

Abstract
The drug discovery and development enterprise, traditionally an industrial juggernaut, has spanned into the academic arena that is partially motivated by the National Institutes of Health Roadmap highlighting translational science and medicine. Since drug discovery and development represents a pipeline of basic to clinical investigations it meshes well with the prime “bench to the bedside” directive of translational medicine. The renewed interest in drug discovery and develpoment in academia provides an opportunity to rethink the hiearchary of studies with the hope to improve the staid approaches that have been critizied for lacking innovation. One area that has received limited attention concerns the use of pharmacokinetic [PK] and pharmacodynamic [PD] studies in the drug development process. Using anticancer drug development as a focus, this review will address past and current deficencies in how PK/PD studies are conducted and offer new strategies that might bridge the gap between preclinical and clinical trials.
Keywords: drug development, pharmacokinetics, pharmacodynamics, anticancer drugs, models
Drug discovery and development are the essential driving forces for drug therapy. These forces have been promoted and illustrated in various “pipeline” formats, yet until recently have been largely stagnant in terms of offering innovation to a process that many claim as onerous, and inefficient. Even with the renewed attention to the drug development process that now as erupted beyond the favored bastion of the pharmaceutical industry into academic centers there are areas still entrenched in old paradigms. This can be appreciated in the anticancer drug development area where efficacy models still dominate and drive the process, and a meaningful connection between preclinical and clinical pharmacology is marginal. In this review the role of pharmacokinetics [PKs] and pharmacodynamics [PDs] in anticancer drug development will be highlighted and a foundation cast that places PK/PD investigations central to the drug development process.
Drug Development and the Government: A Time for Change
The drug development process is intertwined with government regulations and the FDA. The FDA has a primary directive to ensure that drugs are safe and effective, and maintain essential standards in clinical trials, yet there is an expectation and growing demand to accelerate the drug development process. If one considers the drug development pipeline [see Figure 1] and the pharmaceutical industry's mandate to gain regulatory approvals rapidly one can understand why drug development science needs to be rethought. The pipeline starting with early drug discovery has been supported by basic science and its fundamental discoveries in biology, and “omic” technologies that have afforded a rich milieu for disease targets and new chemical entities. This basic science enterprise has traditionally been supported by academic institutions in conjunction with NIH research grants. The data indicate that this paradigm is deficient with R&D expenditures rising and the number of new drug approvals stagnant, and in fact is at a low since 2004 [1]. The FDA in 2004 initiated the Critical Path Initiative to improve the drug development process and incorporate the latest scientific advances in the process, and in mostly focused on biomarkers, bioinformatics and clinical design [2]. In somewhat parallel fashion, the NIH Roadmap was launched as a means to transform biomedical research to ensure basic scientific discovery impacted the treatment of diseases. Thus, both government agencies were trying to grapple with significant advances in scientific discovery and the public demand for tangible benefits that led to cross-fertilization of research disciplines and an emphasis on translational science. The fall-out from these large initiatives did lead to a renewed appreciation of quantitative pharmacology in translational medicine, and terms such as “pharmacometrics”, and “model-based drug development” were ever present, yet these procedures found traction in the clinical domain [3-4], and much of the standard way of doing “business” in preclinical animal testing was unchanged.
Figure 1.

The drug discovery-drug development pipeline showing the major activities of each component. The overall journey for a single drug to gain a new drug application [NDA] requires many years, about 10, and costs hundreds of millions of dollars (adapted from [23]).
Historical Role of Pharmacokinetics/Pharmacodynamics in Drug Development
Most often PKs and PDs operated independently in the drug development arena with PK studies the dominant focus for the purposes of meeting regulatory requirements. This facet is partially responsible for the rather mundane approach to PKs in drug development as the regulatory “bar” was set low. There was little consideration on how to extend and integrate required PK analyses with the PD properties of a drug candidate. The FDA mandated studies to provide PK characteristics, referred to as ADME (Absorption, Distribution, Metabolism and Excretion) of new single agents, issued in the forms of “guidances” emphasized systemic PK characteristics, toxicokinetics and safety rather than establishing PK/PD relationships that define efficacy. Given these guidelines there was little motivation for companies to examine drug distribution in a systematic manner or in particular to understand drug disposition in target tissues.
For anticancer drugs, in which the tumor serves as the target tissue it is alarming that well-defined protocols were not established to characterize tumor-specific PK properties.
