

Abstract
Generalized peeling skin disease is an autosomal-recessive ichthyosiform erythroderma characterized by lifelong patchy peeling of the skin. After genome-wide linkage analysis, we have identified a homozygous nonsense mutation in CDSN in a large consanguineous family with generalized peeling skin, pruritus, and food allergies, which leads to a complete loss of corneodesmosin. In contrast to hypotrichosis simplex, which can be associated with specific dominant CDSN mutations, peeling skin disease is characterized by a complete loss of CDSN expression. The skin phenotype is consistent with a recent murine Cdsn knockout model. Using three-dimensional human skin models, we demonstrate that lack of corneodesmosin causes an epidermal barrier defect supposed to account for the predisposition to atopic diseases, and we confirm the role of corneodesmosin as a decisive epidermal adhesion molecule. Therefore, peeling skin disease will represent a new model disorder for atopic diseases, similarly to Netherton syndrome and ichthyosis vulgaris in the recent past.
Main Text
The skin barrier is important for the control of the transepidermal loss of water and provides protection of the organism against the intrusion of external pathogens. The role of barrier disruption for the development of atopic diseases was made particularly obvious when mutations were identified in FLG (MIM 135940), which can give rise to ichthyosis vulgaris and lead to an increased susceptibility to atopic dermatitis.1,2 Filaggrin, encoded by FLG, is a key constituent of the epidermal barrier. Mice with a mutation in Flg showed elevated IgE levels because of a skin barrier defect after application of allergens.3 Generalized peeling skin disease (PSD, MIM 270300) with pruritus and atopic diseases, also referred to as peeling skin syndrome type B, is an unusual autosomal-recessive ichthyosiform erythroderma characterized by lifelong patchy peeling of the entire skin with an onset at birth or shortly thereafter.4,5 Several patients have been reported who showed PSD with a strong association of pruritus and high IgE levels.4,6–14
PSD shares several clinical features with Netherton syndrome (NS, MIM 256500), such as frequent flares of erythema and scaling and strong association with atopic diseases.5,15 Moreover, both diseases appear very similar at the histological and ultrastructural level, showing an enhanced detachment of corneocytes.4,16 However, individuals with generalized PSD do not show trichorrhexis invaginata (“bamboo hairs”), nor was it possible to demonstrate SPINK5 (MIM 605010) mutations, which are responsible for NS,15 in patients with PSD.9 Moreover, PSD has to be differentiated from the acral peeling skin syndrome (MIM 609796), which is due to autosomal-recessive mutations in the gene of transglutaminase 5 (TGM5, MIM 603805),17 and from peeling skin syndrome type A, which is characterized by asymptomatic and noninflammatory peeling and often starts at 3 to 6 years of age.5,18
We have studied a large consanguineous Roma family from Germany with four individuals with generalized superficial skin peeling since birth (Figure 1), severe pruritus, and atopic manifestations with seasonal variation. The study was approved by the institutional review board of the University Hospital of Münster, and all patients enrolled gave their informed consent. A detailed medical and dermatological history was obtained from all affected persons. The clinical features are summarized in Table 1. The initial clinical presentation was in the first week of life in all four patients. The disease presented as an unusual ichthyosiform erythroderma with white, superficial exfoliation (Figure 1A), with normal birth weight and growth, and without clinical signs for a syndromic form of ichthyosis. However, an initial mild failure to thrive and some periods of Staphylococcus aureus skin infections were noted in the first few years of life. Most strikingly, the affected individuals showed severe pruritus, especially in warm weather, food allergies to nuts and fish, and repeated episodes of angioedema, urticaria, and/or asthma. Overall IgE levels were highly elevated (Table 1). Two children had unusually fine hair in early infancy, and it could be plucked easily (P2 and P3). Patients did not show any hair loss, and hair shaft analysis did not reveal trichorrhexis invaginata, which would have been diagnostic for NS. Punch biopsies (4 mm) for histopathology and immunofluorescence analysis were taken from all patients. In addition, biopsies for ultrastructural analysis and isolation of keratinocytes were taken from patient 3 (P3). Specimens for ultrastructural analysis were proceeded and examined as described previously.16 Similarly to NS, histological and ultrastructural analyses of the skin showed an enhanced detachment of corneocytes (Figure 1B).4,16 However, we have not identified mutations in the genes SPINK5 or TGM5.
Figure 1.
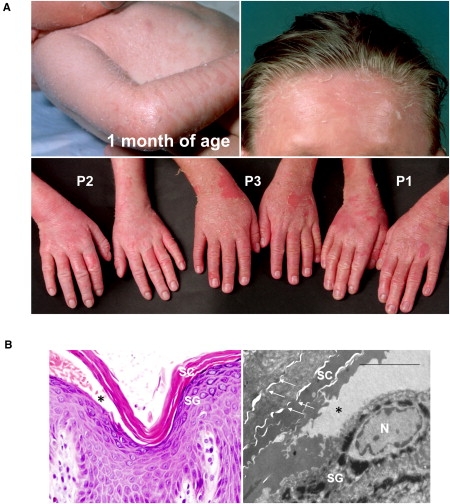
Clinical Appearance of Peeling Skin Disease and Pedigree of the Family Analyzed
(A) Shortly after birth, patient 1 developed an unusual ichthyosiform skin condition with erythema and superficial white scaling. Patient 3 reported seasonal variation of scaling and pruritus, depending on the climate. All patients reported mild to moderate nail changes.
