

Abstract
By promoting cell proliferation, survival and maturation insulin-like growth factor (IGF)-I is essential to the normal growth and development of the central nervous system. It is clear that IGF-I actions are primarily mediated by the type I IGF receptor (IGF1R), and that phosphoinositide 3 (PI3)-Akt kinases and MAP kinases signal many of IGF-I-IGF1R actions in neural cells, including oligodendrocyte lineage cells. The precise downstream targets of these signaling pathways, however, remain to be defined. We studied oligodendroglial cells to determine whether β-catenin, a molecule that is a downstream target of glycogen synthase kinase-3β (GSK3β) and plays a key role in the Wnt canonical signaling pathway, mediates IGF-I actions. We found that IGF-I increases β-catenin protein abundance within an hour after IGF-I-induced phosphorylation of Akt and GSK3β. Inhibiting the PI3-Akt pathway suppressed IGF-I-induced increases in β-catenin and cyclin D1 mRNA, while suppression of GSK3β activity simulated IGF-I actions. Knocking-down β-catenin mRNA by RNA interference suppressed IGF-I-stimulated increases in the abundance of cyclin D1 mRNA, cell proliferation, and cell survival. Our data suggest that β-catenin is an important downstream molecule in the PI3-Akt-GSK3β pathway, and as such it mediates IGF-I upregulation of cyclin D1 mRNA and promotion of cell proliferation and survival in oligodendroglial cells.
Keywords: IGF-I, β-catenin, GSK3β, oligodendrocytes, signaling transduction
INTRODUCTION
Insulin-like growth factor-I (IGF-I) is widely expressed in the developing central nervous system (CNS), and by promoting cell proliferation, survival and maturation it is essential to normal brain growth (see reviews: D’Ercole and Ye, 2008; D’Ercole et al., 1996). In transgenic (Tg) mice, overexpression of IGF-I in brain results in: 1) an increase in neural cell proliferation (Hodge et al., 2004; Popken et al., 2004; Ye et al, 1996), and 2) reduced cell apoptosis (Chrysis et al., 2001; Hodge et al., 2007; Ye et al., 1996), leading to a significant increase in the number of neurons, oligodendrocytes and astrocytes (Dentremont et al., 1999; Hodge et al., 2005; Ye et al., 1995, 2004). In contrast, blunting the expression of IGF-I (Beck et al., 1995; Ye et al., 2002) or the type I IGF receptor (IGF1R, Liu W et al., 2009; Zeger et al., 2007), or reducing IGF-I availability in brain (Ni et al., 1997; Ye, et al., 1995) significantly retards brain growth by decreasing the proliferation of neural precursors and the survival of neurons and oligodendrocytes.
While it is clear that IGF-I actions are primarily, if not entirely, mediated by the cell surface IGF1R (Baker et al., 1993; Efstratiadis, 1998; Liu et al., 1993), IGF-I intracellular signaling mechanisms remain to be completely elucidated. The phosphoinositide-3 (PI3)-Akt kinase and mitogen activated protein (MAP) kinase pathways have been shown to be important to IGF-I-IGF1R signaling in neural cells, including the cells of oligodendrocyte lineage (see review: Ye and D’Ercole, 2006). The precise downstream targets of the kinases that mediate IGF-I actions have yet to be defined.
β-catenin, a key molecule in the Wnt canonical signaling pathway and a downstream target of glycogen synthase kinase-3β (GSK3β), is an important regulator of neural cell growth. Tg mice overexpressing β-catenin exhibit brain overgrowth with a greater proportion of precursors re-entering the cell cycle (Chenn and Walsh, 2002, 2003). Similarly, when β-catenin expression is increased in the subventricle zone (SVZ) of adult mice, precursor number and cell proliferation are increased (Adachi et al., 2007). In contrast, blunting β-catenin expression leads to reduced brain growth (Machon et al., 2003; Schuller and Rowitch, 2007), likely as a result of decreased neural precursor proliferation (Adachi et al., 2007; Machon et al., 2003). Because IGF- I and β-catenin mutant mice exhibit similar phenotypic changes in neural tissues, and because GSK3β, an enzyme that regulates β-catenin abundance, is a downstream substrate of IGF-I-Akt kinase (Romanelli et al., 2007), we postulated that β-catenin is a downstream mediator in the IGF-I-PI3-Akt-GSK3β signaling pathway. To test this hypothesis, we examined oligodendroglial cultures that express β-catenin (Hughson et al., 1998) and respond to IGF-I with an increased growth (Lagarde et al., 2007; McMorris and Dubois-Dalcq, 1988; Ye and D’Ercole, 1999). Here we provide evidence that in oligodendrocytes: 1) β-catenin is a downstream molecule in IGF-I promotion of cyclin D1 mRNA expression, cell proliferation and survival; and 2) the PI3-Akt kinases-GSK3β pathway plays a role in this signaling.