Although the lack of target tissue PK investigations by pharmaceutical companies may be rationalized from a perspective of meeting regulatory obligations (i.e. “get the drug to market”), were academic bodies undertaking such studies? The answer is no, of course, partially explained by the fact that the academic motor for drug discovery and development has only started. A position paper by pediatric oncologists and scientists in 2002 indicated a strong reliance on tumor efficacy models in their preclinical drug development scheme [5]. Although the placement of these studies would have been post-IND (Investigational New Drug), the scheme, which indicated the use of numerous tumor xenograft and orthotopic models in the road to identifying effective agents to test in the clinic, reflected the prevailing attitude that PK/PD studies were tangential to the process, rather than integral. The proposed PK studies were not extensive and were not designed to build PK models or as a means to link to PD endpoints. Regardless, if anticancer drug evaluations were pre- or post-IND or conducted in industrial or academic centers, the emphasis was to demonstrate tumor shrinkage compared to control. Although information can be gained from tumor size-based efficacy trials, the semi-empirical nature of the investigations creates large gaps in our understanding of critical pharmacological components. Drugs deemed ineffective may have failed for PK or PD causes or both, and without detailed measurements the opportunities to improve, for example, by feedback to medicinal chemists in drug discovery, and learn from the deficiency are lost. Drugs found to significantly reduce tumor growth and prolong animal survival would progress through the drug development system yet without a pharmacological foundation. Questions like; what tumor drug concentrations were effective and what was the extent of target inhibition remained elusive. This PK/PD deficiency compounds the semi-empirical approach to drug development in a number of ways. First, since essentially all cancer chemotherapy is a quest to devise effective multi-drug combinations the addition of other drugs to the mix is happenstance since there are no landmark tumor drug concentrations that should be achieved, and no basis to assess drug interactions. Selection of effective doses for compound A and compound B given individually in a combination may be logical but if the combination fails there is no basis to understand the cause, not to mention how the various permutations of a combination of A + B that entail dose levels, sequences and duration of therapy might define an optimal regimen. Second, tumor size-based efficacy trials without PK/PD limit the ability to translate preclinical findings to the clinic. An effective mg/kg dose in a mouse cannot be used as the starting dose in a Phase I trial of an anticancer agent. Starting doses are based on some fraction of doses causing death, such as an LD10. By establishing internal endpoints; tumor drug concentrations or target inhibition, extrapolation procedures can be employed to initiate PK/PD-driven clinical trials. The lack of preclinical PK/PD data is not simply an issue in choosing starting doses for patients, but curtails the usefulness of available PK/PD data obtained from patients. It is understandable that measurements of tumor drug concentrations and PD responses in patients will be sparse, singular timed samples that present a challenge as to how to integrate across subjects into meaningful assessments of drug delivery to the tumor and target inhibition. Preclinical PK/PD models that have been derived from tumor measurements provides a means to scale-up the models to patients and further allows the limited observations in patients to be beneficial, either to revise the scaled models or to validate them [see below].
To summarize, the major pitfalls in the anticancer drug development pipeline are minimal focus on drug disposition and dynamics in tumors and a reliance on tumor-size or survival efficacy studies.
Even though advances in drug discovery continue as is evidenced by an abundance of new drug entities and new cellular targets to pursue, there is a breakdown in the in vivo phase that has been plagued by a lack of integration between PK/PD and efficacy studies [see Figure 2]. As highlighted in Figure 2, early preclinical drug development is viewed as well-integrated and able to produce sufficient lead candidate compounds to a late preclinical drug development that lacks integration of efficacy and PK studies. The crucial junction of the animal investigations has consequences both in the preclinical and clinical drug development phases, and offers opportunities to enhance its quantitative nature that will permit a translational conduit between early preclinical and clinical investigations.
Figure 2.
