

Abstract
Ectodermal dysplasias form a large disease family with more than 200 members. The combination of hair and tooth abnormalities, alopecia, and cutaneous syndactyly is characteristic of ectodermal dysplasia-syndactyly syndrome (EDSS). We used a homozygosity mapping approach to map the EDSS locus to 1q23 in a consanguineous Algerian family. By candidate gene analysis, we identified a homozygous mutation in the PVRL4 gene that not only evoked an amino acid change but also led to exon skipping. In an Italian family with two siblings affected by EDSS, we further detected a missense and a frameshift mutation. PVRL4 encodes for nectin-4, a cell adhesion molecule mainly implicated in the formation of cadherin-based adherens junctions. We demonstrated high nectin-4 expression in hair follicle structures, as well as in the separating digits of murine embryos, the tissues mainly affected by the EDSS phenotype. In patient keratinocytes, mutated nectin-4 lost its capability to bind nectin-1. Additionally, in discrete structures of the hair follicle, we found alterations of the membrane localization of nectin-afadin and cadherin-catenin complexes, which are essential for adherens junction formation, and we found reorganization of actin cytoskeleton. Together with cleft lip and/or palate ectodermal dysplasia (CLPED1, or Zlotogora-Ogur syndrome) due to an impaired function of nectin-1, EDSS is the second known “nectinopathy” caused by mutations in a nectin adhesion molecule.
Main Text
Ectodermal structures, e.g., epidermis, hair, teeth, and sebaceous glands, develop following complex interactions between two adjacent tissue layers, the epithelium and the mesenchyme. A number of signaling molecules, such as fibroblast growth factors, Wnt, bone morphogenetic proteins, and hedgehog, contribute to the fine regulation of epithelial-mesenchymal crosstalk.1 Also, cell-cell adhesion is crucial during epithelial development and morphogenesis.2 Adherens junctions (AJ), tight junctions (TJ), and desmosomes form intercellular junctional complexes that are structurally connected to the cytoskeleton and allow single cells of an epithelial sheet to function as a coordinated tissue.3 Identifying cell adhesion molecules (CAMs) implicated in defective organogenesis may therefore shed light on this complex developmental process.
Ectodermal dysplasias (EDs) are congenital disorders characterized by alterations in two or more ectodermal structures, at least one of these affecting hair, teeth, nails, or sweat glands.4 Clinically, about 200 distinct EDs and ED syndromes (combined with malformations) have been described in the literature, and their number grows constantly.5 A recent debate6 outlined the need for a functional classification system for EDs that integrates both the clinical and molecular knowledge.7,8 However, no more than one third of EDs described so far has been associated to a causative gene, making its pathogenesis largely unknown.9 Hence, identification of molecules underlying EDs is mandatory, not only to improve diagnosis, prognosis, and management but also to establish a more appropriate classification system.
In this study, we investigated two unrelated families displaying hair and teeth abnormalities associated with hands and/or feet cutaneous syndactyly (ectodermal dysplasia-syndactyly syndrome, EDSS) and identified the disease-causing gene.
Family A was originally described by Boudghene-Stambouli and Merad-Boudia10 and consisted of four affected siblings born to first cousin, healthy Algerian parents (Figure 1). In the second family (family B), two siblings, born to nonconsanguineous healthy parents of Italian origin, showed clinical features nearly identical to those observed in family A (see Table S1 available online). All affected individuals manifested partial cutaneous syndactyly variably involving fingers 2-3 and 3-4 and toes 2-3 and 4-5 (Figure 1). In the young patients, hair over the entire scalp was sparse and coarse, with a tendency to break since very early age.10 Eyebrows, eyelashes, and body hair showed identical abnormalities. Progressive hair loss manifested in the second decade of life with patchy areas of alopecia over the scalp and progressed toward complete alopecia, as observed in the oldest subject of family A, aged 40 years. Hair morphological abnormalities included twists at irregular intervals (pili torti) and swellings along the shafts, particularly associated with areas of breakage (Figure 1). Dental findings consisted of abnormally widely spaced teeth, with peg-shaped and conical crowns (Figure 1). All patients had normal sweating.
Figure 1.
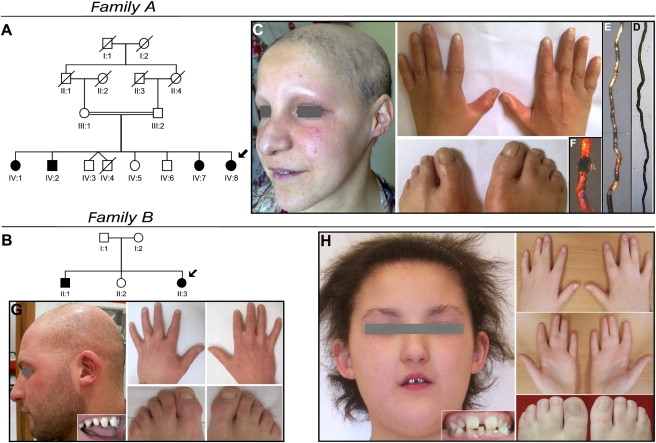
Pedigrees and Clinical Manifestations of Two Families with EDSS
(A and B) Pedigree structure of families A and B. The probands are indicated by arrows, clear symbols represent unaffected individuals, and filled symbols represent affected individuals.
