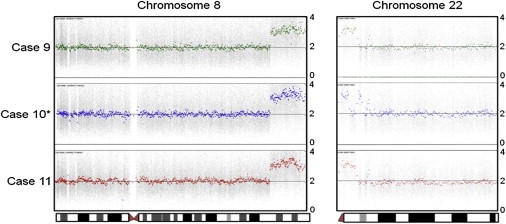
Copy-Number Output for Chromosomes 8 and 22
Three unrelated DNA samples with a supernumerary der(22)t(8;22) are depicted after analysis using the Partek software copy-number analysis suite on data derived from running Affymetrix Genome-Wide Human SNP Arrays 6.0. Output is shown from short-arm telomere (left) to long-arm telomere (right). The y axis indicates copy number from zero to four copies. The three DNA samples demonstrate a duplication of distal 8q and proximal 22q, indicating three copies of the identical chromosomal region. ∗Case 10 has an inversion on 22q.
Figure 2.

t(8;22) Junction-Fragment Sequencing
Sequence of t(8;22) der(8) (A) and der(22) (B) junction fragments from t(8;22) balanced carriers and patients carrying the supernumerary der(22)t(8;22). The sequence is color coded by nucleotide. Stars identify nucleotides that are identical in all sequenced patients. Sequence derived from chromosome 8 is underlined. Superscript a or b indicates a parent-child pair.
A comprehensive search of the literature revealed at least ten prior reports of this translocation (Table 1 and Table 2).26–28,30–35 Several of these reports indicated gross molecular positioning of the 8q and 22q breakpoints consistent with what would be predicted on the basis of our two cases. We subsequently received samples from two patients carrying the supernumerary der(22)t(8;22)26,27 and a father-daughter pair who are t(8;22) balanced carriers28 and have performed additional analyses. As predicted, the breakpoints are identical by FISH (patient 8)26 or SNP array (patient 9; Figure 1).27 PCR plus sequencing of the der(8) and der(22) junction fragments indicates that all of the breakpoints are localized in the PATRRs (Figures 2A and 2B; patients 8, 9, 16, and 17). Interestingly, although the nucleotide sequence of the individual junction fragments is nearly identical, they are each unique. This demonstrates that the t(8;22) has recurred independently at least five times. The junction-fragment sequences did not identify any changes between individuals within the same family (cases 10 and 12, 11 and 13, 16 and 17), indicating that the sequences around the 8q and 22q breakpoints are stable in the derivative chromosomes. Thus, similar to the t(11;22), the t(8;22) recurs and segregates 3:1 to produce unbalanced karyotypes.
Table 1.
Phenotypes of Patients with Supernumerary der(22)t(8;22)
| Case | t(8;22) Inheritance | Sex | Age (Yrs) | Birth Weight | Growth | Ears | Extremities | Genitalia | Development/ IQ | Reference |
|---|---|---|---|---|---|---|---|---|---|---|
| 1a | maternal | M | 18 | normal | normal | prominent | clinodactyly | normal | 50 | Sanchez et al.31 |
| 2a | maternal | F | 29 | normal | normal | prominent, low set | clinodactyly | normal | 59 | Sanchez et al.31 |
| 3b | maternal | M | 12 | - | normal | - | clinodactyly | ectopic testes | 50 | Rethore et al.32 |
| 4b | maternal | M | 10 | - | normal | - | clinodactyly | - | 60 | Rethore et al.32 |
| 5 | paternal | M | 16 | - | normal | - | clinodactyly | ectopic testes | 70 | Rethore et al.32 |
| 6 | maternal | M | - | - | - | - | - | - | - | Rethore et al.32 |
| 7 | maternal | M | 5 | - | - | preauricular pit, low set | - | cryptorchidism | no speech at 5 yrs of age | Rethore et al.32 |
| 8 | paternal | M | 5.5 | normal | weight, length < 3% | preauricular pit, atretic ear | clinodactyly | normal | moderate speech and language delayc | Mark et al.26 |
| 9 | - | M | 43 | - | - | large | - | cryptorchidism | 50–60 | Helbig et al.27 |
| 10 | paternal | F | 10 | - | - | preauricular pit | - | - | educational assistance via math resource room | Gotter et al.23 |
| 11 | maternal | M | 3.5 | normal | normal | large | clinodactyly | - | 63d | This study |
“-” indicates unknown data.
Siblings.
No formal testing.
Mullen Scales of Early Learning.
Table 2.