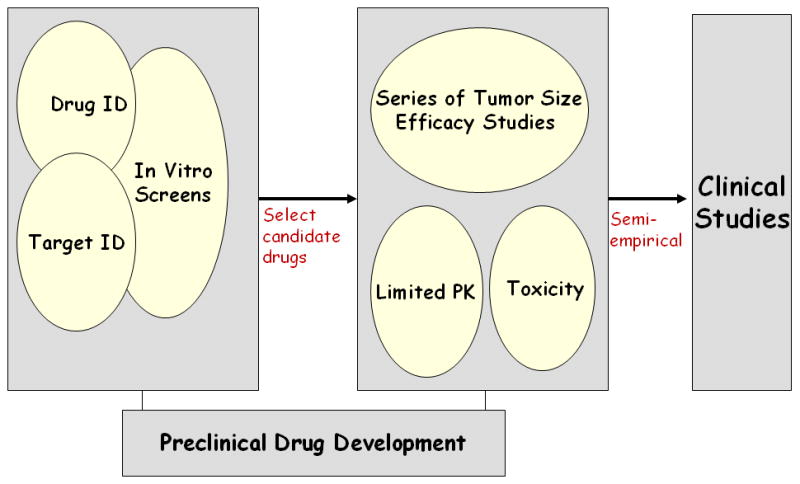
A scheme of traditional anticancer drug development. The early preclinical phase is well-integrated and has applied high-throughput screening tools to the identification [ID] of drugs and targets, and in vitro screens to generate viable lead compounds for the animal study phase. This phase lacks integration and underutilizes PK/PD investigations so that as agents entering clinical trials do so in a semi-empirical manner.
Towards Integration of Pharmacology and Efficacy
There are efforts to improve drug development strategies. A primary deterrent of a drug candidate's suitability to progress in drug development is poor oral bioavailability. As early as 1995, Amidon and coworkers [6] devised the Biopharmaceutical Classification System [BCS] for drugs based on their aqueous solubility and membrane permeability, properties determined in vitro, to predict oral absorption. The BCS that categorized compounds into four classes was adopted by the FDA as a means to assist and by-pass certain in vivo bioavailability studies. Wu and Benet [7] proposed the Biopharmaceutical Drug Disposition Classification System [BDDCS] as an enhancement to the BCS that accounted for routes of drug elimination and active membrane transport processes afforded by numerous gastrointestinal membrane transporters. These efforts coming from academicians highlight an interesting avenue that is increasingly used in drug development efforts; namely, the use of in vitro:in vivo correlations.
It can be appreciated that the ability to reliably predict in vivo ADME properties from in vitro studies can expedite drug development and save resources.
There is considerable interest to employ in vitro methods to predict blood brain barrier [BBB] permeability for drugs considered for neurotherapeutic areas such as Alzheimer's, and multiple sclerosis[8]. Although numerous cell systems have been analyzed, including cells of non-neural origin, there is not a single system that has been adopted, in part, due to the complex nature of the BBB. In deriving a suitable system for in vitro analyses, recapitulation of the tight endothelial cell junctions and electrical resistance, presence of functioning membrane transporters and junctional adhesion proteins are significant hurdles. The challenge of an in vitro system to predict BBB permeability is further compromised by the possibility of needing fresh or low passage endothelial cells. Nonetheless, in vitro models for BBB permeability have been used successfully in certain cases, and future interest is likely to increase as such a tool can be used to as feedback to optimize chemical structure for favorable BBB penetration.
Given the growth of pharmacogenetic-PK relationships that are identified in the clinic after drug approval, there has been a proposal to incorporate in vitro screening of drug metabolism and drug transport characteristics of new chemical entities early in preclinical evaluations [9]. This is not a revolutionary concept as in vitro screens for metabolism and membrane transport are already part of the drug development machinery. What may be new is the use of a broad panel of in vitro metabolic or transport pathway evaluations that for those found to influence drug disposition can be further queried and tested for polymorphic variations to firmly establish pharmacogenetic-PK relationships early in development. The potential benefits not only involve early identification of problem compounds, but improved means to evaluate drug interactions and dosing paradigms for desirable agents.
The aforementioned in vitro screening procedures, and others like these, are likely to be beneficial at least in particular therapeutic areas, however these approaches do not address the real problem of meshing in vivo PK and efficacy studies [see Figure 2]. Similar to the advances in drug discovery that provides ample new chemical entities, the in vitro screens are improved and may alleviate certain bottlenecks in the drug development pipeline, yet the primary culprit of poorly integrated preclinical development is animal testing.