(C) Patient IV:8, aged 25 years, shows diffuse alopecia of the scalp and absent eyebrows and eyelashes. Partial skin syndactyly of fingers 2-3 and toes 2-3 and 4-5 is present bilaterally.
(D–F) Hair shaft abnormalities in this patient observed at light microscopy (D) consist of peculiar repeated twists of the hair (pili torti), whereas polarized microscopy (E) outlines high-frequency bands with consequent alteration of the normal banding pattern of the hair shaft (magnification 40×). Trichoschisis with transverse fracture is evident with polarized microscopy magnification 100× (F).
(G and H) Family B, affected siblings aged 27 and 9 years. The older brother (G) shows alopecia, widely spaced teeth with conical crowns, and partial cutaneous syndactyly of toes 2-3. Fingers 2-3 and 3-4 syndactyly has been surgically corrected. The proband (H) shows short and coarse uncombable hair, sparse eyebrows and eyelashes, widely spaced irregular teeth, and syndactyly of fingers 2-3-4 and of toes 2-3 and 4-5.
Biological specimens from affected and nonaffected members of families A and B were collected after obtaining informed consent from each individual. The study was approved by the local ethics committees and was conducted in accordance with international standards on human research. To unravel the genetic defect of EDSS, we performed a whole-genome homozygosity mapping in four affected and three unaffected siblings of family A with the Affymetrix Genome-Wide Human 6.0 SNP Array (Affymetrix), in accordance with the manufacturer's instructions. Homozygosity was assessed by visual inspection with the Genotyping Console 3.0.1 software and allowed the definition of a 4.18 Mb continuous region of homozygosity shared by the patients on chromosome 1q23.3 between SNPs rs11265404 at 158,686,947 bp and rs16833478 at 162,874,726 bp (Figure S1), according to National Center for Biotechnology Information (NCBI) build 36.3. Six highly informative microsatellite markers spanning this interval were then genotyped in all members of family A and analyzed with GeneMapper V4.0 software (Applied Biosystems). Manually constructed haplotypes confirmed the cosegregation of the disease with the 1q23.3 locus (Figure S1). Two-point linkage analysis, performed with the MLINK program, gave a maximum LOD score of 3.08 at D1S484 (Table S2).
Among 60 annotated RefSeq genes, the PVRL4 gene was of immediate note because the paralog PVRL1 was known to be associated with cleft lip and/or palate ectodermal dysplasia or Zlotogora-Ogur syndrome (CLPED1, MIM 225069).11 Notably, cutaneous syndactyly is also variably observed in patients with CLPED1.12 The coding exons and intron-exon junctions of the PVRL4 gene were thus amplified (primer pairs are available in Table S3), sequenced with the BigDye Terminator v3.1 Sequencing Kit (Applied Biosystems), and run on an ABI 3130XL DNA Analyzer. A single homozygous G-to-A substitution in exon 4 (c.851G>A leading to p.Arg284Gln) was identified in patient IV:8 from family A. This mutation was transmitted from heterozygous parents to all four homozygous patients, and carrier status was demonstrated in three healthy siblings according to haplotype reconstruction (Figure S1 and Figure S2). The mutation was neither found among 250 DNA samples from various geographical regions (including 70 Algerian samples) nor listed as a SNP in public databases.
To confirm the involvement of PVRL4 in EDSS, we tested family B for mutations and identified two distinct alterations in the two affected family members. Both patients were compound heterozygotes for a maternally inherited nucleotide substitution in exon 3 (c.554C>T) leading to p.Thr185Met and a paternally transmitted c.906delT deletion in exon 5, which resulted in a frameshift with premature termination (p.Pro304HisfsX2). Both mutations were absent in a panel of 180 ethnically matched control DNA samples. The mutated Thr185 residue is located in the second Ig-like domain of nectin-4 and is highly conserved among species (Figure 2). Modeling of this domain structure indicated that Thr185 contributes to the formation of a loop between two beta strands and makes contact with Val187 and Leu221 (Figure 2; Figure S3). The replacement of the threonine with the larger methionine residue is predicted to modify these contacts, altering the structure and possibly affecting the interactions with other Ig-like-containing proteins.
Figure 2.
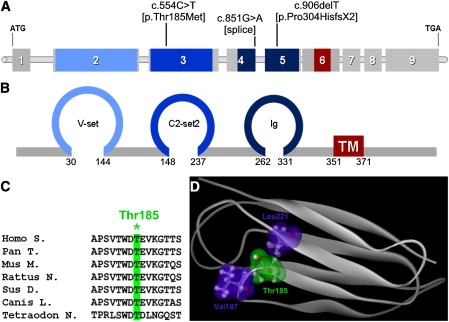
Mutations Identified in the PVRL4 Gene
(A) Schematic view of the human PVRL4 gene and localization of identified mutations.
(B) Schematic representation of nectin-4 exhibiting three extracellular Ig-like domains (blue) and a transmembrane domain (red). The numbers denote the amino acids located at the boundaries of each domain.
(C) Amino acid sequence alignment showing conservation among species of the mutated Thr185 residue (∗).
(D) Modeled structure of nectin-4 in the amino acid range 147–244 encompassing the second Ig-like domain. Site of mutation Thr185 (green cloud) and its interacting amino acids Val187 and Leu221 (blue clouds) are shown.