MATERIALS AND METHODS
OL-1 Oligodendroglial Cell Culture
OL-1 cells were grown in a serum-free defined medium (SFM) supplemented with 30% conditioned medium derived from B104 neuroblastoma cells (B104 CM), as described (Lagarde et al., 2007; Liu H et al., 2009). SFM contains 10 μg/ml insulin (Sigma, St. Louis, MO).
For Western immunoblot analysis, OL-1 cells (4–5 × 105 cells/dish) were plated in 100 mm dishes and expanded by growing in SFM supplemented with 30% B104 CM until cell density reached 3–4 × 106 per dish. After being washed, OL-1 cells were cultured for 3 hr with SFM containing 5 ng/ml of insulin (low-insulin SFM; i.e., a concentration at which insulin does not exert actions through the IGF1R). Experiments were initiated with the addition of IGF-I. For RNA experiments, OL-1 cells (4–5 × 104 cells) were plated into each well of 24-well-plates and cultured with B104 CM-containing SFM overnight to recover from passage shock. OL-1 cells were washed, and cultured with low-insulin SFM for 3 hr before experiments were initiated, as described above.
Primary Oligodendrocyte Precursor (OPC) Culture
Enriched OPC cultures were obtained from brains of Wistar rats at postnatal day of age (P) 1–2, and expanded in DMEM/F12 SFM supplemented with 0.5% FCS and 5 ng/ml basic fibroblast growth factor (bFGF, Invitrogen, Carlsbad, CA), as described (Liu H et al., 2009). Enrichments of OPC were >90%, based on immunostaining of parallel independent cultures with NG2, NeuN and GFAP antibodies. For Western immunoblot analysis, 4 × 105 cells were plated into 100 mm dishes, expanded to 2–3 × 106 cells/dish. After being washed, cells were cultured in low-insulin DMEM/F12 SFM with or without 100 ng/ml of IGF-I.
Western Immunoblot Analysis
Aliquots of 15–20μg protein, extracted as described (Richards et al., 2001), were separated on polyacrylamide gels and transferred onto PVDF membranes. Proteins of interest were detected using specific antibodies and visualized using ECL. Primary antibodies against the following proteins were used: β-actin (1:8,000, Sigma); Akt (1:2,000); phosphorylated Akt at threonine residue (Thr) 308 (pAktThr308, 1:2,000); phosphorylated Akt at serine residue (Ser) 473 (pAktSer473, 1:2,000; Cell Signaling, Danvers, MA), β-catenin (1:5,000, BD Biosciences, Palo Alto, CA); GSK3β (1:1,000, R&D Systems, Minneapolis, MN); phosphorylated GSK3β at Ser 9 (pGSK3βSer9, 1:2,500); Erk1/Erk2 (1:3,000), phosphorylated Erk1/Erk2 (pErk1/pErk2, 1:3,000, Upstate, Temecula, CA); n-Myc (1:4,000, GeneTex, San Antonio, TX), phosphorylated n-myc (pnMyc, 1:2,000, Bethyl Laboratories, Montgomery, TX). Protein abundance was quantified, as described (Ye et al., 2002), after normalizing to either β-actin abundance or the abundance of the respective non-phosphorylated kinase.
Transfection of Small Interfering RNA (siRNA)
OL-1 cells or OPC (5×104 cells) were plated into each well of 24-well-plates. After 6–8 hr in culture, cells were transfected with siRNA duplexes specific for rat β-catenin or a negative control siRNA (pre-designed and synthesized by Qiagen, Valencia, CA), using HiPerFect transfection reagent (Qiagen), as described (Liu H et al., 2009). The target sequences of the β-catenin siRNAs are as follows: 5′ end siRNA: 5′-ctcacttgcaataattacaaa-3′; and 3′ end siRNA: 5′-cagggtgcgatcccacgacta-3′.