Before considering alternate in vivo strategies, it should be appreciated that in vitro PD endpoints have not been adequately incorporated into the drug development hierarchy. This is particularly apparent in the area of in vitro:in vivo correlations, where the effort is focused on in vitro:in vivo relationships of ADME properties. Biomarkers and PDs have receive considerable attention in the clinical domain, and there is controversy as to their utility [10-11]; however what seems to be missing is a concerted effort to translate in vitro cell-based preclinical findings into tumor-based animal investigations. Such an approach was demonstrated for the EGFR inhibitor, gefitinib, whereby phosphorylation of ERK was an essential marker indicative of drug activity, which was further extended to simulate PD profiles in brain tumor patients [12-13]
One screening approach that has been used to accelerate drug development in animals is referred to as cassette or N-in-1 dosing [14-15]. The technique involves the administration of low doses of multiple candidate compounds to single animals and monitoring drug concentrations in plasma and sometimes, in addition, tissues. The method was originally employed to assess systemic PK properties of oral bioavailability and total clearance; however in selected circumstances drug concentration measurements in tissues has been completed to yield a measure of drug distribution. This later scenario is most often applied to characterize BBB permeability for neuropharmacological agents [16]. The method has been criticized for the possibility of providing erroneous data due to competition for transport and elimination mechanisms between chemical analogues. There have been examples that show the PK parameters obtained in a cassette format are relatively comparable to those obtained following single drug administrations [17]. Given the low doses and a trend in using smaller-sized cassettes the likelihood of PK drug interactions is reduced and a valid risk, particularly since the method is aptly known as a screening methodology.
The cassette dosing protocol is a tool that can partially alleviate the bottleneck of in vivo animal testing to provide key PK parameters; however it does not address the reliance on anticancer drug efficacy studies or allow integration between PK/PD studies and tumor sized-based investigations. One approach that we propose is illustrated in Figure 3, tailored to anticancer drugs that may be useful in the treatment of brain tumors. Steps 1-3 represent a cascade of in silico, in vitro and in vivo studies designed to identify PK/PD failures. The proposed studies overlap with existing screening procedures as generically shown in Figure 2, yet Steps 1-3 delineate a progression of investigations that utilize cassette dosing to determine BBB penetration, and a PD phase that seeks to define PD endpoints that respond in a concentration-dependent manner, and can also be applied to in vivo tumor models highlighted in Step 4. The in silico screen depicted in Step 1 utilizes a computer program [i.e. ADMET Predictor] that given the compound's chemical structure can predict numerous ADME parameters that can assist in the selection of compounds for further investigation. The pitfall with such methods is that the accuracy of the predictions is dependent on the similarity between the molecular descriptors of the test compounds and those compounds used to derive the models internal to the program. The program does flag test compounds that are outside the model's chemical space and had to extrapolate to obtain the final output. The cassette dosing studies in Step 2 can provide the data to build predictive models for BBB permeability for chemotypes of interest. The intended Step 3 PD studies are conducted when putative targets have been identified and makes use of antibody-based assays in an intermediate or meso-scale format. For targeted tyrosine kinase inhibitors key signaling molecules in pertinent pathways can be queried in genotyped cells to identify molecular determinants of drug action, which may further serve as PD endpoints in vivo.
Figure 3.
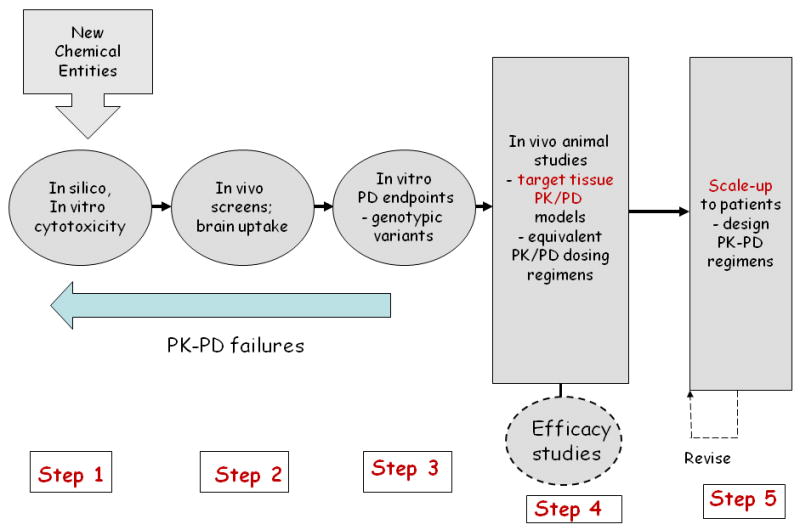
A proposed pharmacokinetic/pharmacodynamic-driven anticancer drug development scheme. Steps 1-3 identify pharmacokinetic/pharmacodynamic failures due to poor systemic properties, brain distribution or insufficient target inhibition. Step 4 studies are initiated with detailed pharmacokinetic/pharmacodynamic investigations that permit the development of physiologically-based pharmacokinetic/pharmacodynamic models that provide a quantitative basis to enter clinical testing.