Phenotypes of t(8;22) Balanced Carriers
| Case | Sex | Age (Yrs) | Other | Phenotype | Reference |
|---|---|---|---|---|---|
| 12 | M | 33 | father of case 10 | none described | Gotter et al.23 |
| 13 | F | 36 | mother of case 11 | none described | This study |
| 14 | M | 54 | - | thrombocytopenia | Gupta et al.34 |
| 15 | F | 26 | - | myasthenia gravis, leukocytosis, thrombocytosis | Keung et al.35 |
| 16 | F | 10 | - | dysgerminoma | Gimelli et al.28 |
| 17 | M | - | father of case 16 | none described | Gimelli et al.28 |
Phenotypic Evaluation
Patients carrying the supernumerary der(22)t(8;22) have a variable phenotype (Table 1), whereas balanced carriers appear to be generally healthy (Table 2). Birth weight and subsequent growth have been normal in all patients, with the exception of patient 8. Dysmorphia has been mild, the majority of patients having ear and extremity abnormalities. All patients presented with mild delay of developmental milestones. Interestingly, patient 9, the oldest patient in this study, was reported to have epilepsy and was diagnosed with large-cell non-Hodgkin lymphoma.27 Thus, although the supernumerary der(22)t(8;22) phenotype is variable between individuals, it tends to include ear and extremity abnormalities in addition to mild mental retardation.
t(8;22) Recurs during Spermatogenesis in Healthy Males
To confirm the recurrence of the t(8;22), we used PCR to detect de novo occurrences of the t(8;22) in sperm from healthy males. PCR was performed with the use of multiple DNA aliquots from 33 sperm samples (eight East Asian and 25 White) and primers specific for the der(8) and/or the der(22) junction fragments. As expected, junction fragments were absent when the genomic DNA of a healthy individual or an 11;22 balanced-translocation carrier was amplified. Conversely, junction fragments were present in the sperm DNA from most, but not all, healthy males (Figure 3A). The translocation frequency was calculated via the methods described in Kurahashi et al.29 The de novo t(8;22) frequency ranges from < 6.38 × 10−7 to 1.00 × 10−5 in the 33 individuals studied (Table 3). This translocation frequency is similar to the t(11;22) frequency derived from the short version of the chromosome 11 PATRR.36 Unlike the t(8;22) and the t(11;22), junction fragments from the t(1;22)22 were not detected in sperm, indicating that not all PATRR22-mediated translocations recur during spermatogenesis.
Figure 3.
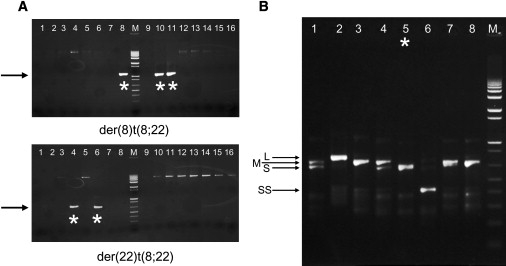
Identification of De Novo t(8;22) in Sperm and PATRR8 Genotyping
(A) Results of der(8)t(8;22) and der(22)t(8;22) PCR on sperm DNA from a healthy male (sperm no.5). PCR primers were used to generate t(8;22)-specific translocation products using 100 ng aliquots of sperm DNA as template (1–16). The arrows indicate the expected size of the junction fragments, and the stars denote positive reactions. All positive reactions were sequenced and correspond to the expected junction-fragment sequences.
(B) Results of PATRR8 PCR from genomic DNA. Arrows indicate the four differently sized products. They are denoted long (L), medium (M), short (S), and super-short (SS). The star indicates genomic DNA from sperm no. 5 (sample shown in A).
Table 3.
Frequency of De Novo t(8;22) in Sperm Samples from Healthy Males
| No. of t(8;22)-Positive PCRs | Total No. of PCRs | t(8;22) Frequency | No. of Sperm Samples | Race |
|---|---|---|---|---|
| 0 | 24 | < 6.38 x10−7 | 10 | Whitea |
| 0 | 48 | < 6.38 x10−7 | 1 | East Asianb |
| 1 | 24 | 1.29 × 10−6 | 8 | Whitea |
| 3 | 48 | 1.96 × 10−6 | 2 | East Asianb |
| 2 | 24 | 2.64 × 10−6 | 4 | Whitea |
| 4 | 48 | 2.64 × 10−6 | 3 | East Asianb |
| 4 | 24 | 5.52 × 10−6 | 2 | Whitea |
| 9 | 48 | 6.29 × 10−6 | 1 | East Asianb |
| 6 | 24 | 8.72 × 10−6 | 1 | Whitea |
| 27 | 96 | 1.0 × 10−5 | 1 | East Asianb |
Based on only der(22)t(8;22) PCR results.
Based on der(8) and der(22)t(8;22) PCR results.
PATRR8 Sequence Analysis
We used sequence from the der(22) and der(8) junction-fragment PCRs from the t(8;22) balanced carriers to reassemble the 8q24.1 breakpoint sequence that was present at the PATRR8 before the translocation event. Analysis of the secondary structures of the sequence showed that each PATRR8 contained a long, undisrupted stem-loop structure ranging in length from 129 to 145 bp and consisting of approximately 97% AT content (Figure S1) (M-Fold sequence analysis package).23 Despite the AT-rich nature of both the PATRR8 and the PATRR22, the longest stretch of homology between the two sequences is 13 nucleotides. The 8q breakpoint is localized at the center of the PATRR, supporting the hypothesis that the center of the palindrome is susceptible to breakage as a prerequisite to translocation. However, because this sequence is based only on reconstructed junction fragments and not the sequence of the pretranslocation PATRR8, it is possible that the actual t(8;22) breakpoint may not be at the center of the palindrome because of nucleotides that have been lost during the translocation event. The amount of sequence loss cannot be determined because the inverted repeats seen in these junction-fragment sequences are not present in “normal” sequence included in the latest genome build or in the Watson, Venter, and YH genomes.37–39 Similarly, the PATRR11 and PATRR22 regions are absent from the human genome reference databases, likely because of the difficulties in cloning and sequencing these repetitive regions.14 The 8q24 PATRR is flanked by two highly homologous Alu repeats that are in an inverted orientation with respect to one another. These Alu elements may also contribute to the formation of hairpin or cruciform structures.