(B) Histopathology (left) revealed an extensive detachment of the stratum corneum (SC) directly above the stratum granulosum (SG) (∗). Ultrastructure (right) showed split formation between keratinocytes of the SG and corneocytes of the SC, as well as weak lateral adhesion of corneocytes. Corneodesmosomes (arrows) are hardly visible; scale bar represents 5 μm.
Table 1.
Clinical Features of Patients P1–P4
| Disease Phenotype |
P1 |
P2 |
P3 |
P4 |
|---|---|---|---|---|
| 10 yrs (male) | 8 yrs (female) | 9 yrs (male) | 1 yr (male) | |
| Onset and initial clinical presentation | Second day of life | Second day of life | Second day of life | Second day of life |
| Superficial exfoliation with unusual ichthyosiform erythroderma | Superficial exfoliation with unusual ichthyosiform erythroderma | Superficial exfoliation with unusual ichthyosiform erythroderma | Superficial exfoliation with unusual ichthyosiform erythroderma | |
| Disease course | Erythematous patches that start scaling after few days, heal, and reoccur; severe exacerbations within the first 4 years of life | Erythematous patches that start scaling after few days, heal and reoccur; severe exacerbations within the first 4 years of life | Erythematous patches that start scaling after few days, heal and reoccur; severe exacerbations within the first 4 years of life | Erythematous patches that start scaling after few days, heal and reoccur; severe exacerbations |
| Seasonal variation | Not specifically | Yes (severe scaling in winter, severe pruritus in summer) | Yes (severe pruritus in summer) | Yes |
| Clinical Features | ||||
| Scaling | ||||
| Distribution | Generalized | Generalized | Generalized | Generalized |
| Type | Exfoliative | Exfoliative | Exfoliative | Exfoliative |
| Color | White | White | White | White |
| Palmoplantar features | Episodically scaling, hyperhidrosis | Mostly uninvolved | Hyperhidrosis | Mostly uninvolved |
| Hair abnormalities | No trichorrhexis invaginata, normal hair growths, brittle hair in early infancy | No trichorrhexis invaginata, easy plugging of the hair, normal hair growths, brittle hair in early infancy | No trichorrhexis invaginata, easy plugging of the hair, normal hair growths | No trichorrhexis invaginata, normal hair growths |
| Nail abnormalities | Mild onychodystrophy, white nail changes | Recurrent onycholysis since sixth year of life | Not specifically, white nail changes | Not specifically, white nail changes |
| S. aureus superinfection | Frequent during the first 4 years of life | Frequent during the first 4 years of life | Frequent during the first 4 years of life | Present |
| Growths | Normal birth weight and size, mild failure to thrive during the first ∼4 years of life | Normal birth weight and size, mild failure to thrive during the first ∼4 years of life | Normal birth weight and size, mild failure to thrive during the first ∼4 years of life | Normal |
| Height (present age) | At 50th percentile | At 50th percentile | At 50th percentile | At 50th percentile |
| Weight (present age) | At 10th percentile | At 10th percentile | At 10th percentile | At 10th percentile |
| Atopic Features | ||||
| Pruritus | Moderate to severe | Moderate to severe | Moderate to severe | Moderate |
| Total IgE level | 2076 IU/ml (<100)a | ND | >2000 IU/ml (<100)a | ND |
| Eosinophils | 0.7 × 103/μl (<0.44)a | ND | 0.79 × 103/μl (<0.60)a | ND |
| Food intolerances | Nuts, fish | Fish | Nuts, fish | Unknown |
| Others | – | History of urticaria with angioedema | History of asthma when in contact with fish | – |
ND, not determined.
Normal values.