The most radical departure from standard drug development approaches is in Step 4 where comprehensive PK/PD studies are completed in orthotopic brain tumor models in mice prior to any efficacy testing. A protocol design of 2 dose levels both as single dose and multiple-dose regimens in brain tumor-bearing mice would permit the development of hybrid physiologically-based[PB] PK/PD models [12, 18]. The number and nature of the brain tumor models would be driven by Step 3 that could lead to definition of molecular signatures for drug activity or more rudimentary sensitive and resistant cell lines. By distilling the drug concentration and PD measurements into PB-PK/PD models, additional mouse tumor models can be analyzed, possibly employing an equivalent PK/PD dosing strategy that attempts to “normalize” PD responses based on the molecular determinants with an overall goal to establish a robust PK/PD signature for activity [12]. Step 4 also contains a provision to conduct standard efficacy studies; however these would be driven by the PK/PD models to select dosing regimens to achieve desired PD profiles, both the extent and duration of inhibition can be simulated via the model. The PD input can be used as input into an efficacy model, most readily done with continuous variables such as tumor size [13], and ultimately serves as confirmation that the chosen PD endpoints have a meaningful link to accepted gross measures of efficacy.
The Big Picture and the Pharmacokinetic/Pharmacodynamic Bridge
The drug development pipeline fails as a translational tool unless there is a mechanism to apply the information gathered in the preclinical stages to the clinic.
The standard approach [Figure 2], discussed above, fails and at best is a semi-empirical leap from the lab to the clinic. The alternate approach [Figure 3], aptly referred to as a PK/PD-driven drug development strategy offers a quantitative foundation to transition from the lab to the clinic. The PB-PK/PD (Physiologically-Based) models can be built in a global or hybrid format depending on the extent of tissue data obtained, but in either case the target tissue is represented physiologically with organ blood volume and volumes [19], and thus, minimizes the “black box” compartmental modeling approach that prevents interspecies scale-up. Normal human and preferably patient values replace the corresponding physiological parameters in the animal-based PB-PK/PD models. The challenge in the scale-up procedure is how to estimate the drug-related parameters that include clearance, drug distribution and transport variables. There is no definitive treatise on how this can be accomplished, but parameters determined from drug distribution studies in animals are clearly applicable [18]. Consistent with the original ideas of PB-PK modeling and the recent resurgence in the approach in the drug development domain, a combination of in silico, and in vitro methods may provide reasonable approximations for drug-dependent parameters that may minimize animal testing or relegate them to validation studies [20]. What has not been rigorously addressed is both the need and method to scale-up parameters associated with the PD model. This is a tantalizing subject and one might argue that for certain drug targets only minor adjustments may be needed in patients since for most targeted drugs, such as tyrosine kinase inhibitors, PD endpoints are represented in the model as a fraction of the baseline rather than in absolute units. The net result of the scale-up procedure is to define a PB-PK or PB-PK/PD model in patients that can simulate drug concentrations in tissues and/or PD response in the tumor as a function of time under any desired conditions [13, 18]. The simulation results can be used to set dosing regimens in early clinical trials, and as more patient data becomes available the model can be revised, and thus, offer a tool that can continually assess the pharmacological aspects of therapy.
We have used a preclinical PB-PK/PD model for gefitinib as a basis to derive an analogous model in brain tumor patients [13]. In this case, there was sufficient PK data in patients to derive a simple blood flow-limited PB-PK model that was subsequently linked to the preclinical PD model that used phosphorylation of ERK as the endpoint in tumors. The study was constructed to contrast gefitinib multiple-dose regimens that would be required in patients to attain the same inhibition profile of pERK in tumors with wild-type EGFR and mutant vIIIEGFR, which is the more sensitive tumor [13]. Figure 4 illustrates the results of the modeling procedure that indicates a 2-fold greater daily dose of gefitinib [1000 mg/day] is needed in patients with wild-type EGFR tumors compared to those with vIIIEGFR tumors. Aforementioned questions on how preclinical PD models may be extrapolated to patients, and how more patient data can be collected in tumors are areas that warrant future investigations, yet this simulation highlights how a tumor-based PK/PD model could be used to tailor therapy to patients.