To account for the differences in t(8;22) frequency between individuals, we genotyped the PATRR region on chromosome 8 in genomic and sperm DNA. Analysis of this region suggests that there are at least four different alleles present in the population that can be distinguished by size through gel electrophoresis (Figure 3B). These can be annotated according to their length: long, medium, short, and super-short. The size of the PATRR8 appears to correlate with the frequency of the t(8;22) in sperm. Interestingly, as of yet, the long and super-short PATRR8 alleles have not been observed to produce de novo translocations in the sperm samples.
We attempted to sequence the PATRR8 region in healthy individuals in order to further characterize this region. We were able to sequence the PATRR8 in two East Asian samples. Analysis of the secondary structure indicates that each contains an undisrupted 150 bp stem-loop structure. Despite some success in sequencing this region, it has proven to be difficult because of the extreme AT-rich nature of the PATRR. In order to reduce the complexity of this region by sequencing only one PATRR8 allele, we conducted analysis of this region in t(8;22) balanced carriers, because they have only one intact PATRR8 allele. We amplified the PATRR8 by PCR in three t(8;22) balanced carriers (cases 13, 16, and 17), plus a hybrid cell line carrying only one copy of human chromosome 8 (GM10156B). The resulting ∼850 bp products were separated by agarose gel electrophoresis, excised, and TA cloned in recombination-deficient Escherichia coli. A minimum of 96 clones were selected from each sample and sequenced. There was significant variability in the resulting sequences, with approximately 70% of clones being unique (Table S2). Most of the clones contained sequence from the ends of the PATRR8 region but lacked the nucleotides at the center. Some appeared to contain two PATRR8s of different lengths, suggesting that perhaps during replication in E.coli the center of the PATRR8 is lost. None of the clones contained an uninterrupted stem-loop structure (M-Fold sequence analysis package). Taken together, these data imply that this region is highly unstable even in cells that are recombination deficient.
The Predicted t(8;11) Can Be Detected in Sperm from Healthy Males
Because the recurrent t(8;22) and t(11;22) translocations occur at PATRRs involving identical regions on 8q, 11q, and 22q, we postulated that a t(8;11)(q24.1,q23) may occur. However, there have been no prior reports of precisely this translocation in the literature. Using an assay similar to that used in the t(8;22) and t(11;22) PCRs, we used previously described primers to amplify the putative der(11)t(8;11) junction fragments in sperm samples from healthy males.13,23 The der(11)t(8;11) junction fragment was detected in three samples (Figure 4), thus demonstrating that the t(8;11) occurs during spermatogenesis in healthy males. Sequence of the PCR products confirms their authenticity. The de novo frequency for the t(8;11) appears to be lower than that of the t(11;22) and t(8;22), with a frequency of < 2.6 × 10−6, based on a limited number of samples. Nonetheless, the identification of a palindrome-mediated rearrangement that does not involve the PATRR22 further implicates PATRR instability as a mechanism for mediating translocations in humans.
Figure 4.
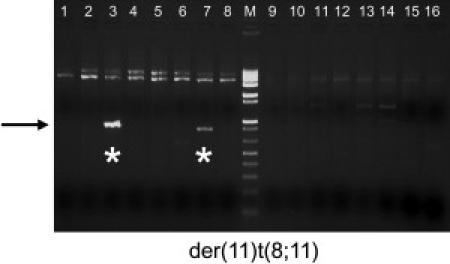
Identification of De Novo t(8;11) in Sperm
Results of der(11)t(8;11) PCR on sperm DNA from a healthy East Asian male. Nested PCR primers were used to generate t(8;11)-specific translocation products using 250 ng aliquots of sperm DNA as template (1–16). The arrow indicates the expected size of the der(11)t(8;11) junction fragment, and the stars denote two der(11)t(8;11)-positive reactions. The positive reactions were sequenced and correspond to the expected der(11)t(8;11) sequences.
Discussion
We have identified another recurrent PATRR-mediated constitutional translocation, the t(8;22). This translocation is capable of segregating 3:1 in meiosis to produce abnormal offspring and occurs during gametogenesis in healthy males. In general, individuals who are balanced carriers of this translocation appear to be healthy. Two of the six balanced carriers in this study were identified only after they had a child who carries the supernumerary der(22)t(8;22). Two carriers have been described with autoimmune disorders, including idiopathic thrombocytopenic purpura,34 myasthenia gravis, leukocytosis, and thrombocytosis.35 Another balanced carrier, a young girl, was reported with a dysgerminoma.28 The report of cancer in a balanced carrier is interesting, given that the PATRR8 is present in the first intron of RNF139 (MIM ∗603046), a ubiquitin ligase that is a potential tumor-suppressor gene.24,40 The RNF139 protein product has been linked to lipid homeostasis and protein-translation initiation.41 This gene was first described in a family with renal cell carcinoma carrying a t(3;8) with a breakpoint in PATRR8 (B.S.E., unpublished data). Thus, although the consequences of haploinsufficiency of RNF139 have not been reported, the observation of cancer in this limited group of t(8;22) balanced carriers is worthy of further investigation.