In order to assess the pathogenic effect of the c.851G>A (p.Arg284Gln) mutation found in family A, we reasoned that, because the nucleotide change affected the last base of exon 4, a role of this mutation on splicing could be envisaged. In fact, several examples of exonic point mutations affecting splicing have been reported in the literature.13 In addition, the Arg284Gln substitution was rated as benign by the PolyPhen and SIFT softwares.14 Conversely, the NetGene2,15 SplicePort,16 and HSF17 prediction tools gave a significantly decreased score for the exon 4 donor splice site, arguing for a possible role in mRNA splicing. To test this hypothesis, we analyzed PVRL4 mRNA products in the skin of patient IV:8 from family A. Amplification of PVRL4 cDNA with primer pairs flanking exon 4 (primers and conditions are listed in Table S3) resulted in abnormal splice products in the patient compared to an unrelated control sample (Figure S4). Direct sequencing of the most abundant of these transcripts confirmed the skipping of exon 4 at the RNA level (Figure S4) with formation of a premature stop codon in this transcript (p.Phe244CysfsX22). The protein resulting from this transcript lacks both its transmembrane and C-terminal afadin-binding consensus motif, making very likely the loss of nectin-4 function.
To understand the potential function of nectin-4 in human tissues, we analyzed mRNA expression in a commercially available human RNA panel (Clontech) and found that it was restricted to placenta, trachea, prostate, lung, and stomach (Figure S5). Because skin and hair were the most severely affected targets of EDSS, we also tested nectin-4 expression by real-time PCR in skin biopsies, plucked hair bulbs, cultured fibroblasts, and cultured epidermal keratinocytes at different stages of differentiation. Total RNA was isolated via a standard TRIzol reagent extraction method.18 Nectin-4 was significantly expressed in skin, hair follicles, and cultured keratinocytes, but not in fibroblasts (Figure 3). Steady-state levels of nectin-4 mRNA from cultured epidermal keratinocytes of patient II:1 (family B) revealed nearly 50% reduced expression (Figure 3), indicating that nonsense mRNA decay is secondary to the frameshift mutation.
Figure 3.
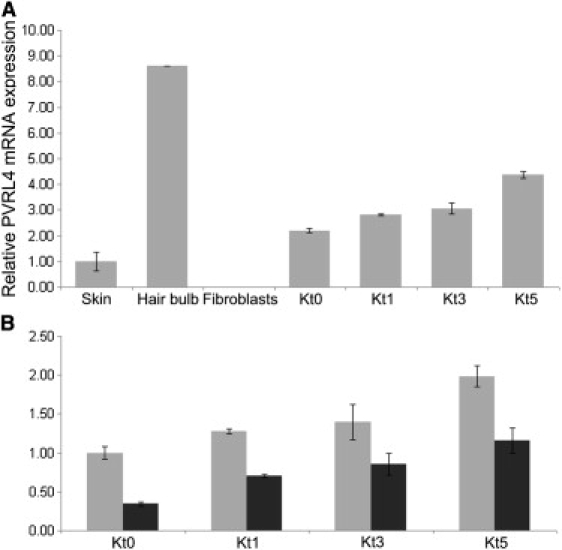
Nectin-4 mRNA Expression in Specimens from Unaffected Individual and EDSS Patient
(A) Relative PVRL4 mRNA expression, as determined by quantitative PCR in skin biopsy, plucked hair bulbs, primary fibroblasts, and proliferating (Kt0) and differentiated (Kt1, Kt3, Kt5) keratinocytes obtained from healthy donors. Keratinocyte differentiation was induced by the addition of 1.2 mM calcium to the culture media for 1 (t1), 3 (t3), or 5 (t5) days. PVRL4 is expressed in skin, keratinocytes, and mainly in hair bulbs, but not in fibroblasts. Expression level increases during keratinocyte differentiation.
(B) Analysis of PVRL4 expression in primary keratinocytes at different stages (Kt0–Kt5) from family B patient II:1 (black column) and from a control individual (gray column). Note the strongly reduced expression in the patient's samples. Experiments were run in duplicate. Error bars are ± standard deviation. The relative expression values were determined via the ΔΔCt method.
Immunohistochemical analysis of normal human skin sections detected nectin-4 at cell-cell junctions of human keratinocytes. In particular, we observed a positive staining in all the suprabasal nucleated layers of epidermis (from the spinous to the granular layer) and in all the nonkeratinized structures of hair with a stronger signal within inner root sheath layers and hair shaft cortex. With increasing keratinization, the expression became lower (Figure 4). Notably, nectin-4 staining was markedly reduced in the interfollicular epidermis from patient II:1 (family B), and only a residual signal was visualized in the hair follicles (Figure 4).
Figure 4.
