

Abstract
Sleep is one of the most pervasive biological phenomena, but one whose function remains elusive. Although many theories of function, indirect evidence, and even common sense suggest sleep is needed for an increase in brain energy, brain energy levels have not been directly measured with modern technology. We here report that ATP levels, the energy currency of brain cells, show a surge in the initial hours of spontaneous sleep in wake-active but not in sleep-active brain regions of rat. The surge is dependent on sleep but not time of day, since preventing sleep by gentle handling of rats for 3 or 6 h also prevents the surge in ATP. A significant positive correlation was observed between the surge in ATP and EEG non-rapid eye movement delta activity (0.5–4.5 Hz) during spontaneous sleep. Inducing sleep and delta activity by adenosine infusion into basal forebrain during the normally active dark period also increases ATP. Together, these observations suggest that the surge in ATP occurs when the neuronal activity is reduced, as occurs during sleep. The levels of phosphorylated AMP-activated protein kinase (P-AMPK), well known for its role in cellular energy sensing and regulation, and ATP show reciprocal changes. P-AMPK levels are lower during the sleep-induced ATP surge than during wake or sleep deprivation. Together, these results suggest that sleep-induced surge in ATP and the decrease in P-AMPK levels set the stage for increased anabolic processes during sleep and provide insight into the molecular events leading to the restorative biosynthetic processes occurring during sleep.
Introduction
The subjective experience of sleep as restorative of energy is a commonsense observation, but one not directly studied physiologically with modern technology in discrete brain regions. The importance of sleep and suggestions about its physiological role have been better documented as a negative, by what happens without sleep, since prolonged sleep deprivation (SD) or sleep restriction adversely influences metabolic processes (Knutson, 2007), general emotional and physical health (Haack and Mullington, 2005), and neurocognitive behavior (Lim and Dinges, 2008). An often postulated, although not directly measured, function of sleep is to restore brain energy expended during active waking (Benington and Heller, 1995). Although constituting only 2% of body mass, brain oxygen and glucose utilization account for ∼20% of those of the whole organism (Magistretti, 1999). Compared with wakefulness, indirect evidence that sleep reduces brain energy demands is a 44% reduction in the cerebral metabolic rate (CMR) of glucose (Maquet, 1995) and a 25% reduction in the CMR of O2 (Madsen et al., 1991). Our previous reports indirectly support a link between wake-related neural activation and energy expenditure, since felines showed an increase in extracellular levels of a metabolic by-product of energy, adenosine (AD), in a wake-active brain region, the basal forebrain (Porkka-Heiskanen et al., 1997), and a decline during spontaneous sleep, a pattern also observed in rodent basal forebrain (McKenna et al., 2003). Moreover, adenosine levels increase markedly if sleep is prevented (SD) (Basheer et al., 1999, 2004).
These adenosine studies prompted us to examine the actual “currency of brain cellular energy” ATP, since adenosine may be an indicator of neuronal activity-dependent energy use, by reflecting ATP breakdown. Steady-state ATP levels were once considered to be stable. However, recent brain studies indicate that electrical stimulation, glucose deprivation, or manipulations of Na+/K+ ATPase activity induce detectable changes in ATP levels (MacLean and Luo, 2004; Bao et al., 2005; Christian et al., 2008).
Here, we report that ATP levels are maintained at a steady-state levels during spontaneous waking but the levels exhibit a surge in the initial hours of sleep in brain regions with predominantly wake-active neuronal activity, a surge abolished by preventing sleep, whereas in the “sleep-active” ventrolateral preoptic (VLPO) region preventing sleep does not change ATP levels. In wake-active brain regions, the spontaneous sleep ATP surge positively correlates with the intensity of non-rapid eye movement (NREM) delta activity (slow-wave delta range, 0.5–4.5 Hz), a marker of homeostatic sleep pressure. This ATP–delta correlation is also confirmed by pharmacologically induced sleep using adenosine perfusion into the basal forebrain. Furthermore, the changes in ATP exhibit reciprocity with the phosphorylated state of the cellular energy sensor, phosphorylated AMP-activated protein kinase (P-AMPK), thus supporting the induction of anabolic processes during sleep.
Materials and Methods
Animal and surgical procedures.
Male Sprague Dawley rats (350–400 g) were used for the present study. The rats were housed in cages with a 12 h light/dark cycle (lights on 7:00 A.M. to 7:00 P.M.), a constant temperature of 23°C, and food and water ad libitum. Animals were treated in accordance with the Association for Assessment and Accreditation of Laboratory Animal Care and Use Committee at Boston Veterans Affairs Healthcare System, Harvard University, and National Institutes of Health. Every effort was made to minimize animal suffering and to reduce the number of animals used. For electroencephalogram (EEG) recordings, the rats were implanted with EEG and electromyogram (EMG) electrodes under general anesthesia (intramuscular, ketamine, 7.5 mg/100 g body weight; xylazine, 0.38 mg/100 g; acepromazine, 0.075 mg/100 g). EEG electrodes (stainless-steel screws) were implanted epidurally over the frontal [primary motor; anteroposterior (AP), +2.0; mediolateral (ML), 2.0] and parietal (retrosplenial; AP, −4.0; ML, 1.0) cortices. EMG recording electrodes (silver wires covered with Teflon) were implanted into neck muscles (Basheer et al., 1999).
In AD- and CSF-treated animals, intracerebral guide cannulas (CMA11 guide; CMA/Microdialysis) were implanted 2 mm above the target. The target coordinates for the probe tip in the basal forebrain were as follows: AP, −0.3; ML, 2.2; dorsoventral, 8.8. After the surgery, the rats were housed in individual cages.
EEG recording and analysis.
Seven days after surgery, the animals were transferred to the recording cages in a sound-attenuated room for habituation with attached EEG cables. Baseline EEG was monitored for a 24 h period starting at 7:00 A.M. The EEG/EMG signals were amplified and sampled at 104 Hz. EEG recordings (acquisition using Grass Gamma, version 4.3) were scored using the Rat Sleep Stager (version 3.2) in 10 s epochs manually for NREM sleep, rapid eye movement (REM) sleep, and wakefulness. Recordings were divided into 1 h bins for the 12 h sleep period (light period, 7:00 A.M. to 7:00 P.M.); the amounts of wake, NREM sleep, REM sleep were calculated for the 12 h light period. To determine the changes in slow-wave EEG power in delta range (0.5–4.5 Hz), each animal's mean delta power during NREM episodes for 12 h light period was first measured. Then, for the NREM delta power of each hour, the percentage change from the mean was determined. For spontaneous sleep, delta power furnishes a reasonable proxy for neuronal activity, increased delta power implying overall decreased neuronal activity in wake-active neuronal regions (Steriade and McCarley, 2005; Vyazovskiy et al., 2009). However, in cortex, the negative delta-neuronal activity correlation breaks down for postdeprivation sleep, when neuronal activity greatly increases during the “on” states of cortical delta activity (Vyazovskiy et al., 2009).
In vivo microdialysis of adenosine.
For the microdialysis experiment, 10 rats, implanted with EEG/EMG electrodes and microdialysis cannula, were used. After habituation in the experiment room, 24 h EEG recordings (7:00 A.M. to 7:00 A.M.) were done on the first day (baseline day). On the second day, microdialysis probes were inserted into the target area via the guide cannula ∼20 h before the start of the experiment. On the third day, the microdialysis lines were connected to the probe and pump at 4:00 P.M., and the artificial CSF (aCSF) (147 mm NaCl, 3 mm KCl, 1.2 mm CaCl, 1.0 mm MgCl, pH 6.6) perfusion was begun at 5:00 P.M. One group of rats (n = 5), indicated as AD group, was unilaterally perfused with aCSF for 2 h (5:00 P.M. to 7:00 P.M.), followed by 3 h (7:00 P.M. to 10:00 P.M.) of perfusion with AD (300 μm; Sigma-Aldrich), whereas the other group, aCSF controls (n = 5), continued to receive aCSF. Previous studies from our laboratory showed that unilateral perfusion with 300 μm AD increased NREM sleep and delta power, when compared with rats perfused with aCSF (Basheer et al., 1999). EEG was recorded throughout the whole infusion period. At the end of the experiment the rats were decapitated, the microdialysis probe removed and frontal cortex (FC), basal forebrain (BF), cingulate cortex (CCX), and hippocampus (HIPP) dissected as described below. We note that the ipsilateral side was used for confirming the position of the tip of the cannula and the contralateral side was used for ATP analysis.