The phenotype in patients carrying the supernumerary der(22)t(8;22) is variable. Birth weight and subsequent growth were normal in all patients except case 8. Patients have not had any life-threatening structural abnormalities. Extremity anomalies were noted in the majority, in addition to mild dysmorphia that includes ear abnormalities. All patients presented with mild delay of developmental milestones and/or mild mental retardation. This phenotype is in stark contrast to the severe phenotype seen in patients with Emanuel syndrome.7 Thus, the incidence of t(8;22) is likely underascertained because of the nonspecific phenotype in the affected offspring. Interestingly, nine of the 11 patients carrying the supernumerary der(22) t(8;22) are male. Males may be overascertained because of the potential overlap of the supernumerary der(22)t(8;22) syndrome phenotype with X-linked mental retardation. As more patients with developmental delays and mild dysmorphia are assessed with aCGH and SNP arrays, perhaps additional individuals with supernumerary der(22)t(8;22) will be identified, allowing for further delineation of the phenotype.
Similar to the t(11;22), nondisjunction of the t(8;22) occurs during both male and female meiosis. The site-specific balanced translocation in the nine families we have described is paternally inherited in three and maternally derived in five (one case unknown). Two families each have two affected siblings who carry the supernumerary der(22)t(8;22). The t(8;22) appears to be prone to 3:1 nondisjunction during meiosis, most likely because the der(22)t(8;22) is small, with a short interstitial segment.42 The only viable, unbalanced t(8;22) karyotype that has been observed is 47,XX or 47,XY, +der(22)t(8;22). This is most likely because the other unbalanced karyotypes are likely to produce almost complete monosomy 8 upon 3:1 malsegregation, and monosomy 22 or trisomy 22 upon 2:2 malsegregation. The same phenomenon has been observed with the t(11;22), with a few notable exceptions. One family was reported as having multiple family members with the karyotype: 45,XX or 45,XY, −11,-22+der(11)t(11;22)(q25;q11).43 However, the chromosome 11 breakpoint of this translocation is located more distally and the 22q11 breakpoint more proximally than the typical t(11;22) (B.S.E., unpublished data). This suggests that the phenotype that emerges from this “variant” t(11;22) rearrangement segregating the der(11) is more compatible with live birth than would be the case for the typical t(11;22). In addition, three patients have been described with 47,XX or 47,XY,t(11;22)(q23;q11),+der(22) t(11;22), the result of nondisjunction in meiosis II.44
The t(8;22) is recurrent and arises in sperm from healthy males at a frequency of approximately 2 × 10−6. According to studies published by Thomas et al.,45,46 there is a propensity for structural chromosome abnormalities to occur in male germ cells. This has been recently confirmed for the t(11;22).3 The fact that a hypothetical t(8;11) rearrangement can be identified in male germ cells adds credence to this hypothesis. However, because none of the t(8;22) rearrangements we have studied to date are de novo, it is difficult to determine whether such PATRR-mediated rearrangements can occur in female as well as male meiosis.
It is striking that the 11;22, 8;22 and 8;11 translocations recur in meiosis to varying degrees whereas the t(1;22) and t(17;22)15 do not. The absence of the t(17;22) is particularly interesting because this translocation has been observed in two independent families. This may be the result of differences in the configurations of the PATRR1 and PATRR17 that make them less susceptible to double-strand breaks than the PATRR8 or PATRR11 during meiosis. Alternatively, the position of chromosome 1 or 17 in relation to chromosome 22 in the interphase meiotic nucleus may preclude frequent interactions between them, making chromosomal rearrangement unlikely.47 As data emerge regarding the position of chromosomal domains during meiosis, this discrepancy might be clarified. Nonetheless, the absence of the t(1;22) and t(17;22) during meiosis indicates that not all PATRR22-mediated translocations occur with a measurable frequency during gametogenesis in healthy males. The identification of the t(8;22) and t(8;11) during meiosis is evidence that these rearrangements recur at a frequency that can be assessed.
The t(8;22) frequency during spermatogenesis is highly variable between individuals. The frequency of the t(11;22) also varies, and this variation is related to the polymorphic sequences at the 11q and 22q breakpoints.36 The PATRR8 is flanked by two highly homologous Alu repeats that are in an inverted orientation with respect to one another. Given the high sequence similarity and inverted orientation, these Alu elements may also induce the formation of hairpin or cruciform structures and may play a role in further destabilization of the PATRR8 region. Genomic variations resulting from the polymorphic presence or absence of one or both of these Alu elements, as well as variability in the PATRR8 itself, may affect the ability of the 8q24 PATRR to undergo rearrangement. We have sequenced this region in a small set of individuals, but we have been unable to obtain the full sequence of the PATRR8 in the majority. The complete PATRR8 sequence from the 8q breakpoint is missing from genomic databases, most likely because of the difficulty of sequencing this and other AT-rich palindromic regions. It is likely that current next-generation sequencing techniques will also be compromised in the analysis of such repetitive regions as a result of short reads and shotgun approaches. Thus, further characterization of the PATRR8 and surrounding sequence will require novel methods, but will hopefully explain the differences in t(8;22) frequency in sperm and provide insights into the mechanism of palindrome-mediated translocations.