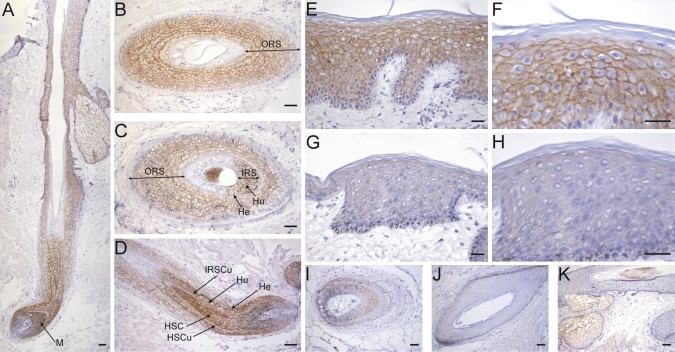
Localization of Nectin-4 in Human Skin and Hair
(A–F) Frozen sections of human skin containing terminal hair obtained from healthy donors were stained with the mouse anti-nectin-4 monoclonal antibody (N4.61), as previously described.42 In the hair follicle (A–D), a pericellular signal is first detected at the level of the hair bulb in suprabasal matrix epithelial cells (M), giving rise to the different specialized hair follicle compartments. The hair shaft cortex and cuticle (HSC and HSCu) (D), as well as all the inner root sheath (IRS) layers (i.e., Henle's [He], Huxley's [Hu], and cuticle [IRSCu]) (C and D) appear strongly positive until the onset of keratinization, which occurs at first in the Henle's layer, then in the IRS and hair shaft cuticle, and finally in the Huxley's layer and hair shaft cortex (A, C, and D). A less intense staining is present in all suprabasal layers of the outer root sheath (ORS) (A–D). The epidermis is also stained, and the signal intensity shows an increasing gradient from the first suprabasal to the granular layer; the signal disappears at the transition zone between the granular layer and stratum corneum (E and F).
(G–J) Nectin-4 staining is strongly reduced in the epidermis of family B patient II:1 (G and H) and in his hair follicles (I and J).
(K) Nonspecific labeling is observed to be limited to the sebaceous glands with the secondary biotin-conjugated antibody alone.
Scale bars represent 100 μm (A, D, and I–K), 25 μm (B and C), and 50 μm (E–H).
Because nectins promote AJ formation through the nectin-afadin and cadherin-catenin complexes,19 we examined the localization of other AJ components and actin cytoskeleton in the patient's skin biopsy. Compared to normal human skin, cell membrane staining of α-catenin, β-catenin, E-cadherin, and afadin appeared highly disorganized. Also, F-actin showed an altered distribution, with loss of pericellular localization (Figure 5; Figure S6). Notably, such alterations were restricted to discrete areas of the hair follicle composing the outer root sheath, whereas no staining differences were appreciable in the interfollicular epidermis and other hair compartments analyzed (Figure S6). Based on previous evidences demonstrating that nectin-4 forms hetero trans-dimers with nectin-1 (encoded by the PVRL1 gene),20 we investigated whether this binding activity was altered in cells from family B patient II:1. Fluorescence-activated cell sorting (FACS) analysis showed, in addition to a highly decreased cell surface expression of nectin-4, a nearly abolished nectin-1 to nectin-4 binding in the patient's keratinocytes (Figure 5).
Figure 5.
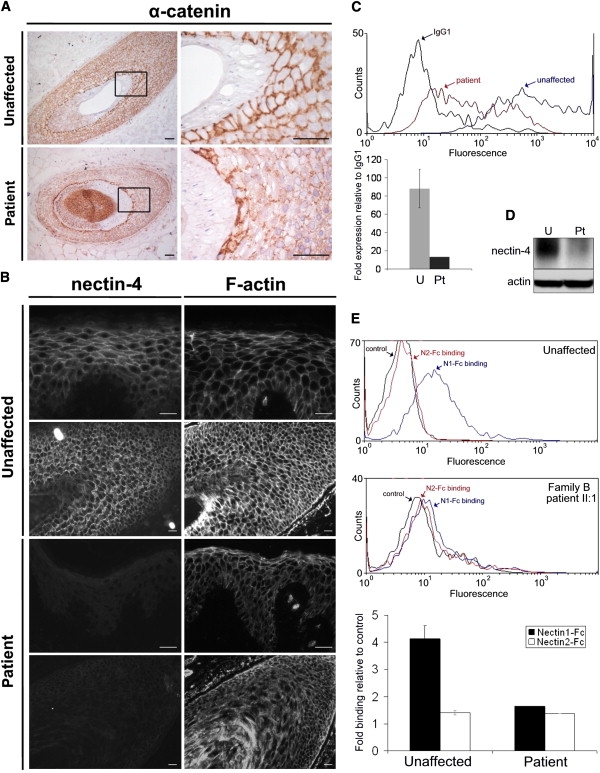
Tissue and Cellular Consequences of Nectin-4 Alteration
(A) Frozen sections of human skin with terminal hair obtained from an unaffected individual and family B patient II:1 were immunostained with the AJ marker α-catenin (clone 7A4, Invitrogen). Higher-magnification images of the boxed regions are shown. In the control sample, α-catenin preferentially stains along cell-cell adhesion sites in all nonkeratinized structures of the hair. In the patient, although a similar staining is observed along the cell-cell junctions at the IRS level, its distribution is altered in the ORS. Scale bars represent 50 μm.
(B) Nectin-4 and F-actin were stained in serial sections of human skin from an unaffected individual and family B patient II:1 with anti-nectin-4 mAb (N4.61) or rhodamine-phalloidin (Invitrogen), respectively. Although strong reduction of nectin-4 expression does not seem to affect the cellular architecture at the interfollicular epidermis level (top), the actin cytoskeleton is strongly disorganized in the hair follicle (bottom). Scale bars represent 25 μm.