Figure 4.
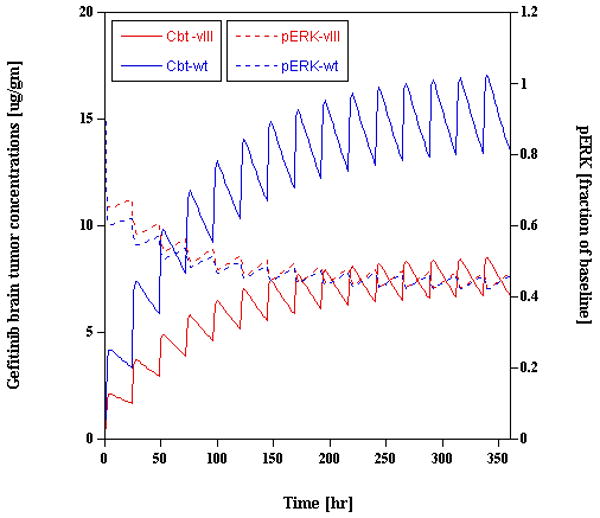
Physiologically-based pharmacokinetic/pharmacodynamic model-predicted gefitinib brain tumor concentrations [left y-axis] and fractional inhibition of pERK [right y-axis] in patients with either wild-type EGFR or mutant vIIIEGFR tumors. To obtain an analogous pERK profile in both tumor types, a regimen of 1000 mg/d or 500 mg/d of gefitinib is predicted in the wild-type EGFR and vIIIEGFR groups, respectively. The simulation used a 15-day duration of gefitinib administration (adapted from reference 13).
The PK/PD-driven approach to drug development is quite consistent with the recent emphasis on individualized therapy [21-22] as there is a merging of patient covariate and pharmacogenetic information with PK/PD parameters.
This area known as population-based PK has been embraced by many clinical pharmacologists and the FDA. The fundamental differences in the proposed PK/PD-driven drug development scheme and the prevailing individualized therapy via population-based methods are two-fold; one, the use of tumor-based PK/PD data, and two, initiation of the model building exercise early in preclinical development. In conclusion, derivation of PB-PK/PD models as a key preclinical goal may limit tedious tumor size-based efficacy studies, and as tumor-based models provide a compelling resource to individualize patient therapy in a rational manner.
Acknowledgments
The author is supported in part by NIH grants CA127963 and CA127963-03S1.
References
- 1.Kaitin KI. Deconstructing the drug development process: the new face of innovation. Clin Pharmacol Ther. 2010;87(3):356–61. doi: 10.1038/clpt.2009.293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Woodcock J, Woosley R. The FDA critical path initiative and its influence on new drug development. Annu Rev Med. 2008;59:1–12. doi: 10.1146/annurev.med.59.090506.155819. [DOI] [PubMed] [Google Scholar]
- 3.Powell JR, Gobburu JV. Pharmacometrics at FDA: evolution and impact on decisions. Clin Pharmacol Ther. 2007;82(1):97–102. doi: 10.1038/sj.clpt.6100234. [DOI] [PubMed] [Google Scholar]
- 4.Danhof M, et al. Mechanism-based pharmacokinetic-pharmacodynamic (PK-PD) modeling in translational drug research. Trends Pharmacol Sci. 2008;29(4):186–91. doi: 10.1016/j.tips.2008.01.007. [DOI] [PubMed] [Google Scholar]
- 5.Houghton PJ, et al. Testing of new agents in childhood cancer preclinical models: meeting summary. Clin Cancer Res. 2002;8(12):3646–57. [PubMed] [Google Scholar]
- 6.Amidon GL, et al. A theoretical basis for a biopharmaceutic drug classification: the correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm Res. 1995;12(3):413–20. doi: 10.1023/a:1016212804288. [DOI] [PubMed] [Google Scholar]
- 7.Wu CY, Benet LZ. Predicting drug disposition via application of BCS: transport/absorption/ elimination interplay and development of a biopharmaceutics drug disposition classification system. Pharm Res. 2005;22(1):11–23. doi: 10.1007/s11095-004-9004-4. [DOI] [PubMed] [Google Scholar]
- 8.Cecchelli R, et al. Modelling of the blood-brain barrier in drug discovery and development. Nat Rev Drug Discov. 2007;6(8):650–61. doi: 10.1038/nrd2368. [DOI] [PubMed] [Google Scholar]