Because the PATRRs on 11q and 8q are both good substrates for double-strand break and rearrangement, we hypothesized that t(8;11)(q24.1,q23) may occur. Indeed, this translocation does occur at a low rate during spermatogenesis in healthy males. Studies including additional sperm samples are necessary to more accurately estimate the frequency of this translocation. However, the frequency appears to be very low and may again be related to the polymorphic characteristics of the PATRR8 and PATRR11 or to the proximity of chromosomes 8 and 11 during meiosis. There are four reports of translocations between chromosomes 8 and 11 in the literature. Two of the four describe translocations involving 11p.48,49 The third reported t(8;11) involves breakpoints in 8q24 and 11q13.50 The fourth reports a translocation with breakpoints in 8q24.3 and 11q23. The breakpoints of this translocation have not been fully characterized, but on the basis of cytogenetic characterization, the 8q breakpoint does not appear to be in the PATRR8.51 Thus, a t(8;11)(q24.1,q23) has not been reported in the literature. There are a number of potential reasons why this specific palindrome-mediated translocation has not been described. It is likely that balanced carriers do not have a phenotype and do not come to the attention of physicians, whereas unbalanced genotypes are likely to be incompatible with life. Furthermore, the translocation would be difficult to detect with standard cytogenetic techniques. Nevertheless, the identification of the t(8;11) in sperm provides evidence that palindrome-mediated genomic instability leading to translocation is a general mechanism that is not dependent on the PATRR22 sequence. The previous report of a t(3;8)24 whose breakpoints have now been identified in PATRR8 and a PATRR on chromosome 3 (B.S.E., unpublished data) add further support for this phenomenon.
In conclusion, we have identified another recurrent, palindrome-mediated translocation. The identification of additional patients who carry the supernumerary der(22)t(8;22) will help to further characterize the phenotype associated with nondisjunction of the t(8;22). In addition, the continued study of the t(8;22) and t(8;11) frequencies and breakpoints as well as the PATRR8 sequence in the general population is likely to provide further insights into the mechanism of palindrome-mediated genomic instability.
Supplemental Data
Supplemental Data include two tables and one figure and can be found with this article online at http://www.cell.com/AJHG/.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
M-Fold sequence analysis package, http://bioweb.pasteur.fr/seqanal/interfaces/mfold-simple.html
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
Acknowledgments
The authors wish to thank C. Coutifaris and E. Wigo for providing sperm samples from healthy males, E. Geiger and the Nucleic Acid and Protein Core at the Children's Hospital of Philadelphia for technical assistance, and the patients and their families for their willingness to participate in this study. This study was supported by grants CA39926 and HD26979 and funds from the Charles E.H. Upham chair in pediatrics (to B.S.E.).
References
- 1.Van Dyke D.L., Weiss L., Roberson J.R., Babu V.R. The frequency and mutation rate of balanced autosomal rearrangements in man estimated from prenatal genetic studies for advanced maternal age. Am. J. Hum. Genet. 1983;35:301–308. [PMC free article] [PubMed] [Google Scholar]
- 2.Warburton D. De novo balanced chromosome rearrangements and extra marker chromosomes identified at prenatal diagnosis: clinical significance and distribution of breakpoints. Am. J. Hum. Genet. 1991;49:995–1013. [PMC free article] [PubMed] [Google Scholar]
- 3.Ohye T., Inagaki H., Kogo H., Tsutsumi M., Kato T., Tong M., Macville M.V., Medne L., Zackai E.H., Emanuel B.S., Kurahashi H. Paternal origin of the de novo constitutional t(11;22)(q23;q11) Eur. J. Hum. Genet. 2010;18:783–787. doi: 10.1038/ejhg.2010.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Petit P., Vermeersch J.R., Fryns J.P. Identification of a de novo 46, XY,4p+ with incomplete Wolf-Hirschhorn syndrome as 46,XY,der(4)t(4;8)(p16.3;p23.1) Genet. Couns. 1998;9:157–158. [PubMed] [Google Scholar]