Quantitative Real Time-PCR (qRT-PCR)
Total RNA isolation and cDNA reverse transcription were performed as described (Liu H et al., 2009). The resultant mRNA-derived cDNA was quantified by qRT-PCR using primers specific for genes of interest (see supplemental Table S1 for sequences). Primers for 18S rRNA were obtained from Applied Biosystems (Austin, TX). The abundance of mRNA was determined, based on a standard curve for each target mRNA and 18S rRNA, as described (Liu H et al., 2009).
Propidium Iodide (PI) Labeling and Ki67 Immunostaining
To identify apoptotic cells with permeable cell membranes, PI (Sigma, 0.3 mg/ml) was added to cultures for 5 minutes at the end of experiments. Under a florescent inverted microscope equipped with Hoffman module, apoptotic cells with a PI labeled nucleus or intact cell with an unlabeled nucleus were scored. For each well (sample), the number of PI-labeled cells and unlabeled cells was counted in randomly selected 10 high power view fields (40×), and the percentage of PI-labeled cells was calculated.
To identify proliferating cells, cultures were fixed with 4% paraformaldehyde for 10 minutes at room temperature, and subjected to immunostaining with an antibody against Ki67 (1:3,000, Vector Laboratories, Burlingame, CA). As described above, Ki67-positive cells and Ki67-negative cells were counted in 10 randomly selected fields, and the percentage of Ki67-positive cells was calculated.
Statistics
All experiments were repeated at least two times. Student-t test or one-way ANOVA was used to test statistical significance among the groups, followed by comparison of each group mean with the Newman-Keuls-Student test assisted with the software SigmaStat for Windows (SPSS, Inc., Chicago, IL).
RESULTS
Consistent with a previous report showing β-catenin expression in O2A OPC and CG4 cells (Hughson et al., 1998), β-catenin was readily detected in OL-1 oligodendroglial cells by Western immunoblot analysis. IGF-I treatment of OL-1 cells resulted in a 5 to 8 fold increase in the abundance of β-catenin protein that was first observed 1 hr after IGF-I treatment; and this increase persisted for the remainder of the 24 hr period of IGF-I treatment (Figure 1A and 1B). Accompanying the increase in β-catenin abundance, IGF-I treatment also significantly increased the abundance of mRNA for cyclin D1 (Figure 1C), a protein that is involved in the regulation of cell proliferation and whose mRNA expression is known to be regulated by β-catenin. In contrast during same time period, the abundance of nMyc, a known β-catenin target gene in certain cell types, and its phosphorylated form (pnMyc), was similar in IGF-I treated and non-treated cultures. Representative Western immunoblots of nMyc and pnMyc in cells treated with IGF-I for 0.5 hr to 3 hr are shown in Figure 1D.
Figure 1.

IGF-I increase of β-catenin protein and cyclin D1 mRNA abundance in OL-1 cells. Panel A. Representative Western immunoblot of β-catenin in OL-1 cells treated with IGF-I (100 ng/ml). The duration of treatment is indicated at the top of each lane. Panel B. Quantification of β-catenin protein abundance in OL-1 cells treated with 100 ng/ml IGF-I. Protein obtained from cell cultures immediately before the addition of IGF-I served as 0 hr controls. Panel C. Quantification of cyclin D1 mRNA expression in OL-1 cells treated with or without (Control) 100 ng/ml IGF-I for the duration indicated. Panel D. Representative Western immunoblot of phosphorylated nMyc (pnMyc) and total nMyc. The duration of IGF-I (100 ng/ml) treatment is indicated at the top of each lane. In both panels B and C, values represent mean ± SE of 3–6 samples. *, P < 0.05; **, P < 0.01; ***, P < 0.001, compared to 0 hr controls.