(C) FACS analysis on dissociated keratinocytes showing a decrease of cell surface expression of nectin-4 in family B patient II:1 (red) compared to a control sample (blue). The relative histogram documents the strong reduction of expression (relative to IgG1) between mean values of three unaffected control samples (U, light gray) and the patient (Pt, dark gray). Cells were incubated with a mouse IgG1 anti-nectin-4 IgV domain monoclonal antibody (N4.61)42 or an irrelevant mouse IgG1 monoclonal antibody (5 μg/ml). After washes, cells were incubated with a phycoerythrin-labeled goat anti-mouse secondary antibody (Immunotech) (1/100). Incubation conditions were 60 min at +4°C.
(D) Immunoblot analysis of nectin-4, performed with monoclonal antibody (BAF2659; R&D Systems) on protein extracts of differentiated primary keratinocytes from an unaffected individual and family B patient II:1, confirming strong reduction of protein expression.
(E) Trans-heterophilic interaction between nectin-1 and nectin-4 was determined by FACS analysis by using binding of nectin-1-Fc soluble recombinant protein, as previously described.27 A nectins trans-interactive network exists with nectin-1 binding nectin-3 and nectin-4 and with nectin-2 binding nectin-3.20 We found the absence of nectin-3 expression in control keratinocytes (data not shown) and thus used nectin-2-Fc as a negative binding control. The histogram represents nectin-1-Fc (black) and nectin-2-Fc (white) binding expression (relative to control) in keratinocytes from family B patient II:1 compared to three unaffected individuals. In patient's keratinocytes, nectin-1 to nectin-4 binding was dramatically reduced.
Cutaneous syndactyly was a consistent finding in our patients. Therefore, we assessed by real-time PCR nectin-4 mRNA expression during different stages of limb development in the mouse embryo. Nectin-4 was not significantly expressed until stage E14.5; it reached maximum expression at E16.5 and decreased at later stages (Figure 6). Whole-mount in situ hybridization was performed as previously described,21 with a probe specific for murine Pvrl4 generated by PCR (Table S3). Strikingly, this confirmed an interdigital nectin-4 expression at embryonic stage E15.5 (Figure 6). In addition, a strong expression at the roots of vibrissae was observed starting from E13.5 (Figure S7).
Figure 6.

Nectin-4 Expression during Mouse Limb Development
(A) Relative Pvrl4 mRNA expression in whole-limb mRNA at different embryonic stages determined by quantitative PCR. Three to five limbs were pooled for mRNA isolation. The relative expression values were determined via the ΔΔCt method. Experiments were run in duplicate. Error bars are ± standard deviation.
(B) Whole-mount in situ hybridization for Pvrl4 mRNA expression in E15.5 mouse limb denotes strong expression in areas where digit separation is ongoing.
Nectins (from the Latin word necto, meaning “to connect”)22 are calcium-independent immunoglobulin (Ig)-like CAMs functioning in cell-cell junctions in cooperation with or independently of cadherins (reviewed in 23). Four distinct nectins (nectin-1 to nectin-4) are known and contribute to cell adhesion through both homophilic and heterophilic cis and trans interactions mediated by their extracellular Ig-like domains.24 Nectins are connected to the actin cytoskeleton through afadin, an F-actin-binding protein,22 and, in a complex interplay with other CAMs and signal transduction molecules, they regulate several cellular activities, ranging from movement to polarization, differentiation, and entry of viruses.25
So far, only nectin-1 has been linked to human hereditary disorders. Mutations in PVRL1, the nectin-1-encoding gene, cause cleft lip and/or palate ectodermal dysplasia or Zlotogora-Ogur syndrome (CLPED1, MIM 225069) and nonsyndromic orofacial cleft (OFC7).11,26 There are similarities between the nectin-4 expression pattern we observed and that reported for nectin-1. In particular, in mouse embryos, nectin-1 was expressed from E8.5 to E16.5 and decreased after E18.5, with staining at E15 in various ectodermal tissues, including skin, tooth, and hair.27 Detailed mouse embryo and adult human skin immunohistochemistry studies further showed positive nectin-1 expression in the epidermis, mainly in the spinous layer.28 Notably, a trans-heterophilic interaction between nectin-1 and nectin-4 was previously demonstrated,20 further supporting a common mode of action of these molecules in regulating ectodermal organogenesis. The impairment of the binding of the EDSS patient's keratinocytes to nectin-1-Fc that we observed further corroborates this hypothesis. Indeed, clinically, CLPED1 and EDSS show several overlapping features, e.g., alopecia, abnormal teeth, and syndactyly, although cleft lip and/or palate appears to be characteristic of CLPED1.12 The spatiotemporal expression of other nectins (i.e., nectin-2 and nectin-3) has been extensively investigated in the mouse embryo. Interestingly, in addition to nectin-1, nectin-2 was also expressed in the hair matrix, whereas nectin-3 was nearly absent in the epidermis and vibrissae. These data also indicate that nectins are differentially expressed during embryonic development, thus suggesting a role in the morphogenesis of distinct epithelial tissues.27