SD.
SD was done by “gentle handling,” which, according to standard protocols (Franken et al., 1991), involved presentation of new objects into the cage or gentle touching by a brush or hands when new objects did not keep awake. SD either began at 7:00 A.M. (ending at 10:00 A.M. for 3 h SD or 1:00 P.M. for 6 h SD) or began at 10:00 A.M., ending at 1:00 P.M. During SD, the rats continued to have access to food and water ad libitum. During this period, rats consumed twice the usual amount of food compared with sleeping controls.
Tissue collection and measurement of ATP content.
The rats were killed by decapitation, and brains were removed. Coronal slices (2 mm thick) were carefully placed on a dry ice (−78.5°C)-containing covered Petri dish for rapid freezing and subsequent dissection. The following six brain regions were dissected: FC (tissue volume, ∼2 × 2 × 1 mm; bregma, 4.2 to 2.2), BF (∼2 × 1 × 1 mm; bregma, −0.26 to −1.2), CCX (∼2 × 2 × 1 mm; bregma, −0.26 to −1.2), lateral hypothalamus (LH) (∼1 × 1.0 × 0.2 mm; bregma, −1.3 to −1.5), VLPO (∼0.2 × 0.2 × 0.2 mm; bregma, −0.3 to −0.5), and the entire HIPP. Extreme care was exercised to complete this process rapidly, with an average time of 80 ± 9 s for tissue collection, both during dark and light periods for all the animals; collection times did not differ between dark and light periods. The dissected regions were kept frozen on dry ice and stored at −80°C until used for biochemical measurements.
Determination of ATP was performed by a luciferin–luciferase-based assay (Lundin and Thore, 1975; McElroy and DeLuca, 1983) using a commercial ATP assay system with bioluminescence detection kit (Enliten; Promega). The assay principle is that, in the presence of ATP and oxygen, luciferase from Photinus pyralis catalyzes d-luciferin to oxyluciferin, Pi, AMP, carbon dioxide, and light. The light intensity is measured by luminometry. This technique has been widely used for ATP measurement in cell cultures, slices, and also to measure in vivo changes using frozen dissected tissue including brain tissue (Bao et al., 2005; Park et al., 2006; Chang et al., 2008; Christian et al., 2008). ATP was measured according to the manufacturer's protocol. Briefly, weighed tissue samples were homogenized in 5% trichloroacetic acid (TCA) and transferred to 1.5 ml Eppendorf tubes. The samples were centrifuged at 5000 rpm in cold for 5 min, and the supernatant were transferred to a fresh tube. Samples (10 μl) were neutralized with Tris acetate buffer (490 μl) adjusted to a pH value of 7.75. The luciferase reagent was added immediately before measurement in the luminometer (Flexstation III; Molecular Devices), as described by the supplier. A new standard curve was made daily before each measurement using known standards and ATP-free water. The absolute concentration was calculated per milligram of wet tissue weight using known ATP standards provided in the kit. ATP concentrations are expressed as 10−8 m/mg tissue. This method gave highly reproducible results. The mean coefficient of variation (SD/mean) at the same time of day 10:00 P.M. in the brain regions was 0.13.
Reversed-phase HPLC.
The concentrations of phosphocreatine (PCr), creatine (Cr), and AMP were measured using reversed-phase HPLC and UV detection as described previously (Helzberg et al., 1987; Dworak et al., 2007). A C-18, 150 × 3.9 mm, 4 μm particle size column (Waters) was equilibrated with a mobile phase (buffer A) containing 14 mm H2KO4P and 3 mm tetrabutylammonium bisulfate (Sigma-Aldrich) adjusted to pH 5.4 with 2 m KOH. A step gradient was obtained with the second mobile phase (buffer B) containing 70% methanol. The gradient was formed as follows: 0–4 min 100% buffer A; 4–20 min up to 40% buffer B; 20–25 min 40% buffer B; 25–35 min up to 100% buffer A. The flow rate throughout chromatographic runs was 1.0 ml/min. A new standard curve was made daily. All reagents were of the highest purity available. PCr and Cr were purchased from Fluka. AMP was purchased from Sigma-Aldrich. TCA-supernatants were neutralized with 2.0 m K2HPO4. The compounds (PCr, Cr, AMP) were identified on the basis of the retention time. Quantitative analysis of both standards and samples was performed at 210 nm wavelengths for PCr and Cr and 254 nm wavelength for AMP.
Determination of P-AMPK.
The changes in P-AMPK levels were examined in frontal cortex and basal forebrain only. These two regions exhibit the highest surge in ATP during sleep. In BF, tissue samples (n = 4 each group/region) were collected at 7:00 A.M. (baseline ATP levels), 10:00 A.M. (highest surge in ATP), 1:00 P.M., and 7:00 P.M. Samples were also collected after 3 h of SD (7:00 A.M. to 10:00 A.M.; no surge in ATP; levels equal to baseline levels) and after 3 h SD followed by 3 h recovery sleep (RS) (ATP levels rise with recovery sleep). In FC, the samples were collected at 7:00 A.M., 10:00 A.M., and after 3 h of SD. The samples were collected with extreme rapidity on dry ice as described above. Samples were homogenized on ice in a lysis buffer containing a RIPA buffer, okadaic acid (25 μm), staurosporine (25 μm), and a protease inhibitor and transferred to an Eppendorf tube. Samples were centrifuged at 5k in cold for 5 min, and the supernatant was transferred to a fresh Eppendorf tube. Protein concentration was estimated using BCA methods (Pierce) and 20 μg protein samples were electrophoresed in 12% SDS-PAGE gels. Proteins were electroblotted onto nylon membranes for Western blotting. P-AMPKα1 rabbit polyclonal antibody (1:1000 dilution; sc-33524; Santa Cruz Biotechnology) was used for the detection of Thr172 P-AMPKα1 by Western blotting. HRP-conjugated goat anti-rabbit (1:5000) was used to detect the Western blot using Western Lightning Chemiluminescence Reagent Plus (PerkinElmer). The membranes were further probed with antibody against β-actin, and the band densities for P-AMPK were normalized using β-actin. We did not repeatedly freeze/thaw samples, since previous work found that P-AMPK levels increase in homogenates because of freeze/thawing (Scharf et al., 2008a).
Results
Diurnal change in ATP levels in rat brain
Male Sprague Dawley rats were maintained under 12 h light/dark periods. Every 3 h for 24 h, starting from 7:00 A.M. (lights on, onset of normal sleep period), ATP levels were determined using a validated luciferin–luciferase ATP detection assay in FC, BF, CCX, and HIPP. Within each of the four brain regions, the steady-state ATP level was stable during the wake period (7:00 P.M. to 7:00 A.M., lights off), but ATP levels dramatically altered values in all four regions during the sleep period (7:00 A.M. to 7:00 P.M., lights on). The average ATP molar values during the wake (dark) period calculated per milligram of tissue wet weight varied between regions (n = 6) but were relatively stable within a region. In contrast, during the light period, when rats were asleep most of the time (66.7 ± 6.3%), ATP levels in each brain region generally were elevated and, most importantly, were not constant, but showed significant alterations in values within each brain region compared with waking (Fig. 1).
Figure 1.