We carried out a whole-genome linkage analysis via chip-based SNP analysis. Peripheral blood was collected in EDTA from all available family members, and DNA was extracted from leukocytes via standard procedures. DNA samples of three affected children and their parents, representing one family branch each (see Figure S1 available online), were genotyped via the Affymetrix GeneChip Human Mapping 250K Sty Array (Affymetrix). Genotypes were called by the GeneChip DNA Analysis Software (Affymetrix). Data were checked using the program Graphical Representation of Relationships.19 Parametric linkage analysis was done with the programs Allegro20 and MERLIN.21 All data handling was done via the graphical user interface ALOHOMORA.22 Linkage analysis identified a candidate region on chromosome 6p (Figure 2A). Refined mapping localized the gene to a 3.3 cM interval with a maximum LOD score of 5.4, corresponding to 5.7 Mb in length, assuming two separate pedigrees with two affected individuals each (Figure S1). Because an undefined connection exists between both branches of the family and because they originated from the same tribe, we then searched for identical homozygous intervals between all four patients by using SNPs and microsatellites in the linked region. The largest homozygous interval was 3.0 Mb in length and contained 195 genes. CDSN (MIM 602593), located in the minimal interval, was chosen as a functional candidate gene because it encodes a component of the corneodesmosomes in the epidermal stratum corneum, and it was analyzed for mutations. The coding region was sequenced in the patients and in their parents and siblings (NM_001264.3; Table S1). All four affected children showed the same homozygous nonsense mutation in CDSN, c.175A>T, resulting in a premature termination codon, p.Lys59X (Figures 2B and 2C). We observed full cosegregation of the mutation in the entire family (Figure S1) and did not detect the mutation among 220 chromosomes from ethnically matching control persons.
Figure 2.

Mapping of the Gene and Identification of the Underlying Genetic Defect
(A) Multipoint linkage analysis revealed a locus on chromosome 6p. For reasons of clarity, LOD scores below –3 are not shown. Vertical lines denote boundaries of the chromosomes, which are labeled above the graph.
(B) The mutation was found in exon 2 of CDSN. The position within the proposed protein structure is shown.
(C) Sequencing analysis of CDSN showed a homozygous transversion (indicated by an arrow) at c.175 in all affected individuals, resulting in a termination codon.
(D) Protein extracts from normal keratinocytes and keratinocytes of patient 3 were subjected to immunoblot analysis. Polypeptides of ∼54 kDa and 45 kDa correspond to full-length and proteolytically processed forms of corneodesmosin. Preparations from differentiated patient keratinocytes (+Ca2+) did not show any corneodesmosin signal. Transglutaminase 1 (TGase1) served as a late differentiation control.
To investigate CDSN expression, we isolated total RNA from primary keratinocytes cultured with 1.2 mM Ca2+. RNA was adjusted and quantified via one-step qRT-PCR (Table S1) with SYBR GreenER labeling (Invitrogen) on a LightCycler 480 system (Roche Applied Sciences). Normalization was done with ACTB, RPS18, and 18S RNA. Relative quantification showed a reduction of CDSN mRNA amounts by 75%, pointing to nonsense-mediated CDSN mRNA decay (Figure S2). Equal quantities of protein isolated from primary keratinocytes were separated by 10% SDS-PAGE under reducing conditions and transferred to polyvinylidene fluoride membrane (Millipore). The blots were probed with primary antibodies directed against corneodesmosin, transglutaminase 1, and actin. Immunoblot analysis demonstrated the absence of corneodesmosin in the patient, whereas differentiated keratinocytes of healthy controls showed a strong signal (Figure 2D). Transglutaminase 1 was detected as a late differentiation control in differentiated keratinocytes from both patients and unaffected persons.
Immunohistochemistry on skin biopsy samples was performed with antibodies against involucrin, filaggrin, loricrin, and keratins 2, 10, and 14. Direct immunofluorescenceanalysis was performed on cryosections for corneodesmosin, desmocollin 1, transglutaminase 1, LEKTI, desmoglein 1, transglutaminase 5, and elafin. Antigen mapping of corneodesmosin was negative for all skin biopsies of the patients (Figure 3A). In contrast, other components of the desmosomes and corneodesmosomes, i.e., desmocollin 1 and desmoglein 1, showed an almost regular expression. LEKTI, missing in NS, presented a broadened signal zone in the upper epidermal layers of our patients, transglutaminase 5 was normal, and further late epidermal differentiation markers, e.g., filaggrin, involucrin, loricrin, transglutaminase 1, and keratin 2, likewise showed an enhanced immunohistochemical staining (Figure 3B; Figure S3). These proteins are important for the cornified cell envelope formation, and the changes probably reflect homeostatic responses to epidermal stress or inflammation.
Figure 3.

Characterization of Patient Epidermis
(A) Immunofluorescence study revealed a pericellular corneodesmosin signal within the upper SG and lower SC. In contrast, epidermis of patients 1 and 3 showed no signal. Scale bars represent 20 μm.
(Ba–Bf) LEKTI, deficient in Netherton syndrome, showed a broad and strong expression in the skin of the PSD patients (Ba and Bd). Further desmosomal components, i.e., desmocollin 1 (Dsc1, Bb and Be) and desmoglein 1 (Dsg1, Bc and Bf), showed a strong presence in the skin of the patients (only P3 is shown). Scale bars represent 20 μm.