The combination of defective hair morphogenesis and limb abnormalities has been previously reported in human disorders determined by defective CAMs. An interesting example is CDH3, a gene encoding P-cadherin, in which biallelic mutations cause hypotrichosis with juvenile macular dystrophy (HJMD, MIM 601553) and ectodermal dysplasia, ectrodactyly, and macular dystrophy (EEM, MIM 225280).29,30 It is noteworthy that the hair abnormalities we observed in EDSS patients (i.e., pili torti and alopecia) are almost identical to those reported in HJMD patients.29 Of further interest, recent evidence suggests that P-cadherin is a transcriptional target of p63, with a crucial role in hair follicle development and limb bud outgrowth.31 Indeed, TP63 mutations cause human developmental disorders that are characterized by various degrees of ectodermal dysplasia, limb abnormalities, and facial clefts.32 Nectins cooperate with cadherins at AJ binding to the actin cytoskeleton after the recruitment of the E-cadherin-β-catenin complex.3,33 Our data showed that an impairment of nectin-4 not only alters the nectin-afadin complex but also affects the classical cadherin-catenin unit, which is crucial for AJ formation.19 It is noteworthy that, although these effects were evident in the outer root sheath of the hair follicle, no gross morphological abnormalities were seen within the epidermis, suggesting a critical role for nectin-4 in hair morphogenesis and cycling. Although further studies are warranted to elucidate the function of nectin-4 in ectodermal derivatives and limb bud development, it is tempting to speculate that this molecule participates in reciprocal and sequential interactions between epithelium and mesenchyme for tissue formation and morphogenesis, as seen for other CAMs such as cadherins.34
Cutaneous syndactyly results from an impairment of apoptotic cell death in the tissue lying between the developing digits.35 Different signaling pathways are involved in the regulation of cell death in the interdigital tissue.36 Best documented is the role of the bone morphogenetic proteins (BMPs) family in inducing cell death in the interdigital mesenchyme by its downstream targets Msx1 and Msx2. Interestingly, inactivation of Rac in the interdigital mesenchyme evoked defective BMP signaling and cutaneous syndactyly.37 Like the nectin-4 interaction partner afadin, Rac has a modifying function for the actin cytoskeleton. Moreover, the interaction with cadherins also implies an influence of nectin-4 on Wnt signaling, which plays a relevant role in limb development. Dkk1-mediated inhibition of Wnt signaling was shown to be important for digit separation in chicken and mouse.38 In addition, mutations in the SOST gene, encoding the Wnt antagonist sclerostin, also cause cutaneous syndactyly.39 However, interdigital cell death is most prominent in the mouse limbs between E12.5 and E14.5,40 thus before nectin-4 is strongly expressed, indicating that other mechanisms beyond apoptosis might be involved.
A growing number of CAMs are being implicated in the pathogenesis of ED syndromes. In addition to nectin-1 and P-cadherin defects, leading to CLPED1 and HJMD/EEM syndromes, respectively,11,29,30 mutations in PKP1 gene encoding the desmosomal protein plakophilin-1 cause ED/skin fragility syndrome (MIM 604536).41 The identification of nectin-4 in the pathogenesis of EDSS strengthens the role of defective CAMs in a discrete group of syndromic EDs and warrants the investigation of this class of molecules in other, molecularly uncharacterized, disorders of ectodermal derivates.
Acknowledgments
We are very grateful to the family members for their generous participation in this study. We thank Sheila Pierce for critically revising the manuscript, Enza Maria Valente for LOD score calculations, Sara Loddo, Naomi De Luca, and Massimo Teson for technical support, and Björn Fischer for quantitative PCR in mouse. This study was supported in part by funding from the Italian Ministry of Health, Ricerca Corrente 2009–2010 (F.B., G.Z., and B.D.).
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Human Splicing Finder (HSF), http://www.umd.be/HSF/
NCBI browser, http://www.ncbi.nlm.nih.gov/Genbank/
NetGene2, http://www.cbs.dtu.dk/services/NetGene2/
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
Polyphen, http://genetics.bwh.harvard.edu/pph/
Sorting Intolerant from Tolerant (SIFT), http://sift.jcvi.org/
SplicePort, http://www.cs.umd.edu/projects/SplicePort
UCSC Genome Browser, http://www.genome.ucsc.edu/
Accession Number
The RefSeq accession number for the PVRL4 gene sequence reported in this paper is NM_030916.2.