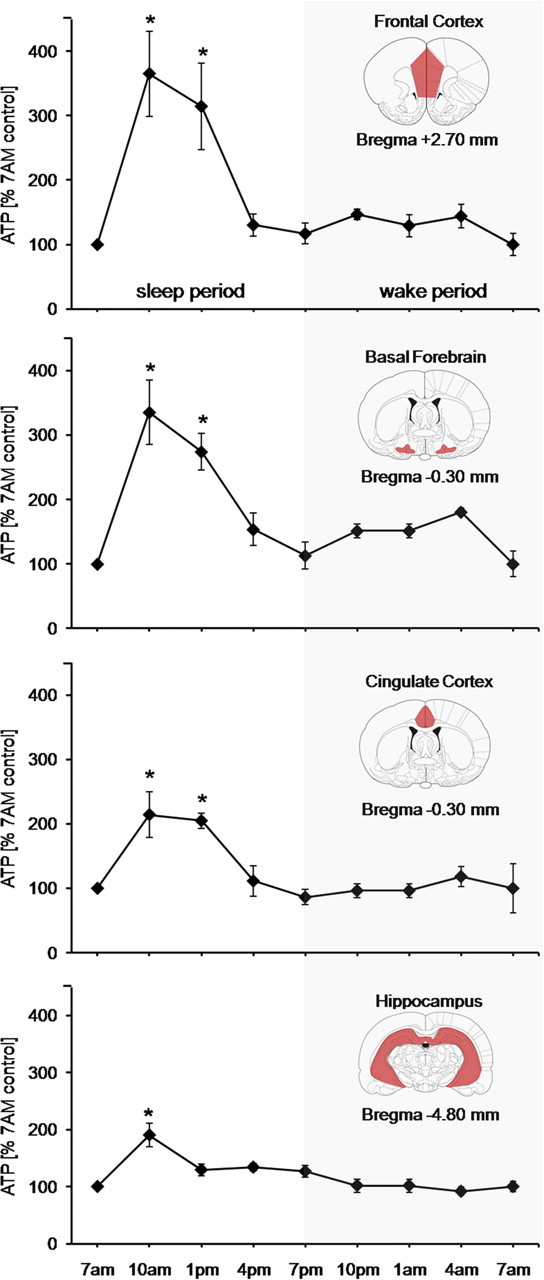
Diurnal variation in steady-state ATP levels in brain regions. Mean ATP levels (±SEM) normalized to 7:00 A.M. values in frontal cortex, basal forebrain, cingulate cortex, and hippocampus. Average ATP values during the wake (dark) period (dark background) remained stable, whereas during the sleep (lights-on) period the ATP levels showed significant alterations in values within each brain region [Kruskal–Wallis ANOVAs, n = 6 in each region; FC (H = 21.622; df = 4; p = 0.001); BF (H = 17.123; df = 4; p = 0.002); CCX (H = 15.449; df = 4; p = 0.004); HIPP (H = 14.439; df = 4; p = 0.006)] compared with waking. The levels surged significantly (n = 6; *p = 0.01) for 10:00 A.M. and 1:00 A.M. compared with other time points. Coronal anatomical plates are from the atlas of Paxinos and Watson (2007).
The increase in ATP during sleep followed a distinct pattern. The lowest levels were seen at 7:00 A.M. and were used as a baseline for comparison of light (sleep) period values and were comparable with the average ATP levels during the wake period (7:00 P.M. to 7:00 A.M.). In the initial sleep period in all four brain regions, ATP levels surged significantly, showing highest values at 10:00 A.M. (n = 6; p < 0.01) that declined slightly by 1:00 P.M., although they remained significantly higher than the baseline values (n = 6; p < 0.01) (Fig. 1). Using the 7:00 A.M. values as baseline for diurnal comparisons, the percentage increases at 10:00 A.M. and 1:00 P.M. were comparable in FC (265 ± 65 and 214 ± 67%) and BF (235 ± 49% and 174 + 28%), but greater than the increases noted in CCX (114 ± 35 and 104 ± 11%) and HIPP (90 ± 20 and 29 ± 10%) (Fig. 1). The levels declined to baseline values by 4:00 P.M. This initial ATP surge follows the initial sleep period surge of EEG NREM delta activity (delta activity, 0.5–4.5 Hz). Delta activity, like ATP, showed significant variability (n = 5; p < 0.013) during sleep, declining toward the end of the sleep period (Fig. 2A,B).
Figure 2.

NREM delta activity for entire lights-on (sleep) period. A, Delta activity values are calculated as percentage of mean NREM delta activity (±SEM) for each hour of NREM sleep, with values placed at midpoint of respective hour. NREM delta activity was significantly higher in the first 4 h compared with later time points (12:00 P.M. to 7:00 P.M.) (F, 2.538; df = 11; *p < 0.05). B, Correlation of the average NREM delta activity and ATP. NREM delta was averaged for 3 h bins (solid line with black diamond) and compared with the ATP levels for basal forebrain (dashed line with open triangle) calculated at the end of corresponding 3 h. Note that the increases in ATP levels closely follow the changes in 3 h average NREM delta activity.
Correlation between ATP and NREM delta activity (0.5–4.5 Hz) during sleep
To furnish an experimental manipulation of EEG NREM delta activity that would further test our hypothesis of NREM delta–ATP association and to rule out potentially confounding diurnal effects, such as light, we used microdialysis to perfuse adenosine (300 μm) unilaterally into the BF at the onset of the active (dark) period, from 7:00 P.M. to 10:00 P.M., a procedure previously shown by us to increase NREM delta (Basheer et al., 1999). Indeed, during microdialysis perfusion, compared with the rats that were perfused with aCSF, adenosine-perfused animals showed higher increases in percentage time in NREM sleep and NREM delta activity when compared with the same time period on a baseline day with no perfusion (Fig. 3A). There was a significant increase in ATP at the end of adenosine perfusion in all four brain regions compared with aCSF controls (Fig. 3B), whereas AMP concentrations showed a tendency toward decrease with statistically significant decrease in BF only [FC, −8.91 ± 3.99% (p = 0.137); BF, −18.19 ± 6.01% (p = 0.032); CCX, −13.18 ± 8.54% (p = 0.343); HIPP, −18.34 ± 7.90% (p = 0.072); t test]. An association between NREM delta and ATP over the 3 h of perfusion was observed, as shown by their strong correlation in FC (ρ = 0.830; p = 0.0006), the site of EEG recording, and BF (ρ = 0.636; p = 0.04), a site known to be related to delta activity (Basheer et al., 1999) (Fig. 3C,D).
Figure 3.

Unilateral AD perfusion into basal forebrain causes highly correlated delta and ATP increases. A, Compared with no treatment (dotted line, 100%) unilateral AD perfusion into basal forebrain (black bars) during 3 h (7:00–10:00 P.M.) in the dark period significantly increased both NREM sleep duration (240.8 ± 38.8%) and NREM delta activity (167.6 ± 27%) (within animal comparisons; n = 5; Student's t test, *p = 0.008). In contrast, aCSF perfusion values (white bars) were not significantly different from no treatment. B, ATP concentrations measured in FC, BF, CCX, and HIPP at the end of 3 h of AD or aCSF infusion into basal forebrain. AD-perfused rats showed significant increases in ATP concentrations in all brain regions when compared with aCSF-perfused controls (n = 5/region for AD and aCSF groups; Kruskal–Wallis ANOVA on ranks, H = 28.455, df = 7, p = 0.001; Student–Newman–Keul pairwise comparison for each region, *p = 0.05). C, D, Correlation analysis of ATP levels and NREM delta activity in AD- and aCSF-perfused rats. In all animals (adenosine, black diamonds; aCSF, white diamonds), a positive correlation was observed between ATP levels and NREM delta in FC (C) (n = 5/group; Spearman's ρ = 0.83; p = 0.0006) and in BF (D) (n = 5/group; Spearman's ρ = 0.64; p = 0.04). Error bars indicate SEM.
Sleep deprivation prevents the ATP surge
To further distinguish whether the surge in ATP was associated with the time of day (diurnal) variation or with sleep behavior, we subjected rats to 3 h SD by gentle handling starting at 7:00 A.M., and examined the ATP levels at 10:00 A.M., the time when ATP levels were highest in diurnal control animals. This SD blocked the ATP surge seen in controls in each of the four brain regions in the SD animals (Fig. 4A–D). When the rats were allowed 3 h RS after the 3 h of SD, only in BF did the ATP levels surge to match the levels of the diurnal controls (1:00 P.M.). In the other brain regions, 6 h of RS was needed to induce the surge in ATP levels. Thus, SD for 3 h postponed the ATP surge in all four regions, although with variable time lags. Interestingly, ATP levels did not decline below the baseline (7:00 A.M. values), suggesting a balance between the ATP synthesis and usage during short-duration SD.