To further study the lack of corneodesmosin in PSD that is supposed to lead to an increased desquamation of the corneocytes, we generated three-dimensional skin models by using organotypic tissue coculture systems. Primary human keratinocytes (NHEKs) were isolated from foreskin obtained after juvenile circumcision in an age-matched, 9-year-old boy and from patient skin biopsy samples (P3) and were cultivated until second passage in the presence of a feeder layer of irritated 3T3 mouse fibroblasts, as described.23 Tissue engineering was performed as reported elsewhere in more detail (K.-M.E. and H.C.H. et al., unpublished data). In brief, NHEKs were isolated from foreskin obtained after juvenile circumcision in an age-matched boy and from patient skin punch biopsy samples (P3) and cultivated in the presence of a feeder layer of irritated 3T3 mouse fibroblasts.23 Passage 2 keratinocytes were cultivated in serum-free, defined cultivation media without feeder cells with 0.15 mM Ca2+. Normal human dermal fibroblasts (NHDF) were isolated from neonatal foreskin and patient skin biopsy samples and were cultivated in DMEM with 10% fetal calf serum (FCS), 100 IU/ml penicillin, and 100 μg/ml streptomycin. Six-well plates with 3 μm inserts with 4.22 cm2 growth area (BD Biosciences) were used. For each well, a mixture of collagen I (bovine, Nutacon) and 10× HBSS was brought to neutral pH with NaOH, 2.5 × 105 NHDF in FCS was added, and the mixture was poured into the filter insert. The matrix was stored for 2–4 hr in a CO2-free incubator before keratinocyte growth medium was added. The system was transferred to an incubator with 5% CO2 and 95% humidity. After removing the medium from the top, 4.5 × 106 to 5.2 × 106 NHEK was seeded onto the matrices. On the next day, the medium was changed to defined cultivation medium.24 Models were cultivated for 8 days and then harvested with biopsy punches (5 to 8 mm). Either samples were processed and embedded in paraffin or RNA and protein were isolated.
Skin equivalents with patient fibroblasts and keratinocytes (Figure 4A) demonstrated formation of the basal keratinocyte layer and the dermal-epidermal junction zone and terminal keratinocyte differentiation in the epidermis models, as shown by hematoxylin and eosin (H & E) staining (Figure 4A) and immunofluorescence staining of markers such as integrin β1, collagen IV, keratin 14, and keratin 10 (data not shown). Less well-organized epidermal structure and extended expression of late differentiation markers such as involucrin and filaggrin (Figure 4B) compared to skin equivalents with normal keratinocytes resembled our findings in patient skin. Consistent with the findings in patient skin biopsy specimens, we showed a complete loss of corneodesmosin (Figure 4B). We furthermore analyzed the presence of secreted serine proteases of the epidermis. Staining of kallikrein 5 was elevated in patient models and the expression was broadened in the epidermis, consistent with the results from a recent study.9 These results pointed to a disturbance of terminal epidermal differentiation upon loss of corneodesmosin and a deregulation of proteins playing a role for the cornified cell envelope and corneodesmosome degradation.
Figure 4.

Three-Dimensional Epidermis Models and Analysis of the Barrier Defect
(A) H & E staining of reconstructed epidermis with keratinocytes from healthy probands demonstrated an even structure of the epidermis. Models with keratinocytes from patient 3 (P3) showed a loose and increased stratum corneum and a more irregular organization of differentiating keratinocytes.
(B) Immunofluorescence analysis confirmed the lack of corneodesmosin in models generated from patient keratinocytes, in contrast to normal epidermis equivalents. Normal epidermis equivalents showed regular terminal differentiation with a synthesis of kallikrein 5 and filaggrin. The expression patterns of KLK5 and FLG appeared strong and broadened in patient models.
(C) Models with normal keratinocytes exhibited a barrier function comparable to standardized skin models. Clearly increased testosterone permeation into models with patient keratinocytes demonstrated a severe epidermal barrier defect (mean values ± SD for each time point).
The generation of three-dimensional models precisely replicating the epidermal skin enabled us to further analyze the epidermal barrier integrity. Test substances were the Organisation for Economic Cooperation and Development-proposed standard compounds for cutaneous absorption studies, caffeine and testosterone (Sigma-Aldrich). Experiments were performed according to a validated protocol,25 which was proved to be applicable to the use of reconstructed full-thickness skin.26 Briefly, stock solutions of testosterone (40 μg/ml) and caffeine (1000 μg/ml) were spiked with an appropriate amount of the radiolabeled agent to achieve a total radioactivity of 2 μCi/ml. Special inserts constructed for the EPISKIN model27 were used with an assayed surface area of 0.357 cm2. Skin models were mounted into Franz cells (diameter 15 mm, volume 12 ml; PermeGear) with the stratum corneum facing the air. The system was allowed to equilibrate for 30 min before a sample of receptor fluid was collected. Subsequently, 110 μl of spiked caffeine and testosterone solutions was applied on the reconstructed skin model, resulting in a dose of 284.1 μg/cm2 of caffeine and a dose of 11.36 μg/cm2 of testosterone, respectively. Receptor fluid was sampled repeatedly every half hour and replaced by fresh PBS. Permeation tests were performed at least in quadruplicates except for the first experiment with patient cells, which was performed in duplicate. Two independent experiments were performed with control cells and patient cells. For each experiment, cumulative amounts of the permeated compounds in the receptor medium were plotted versus time (mean values ± standard deviation [SD]).