- 5.Tönnies H., Stumm M., Neumann L., Volleth M., Grumpelt U., Müsebeck J., Annuss G., Neitzel H. Two further cases of WHS with unbalanced de novo translocation t(4;8) characterised by CGH and FISH. J. Med. Genet. 2001;38:E21. doi: 10.1136/jmg.38.6.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kurahashi H., Inagaki H., Ohye T., Kogo H., Kato T., Emanuel B.S. Palindrome-mediated chromosomal translocations in humans. DNA Repair (Amst.) 2006;5:1136–1145. doi: 10.1016/j.dnarep.2006.05.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carter M.T., St Pierre S.A., Zackai E.H., Emanuel B.S., Boycott K.M. Phenotypic delineation of Emanuel syndrome (supernumerary derivative 22 syndrome): Clinical features of 63 individuals. Am. J. Med. Genet. A. 2009;149A:1712–1721. doi: 10.1002/ajmg.a.32957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tranebjaerg L., Petersen A., Hove K., Rehder H., Mikkelsen M. Clinical and cytogenetic studies in a large (4;8) translocation family with pre- and postnatal Wolf syndrome. Ann. Genet. 1984;27:224–229. [PubMed] [Google Scholar]
- 9.Giglio S., Calvari V., Gregato G., Gimelli G., Camanini S., Giorda R., Ragusa A., Guerneri S., Selicorni A., Stumm M. Heterozygous submicroscopic inversions involving olfactory receptor-gene clusters mediate the recurrent t(4;8)(p16;p23) translocation. Am. J. Hum. Genet. 2002;71:276–285. doi: 10.1086/341610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Maas N.M., Van Vooren S., Hannes F., Van Buggenhout G., Mysliwiec M., Moreau Y., Fagan K., Midro A., Engiz O., Balci S. The t(4;8) is mediated by homologous recombination between olfactory receptor gene clusters, but other 4p16 translocations occur at random. Genet. Couns. 2007;18:357–365. [PubMed] [Google Scholar]
- 11.Edelmann L., Spiteri E., Koren K., Pulijaal V., Bialer M.G., Shanske A., Goldberg R., Morrow B.E. AT-rich palindromes mediate the constitutional t(11;22) translocation. Am. J. Hum. Genet. 2001;68:1–13. doi: 10.1086/316952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tapia-Páez I., Kost-Alimova M., Hu P., Roe B.A., Blennow E., Fedorova L., Imreh S., Dumanski J.P. The position of t(11;22)(q23;q11) constitutional translocation breakpoint is conserved among its carriers. Hum. Genet. 2001;109:167–177. doi: 10.1007/s004390100560. [DOI] [PubMed] [Google Scholar]
- 13.Kurahashi H., Shaikh T.H., Zackai E.H., Celle L., Driscoll D.A., Budarf M.L., Emanuel B.S. Tightly clustered 11q23 and 22q11 breakpoints permit PCR-based detection of the recurrent constitutional t(11;22) Am. J. Hum. Genet. 2000;67:763–768. doi: 10.1086/303054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kurahashi H., Shaikh T.H., Hu P., Roe B.A., Emanuel B.S., Budarf M.L. Regions of genomic instability on 22q11 and 11q23 as the etiology for the recurrent constitutional t(11;22) Hum. Mol. Genet. 2000;9:1665–1670. doi: 10.1093/hmg/9.11.1665. [DOI] [PubMed] [Google Scholar]
- 15.Kurahashi H., Shaikh T., Takata M., Toda T., Emanuel B.S. The constitutional t(17;22): another translocation mediated by palindromic AT-rich repeats. Am. J. Hum. Genet. 2003;72:733–738. doi: 10.1086/368062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lisnić B., Svetec I.K., Stafa A., Zgaga Z. Size-dependent palindrome-induced intrachromosomal recombination in yeast. DNA Repair (Amst.) 2009;8:383–389. doi: 10.1016/j.dnarep.2008.11.017. [DOI] [PubMed] [Google Scholar]
- 17.Cunningham L.A., Coté A.G., Cam-Ozdemir C., Lewis S.M. Rapid, stabilizing palindrome rearrangements in somatic cells by the center-break mechanism. Mol. Cell. Biol. 2003;23:8740–8750. doi: 10.1128/MCB.23.23.8740-8750.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lewis S.M., Coté A.G. Palindromes and genomic stress fractures: bracing and repairing the damage. DNA Repair (Amst.) 2006;5:1146–1160. doi: 10.1016/j.dnarep.2006.05.014. [DOI] [PubMed] [Google Scholar]
- 19.Spiteri E., Babcock M., Kashork C.D., Wakui K., Gogineni S., Lewis D.A., Williams K.M., Minoshima S., Sasaki T., Shimizu N. Frequent translocations occur between low copy repeats on chromosome 22q11.2 (LCR22s) and telomeric bands of partner chromosomes. Hum. Mol. Genet. 2003;12:1823–1837. doi: 10.1093/hmg/ddg203. [DOI] [PubMed] [Google Scholar]