Figure 4.

SD postpones the sleep-induced surge in ATP. A–D, SD (hatched bar) at the 7:00 A.M. onset of light period until 10:00 A.M. significantly attenuated the ATP increase in all brain regions compared with the sleeping diurnal controls (n = 6; *p = 0. 01). When RS (open bars) was permitted after 3 h SD, a surge in ATP occurred after a 3 h lag in basal forebrain and after a 6 h lag in frontal and cingulate cortex. E–H, Three hour SD beginning at 10:00 A.M., when the ATP surge was maximal, caused a decline in ATP to levels lower than sleeping controls at 1:00 P.M. (BF, p = 0.022; FC, p = 0.006; CCX, p = 0.004; HIPP, p = 0.004). When the 3 h SD was followed by 3 h of RS (1:00 P.M. to 4:00 P.M.), ATP levels increased to levels significantly higher (*p < 0.01) than in diurnal controls (4:00 P.M.) in basal forebrain and cingulate cortex, and equal to the diurnal control levels in frontal cortex and hippocampus. I–L, Six hour SD (hatched box) markedly decreased ATP levels compared with diurnal controls in all brain regions (each *p < 0.05). In addition, a small but statistically significant decrease occurred to below the baseline (7:00 A.M.) level in all brain regions (each **p < 0.05) except for FC, where the levels matched the baseline. In all brain regions, 3 h of RS (open bar) produced an ATP increase to levels that matched those of diurnal controls at 4:00 P.M. Error bars indicate SEM.
Next, we addressed whether interrupting the sleep period would also influence the pattern of ATP change observed in normally sleeping animals. Rats were allowed to sleep during the first 3 h of the light period (7:00–10:00 A.M.), allowing the initial ATP surge to occur, and then received 3 h SD (10:00 A.M. to 1:00 P.M.). In normally sleeping control animals, the ATP levels during this period begin to exhibit a slow decline, although still considerably higher than the baseline (Fig. 1). SD at this point in the sleep period increased the rate of decline in ATP, so that ATP levels reached baseline by the end of the 3 h SD, whereas this transition took 6 h in the normally sleeping animals. ATP levels after 3 h SD were significantly lower in all four regions (BF, p = 0.022; FC, p = 0.006; CCX, p = 0.004; HIPP, p = 0.004; n = 6/region) compared with their respective diurnal controls (Fig. 4E–H). When 3 h SD was followed by 3 h of RS (1:00 P.M. to 4:00 P.M.), ATP levels in all brain regions increased during the RS to levels higher than those in their diurnal controls (4:00 P.M.), again demonstrating the sleep dependence of the ATP increase (Fig. 4E–H).
We then examined the effect of a longer duration of SD (6 h; 7:00 A.M. to 1:00 P.M.) on ATP. In contrast to 3 h SD, 6 h SD produced a small but statistically significant decrease below the baseline (7:00 A.M.) level in all brain regions (each p < 0.05) except for FC, where the levels matched the baseline (Fig. 4I–L). At 4:00 P.M., after 6 h SD and 3 h RS, ATP levels in all four brain regions were comparable with the diurnal control values, which were near the baseline levels. Together, these results indicate that, during waking (either spontaneous or caused by SD), ATP levels remain close to the 7:00 A.M. baseline values (3 h SD) or even slightly lower (6 h SD), whereas sleep onset results in a rapid and significant surge in ATP levels. These effects of sleep and wake behavior on brain ATP levels are thus independent of the time of day.
Differential regulation of ATP between sleep-active and wake-active brain regions
To determine whether the increase in ATP is related to local neuronal activity patterns associated with sleep and wake, in a separate experiment (n = 5/group), we compared the 3 h SD-induced changes in the levels of ATP in two functionally diverse regions of hypothalamus, namely the LH known to predominantly contain wake- and REM-active neurons (Szymusiak and McGinty, 2008; Hassani et al., 2009), and the VLPO, which predominantly contains sleep-active neurons (Sherin et al., 1996). In the same rats, we also reexamined FC. After 3 h SD, ATP concentrations were significantly reduced in FC (−53.36 ± 8.01%; p = 0.007) when compared with sleeping controls as was observed in the previous group of rats. ATP also showed significant decrease in LH (−40.17 ± 19.8%; p = 0.048), whereas no significant change was observed in the sleep-active VLPO (+10.69 ± 20.36%; p = 0.719) (Fig. 5).
Figure 5.

Effect of sleep deprivation on the levels of ATP in LH and VLPO. A, Coronal sections of the brain showing bregma coordinates of dissected brain regions: FC, LH, VLPO. B, Rats (N = 5/group), sleep deprived for 3 h (7:00–10:00 A.M.; open bar) showed significant decrease in ATP in FC (−53.00 ± 8.01%; **p < 0.01) and LH (−40.17 ± 19.8%; *p < 0.05), whereas no significant change was observed in VLPO (+10.69 ± 20.36%; p = 0.719) when compared with undisturbed time matched sleeping controls (black bars). Error bars indicate SEM.
SD-induced changes in phosphocreatine and creatine
To further examine the SD-induced changes in the energy status, we measured the levels of PCr and Cr in FC, BF, CCX, and HIPP. PCr serves as a rapidly mobilizable reserve of high-energy phosphates in brain. In the event of increased ATP use, PCr can anaerobically donate a phosphate group to ADP to form ATP. The levels of PCr showed a trend increase at 10:00 A.M. when ATP levels surge. The levels of PCr decreased significantly in FC (−57 ± 17%; p < 0.001), BF (−58 ± 23%; p < 0.01), and CCX (−45 ± 20%; p = 0.044) after 3 h SD (7:00 A.M. to 10:00 A.M.) when compared with undisturbed time matched (10:00 A.M.) controls. HIPP did not show significant change (−24 ± 27; p = 0.228). The Cr levels showed a significant increase in HIPP (+42 ± 5%; p = 0.002), whereas the other three regions showed trend increases (Fig. 6).
Figure 6.

Changes in the phosphocreatine and creatine levels. After 3 h of sleep (10:00 A.M.), the levels of PCr (solid bars) showed a trend increase when compared with the levels observed at 7:00 A.M. in all four regions (n = 6/group), with no change in Cr (open bars) levels. The levels of PCr decreased significantly in FC (−57 ± 17%; *p < 0.001), BF (−58 ± 23%; *p < 0.01), and CCX (−45 ± 20%; *p = 0.044) after 3 h SD (7:00 A.M. to 10:00 A.M.) when compared with undisturbed time-matched sleeping controls. HIPP did not show significant change (−24 ± 27; *p = 0.228). The Cr levels showed a significant increase in HIPP (+42 ± 5%; *p = 0.002), whereas the other three regions showed trend increases. Error bars indicate SEM.
Reciprocal changes in ATP and P-AMPK
We next addressed the mechanism causing the sleep-associated initial surge in ATP to return to baseline levels during the later hours in the light (sleep) period. We investigated whether the AMPK has a role in detecting and responding to ATP changes during sleep and SD. This phylogenetically conserved kinase monitors changes in cellular concentrations of ATP and AMP. Increased ATP usage (higher AMP/ATP ratio) activates AMPK by preventing the dephosphorylation of this kinase (Hardie, 2007). P-AMPK, in turn, phosphorylates multiple downstream target proteins, regulating cellular energy metabolism by inhibition of ATP-consuming anabolic pathways and activation of ATP-generating catabolic pathways (Hardie, 2007). We hypothesized that, at 7:00 A.M., when the AMP/ATP ratio is presumably high after the 12 h of predominantly active period, a more elevated level of P-AMPK would be detected than at 10:00 A.M., at the peak of the sleep-induced ATP surge, accompanied by a decreased AMP/ATP ratio. In contrast, if sleep were to be prevented by 3 h SD between 7:00 A.M. to 10:00 A.M., P-AMPK levels would be expected to remain high until recovery sleep is allowed.