The apparent permeability coefficient Papp and lag time were calculated from a regression line based on mean values of the experiment.28 Normal skin models showed significantly lower permeation values (Figure 4C), though they slightly exceeded those obtained with a commercially available reconstructed skin model.26 The Papp values with testosterone were 8.58 × 10−6 ± 1.47 × 10−6 cm/s and 5.29 × 10−6 ± 0.72 × 10−6 cm/s, respectively, in patient models compared to models with normal keratinocytes (mean values ± SD; p = 0.0039) and 13.79 × 10−6 ± 1.84 × 10−6 cm/s and 10.78 × 10−6 ± 0.82 × 10−6 cm/s, respectively, with caffeine (p = 0.017). Thus, we identified a severe barrier defect in patient models with the lack of corneodesmosin.
Corneodesmosin is a phosphorylated basic keratinocyte adhesion glycoprotein located in the extracellular part of the desmosomes and corneodesmosomes at the transition from the stratum granulosum (SG) to the stratum corneum (SC) and in the inner root sheath of the hair follicles.29 The 52–56 kDa precursor form is synthesized in the middle epidermis, transported via lamellar bodies, and secreted into the extracellular space, where it associates to the desmosome core and the cornified cell envelope. Two glycine- and serine-rich domains, from amino acids 65–175 and 375–450, are responsible for forming adhesive secondary structures similar to glycine loops. Functionally, corneodesmosin reinforces cell-cell cohesion within the upper epidermis and the stratum corneum. Kallikrein 5 (stratum corneum tryptic enzyme) and kallikrein 7 (stratum corneum chymotryptic enzyme) cooperate to progressively proteolyse corneodesmosin. Its degradation is necessary for desquamation.30,31
An uncommon human hair disease was described earlier that was caused by autosomal-dominant mutations in CDSN: heterozygous nonsense mutations, namely p.Gln200X, p.Gln215X, and p.Tyr239X, were shown in a specific type of hypotrichosis simplex (MIM 146520).32,33 Affected individuals have normal hair in early childhood but experience progressive loss of scalp hair beginning in the middle of the first decade of life and almost complete baldness by the third decade; they do not show any signs of skin disease. It was assumed that abnormal corneodesmosin aggregates could exert a toxic effect to the hair follicle cells, thereby contributing to permanent hair loss.32 Of note, the phenotype of hypotrichosis simplex does not include skin peeling or ichthyosis, which could be explained by the presence of a normal allele in these patients. In our families, heterozygous carriers of the recessive mutation p.Lys59X did not show any skin or hair phenotype. Hence, hypotrichosis simplex and PSD can be assumed to involve different pathomechanisms.
Review of the literature,4,6,10,13 dermatological questioning, and clinical examination revealed that PSD is associated with a clinically asymptomatic hair phenotype, namely a mild hair-anchoring deficiency. A recent mouse model with targeted deletion of the murine corneodesmosin gene underlined the essential role of the protein for the hair physiology.34 In mice, Cdsn-deficient skin showed rapid hair loss. The strong expression of CDSN in the inner root sheath of human hair follicles, including the innermost cuticle layer,34 points to an important role of corneodesmosin for the hair-anchoring structure. The rather mild hair phenotype in PSD patients in contrast to mice with Cdsn-deficient skin, however, might reflect the differences in structure and size of mouse skin and mouse hair growth pattern.
Inactivation of corneodesmosin in mouse skin resulted in early postnatal death because of a breakdown of the epidermal barrier shortly after birth, when mechanical stress of a normal postnatal environment was exerted onto the skin.34,35 The defect of the epidermal barrier was accompanied by a 10-fold increase in transepidermal water loss.35 Here we have shown the impaired epidermal barrier caused by corneodesmosin deficiency in humans, which is evident clinically, e.g., by failure to thrive in early infancy,36 by ultrastructure analysis, which showed an extremely reduced coherence of corneocytes (Figure 1B), and by increased permeability of reagents into the epidermis (Figure 4C). The less-developed barrier is well in accordance with the sensitivity for irritants in patients with atopic diseases. We assume that the compromised epidermal barrier of corneodesmosin-deficient skin may facilitate the increased penetration of allergens into the epidermis, which could explain the development of atopic manifestations. A similar example is filaggrin deficiency, which can give rise to ichthyosis vulgaris and lead to an increased susceptibility to atopic dermatitis.2,3,37–39 Interestingly, our patients also showed manifest food allergies, i.e., to fish and nuts.
We conclude that a nonsense mutation in CDSN, which leads to the complete loss of corneodesmosin, causes peeling skin disease. Corneodesmosin is vastly important for the epidermal barrier integrity, and its absence may give rise to a strong predisposition to atopic manifestations. Corneodesmosin deficiency may therefore function as a novel human model disorder for atopic diseases, as did Netherton syndrome and ichthyosis vulgaris in the recent past.