To determine whether the increase in IGF-I-stimulated β-catenin mediated IGF-I actions, we utilized siRNA to blunt the expression of β-catenin and examined its impact on IGF-I-stimulated actions. We first examined the specificity of β-catenin siRNA inhibition on β-catenin mRNA expression. Twenty-four hours after transfection, the abundance of β-catenin mRNA and cyclin D1 mRNA in OL-1 cells transfected with β-catenin siRNAs was reduced by ~40% and ~58%, respectively, as compared to cells transfected with control siRNA (supplemental Figure S1). The abundance of mRNA for myelin basic protein (MBP) and proteolipid protein (PLP), two oligodendrocyte/myelin-specific proteins, as well as that of histone deacetylase 11 (HDAC11) mRNA, did not change (supplemental Figure S1), suggesting that blunting β-catenin mRNA expression had specific consequences on cyclin D1 mRNA expression, rather than altering the expression of mRNA non-specifically.
Next we determined whether suppression of β-catenin mRNA expression blocks IGF-I stimulated effects on cyclin D1 mRNA expression and cell proliferation. OL-1 cells were transfected with β-catenin siRNAs or a negative control siRNA for 24 hr, as previously performed. Culture medium then was replaced with low-insulin SFM, and cells were treated with IGF-I for 24 hr (Figure 2A provides a schematic of this protocol). Consistent with the results reported above, the abundance of β-catenin mRNA, but not MBP, PLP and HDAC11 mRNA, was decreased by ~34% in the cells that were previously treated with β-catenin siRNAs for 24 hr (Figure 2B).
Figure 2.

β-catenin siRNA suppression of IGF-I-promoted β-catenin and cyclin D1 mRNA expression in OL-1 cells. Panel A. Schematic of the time course of the β-catenin siRNA experiment. OL-1 cells were treated with β-catenin siRNA (5 nM of each 5′ and 3′ end β-catenin siRNA) or a negative control siRNA (10 nM) for 24 hr. Medium was then replaced with low insulin SFM, and cells were incubated with or without 100 ng/ml of IGF-I for an additional 24 hr. Panel B. Quantification of β-catenin (βCat) mRNA abundance, as well as that of MBP, PLP, HDAC11 (HD11) and cyclin D1 (cycD1), in cells pre-treated with β-catenin siRNA. *, P < 0.05, compared to cells treated with control siRNA (neg siRNA). Panel C. Quantification of β-catenin and cyclin D1 mRNA abundance in IGF-I-treated cells that were pre-transfected with β-catenin siRNA or control siRNA (neg siRNA). mRNA abundance is expressed as a percentage of their non-IGF-I-treated controls (open columns), and values represent mean ± SE of 3–5 samples. *, P < 0.01; **, P < 0.001; compared to non-IGF-I treated controls. ^, P < 0.001; compared to IGF-I treated cells that were pre-transfected with negative control siRNA. Panel D. The number of Ki67+ proliferating cells in IGF-I-treated cells that were pre-transfected with β-catenin siRNA or control siRNA (neg siRNA). Values represent mean ± SE from 3–5 samples. *, P<0.02; **, P<0.001, compared to their respective non-IGF-I treated controls; ^, P<0.01, compared to IGF-I treated cells that were pre-transfected with control siRNA.
When compared to non-IGF-I treated controls, cells treated with IGF-I for 24 hr exhibited significant increases in β-catenin mRNA abundance even with β-catenin siRNA pre-treatment (Figure 2C); however, the magnitude of the IGF-I stimulated increases was blunted with β-catenin siRNA in a dose-dependent manner. More specifically, in cells transfected with control siRNA, IGF-I increased β-catenin mRNA abundance by ~500%, while in cells treated with 2.5 nM or 5 nM β-catenin siRNA, IGF-I increased the β-catenin mRNA abundance by ~250% and ~100%, respectively (Figure 2C). β-catenin siRNA also blocked IGF-I stimulation of cyclin D1 mRNA expression in a dose-dependent manner (by ~50% with 2.5 nM catenin siRNA and by~75% with 5.0 nM catenin siRNA) (Figure 2C).