To test this hypothesis, first we examined the changes in P-AMPK levels in BF at 7:00 A.M., 10:00 A.M., 1:00 P.M., and 7:00 P.M. during lights on period and compared them with 3 h of SD (10:00 A.M.) followed by 3 h of RS (1:00 P.M.). As shown in Figure 7, A and B, the levels of P-AMPK decrease at 10:00 A.M. (n = 4; p = 0.02) and 1:00 P.M. and show a tend increase at 7:00 P.M. If sleep is prevented for 3 h, the level of P-AMPK increases significantly (n = 4; p = 0.01) at 10:00 A.M. when compared with the undisturbed sleeping time matched controls and the rats that were allowed 3 h recovery sleep after SD. To further confirm the inverse changes in P-AMPK and ATP, we compared the levels of P-AMPK in BF and FC at two diurnal time points, 7:00 A.M. and 10:00 A.M., with that of the changes in ATP levels at the same time point (ATP levels are highest at 10:00 A.M. when compared with 7:00 A.M. baseline). As was the case with BF, a reciprocal relationship between P-AMPK and ATP was observed in FC (trend level, 7:00 A.M. vs 10:00 A.M.; p = 0.13). Also, in accord with this hypothesis, similar reciprocal relationships between P-AMPK and ATP levels were also observed when 3 h SD rats were compared with 10:00 A.M. diurnal controls (n = 4; BF, p < 0.02; FC showed a similar trend, p < 0.08) (Fig. 7C–F).
Figure 7.

Sleep–wake and SD-associated changes in P-AMPK. A, Western blots show the P-AMPK band intensity and the corresponding band for the normalizer β-actin. B, A graph of the levels of P-AMPK protein in BF show a decline with sleep, with lower values at 10:00 A.M. and 1:00 P.M. when compared with 7:00 A.M. (open triangles; solid line), whereas 3 h of SD (7:00 A.M. to 10:00 A.M.) produces significantly higher levels (n = 4; *p = 0.02) of P-AMPK when compared with the 10:00 A.M. diurnal control and rats that were allowed 3 h of RS (closed squares, dotted line). C–F, Reciprocal relationship between ATP and P-AMPK. C, Western blots show that, in BF, the levels of P-AMPK protein are higher at 7:00 A.M. and after 3 h SD when compared with 10:00 A.M. values in sleeping animals. D, A reciprocal relationship is observed between P-AMPK (open bar) and ATP (black bar) levels in basal forebrain. Compared with 10:00 A.M. values, P-AMPK levels are significantly higher (n = 4; *p = 0.02), whereas the ATP levels are significantly lower (n = 6; p = 0.01) at 7:00 A.M. Compared with 10:00 A.M. values in sleeping controls (C), 10:00 A.M. values in animals not allowed to sleep after 7:00 A.M. (SD) show that the P-AMPK levels remain high (n = 4; *p = 0.02) and ATP values remain low (n = 4; *p = 0.02). E, F, In frontal cortex, a similar reciprocal relationship between P-AMPK (trend-level, 7:00 A.M. vs 10:00 A.M., p = 0.13; 10:00 A.M. vs 3 h SD, p = 0.08) and ATP is present (*p = 0.02). Error bars indicate SEM.
Discussion
Our results provide molecular evidence that ATP, the energy currency of brain cells, shows a surge in the initial hours of sleep, which is tightly correlated with EEG NREM delta activity during spontaneous sleep, and is accompanied by reciprocal changes in phosphorylated AMPK, a main regulator of anabolic and catabolic pathways. Our results are consistent with previous work on whole-brain energy metabolism during sleep–wake, showing an overall increase of high-energy phosphates during sleep (Van den Noort and Brine, 1970; Durie et al., 1978; Dworak et al., 2007). However, early work on brain energetics presented contradictory views on energy changes during sleep (Reich et al., 1972; Durie et al., 1978). Moreover, these studies did not parcellate brain into individual regions, did not measure ATP during a 24 h light/dark cycle, and did not record sufficient electroencephalographic activity to distinguish wake, NREM, and REM sleep. Thus, to our knowledge, this is the first report of ATP changes during sleep in discrete brain regions and its direct relationship to NREM delta activity and P-AMPK.
The diurnal pattern of ATP rise and fall is strikingly similar to the rise and fall of slow-wave delta activity, a marker of sleep homeostasis (Borbély, 2001) during the sleep period (Figs. 1, 2A,B). A similar profile of slow-wave delta activity during the sleep period was also described by Dash et al. (2009). Preventing sleep by keeping the rats awake by gentle handling (SD) prevents the surge in ATP. During recovery sleep that followed SD, the apparent association between the ATP surge and slow-wave delta activity observed during spontaneous sleep, was limited to BF only and not seen for cortical ATP, which increased only after 6 h of RS (Fig. 4A–D). BF has been implicated in RS delta activity (Kalinchuk et al., 2008; Kaur et al., 2008). Our previous work has implicated the adenosine A1 receptor in postsynaptic inhibition of wake–active neurons and the promotion of delta activity (Rainnie et al., 1994; Arrigoni et al., 2006). Importantly, the inhibitory effects of adenosine, generated from ATP released by gliotransmisson, acting on the A1 receptor have also been shown to play a role in both the delta activity and the slow oscillation (<1 Hz) of NREM sleep, and thus gliotransmission may contribute to our ATP findings (Fellin et al., 2009; Halassa et al., 2009). Thus, the net surge in ATP levels during the initial period of sleep is likely attributable to a decreased brain energy use during NREM, when neuronal activity and accompanied energy consumption are low.
Although it is known that SD evokes an increase in delta activity during initial hours of subsequent recovery slow-wave sleep that is proportional to the sleep loss (Tobler and Borbély, 1986; Steriade and McCarley, 2005), the uncoupling between cortical ATP surge and recovery sleep delta (data not shown) that occurs in the initial hours of RS is intriguing. The absence of ATP surge in cortex suggests continued ATP utilization during initial periods of RS. Indeed, the findings of Vyazovskiy et al. (2009) indicate that, although delta is increased in RS after SD, there is a simultaneous increase in neuronal activity in cortex, because of the increased firing during the “on” states of delta activity compared with nondeprived control animals. Thus, although postdeprivation delta is an imperfect indicator of neuronal activity, this direct measurement of neuronal activity supports our main contention that decreased neuronal activity promotes ATP increases and increased activity leads to ATP decreases.
A direct relationship between neuronal activity and energy consumption was suggested as early as 1890 by Roy and Sherrington and confirmed by numerous studies (Attwell and Laughlin, 2001; Dhar and Wong-Riley, 2009; Magistretti, 2009). More recent work indicates that some 87% of brain energy consumption is proportional to neuronal firing rates, predominantly consumed in the restoration of membrane potential after postsynaptic potentials, whereas 13% is used to maintain resting potentials, indicating that excitatory neurotransmission and Na+-K+-ATPase activity consumes most of the energy expended by the brain (Alle et al., 2009; Magistretti, 2009). In accordance with this is our observation on VLPO, an area known to predominantly contain sleep-active neurons (Sherin et al., 1996), in which SD did not have any effect on its ATP levels (Fig. 5). On the contrary, in other wake-active brain areas, SD-induced continued neuronal activation resulted in significant decrease in phosphocreatine levels (Fig. 6), suggesting rapid mobilization of high-energy phosphates from phosphocreatine to prevent ATP depletion.
The observed high ATP levels during sleep might have multiple functional consequences. ATP acts as an activity-dependent metabolite and signaling molecule between neurons and glia and modifies membrane potentials through ATP-modulated potassium (KATP) channels (Fields and Stevens, 2000; Peters et al., 2004; Pascual et al., 2005), thus strongly influencing neuronal information processing, synaptic strength, gene expression, protein, fatty acid, and glycogen synthesis (Fields and Stevens, 2000; Peters et al., 2004; Pascual et al., 2005). One of the supporting pieces of evidence is from the data showing increased glycogen biosynthesis during sleep (Karnovsky et al., 1983; Kong et al., 2002; Petit et al., 2002). However, other studies showed that SD did not affect brain glycogen (Franken et al., 2006) and that the changes are dependent on the age and strain of the animals (Gip et al., 2002). Because of these variable results, and its limited role in total brain energy contribution (Gruetter, 2003), the significance of glycogen metabolism during sleep is not clear.