Acknowledgments
We would like to thank our patients and their families, as well as J. Bückmann, P. Wissel, D. Presser, R. Casper, O. Kawaletz, J. Kurtenbach, and B. Willis. Our work is supported by the German Federal Ministry for Education and Research as part of the Network for Rare Diseases (NIRK, GFGM01143901), the Ministry for Innovation, Science, Research, and Technology of the Land Nordrhein Westfalen, the Selbsthilfe Ichthyose e.V. (http://www.ichthyose.de), the Interdisciplinary Center of Clinical Research (Lo2/017/07), and the Medical Faculty (OJ120817) of the University of Münster.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
BLAST Assembled RefSeq Genomes, http://blast.ncbi.nlm.nih.gov/Blast.cgi
Ensembl Genome Browser, http://www.ensembl.org/
Expasy, http://www.expasy.org/
GenBank, http://www.ncbi.nlm.nih.gov/Genbank/
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
Primer3, http://frodo.wi.mit.edu/primer3/
The National Center for Biotechnology Information (NCBI), http://www.ncbi.nlm.nih.gov/
UCSC Genome Browser, http://genome.ucsc.edu/
References
- 1.Smith F.J., Irvine A.D., Terron-Kwiatkowski A., Sandilands A., Campbell L.E., Zhao Y., Liao H., Evans A.T., Goudie D.R., Lewis-Jones S. Loss-of-function mutations in the gene encoding filaggrin cause ichthyosis vulgaris. Nat. Genet. 2006;38:337–342. doi: 10.1038/ng1743. [DOI] [PubMed] [Google Scholar]
- 2.Palmer C.N., Irvine A.D., Terron-Kwiatkowski A., Zhao Y., Liao H., Lee S.P., Goudie D.R., Sandilands A., Campbell L.E., Smith F.J. Common loss-of-function variants of the epidermal barrier protein filaggrin are a major predisposing factor for atopic dermatitis. Nat. Genet. 2006;38:441–446. doi: 10.1038/ng1767. [DOI] [PubMed] [Google Scholar]
- 3.Fallon P.G., Sasaki T., Sandilands A., Campbell L.E., Saunders S.P., Mangan N.E., Callanan J.J., Kawasaki H., Shiohama A., Kubo A. A homozygous frameshift mutation in the mouse Flg gene facilitates enhanced percutaneous allergen priming. Nat. Genet. 2009;41:602–608. doi: 10.1038/ng.358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Levy S.B., Goldsmith L.A. The peeling skin syndrome. J. Am. Acad. Dermatol. 1982;7:606–613. doi: 10.1016/s0190-9622(82)70140-9. [DOI] [PubMed] [Google Scholar]
- 5.Traupe H. Springer; Berlin: 1989. The Ichthyoses. A Guide to Clinical Diagnosis, Genetic Counseling, and Therapy. [Google Scholar]
- 6.Aras N., Sutman K., Tastan H.B., Baykal K., Can C. Peeling skin syndrome. J. Am. Acad. Dermatol. 1994;30:135–136. doi: 10.1016/s0190-9622(08)81903-2. [DOI] [PubMed] [Google Scholar]
- 7.Dicken C.H. Peeling skin syndrome. J. Am. Acad. Dermatol. 1985;13:158–160. doi: 10.1016/s0190-9622(85)80339-x. [DOI] [PubMed] [Google Scholar]
- 8.Hacham-Zadeh S., Holubar K. Skin peeling syndrome in a Kurdish family. Arch. Dermatol. 1985;121:545–546. [PubMed] [Google Scholar]
- 9.Komatsu N., Suga Y., Saijoh K., Liu A.C., Khan S., Mizuno Y., Ikeda S., Wu H.K., Jayakumar A., Clayman G.L. Elevated human tissue kallikrein levels in the stratum corneum and serum of peeling skin syndrome-type B patients suggests an over-desquamation of corneocytes. J. Invest. Dermatol. 2006;126:2338–2342. doi: 10.1038/sj.jid.5700379. [DOI] [PubMed] [Google Scholar]
- 10.Mevorah B., Frenk E., Saurat J.H., Siegenthaler G. Peeling skin syndrome: A clinical, ultrastructural and biochemical study. Br. J. Dermatol. 1987;116:117–125. doi: 10.1111/j.1365-2133.1987.tb05799.x. [DOI] [PubMed] [Google Scholar]
- 11.Mevorah B., Salomon D., Siegenthaler G., Hohl D., Meier M.L., Saurat J.H., Frenk E. Ichthyosiform dermatosis with superficial blister formation and peeling: Evidence for a desmosomal anomaly and altered epidermal vitamin A metabolism. J. Am. Acad. Dermatol. 1996;34:379–385. doi: 10.1016/s0190-9622(07)80013-2. [DOI] [PubMed] [Google Scholar]