References
- 1.Fuchs E. Scratching the surface of skin development. Nature. 2007;445:834–842. doi: 10.1038/nature05659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gumbiner B.M. Cell adhesion: The molecular basis of tissue architecture and morphogenesis. Cell. 1996;84:345–357. doi: 10.1016/s0092-8674(00)81279-9. [DOI] [PubMed] [Google Scholar]
- 3.Perez-Moreno M., Jamora C., Fuchs E. Sticky business: Orchestrating cellular signals at adherens junctions. Cell. 2003;112:535–548. doi: 10.1016/s0092-8674(03)00108-9. [DOI] [PubMed] [Google Scholar]
- 4.Freire-Maia N. Ectodermal dysplasias. Hum. Hered. 1971;21:309–312. doi: 10.1159/000152419. [DOI] [PubMed] [Google Scholar]
- 5.Freire-Maia N., Lisboa-Costa T., Pagnan N.A. Ectodermal dysplasias: How many? Am. J. Med. Genet. 2001;104:84. doi: 10.1002/ajmg.1586. [DOI] [PubMed] [Google Scholar]
- 6.Salinas C.F., Jorgenson R.J., Wright J.T., DiGiovanna J.J., Fete M.D. 2008 international conference on ectodermal dysplasias classification: Conference report. Am. J. Med. Genet. A. 2009;149A:1958–1969. doi: 10.1002/ajmg.a.32882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Irvine A.D. Towards a unified classification of the ectodermal dysplasias: Opportunities outweigh challenges. Am. J. Med. Genet. A. 2009;149A:1970–1972. doi: 10.1002/ajmg.a.32852. [DOI] [PubMed] [Google Scholar]
- 8.Itin P.H. Rationale and background as basis for a new classification of the ectodermal dysplasias. Am. J. Med. Genet. A. 2009;149A:1973–1976. doi: 10.1002/ajmg.a.32739. [DOI] [PubMed] [Google Scholar]
- 9.Visinoni A.F., Lisboa-Costa T., Pagnan N.A., Chautard-Freire-Maia E.A. Ectodermal dysplasias: Clinical and molecular review. Am. J. Med. Genet. A. 2009;149A:1980–2002. doi: 10.1002/ajmg.a.32864. [DOI] [PubMed] [Google Scholar]
- 10.Boudghene-Stambouli O., Merad-Boudia A. [Association of ectodermal dysplasia and syndactylia] Ann. Dermatol. Venereol. 1991;118:107–110. [PubMed] [Google Scholar]
- 11.Suzuki K., Hu D., Bustos T., Zlotogora J., Richieri-Costa A., Helms J.A., Spritz R.A. Mutations of PVRL1, encoding a cell-cell adhesion molecule/herpesvirus receptor, in cleft lip/palate-ectodermal dysplasia. Nat. Genet. 2000;25:427–430. doi: 10.1038/78119. [DOI] [PubMed] [Google Scholar]
- 12.Zlotogora J. Syndactyly, ectodermal dysplasia, and cleft lip/palate. J. Med. Genet. 1994;31:957–959. doi: 10.1136/jmg.31.12.957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cartegni L., Chew S.L., Krainer A.R. Listening to silence and understanding nonsense: Exonic mutations that affect splicing. Nat. Rev. Genet. 2002;3:285–298. doi: 10.1038/nrg775. [DOI] [PubMed] [Google Scholar]
- 14.Ng P.C., Henikoff S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31:3812–3814. doi: 10.1093/nar/gkg509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hebsgaard S.M., Korning P.G., Tolstrup N., Engelbrecht J., Rouzé P., Brunak S. Splice site prediction in Arabidopsis thaliana pre-mRNA by combining local and global sequence information. Nucleic Acids Res. 1996;24:3439–3452. doi: 10.1093/nar/24.17.3439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dogan R.I., Getoor L., Wilbur W.J., Mount S.M. SplicePort - An interactive splice-site analysis tool. Nucleic Acids Res. 2007;35:W285–W291. doi: 10.1093/nar/gkm407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Desmet F.O., Hamroun D., Lalande M., Collod-Béroud G., Claustres M., Béroud C. Human Splicing Finder: An online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 2009;37:e67. doi: 10.1093/nar/gkp215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chomczynski P., Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal. Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 19.Niessen C.M. Tight junctions/adherens junctions: Basic structure and function. J. Invest. Dermatol. 2007;127:2525–2532. doi: 10.1038/sj.jid.5700865. [DOI] [PubMed] [Google Scholar]
- 20.Reymond N., Fabre S., Lecocq E., Adelaïde J., Dubreuil P., Lopez M. Nectin4/PRR4, a new afadin-associated member of the nectin family that trans-interacts with nectin1/PRR1 through V domain interaction. J. Biol. Chem. 2001;276:43205–43215. doi: 10.1074/jbc.M103810200. [DOI] [PubMed] [Google Scholar]
- 21.Stricker S., Verhey van Wijk N., Witte F., Brieske N., Seidel K., Mundlos S. Cloning and expression pattern of chicken Ror2 and functional characterization of truncating mutations in Brachydactyly type B and Robinow syndrome. Dev. Dyn. 2006;235:3456–3465. doi: 10.1002/dvdy.20993. [DOI] [PubMed] [Google Scholar]
- 22.Takahashi K., Nakanishi H., Miyahara M., Mandai K., Satoh K., Satoh A., Nishioka H., Aoki J., Nomoto A., Mizoguchi A., Takai Y. Nectin/PRR: An immunoglobulin-like cell adhesion molecule recruited to cadherin-based adherens junctions through interaction with Afadin, a PDZ domain-containing protein. J. Cell Biol. 1999;145:539–549. doi: 10.1083/jcb.145.3.539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Takai Y., Ikeda W., Ogita H., Rikitake Y. The immunoglobulin-like cell adhesion molecule nectin and its associated protein afadin. Annu. Rev. Cell Dev. Biol. 2008;24:309–342. doi: 10.1146/annurev.cellbio.24.110707.175339. [DOI] [PubMed] [Google Scholar]