High ATP levels would also provide the necessary energy for subsequent plasticity changes (Dennis et al., 2001; Hoeffer and Klann, 2010). Higher energy utilization during wake and its reversal during sleep is also congruent with a current hypothesis of synaptic homeostasis (Tononi and Cirelli, 2006), since increased synaptic potentiation (synaptic weight) during wake would contribute to high energy consumption, and the surge in ATP during sleep would reflect the energy-conserving processes such as synaptic downscaling during sleep. Also of note, the availability of ATP in sleep could promote the changes related to synaptic plasticity and memory consolidation (Huber et al., 2004, 2006; Stickgold, 2005; Landsness et al., 2009).
Our results provide direct evidence for an energy-dependent regulation of sleep-related anabolic processes by the reciprocal relationship of P-AMPK and ATP. We observed that P-AMPK levels are high during wake and decrease during sleep, whereas SD induces a significant increase in P-AMPK (Fig. 7). AMPK is known as a major sensor and regulator of cellular energy status through the regulation of multiple biochemical pathways (Hardie, 2007). The phosphorylation status of this protein is changed by the intracellular AMP/ATP ratios. During conditions of high ATP use (high AMP/ATP ratio) as observed during spontaneous waking and SD, AMPK is phosphorylated. Higher P-AMPK switch on catabolic processes that provide alternative routes to generate ATP, such as glycolysis, fatty acid oxidation, and mitochondrial biogenesis, while switching off ATP-consuming anabolic processes (Hardie, 2007). In contrast, when ATP utilization is at its minimum (low AMP/ATP ratio) as observed during sleep, decreased levels of P-AMPK promote anabolic processes via multiple downstream pathways, including the synthesis of fatty acids, glycogen, and protein (Hardie, 2007). P-AMPK regulates protein synthesis at multiple points, including the mammalian target of rapamycin (mTOR) pathway or by activating eEF2 (eukaryote elongation factor 2) (Hardie, 2007). Increased P-AMPK represses the mTOR signal cascades and prevents the maintenance of late-phase long-term-potentiation (Potter et al., 2010), thus directly linking energy metabolism and synaptic plasticity in the mammalian brain (Potter et al., 2010). A decrease in P-AMPK observed at 10:00 A.M. when the ATP levels are high is suggestive of potentiation of anabolic process during sleep. This is also consistent with the recent findings on the role of sleep in memory consolidation, synaptic plasticity and remodeling. Our findings of an increased P-AMPK during waking and SD and a decrease in P-AMPK during sleep are in agreement with previous reports demonstrating an increase in the transcription of genes involved in protein synthesis and synaptic plasticity (Cirelli et al., 2004; Mackiewicz et al., 2007), increased translation of proteins during sleep (Ramm and Smith, 1990; Nakanishi et al., 1997), and, after SD, an overall decrease in protein synthesis (O'Hara et al., 2007).
In summary, our data showing an increase in ATP levels during sleep provide molecular evidence in support of the long-standing view that an important function of sleep is related to providing the brain with increased energy stores (Benington and Heller, 1995; Scharf et al., 2008b). Our data, however, significantly recast the sleep and energy restoration hypothesis. Instead of speaking of energy “restoration,” since ATP levels at the end of the wake period are not strikingly lower than at wake period onset, we restate the hypothesis as “sleep is for an energy surge,” a surge that permits energy-consuming anabolic processes, such as protein and fatty acid synthesis, to occur. Short-term SD delays and longer-term SD both delays and limits the extent of this ATP surge, and this decrease in the surge may impair energy-requiring biosynthetic processes. We summarize our hypotheses and data about the association between sleep–wake-dependent changes in ATP, AMPK, and accompanied anabolic and catabolic pathways in a figure (Fig. 8).
Figure 8.

Model for sleep–wake-dependent changes in ATP, AMPK, and AMPK-regulated anabolic and catabolic pathways. This figure summarizes our hypotheses about regulation of AMPK-dependent anabolic and catabolic processes during sleep and wake. During spontaneous waking and sleep deprivation, both characterized by increased neuronal activity, ATP use is increased and the resulting higher AMP/ATP ratio leads to higher levels of phosphorylated AMPK (P-AMPK). This active form facilitates catabolic processes and inhibits energy requiring anabolic processes. In contrast, during the initial hours of sleep and during recovery sleep after sleep deprivation, states characterized by high NREM delta activity/low neuronal activity, ATP levels surge. This leads to a lower AMP/ATP ratio and a decrease in phosphorylated AMPK. The inactive AMPK allows anabolic processes to proceed including biosynthesis of protein, glycogen, fatty acid, etc., and thereby facilitates restorative biosynthetic processes occurring during sleep.
In conclusion, our data suggest that an initial ATP surge nourishes the anabolic, restorative biosynthetic processes occurring during sleep, in accord with Shakespeare's intuitive phrasing, “Sleep… great nature's second course, Chief nourisher in life's feast” (Macbeth, Act II, Scene II).
Footnotes
This work was supported by a Department of Veterans Affairs Medical Research Service Award (R.B.), Deutsche Forschungsgemeinschaft Fellowship DW66/1-1 (M.D.), and National Institute of Mental Health Grant NIMH39683 (R.W.M.). We gratefully acknowledge Farzana Pervin Nipa for technical assistance, and Diane Ghera and Dewayne Williams for help with animal care.
References
- Alle H, Roth A, Geiger JR. Energy-efficient action potentials in hippocampal mossy fibers. Science. 2009;325:1405–1408. doi: 10.1126/science.1174331. [DOI] [PubMed] [Google Scholar]
- Arrigoni E, Chamberlin NL, Saper CB, McCarley RW. Adenosine inhibits basal forebrain cholinergic and noncholinergic neurons in vitro. Neuroscience. 2006;140:403–413. doi: 10.1016/j.neuroscience.2006.02.010. [DOI] [PubMed] [Google Scholar]
- Attwell D, Laughlin SB. An energy budget for signaling in the grey matter of the brain. J Cereb Blood Flow Metab. 2001;21:1133–1145. doi: 10.1097/00004647-200110000-00001. [DOI] [PubMed] [Google Scholar]
- Bao L, Avshalumov MV, Rice ME. Partial mitochondrial inhibition causes striatal dopamine release suppression and medium spiny neuron depolarization via H2O2 elevation, not ATP depletion. J Neurosci. 2005;25:10029–10040. doi: 10.1523/JNEUROSCI.2652-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basheer R, Porkka-Heiskanen T, Stenberg D, McCarley RW. Adenosine and behavioral state control: adenosine increases c-Fos protein and AP1 binding in basal forebrain of rats. Brain Res Mol Brain Res. 1999;73:1–10. doi: 10.1016/s0169-328x(99)00219-3. [DOI] [PubMed] [Google Scholar]
- Basheer R, Strecker RE, Thakkar MM, McCarley RW. Adenosine and sleep-wake regulation. Prog Neurobiol. 2004;73:379–396. doi: 10.1016/j.pneurobio.2004.06.004. [DOI] [PubMed] [Google Scholar]