- 20.Kehrer-Sawatzki H., Häussler J., Krone W., Bode H., Jenne D.E., Mehnert K.U., Tümmers U., Assum G. The second case of a t(17;22) in a family with neurofibromatosis type 1: sequence analysis of the breakpoint regions. Hum. Genet. 1997;99:237–247. doi: 10.1007/s004390050346. [DOI] [PubMed] [Google Scholar]
- 21.Nimmakayalu M.A., Gotter A.L., Shaikh T.H., Emanuel B.S. A novel sequence-based approach to localize translocation breakpoints identifies the molecular basis of a t(4;22) Hum. Mol. Genet. 2003;12:2817–2825. doi: 10.1093/hmg/ddg301. [DOI] [PubMed] [Google Scholar]
- 22.Gotter A.L., Shaikh T.H., Budarf M.L., Rhodes C.H., Emanuel B.S. A palindrome-mediated mechanism distinguishes translocations involving LCR-B of chromosome 22q11.2. Hum. Mol. Genet. 2004;13:103–115. doi: 10.1093/hmg/ddh004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gotter A.L., Nimmakayalu M.A., Jalali G.R., Hacker A.M., Vorstman J., Conforto Duffy D., Medne L., Emanuel B.S. A palindrome-driven complex rearrangement of 22q11.2 and 8q24.1 elucidated using novel technologies. Genome Res. 2007;17:470–481. doi: 10.1101/gr.6130907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gemmill R.M., West J.D., Boldog F., Tanaka N., Robinson L.J., Smith D.I., Li F., Drabkin H.A. The hereditary renal cell carcinoma 3;8 translocation fuses FHIT to a patched-related gene, TRC8. Proc. Natl. Acad. Sci. USA. 1998;95:9572–9577. doi: 10.1073/pnas.95.16.9572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Poland K.S., Azim M., Folsom M., Goldfarb R., Naeem R., Korch C., Drabkin H.A., Gemmill R.M., Plon S.E. A constitutional balanced t(3;8)(p14;q24.1) translocation results in disruption of the TRC8 gene and predisposition to clear cell renal cell carcinoma. Genes Chromosomes Cancer. 2007;46:805–812. doi: 10.1002/gcc.20466. [DOI] [PubMed] [Google Scholar]
- 26.Mark H.F., Wyandt H., Huang X.L., Milunsky J.M. Delineation of a supernumerary marker chromosome utilizing a multimodal approach of G-banding, fluorescent in situ hybridization, confirmatory P1 artificial chromosome fluorescent in situ hybridization, and high-resolution comparative genomic hybridization. Clin. Genet. 2005;68:146–151. doi: 10.1111/j.1399-0004.2005.00466.x. [DOI] [PubMed] [Google Scholar]
- 27.Helbig I., Wirtenberger M., Jauch A., Hager H.D., Tariverdian G., Hemminki K., Burwinkel B., Klaes R. Trisomy 8q and partial trisomy 22 in a 43-year-old man with moderate intellectual disability, epilepsy and large cell non-Hodgkin lymphoma. Am. J. Med. Genet. A. 2006;140:1658–1662. doi: 10.1002/ajmg.a.31350. [DOI] [PubMed] [Google Scholar]
- 28.Gimelli S., Beri S., Drabkin H.A., Gambini C., Gregorio A., Fiorio P., Zuffardi O., Gemmill R.M., Giorda R., Gimelli G. The tumor suppressor gene TRC8/RNF139 is disrupted by a constitutional balanced translocation t(8;22)(q24.13;q11.21) in a young girl with dysgerminoma. Mol. Cancer. 2009;8:52. doi: 10.1186/1476-4598-8-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kurahashi H., Emanuel B.S. Unexpectedly high rate of de novo constitutional t(11;22) translocations in sperm from normal males. Nat. Genet. 2001;29:139–140. doi: 10.1038/ng1001-139. [DOI] [PubMed] [Google Scholar]
- 30.Lejeune J., Rethore M.O., Dutrillaux B., Martin G. Translocation 8-22 with no length change and partial trisomy for 8q. Detection by heat denaturation. Exp. Cell Res. 1972;74:293–295. doi: 10.1016/0014-4827(72)90510-1. [DOI] [PubMed] [Google Scholar]
- 31.Sánchez O., Yunis J.J. Partial trisomy 8 (8q24) and the trisomy-8 syndrome. Humangenetik. 1974;23:297–303. doi: 10.1007/BF00272513. [DOI] [PubMed] [Google Scholar]
- 32.Rethoré M.O., Aurias A., Couturier J., Dutrillaux B., Prieur M., Lejeune J. Chromosome 8 : complete trisomy and segmental trisomies. Ann. Genet. 1977;20:5–11. [PubMed] [Google Scholar]
- 33.Gödde-Salz E., Oesinghaus S., Grote W. Meiotic segregation in familial reciprocal translocation t(8q;22q) Am. J. Med. Genet. 1982;11:241–247. doi: 10.1002/ajmg.1320110209. [DOI] [PubMed] [Google Scholar]
- 34.Gupta, S., Joshi, J., Perrone, R., Gallo, J., Allen, S.L., and Koduru, P. (2004) Constitutional t(8;22)(q24.1;q11.2): Prevalance and clinical significance. Presented at the annual meeting of The American Society of Human Genetics, 2004, Toronto, Canada. Abstract Program #988.