To determine the consequences of suppression of β-catenin and cyclin D1 mRNA expression by β-catenin siRNA on IGF-I-stimulated cell proliferation, we quantified the number of cells positive for Ki67, a protein that is expressed in the G1, G2 and M phases of the cell cycle. When cultured in low-insulin SFM, only ~7% of OL-1 cells were Ki67 positive (Figure 2D), a finding that is similar to our previous report (Lagarde et al., 2007), and treatment with β-catenin siRNAs did not significantly influence the number of Ki67+ cells (Figure 2D). IGF-I treatment significantly increased the number of Ki67+ cells, even in the presence of β-catenin siRNA pre-treatment. Similar to the changes in cyclin D1 mRNA abundance, however, the magnitude of the IGF-I stimulated increases was blunted by β-catenin siRNA pre-treatment in a dose-dependent manner (Figure 2D).
To determine whether the PI3-Akt-GSK3β pathway mediates IGF-I influence on β-catenin abundance and cyclin D1 mRNA expression, we first assessed the time course of IGF-I actions on phosphorylation of AktSer473 and GSK3βSer9. While phosphorylation of AktSer473 or AktThr308 activates Akt, GSK3β becomes inactivated when its Ser9 residue is phosphorylated. IGF-I treatment rapidly increased the abundance of pAktSer473, which neared its peak of ~1,200% of controls as early as 1 minute after treatment (Figure 3). The increase in AktSer473 phosphorylation remained at high levels for the next 23 hrs. These findings are consistent with previously reported data showing a sustained elevation of pAkt in primary oligodendrocyte cultures in response to IGF-I treatment (Romanelli, et al, 2007).
Figure 3.

PI3 kinase inhibitor wortmannin (Wort) suppression of IGF-I-induced increases in the abundance of pAktSer473, pGSK3βSer9, β-catenin protein, and cyclin D1 mRNA in OL-1 cells. Panel A. Representative Western immunoblot of pAktSer473 and pGSK3βSer9, as well as total Akt and GSK3β. The duration of IGF-I (100 ng/ml) treatment is indicated at the top of each lane. An arrowhead points to pAktSer473 bands. Panel B. Quantification of pAktSer473 and pGSK3βSer9 in OL-1 cells treated with 100 ng/ml IGF-I. Cultures harvested immediately before addition of IGF-I serve as 0 minute controls. Values represent mean ± SE of 3–5 samples. ^, P=0.062; *, P < 0.05; **, P < 0.01; ***, P < 0.001, compared to 0 minute controls. Panel C. pAktSer473 and pGSK3βSer9 abundance in cells treated with IGF-I for 24 hr, which were pretreated with Wort for 1 hr. Panel D. The abundance of β-catenin protein and Cyclin D1 mRNA in cells treated with IGF-I and Wort. In both panels C and D, values represent mean ± SE of 3–5 samples. *, P < 0.05; **, P < 0.01; ***, P < 0.001, compared to non-treated controls. ^, P < 0.05; ^^, P < 0.01; ^^^, P < 0.001, compared to cells treated with IGF-I alone.
As expected, phosphorylation of GSK3βSer9, a known Akt downstream target that is inactivated when phosphorylated, also was increased in a pattern similar to IGF-I induced AktSer473 phosphorylation (Figure 3). The peak of the increase, however, appeared to be one minute later than that of AktSer473. The IGF-I-induced peak increases in the abundance of both pAktSer473 and pGSK3βSer9, however, were ~1 hr and ~4 hr earlier, respectively, than the peak increases in β-catenin protein and cyclin D1 mRNA induced by IGF-I.
The role of PI3-Akt kinases in IGF-I regulation of β-catenin abundance was further determined using wortmannin, an inhibitor of the Akt activating enzyme PI3 kinase. Incubation with wortmannin for 1 hr before IGF-I treatment blunted IGF-I-stimulated AktSer473 and GSK3βSer9 phosphorylation in a dose-dependent manner (Figure 3C, supplemental Figure S2). Consistent with a role for the PI3-Akt kinase pathway in mediating IGF-I actions on β-catenin expression, wortmannin also blocked IGF-I-increased abundance of β-catenin protein, as well as that of cyclin D mRNA after 24 hr in culture (Figure 3D). The former finding is in line with previously reported data showing that PI3 kinase inhibitors block IGF-I-induced accumulation of β-catenin in cultured stem cells (Gehmert et al., 2008). Similar results also were observed in cultures treated with wortmannin and IGF-I for 3 hr (data not shown). In contrast, pre-incubation with PD98059, an inhibitor of Mek (an Erk upstream activating enzyme) did not affect IGF-Istimulation of β-catenin expression, although it partially blocked the IGF-I-induced increase in cyclin D mRNA expression (Figure 4).