- 12.Mizuno Y., Suga Y., Hasegawa T., Haruna K., Kohroh K., Ogawa H., Ikeda S., Shimizu T., Komatsu N. A case of peeling skin syndrome successfully treated with topical calcipotriol. J. Dermatol. 2006;33:430–432. doi: 10.1111/j.1346-8138.2006.00102.x. [DOI] [PubMed] [Google Scholar]
- 13.Tsai K., Valente N.Y., Nico M.M. Inflammatory peeling skin syndrome studied with electron microscopy. Pediatr. Dermatol. 2006;23:488–492. doi: 10.1111/j.1525-1470.2006.00290.x. [DOI] [PubMed] [Google Scholar]
- 14.Wile U.J. Familial study of three unusual cases of congenital ichthyosiform erythrodermia. Arch. Dermatol. Syph. 1924;10:487–498. [Google Scholar]
- 15.Chavanas S., Bodemer C., Rochat A., Hamel-Teillac D., Ali M., Irvine A.D., Bonafé J.L., Wilkinson J., Taïeb A., Barrandon Y. Mutations in SPINK5, encoding a serine protease inhibitor, cause Netherton syndrome. Nat. Genet. 2000;25:141–142. doi: 10.1038/75977. [DOI] [PubMed] [Google Scholar]
- 16.Hausser I., Anton-Lamprecht I. Severe congenital generalized exfoliative erythroderma in newborns and infants: A possible sign of Netherton syndrome. Pediatr. Dermatol. 1996;13:183–199. doi: 10.1111/j.1525-1470.1996.tb01202.x. [DOI] [PubMed] [Google Scholar]
- 17.Cassidy A.J., van Steensel M.A., Steijlen P.M., van Geel M., van der Velden J., Morley S.M., Terrinoni A., Melino G., Candi E., McLean W.H. A homozygous missense mutation in TGM5 abolishes epidermal transglutaminase 5 activity and causes acral peeling skin syndrome. Am. J. Hum. Genet. 2005;77:909–917. doi: 10.1086/497707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kurban A.K., Azar H.A. Familial continual skin peeling. Br. J. Dermatol. 1969;81:191–195. doi: 10.1111/j.1365-2133.1969.tb16005.x. [DOI] [PubMed] [Google Scholar]
- 19.Abecasis G.R., Cherny S.S., Cookson W.O., Cardon L.R. GRR: Graphical representation of relationship errors. Bioinformatics. 2001;17:742–743. doi: 10.1093/bioinformatics/17.8.742. [DOI] [PubMed] [Google Scholar]
- 20.Gudbjartsson D.F., Thorvaldsson T., Kong A., Gunnarsson G., Ingolfsdottir A. Allegro version 2. Nat. Genet. 2005;37:1015–1016. doi: 10.1038/ng1005-1015. [DOI] [PubMed] [Google Scholar]
- 21.Abecasis G.R., Cherny S.S., Cookson W.O., Cardon L.R. Merlin—rapid analysis of dense genetic maps using sparse gene flow trees. Nat. Genet. 2002;30:97–101. doi: 10.1038/ng786. [DOI] [PubMed] [Google Scholar]
- 22.Rüschendorf F., Nürnberg P. ALOHOMORA: A tool for linkage analysis using 10K SNP array data. Bioinformatics. 2005;21:2123–2125. doi: 10.1093/bioinformatics/bti264. [DOI] [PubMed] [Google Scholar]
- 23.Leigh I., Watt F. Cambridge University Press; Cambridge: 1994. Keratinocyte Methods. [Google Scholar]
- 24.Allen-Hoffmann B.L., Rheinwald J.G. Polycyclic aromatic hydrocarbon mutagenesis of human epidermal keratinocytes in culture. Proc. Natl. Acad. Sci. USA. 1984;81:7802–7806. doi: 10.1073/pnas.81.24.7802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schäfer-Korting M., Bock U., Diembeck W., Düsing H.J., Gamer A., Haltner-Ukomadu E., Hoffmann C., Kaca M., Kamp H., Kersen S. The use of reconstructed human epidermis for skin absorption testing: Results of the validation study. Altern. Lab. Anim. 2008;36:161–187. doi: 10.1177/026119290803600207. [DOI] [PubMed] [Google Scholar]
- 26.Ackermann K., Borgia S.L., Korting H.C., Mewes K.R., Schäfer-Korting M. The Phenion full-thickness skin model for percutaneous absorption testing. Skin Pharmacol. Physiol. 2010;23:105–112. doi: 10.1159/000265681. [DOI] [PubMed] [Google Scholar]
- 27.Schäfer-Korting M., Bock U., Gamer A., Haberland A., Haltner-Ukomadu E., Kaca M., Kamp H., Kietzmann M., Korting H.C., Krächter H.U. Reconstructed human epidermis for skin absorption testing: Results of the German prevalidation study. Altern. Lab. Anim. 2006;34:283–294. doi: 10.1177/026119290603400312. [DOI] [PubMed] [Google Scholar]
- 28.Moss G.P., Dearden J.C., Patel H., Cronin M.T. Quantitative structure-permeability relationships (QSPRs) for percutaneous absorption. Toxicol. In Vitro. 2002;16:299–317. doi: 10.1016/s0887-2333(02)00003-6. [DOI] [PubMed] [Google Scholar]