- Benington JH, Heller HC. Restoration of brain energy metabolism as the function of sleep. Prog Neurobiol. 1995;45:347–360. doi: 10.1016/0301-0082(94)00057-o. [DOI] [PubMed] [Google Scholar]
- Borbély AA. From slow waves to sleep homeostasis: new perspectives. Arch Ital Biol. 2001;139:53–61. [PubMed] [Google Scholar]
- Chang EJ, Ha J, Oerlemans F, Lee YJ, Lee SW, Ryu J, Kim HJ, Lee Y, Kim HM, Choi JY, Kim JY, Shin CS, Pak YK, Tanaka S, Wieringa B, Lee ZH, Kim HH. Brain-type creatine kinase has a crucial role in osteoclast-mediated bone resorption. Nat Med. 2008;14:966–972. doi: 10.1038/nm.1860. [DOI] [PubMed] [Google Scholar]
- Christian SL, Ross AP, Zhao HW, Kristenson HJ, Zhan X, Rasley BT, Bickler PE, Drew KL. Arctic ground squirrel (Spermophilus parryii) hippocampal neurons tolerate prolonged oxygen-glucose deprivation and maintain baseline ERK1/2 and JNK activation despite drastic ATP loss. J Cereb Blood Flow Metab. 2008;28:1307–1319. doi: 10.1038/jcbfm.2008.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirelli C, Gutierrez CM, Tononi G. Extensive and divergent effects of sleep and wakefulness on brain gene expression. Neuron. 2004;41:35–43. doi: 10.1016/s0896-6273(03)00814-6. [DOI] [PubMed] [Google Scholar]
- Dash MB, Douglas CL, Vyazovskiy VV, Cirelli C, Tononi G. Long-term homeostasis of extracellular glutamate in the rat cerebral cortex across sleep and waking states. J Neurosci. 2009;29:620–629. doi: 10.1523/JNEUROSCI.5486-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis PB, Jaeschke A, Saitoh M, Fowler B, Kozma SC, Thomas G. Mammalian TOR: a homeostatic ATP sensor. Science. 2001;294:1102–1105. doi: 10.1126/science.1063518. [DOI] [PubMed] [Google Scholar]
- Dhar SS, Wong-Riley MT. Coupling of energy metabolism and synaptic transmission at the transcriptional level: role of nuclear respiratory factor 1 in regulating both cytochrome c oxidase and NMDA glutamate receptor subunit genes. J Neurosci. 2009;29:483–492. doi: 10.1523/JNEUROSCI.3704-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durie BDJ, Adam K, Oswald I, Flynn IW. Sleep: cellular energy charge and protein synthetic capability. IRCS Med Sci. 1978;6:351. [Google Scholar]
- Dworak M, Diel P, Voss S, Hollmann W, Strüder HK. Intense exercise increases adenosine concentrations in rat brain: implications for a homeostatic sleep drive. Neuroscience. 2007;150:789–795. doi: 10.1016/j.neuroscience.2007.09.062. [DOI] [PubMed] [Google Scholar]
- Fellin T, Halassa MM, Terunuma M, Succol F, Takano H, Frank M, Moss SJ, Haydon PG. Endogenous nonneuronal modulators of synaptic transmission control cortical slow oscillations in vivo. Proc Natl Acad Sci U S A. 2009;106:15037–15042. doi: 10.1073/pnas.0906419106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fields RD, Stevens B. ATP: an extracellular signaling molecule between neurons and glia. Trends Neurosci. 2000;23:625–633. doi: 10.1016/s0166-2236(00)01674-x. [DOI] [PubMed] [Google Scholar]
- Franken P, Dijk DJ, Tobler I, Borbély AA. Sleep deprivation in rats: effects on EEG power spectra, vigilance states, and cortical temperature. Am J Physiol. 1991;261:R198–R208. doi: 10.1152/ajpregu.1991.261.1.R198. [DOI] [PubMed] [Google Scholar]
- Franken P, Gip P, Hagiwara G, Ruby NF, Heller HC. Glycogen content in the cerebral cortex increases with sleep loss in C57BL/6J mice. Neurosci Lett. 2006;402:176–179. doi: 10.1016/j.neulet.2006.03.072. [DOI] [PubMed] [Google Scholar]
- Gip P, Hagiwara G, Ruby NF, Heller HC. Sleep deprivation decreases glycogen in the cerebellum but not in the cortex of young rats. Am J Physiol Regul Integr Comp Physiol. 2002;283:R54–R59. doi: 10.1152/ajpregu.00735.2001. [DOI] [PubMed] [Google Scholar]
- Gruetter R. Glycogen: the forgotten cerebral energy store. J Neurosci Res. 2003;74:179–183. doi: 10.1002/jnr.10785. [DOI] [PubMed] [Google Scholar]
- Haack M, Mullington JM. Sustained sleep restriction reduces emotional and physical well-being. Pain. 2005;119:56–64. doi: 10.1016/j.pain.2005.09.011. [DOI] [PubMed] [Google Scholar]
- Halassa MM, Florian C, Fellin T, Munoz JR, Lee SY, Abel T, Haydon PG, Frank MG. Astrocytic modulation of sleep homeostasis and cognitive consequences of sleep loss. Neuron. 2009;61:213–219. doi: 10.1016/j.neuron.2008.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardie DG. AMP-activated/SNF1 protein kinases: conserved guardians of cellular energy. Nat Rev Mol Cell Biol. 2007;8:774–785. doi: 10.1038/nrm2249. [DOI] [PubMed] [Google Scholar]
- Hassani OK, Lee MG, Henny P, Jones BE. Discharge profiles of identified GABAergic in comparison to cholinergic and putative glutamatergic basal forebrain neurons across the sleep–wake cycle. J Neurosci. 2009;29:11828–11840. doi: 10.1523/JNEUROSCI.1259-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helzberg JH, Brown MS, Smith DJ, Gore JC, Gordon ER. Metabolic state of the rat liver with ethanol: comparison of in vivo 31phosphorus nuclear magnetic resonance spectroscopy with freeze clamp assessment. Hepatology. 1987;7:83–88. doi: 10.1002/hep.1840070118. [DOI] [PubMed] [Google Scholar]
- Hoeffer CA, Klann E. mTOR signaling: at the crossroads of plasticity, memory and disease. Trends Neurosci. 2010;33:67–75. doi: 10.1016/j.tins.2009.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber R, Ghilardi MF, Massimini M, Tononi G. Local sleep and learning. Nature. 2004;430:78–81. doi: 10.1038/nature02663. [DOI] [PubMed] [Google Scholar]
- Huber R, Ghilardi MF, Massimini M, Ferrarelli F, Riedner BA, Peterson MJ, Tononi G. Arm immobilization causes cortical plastic changes and locally decreases sleep slow wave activity. Nat Neurosci. 2006;9:1169–1176. doi: 10.1038/nn1758. [DOI] [PubMed] [Google Scholar]
- Kalinchuk AV, McCarley RW, Stenberg D, Porkka-Heiskanen T, Basheer R. The role of cholinergic basal forebrain neurons in adenosine-mediated homeostatic control of sleep: lessons from 192 IgG-saporin lesions. Neuroscience. 2008;157:238–253. doi: 10.1016/j.neuroscience.2008.08.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karnovsky ML, Reich P, Anchors JM, Burrows BL. Changes in brain glycogen during slow-wave sleep in the rat. J Neurochem. 1983;41:1498–1501. doi: 10.1111/j.1471-4159.1983.tb00853.x. [DOI] [PubMed] [Google Scholar]
- Kaur S, Junek A, Black MA, Semba K. Effects of ibotenate and 192IgG-saporin lesions of the nucleus basalis magnocellularis/substantia innominata on spontaneous sleep and wake states and on recovery sleep after sleep deprivation in rats. J Neurosci. 2008;28:491–504. doi: 10.1523/JNEUROSCI.1585-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knutson KL. Impact of sleep and sleep loss on glucose homeostasis and appetite regulation. Sleep Med Clin. 2007;2:187–197. doi: 10.1016/j.jsmc.2007.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong J, Shepel PN, Holden CP, Mackiewicz M, Pack AI, Geiger JD. Brain glycogen decreases with increased periods of wakefulness: implications for homeostatic drive to sleep. J Neurosci. 2002;22:5581–5587. doi: 10.1523/JNEUROSCI.22-13-05581.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landsness EC, Crupi D, Hulse BK, Peterson MJ, Huber R, Ansari H, Coen M, Cirelli C, Benca RM, Ghilardi MF, Tononi G. Sleep-dependent improvement in visuomotor learning: a causal role for slow waves. Sleep. 2009;32:1273–1284. doi: 10.1093/sleep/32.10.1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim J, Dinges DF. Sleep deprivation and vigilant attention. Ann N Y Acad Sci. 2008;1129:305–322. doi: 10.1196/annals.1417.002. [DOI] [PubMed] [Google Scholar]