- 24.Irie K., Shimizu K., Sakisaka T., Ikeda W., Takai Y. Roles and modes of action of nectins in cell-cell adhesion. Semin. Cell Dev. Biol. 2004;15:643–656. doi: 10.1016/j.semcdb.2004.09.002. [DOI] [PubMed] [Google Scholar]
- 25.Sakisaka T., Ikeda W., Ogita H., Fujita N., Takai Y. The roles of nectins in cell adhesions: Cooperation with other cell adhesion molecules and growth factor receptors. Curr. Opin. Cell Biol. 2007;19:593–602. doi: 10.1016/j.ceb.2007.09.007. [DOI] [PubMed] [Google Scholar]
- 26.Sözen M.A., Suzuki K., Tolarova M.M., Bustos T., Fernández Iglesias J.E., Spritz R.A. Mutation of PVRL1 is associated with sporadic, non-syndromic cleft lip/palate in northern Venezuela. Nat. Genet. 2001;29:141–142. doi: 10.1038/ng740. [DOI] [PubMed] [Google Scholar]
- 27.Okabe N., Ozaki-Kuroda K., Nakanishi H., Shimizu K., Takai Y. Expression patterns of nectins and afadin during epithelial remodeling in the mouse embryo. Dev. Dyn. 2004;230:174–186. doi: 10.1002/dvdy.20033. [DOI] [PubMed] [Google Scholar]
- 28.Matsushima H., Utani A., Endo H., Matsuura H., Kakuta M., Nakamura Y., Matsuyoshi N., Matsui C., Nakanishi H., Takai Y., Shinkai H. The expression of nectin-1alpha in normal human skin and various skin tumours. Br. J. Dermatol. 2003;148:755–762. doi: 10.1046/j.1365-2133.2003.05225.x. [DOI] [PubMed] [Google Scholar]
- 29.Sprecher E., Bergman R., Richard G., Lurie R., Shalev S., Petronius D., Shalata A., Anbinder Y., Leibu R., Perlman I. Hypotrichosis with juvenile macular dystrophy is caused by a mutation in CDH3, encoding P-cadherin. Nat. Genet. 2001;29:134–136. doi: 10.1038/ng716. [DOI] [PubMed] [Google Scholar]
- 30.Kjaer K.W., Hansen L., Schwabe G.C., Marques-de-Faria A.P., Eiberg H., Mundlos S., Tommerup N., Rosenberg T. Distinct CDH3 mutations cause ectodermal dysplasia, ectrodactyly, macular dystrophy (EEM syndrome) J. Med. Genet. 2005;42:292–298. doi: 10.1136/jmg.2004.027821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shimomura Y., Wajid M., Shapiro L., Christiano A.M. P-cadherin is a p63 target gene with a crucial role in the developing human limb bud and hair follicle. Development. 2008;135:743–753. doi: 10.1242/dev.006718. [DOI] [PubMed] [Google Scholar]
- 32.van Bokhoven H., Brunner H.G. Splitting p63. Am. J. Hum. Genet. 2002;71:1–13. doi: 10.1086/341450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Takai Y., Nakanishi H. Nectin and afadin: Novel organizers of intercellular junctions. J. Cell Sci. 2003;116:17–27. doi: 10.1242/jcs.00167. [DOI] [PubMed] [Google Scholar]
- 34.Jamora C., DasGupta R., Kocieniewski P., Fuchs E. Links between signal transduction, transcription and adhesion in epithelial bud development. Nature. 2003;422:317–322. doi: 10.1038/nature01458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Montero J.A., Hurlé J.M. Sculpturing digit shape by cell death. Apoptosis. 2010;15:365–375. doi: 10.1007/s10495-009-0444-5. [DOI] [PubMed] [Google Scholar]
- 36.Zuzarte-Luis V., Hurlé J.M. Programmed cell death in the embryonic vertebrate limb. Semin. Cell Dev. Biol. 2005;16:261–269. doi: 10.1016/j.semcdb.2004.12.004. [DOI] [PubMed] [Google Scholar]
- 37.Suzuki D., Yamada A., Amano T., Yasuhara R., Kimura A., Sakahara M., Tsumaki N., Takeda S., Tamura M., Nakamura M. Essential mesenchymal role of small GTPase Rac1 in interdigital programmed cell death during limb development. Dev. Biol. 2009;335:396–406. doi: 10.1016/j.ydbio.2009.09.014. [DOI] [PubMed] [Google Scholar]
- 38.Grotewold L., Rüther U. The Wnt antagonist Dickkopf-1 is regulated by Bmp signaling and c-Jun and modulates programmed cell death. EMBO J. 2002;21:966–975. doi: 10.1093/emboj/21.5.966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Balemans W., Ebeling M., Patel N., Van Hul E., Olson P., Dioszegi M., Lacza C., Wuyts W., Van Den Ende J., Willems P. Increased bone density in sclerosteosis is due to the deficiency of a novel secreted protein (SOST) Hum. Mol. Genet. 2001;10:537–543. doi: 10.1093/hmg/10.5.537. [DOI] [PubMed] [Google Scholar]
- 40.Zakeri Z., Quaglino D., Ahuja H.S. Apoptotic cell death in the mouse limb and its suppression in the hammertoe mutant. Dev. Biol. 1994;165:294–297. doi: 10.1006/dbio.1994.1255. [DOI] [PubMed] [Google Scholar]
- 41.McGrath J.A., McMillan J.R., Shemanko C.S., Runswick S.K., Leigh I.M., Lane E.B., Garrod D.R., Eady R.A. Mutations in the plakophilin 1 gene result in ectodermal dysplasia/skin fragility syndrome. Nat. Genet. 1997;17:240–244. doi: 10.1038/ng1097-240. [DOI] [PubMed] [Google Scholar]
- 42.Fabre-Lafay S., Monville F., Garrido-Urbani S., Berruyer-Pouyet C., Ginestier C., Reymond N., Finetti P., Sauvan R., Adélaïde J., Geneix J. Nectin-4 is a new histological and serological tumor associated marker for breast cancer. BMC Cancer. 2007;7:73. doi: 10.1186/1471-2407-7-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.