- 9.Katz DA, et al. Defining drug disposition determinants: a pharmacogenetic-pharmacokinetic strategy. Nat Rev Drug Discov. 2008;7(4):293–305. doi: 10.1038/nrd2486. [DOI] [PubMed] [Google Scholar]
- 10.Glassman RH, Ratain MJ. Biomarkers in early cancer drug development: limited utility. Clin Pharmacol Ther. 2009;85(2):134–5. doi: 10.1038/clpt.2008.231. [DOI] [PubMed] [Google Scholar]
- 11.Carden CP, et al. From darkness to light with biomarkers in early clinical trials of cancer drugs. Clin Pharmacol Ther. 2009;85(2):131–3. doi: 10.1038/clpt.2008.223. [DOI] [PubMed] [Google Scholar]
- 12.Wang S, et al. Preclinical pharmacokinetic/pharmacodynamic models of gefitinib and the design of equivalent dosing regimens in EGFR wild-type and mutant tumor models. Mol Cancer Ther. 2008;7(2):407–17. doi: 10.1158/1535-7163.MCT-07-2070. [DOI] [PubMed] [Google Scholar]
- 13.Wang S, Zhou Q, Gallo JM. Demonstration of the equivalent pharmacokinetic/pharmacodynamic dosing strategy in a multiple-dose study of gefitinib. Mol Cancer Ther. 2009;8(6):1438–47. doi: 10.1158/1535-7163.MCT-09-0089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.White RE, Manitpisitkul P. Pharmacokinetic theory of cassette dosing in drug discovery screening. Drug Metab Dispos. 2001;29(7):957–66. [PubMed] [Google Scholar]
- 15.Smith NF, Raynaud FI, Workman P. The application of cassette dosing for pharmacokinetic screening in small-molecule cancer drug discovery. Mol Cancer Ther. 2007;6(2):428–40. doi: 10.1158/1535-7163.MCT-06-0324. [DOI] [PubMed] [Google Scholar]
- 16.Zhang MY, et al. Brain and plasma exposure profiling in early drug discovery using cassette administration and fast liquid chromatography-tandem mass spectrometry. J Pharm Biomed Anal. 2004;34(2):359–68. doi: 10.1016/S0731-7085(03)00523-5. [DOI] [PubMed] [Google Scholar]
- 17.He K, et al. N-in-1 dosing pharmacokinetics in drug discovery: experience, theoretical and practical considerations. J Pharm Sci. 2008;97(7):2568–80. doi: 10.1002/jps.21196. [DOI] [PubMed] [Google Scholar]
- 18.Zhou Q, et al. Predicting human tumor drug concentrations from a preclinical pharmacokinetic model of temozolomide brain disposition. Clin Cancer Res. 2007;13(14):4271–9. doi: 10.1158/1078-0432.CCR-07-0658. [DOI] [PubMed] [Google Scholar]
- 19.Gallo JM, et al. Pharmacokinetic model-predicted anticancer drug concentrations in human tumors. Clin Cancer Res. 2004;10(23):8048–58. doi: 10.1158/1078-0432.CCR-04-0822. [DOI] [PubMed] [Google Scholar]
- 20.De Buck SS, Mackie CE. Physiologically based approaches towards the prediction of pharmacokinetics: in vitro-in vivo extrapolation. Expert Opin Drug Metab Toxicol. 2007;3(6):865–78. doi: 10.1517/17425255.3.6.865. [DOI] [PubMed] [Google Scholar]
- 21.Gerretsen P, et al. The intersection of pharmacology, imaging, and genetics in the development of personalized medicine. Dialogues Clin Neurosci. 2009;11(4):363–76. doi: 10.31887/DCNS.2009.11.4/pgerretsen. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ma BB, Hui EP, Mok TS. Population-based differences in treatment outcome following anticancer drug therapies. Lancet Oncol. 2010;11(1):75–84. doi: 10.1016/S1470-2045(09)70160-3. [DOI] [PubMed] [Google Scholar]
- 23.O'Driscoll C. A Virtual Sapce Odessey. Horizon Symposium Charting Chemical Space. 2004:1–4. [Google Scholar]