- Lundin A, Thore A. Analytical information obtainable by evaluation of the time course of firefly bioluminescence in the assay of ATP. Anal Biochem. 1975;66:47–63. doi: 10.1016/0003-2697(75)90723-x. [DOI] [PubMed] [Google Scholar]
- Mackiewicz M, Shockley KR, Romer MA, Galante RJ, Zimmerman JE, Naidoo N, Baldwin DA, Jensen ST, Churchill GA, Pack AI. Macromolecule biosynthesis: a key function of sleep. Physiol Genomics. 2007;31:441–457. doi: 10.1152/physiolgenomics.00275.2006. [DOI] [PubMed] [Google Scholar]
- MacLean DB, Luo LG. Increased ATP content/production in the hypothalamus may be a signal for energy-sensing of satiety: studies of the anorectic mechanism of a plant steroidal glycoside. Brain Res. 2004;1020:1–11. doi: 10.1016/j.brainres.2004.04.041. [DOI] [PubMed] [Google Scholar]
- Madsen PL, Schmidt JF, Wildschiødtz G, Friberg L, Holm S, Vorstrup S, Lassen NA. Cerebral O2 metabolism and cerebral blood flow in humans during deep and rapid-eye-movement sleep. J Appl Physiol. 1991;70:2597–2601. doi: 10.1152/jappl.1991.70.6.2597. [DOI] [PubMed] [Google Scholar]
- Magistretti PJ. Brain energy metabolism. In: Zigmond M, Bloom FE, Landis S, Roberts J, Squire L, editors. Fundamental neuroscience. San Diego: Academic; 1999. pp. 389–413. [Google Scholar]
- Magistretti PJ. Neuroscience. Low-cost travel in neurons. Science. 2009;325:1349–1351. doi: 10.1126/science.1180102. [DOI] [PubMed] [Google Scholar]
- Maquet P. Sleep function(s) and cerebral metabolism. Behav Brain Res. 1995;69:75–83. doi: 10.1016/0166-4328(95)00017-n. [DOI] [PubMed] [Google Scholar]
- McElroy WD, DeLuca MA. Firefly and bacterial luminescence: basic science and applications. J Appl Biochem. 1983;5:197–209. [PubMed] [Google Scholar]
- McKenna JT, Dauphin LJ, Mulken KJ, Stronge AM, McCarley RW, Strecker RE. Nocturnal elevation of extracellular adenosine in the rat basal forebrain. Sleep Res Online. 2003;5:155–160. [Google Scholar]
- Nakanishi H, Sun Y, Nakamura RK, Mori K, Ito M, Suda S, Namba H, Storch FI, Dang TP, Mendelson W, Mishkin M, Kennedy C, Gillin JC, Smith CB, Sokoloff L. Positive correlations between cerebral protein synthesis rates and deep sleep in Macaca mulatta. Eur J Neurosci. 1997;9:271–279. doi: 10.1111/j.1460-9568.1997.tb01397.x. [DOI] [PubMed] [Google Scholar]
- O'Hara BF, Ding J, Bernat RL, Franken P. Genomic and proteomic approaches towards an understanding of sleep. CNS Neurol Disord Drug Targets. 2007;6:71–81. doi: 10.2174/187152707779940745. [DOI] [PubMed] [Google Scholar]
- Park J, Lee SB, Lee S, Kim Y, Song S, Kim S, Bae E, Kim J, Shong M, Kim JM, Chung J. Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature. 2006;441:1157–1161. doi: 10.1038/nature04788. [DOI] [PubMed] [Google Scholar]
- Pascual O, Casper KB, Kubera C, Zhang J, Revilla-Sanchez R, Sul JY, Takano H, Moss SJ, McCarthy K, Haydon PG. Astrocytic purinergic signaling coordinates synaptic networks. Science. 2005;310:113–116. doi: 10.1126/science.1116916. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The rat brain in stereotaxic coordinates. Ed 6. New York: Academic; 2007. [DOI] [PubMed] [Google Scholar]
- Peters A, Schweiger U, Pellerin L, Hubold C, Oltmanns KM, Conrad M, Schultes B, Born J, Fehm HL. The selfish brain: competition for energy resources. Neurosci Biobehav Rev. 2004;28:143–180. doi: 10.1016/j.neubiorev.2004.03.002. [DOI] [PubMed] [Google Scholar]
- Petit JM, Tobler I, Allaman I, Borbély AA, Magistretti PJ. Sleep deprivation modulates brain mRNAs encoding genes of glycogen metabolism. Eur J Neurosci. 2002;16:1163–1167. doi: 10.1046/j.1460-9568.2002.02145.x. [DOI] [PubMed] [Google Scholar]
- Porkka-Heiskanen T, Strecker RE, Thakkar M, Bjorkum AA, Greene RW, McCarley RW. Adenosine: a mediator of the sleep-inducing effects of prolonged wakefulness. Science. 1997;276:1265–1268. doi: 10.1126/science.276.5316.1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potter WB, O'Riordan KJ, Barnett D, Osting SM, Wagoner M, Burger C, Roopra A. Metabolic regulation of neuronal plasticity by the energy sensor AMPK. PLoS One. 2010;5:e8996. doi: 10.1371/journal.pone.0008996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rainnie DG, Grunze HC, McCarley RW, Greene RW. Adenosine inhibition of mesopontine cholinergic neurons: implications for EEG arousal. Science. 1994;263:689–692. doi: 10.1126/science.8303279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramm P, Smith CT. Rates of cerebral protein synthesis are linked to slow wave sleep in the rat. Physiol Behav. 1990;48:749–753. doi: 10.1016/0031-9384(90)90220-x. [DOI] [PubMed] [Google Scholar]
- Reich P, Geyer SJ, Karnovsky ML. Metabolism of brain during sleep and wakefulness. J Neurochem. 1972;19:487–497. doi: 10.1111/j.1471-4159.1972.tb01358.x. [DOI] [PubMed] [Google Scholar]
- Scharf MT, Mackiewicz M, Naidoo N, O'Callaghan JP, Pack AI. AMP-activated protein kinase phosphorylation in brain is dependent on method of killing and tissue preparation. J Neurochem. 2008a;105:833–841. doi: 10.1111/j.1471-4159.2007.05182.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharf MT, Naidoo N, Zimmerman JE, Pack AI. The energy hypothesis of sleep revisited. Prog Neurobiol. 2008b;86:264–280. doi: 10.1016/j.pneurobio.2008.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherin JE, Shiromani PJ, McCarley RW, Saper CB. Activation of ventrolateral preoptic neurons during sleep. Science. 1996;271:216–219. doi: 10.1126/science.271.5246.216. [DOI] [PubMed] [Google Scholar]
- Steriade M, McCarley RW. Brain control of wakefulness and sleep. Ed 2. New York: Kluwer Academic/Plenum; 2005. [Google Scholar]
- Stickgold R. Sleep-dependent memory consolidation. Nature. 2005;437:1272–1278. doi: 10.1038/nature04286. [DOI] [PubMed] [Google Scholar]
- Szymusiak R, McGinty D. Hypothalamic regulation of sleep and arousal. Ann N Y Acad Sci. 2008;1129:275–286. doi: 10.1196/annals.1417.027. [DOI] [PubMed] [Google Scholar]
- Tobler I, Borbély AA. Sleep EEG in the rat as a function of prior waking. Electroencephalogr Clin Neurophysiol. 1986;64:74–76. doi: 10.1016/0013-4694(86)90044-1. [DOI] [PubMed] [Google Scholar]
- Tononi G, Cirelli C. Sleep function and synaptic homeostasis. Sleep Med Rev. 2006;10:49–62. doi: 10.1016/j.smrv.2005.05.002. [DOI] [PubMed] [Google Scholar]
- Van den Noort S, Brine K. Effect of sleep on brain labile phosphates and metabolic rate. Am J Physiol. 1970;218:1434–1439. doi: 10.1152/ajplegacy.1970.218.5.1434. [DOI] [PubMed] [Google Scholar]
- Vyazovskiy VV, Olcese U, Lazimy YM, Faraguna U, Esser SK, Williams JC, Cirelli C, Tononi G. Cortical firing and sleep homeostasis. Neuron. 2009;63:865–878. doi: 10.1016/j.neuron.2009.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]