- 35.Keung Y.K., Knovich M.A., Molnar I., Pettenati M. Constitutional t(8;22) in a patient with myasthenia gravis, leukocytosis, and thrombocytosis. Cancer Genet. Cytogenet. 2004;148:87–88. doi: 10.1016/s0165-4608(03)00210-3. [DOI] [PubMed] [Google Scholar]
- 36.Kato T., Inagaki H., Yamada K., Kogo H., Ohye T., Kowa H., Nagaoka K., Taniguchi M., Emanuel B.S., Kurahashi H. Genetic variation affects de novo translocation frequency. Science. 2006;311:971. doi: 10.1126/science.1121452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wheeler D.A., Srinivasan M., Egholm M., Shen Y., Chen L., McGuire A., He W., Chen Y.J., Makhijani V., Roth G.T. The complete genome of an individual by massively parallel DNA sequencing. Nature. 2008;452:872–876. doi: 10.1038/nature06884. [DOI] [PubMed] [Google Scholar]
- 38.Levy S., Sutton G., Ng P.C., Feuk L., Halpern A.L., Walenz B.P., Axelrod N., Huang J., Kirkness E.F., Denisov G. The diploid genome sequence of an individual human. PLoS Biol. 2007;5:e254. doi: 10.1371/journal.pbio.0050254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang J., Wang W., Li R., Li Y., Tian G., Goodman L., Fan W., Zhang J., Li J., Zhang J. The diploid genome sequence of an Asian individual. Nature. 2008;456:60–65. doi: 10.1038/nature07484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gemmill R.M., Bemis L.T., Lee J.P., Sozen M.A., Baron A., Zeng C., Erickson P.F., Hooper J.E., Drabkin H.A. The TRC8 hereditary kidney cancer gene suppresses growth and functions with VHL in a common pathway. Oncogene. 2002;21:3507–3516. doi: 10.1038/sj.onc.1205437. [DOI] [PubMed] [Google Scholar]
- 41.Lee J.P., Brauweiler A., Rudolph M., Hooper J.E., Drabkin H.A., Gemmill R.M. The TRC8 ubiquitin ligase is sterol regulated and interacts with lipid and protein biosynthetic pathways. Mol. Cancer Res. 2010;8:93–106. doi: 10.1158/1541-7786.MCR-08-0491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lindenbaum R.H., Bobrow M. Reciprocal translocations in man. 3:1 Meiotic disjunction resulting in 47- or 45-chromosome offspring. J. Med. Genet. 1975;12:29–43. doi: 10.1136/jmg.12.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fu W., Borgaonkar D.S., Ladewig P.P., Weaver J., Pomerance H.H. Structural aberrations of the long arm of chromosome no. 22. Report fo a family with translocation t(11;22) (q25;q11) Clin. Genet. 1976;10:329–336. doi: 10.1111/j.1399-0004.1976.tb00057.x. [DOI] [PubMed] [Google Scholar]
- 44.Petković I., de Capoa A., Giancotti P., Barisić I. Unusual segregation of t(11;22) resulting from crossing-over followed by 3:1 disjunction at meiosis I. Clin. Genet. 1996;50:515–519. doi: 10.1111/j.1399-0004.1996.tb02725.x. [DOI] [PubMed] [Google Scholar]
- 45.Thomas N.S., Morris J.K., Baptista J., Ng B.L., Crolla J.A., Jacobs P.A. De novo apparently balanced translocations in man are predominantly paternal in origin and associated with a significant increase in paternal age. J. Med. Genet. 2010;47:112–115. doi: 10.1136/jmg.2009.069716. [DOI] [PubMed] [Google Scholar]
- 46.Thomas N.S., Durkie M., Van Zyl B., Sanford R., Potts G., Youings S., Dennis N., Jacobs P. Parental and chromosomal origin of unbalanced de novo structural chromosome abnormalities in man. Hum. Genet. 2006;119:444–450. doi: 10.1007/s00439-006-0157-6. [DOI] [PubMed] [Google Scholar]
- 47.Ashley T., Gaeth A.P., Inagaki H., Seftel A., Cohen M.M., Anderson L.K., Kurahashi H., Emanuel B.S. Meiotic recombination and spatial proximity in the etiology of the recurrent t(11;22) Am. J. Hum. Genet. 2006;79:524–538. doi: 10.1086/507652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fert-Ferrer S., Guichet A., Tantau J., Delezoide A.L., Ozilou C., Romana S.P., Gosset P., Viot G., Loison S., Moraine C. Subtle familial unbalanced translocation t(8;11)(p23.2;p15.5) in two fetuses with Beckwith-Wiedemann features. Prenat. Diagn. 2000;20:511–515. doi: 10.1002/1097-0223(200006)20:6<511::aid-pd849>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 49.Sato H., Takaya K., Nihira S., Fujita H. Familial mental retardation associated with balanced chromosome rearrangement rcp t(8;11)(q24.3;p15.1) J. Med. Genet. 1989;26:642–644. doi: 10.1136/jmg.26.10.642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Soh K., Yajima A., Ozawa N., Abe Y., Takabayashi T., Sato S., Sou S., Suzuki M. Chromosome analysis in couples with recurrent abortions. Tohoku J. Exp. Med. 1984;144:151–163. doi: 10.1620/tjem.144.151. [DOI] [PubMed] [Google Scholar]
- 51.Ounap K., Bartsch O., Uibo O., Laidre P. Girl with combined cellular immunodeficiency, pancytopenia, malformations, deletion 11q23.3 —> qter, and trisomy 8q24.3 —> qter. Am. J. Med. Genet. 2002;108:322–326. doi: 10.1002/ajmg.10284. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.