- 29.Lundström A., Serre G., Haftek M., Egelrud T. Evidence for a role of corneodesmosin, a protein which may serve to modify desmosomes during cornification, in stratum corneum cell cohesion and desquamation. Arch. Dermatol. Res. 1994;286:369–375. doi: 10.1007/BF00371795. [DOI] [PubMed] [Google Scholar]
- 30.Simon M., Jonca N., Guerrin M., Haftek M., Bernard D., Caubet C., Egelrud T., Schmidt R., Serre G. Refined characterization of corneodesmosin proteolysis during terminal differentiation of human epidermis and its relationship to desquamation. J. Biol. Chem. 2001;276:20292–20299. doi: 10.1074/jbc.M100201200. [DOI] [PubMed] [Google Scholar]
- 31.Caubet C., Jonca N., Brattsand M., Guerrin M., Bernard D., Schmidt R., Egelrud T., Simon M., Serre G. Degradation of corneodesmosome proteins by two serine proteases of the kallikrein family, SCTE/KLK5/hK5 and SCCE/KLK7/hK7. J. Invest. Dermatol. 2004;122:1235–1244. doi: 10.1111/j.0022-202X.2004.22512.x. [DOI] [PubMed] [Google Scholar]
- 32.Levy-Nissenbaum E., Betz R.C., Frydman M., Simon M., Lahat H., Bakhan T., Goldman B., Bygum A., Pierick M., Hillmer A.M. Hypotrichosis simplex of the scalp is associated with nonsense mutations in CDSN encoding corneodesmosin. Nat. Genet. 2003;34:151–153. doi: 10.1038/ng1163. [DOI] [PubMed] [Google Scholar]
- 33.Dávalos N.O., García-Vargas A., Pforr J., Dávalos I.P., Picos-Cárdenas V.J., García-Cruz D., Kruse R., Figuera L.E., Nöthen M.M., Betz R.C. A non-sense mutation in the corneodesmosin gene in a Mexican family with hypotrichosis simplex of the scalp. Br. J. Dermatol. 2005;153:1216–1219. doi: 10.1111/j.1365-2133.2005.06958.x. [DOI] [PubMed] [Google Scholar]
- 34.Matsumoto M., Zhou Y., Matsuo S., Nakanishi H., Hirose K., Oura H., Arase S., Ishida-Yamamoto A., Bando Y., Izumi K. Targeted deletion of the murine corneodesmosin gene delineates its essential role in skin and hair physiology. Proc. Natl. Acad. Sci. USA. 2008;105:6720–6724. doi: 10.1073/pnas.0709345105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Leclerc E.A., Huchenq A., Mattiuzzo N.R., Metzger D., Chambon P., Ghyselinck N.B., Serre G., Jonca N., Guerrin M. Corneodesmosin gene ablation induces lethal skin-barrier disruption and hair-follicle degeneration related to desmosome dysfunction. J. Cell Sci. 2009;122:2699–2709. doi: 10.1242/jcs.050302. [DOI] [PubMed] [Google Scholar]
- 36.Moskowitz D.G., Fowler A.J., Heyman M.B., Cohen S.P., Crumrine D., Elias P.M., Williams M.L. Pathophysiologic basis for growth failure in children with ichthyosis: An evaluation of cutaneous ultrastructure, epidermal permeability barrier function, and energy expenditure. J. Pediatr. 2004;145:82–92. doi: 10.1016/j.jpeds.2004.03.052. [DOI] [PubMed] [Google Scholar]
- 37.Sandilands A., Terron-Kwiatkowski A., Hull P.R., O'Regan G.M., Clayton T.H., Watson R.M., Carrick T., Evans A.T., Liao H., Zhao Y. Comprehensive analysis of the gene encoding filaggrin uncovers prevalent and rare mutations in ichthyosis vulgaris and atopic eczema. Nat. Genet. 2007;39:650–654. doi: 10.1038/ng2020. [DOI] [PubMed] [Google Scholar]
- 38.Weidinger S., Illig T., Baurecht H., Irvine A.D., Rodriguez E., Diaz-Lacava A., Klopp N., Wagenpfeil S., Zhao Y., Liao H. Loss-of-function variations within the filaggrin gene predispose for atopic dermatitis with allergic sensitizations. J. Allergy Clin. Immunol. 2006;118:214–219. doi: 10.1016/j.jaci.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 39.Marenholz I., Nickel R., Rüschendorf F., Schulz F., Esparza-Gordillo J., Kerscher T., Grüber C., Lau S., Worm M., Keil T. Filaggrin loss-of-function mutations predispose to phenotypes involved in the atopic march. J. Allergy Clin. Immunol. 2006;118:866–871. doi: 10.1016/j.jaci.2006.07.026. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.