Figure 4.

MAP kinase inhibitor PD98059 suppression of Erk phosphorylation and cyclin D1 mRNA expression stimulated by IGF-I in OL-1 cells. Panel A. Representative Western immunoblot of phosphorylated Erk2/1 (pErk2/1) and total Erk2 in cells treated with 100 ng/ml IGF-I. Treatment duration is indicated at the top of each lane. Panel B. Representative Western immunoblot of phosphorylated Erk2/1 (pErk2/1) and total Erk2 in cells treated with 100 ng/ml IGF-I (I) in the presence or absence of 10 or 20 nM PD98059 (P). Panel C. Quantification of β-catenin protein and cyclin D1 mRNA abundance in cells treated with 100 ng/ml IGF-I and 20 nM PD98059 for 24 hr. Values represent mean ± SE of 4–6 samples. *, P < 0.05, **, P < 0.01, compared to control cells. ^, P < 0.05, compared to cells treated with only IGF-I.
IGF-I treatment rapidly increased pGSK3βSer9 and inhibition of PI3 kinase activity suppressed IGF-I actions on GSK3β phosphorylation and β-catenin abundance (Figure 3). These findings are consistent with the concepts that GSK3β is a downstream target of PI3-Akt kinase and has a role in mediating IGF-I actions on β-catenin. To confirm a role for GSK3β in IGF-I signaling, we treated OL-1 cells with LiCl, a compound that induces GSK3Ser9 phosphorylation and in turn suppresses its activity. Without affecting Akt phosphorylation, treatment with 2 mM or 20 mM LiCl for 24 hr significantly increased the abundance of pGSK3Ser9, being ~290% and ~800% of non-treated controls, respectively (Figure 5). Treatment with similar concentration of NaCl (serving as an ion control) did not alter the abundance of pGSK3Ser9. Following a similar pattern, LiCl treatment significantly increased the abundance of β-catenin protein (Figure 5B).
Figure 5.

The abundance of phosphorylated GSK3βSer9 and β-catenin protein, and the number of Ki67+ proliferating cells in LiCl treated OL-1 cultures. Panel A. Representative Western immunoblot analyses of pGSK3βSer9, GSK3β, pAktSer473, Akt and β-catenin. Cells were cultured in B104 CM-free SFM and were treated with or without NaCl and LiCl for 24 hr at the concentration indicated at the top of each line. An arrowhead at the left points to pAktSer473 bands. Panel B. Quantification of pGSK3βSer9 and β-catenin protein abundance. Values represent mean ± SE of 3–5 samples. *, P < 0.01, compared to untreated controls. ^, P < 0.05, compared to control cells treated with 20 mM NaCl. Panel C. The number of Ki67+ proliferating OL-1 cells in cultures treated for 24 hr with 20 nM NaCl, 20 nM LiCl, 100 ng/ml IGF-I, or a combination of LiCl and IGF-I. Values represent mean ± SE of 3–5 samples. *, P < 0.05; **, P<0.01, compared to untreated controls. ^, P < 0.05, compared to cells treated with LiCl alone.
Similar to IGF-I treatment, suppression of GSK3β activity significantly increased the number of Ki67+ proliferating cells in OL-1 cultures. While treatment with NaCl did not alter the number of proliferating cells, treatment with LiCl more than doubled the number of Ki67+ proliferating OL-1 cells (Figure 5C). In IGF-I treated cultures, the number of Ki67+ proliferating cells was further increased by ~20%, as compared to LiCl-treated cultures, and the increases were similar to those in cultures treated with a combination of IGF-I and LiCl (Figure 5C).
As reported above, IGF-I rapidly increases β-catenin protein in OL-1 cells as early as 1 hr after treatment (Figure 1). In addition, as found in siRNA experiments, IGF-I treatment also stimulates an increase in β-catenin mRNA abundance after 24 hr (Figure 2). To further define IGF-I actions on β-catenin mRNA expression, we examined the time course of β-catenin mRNA expression. Unlike the rapid increases in β-catenin protein abundance, β-catenin mRNA abundance was not altered in IGF-I-treated cultures during the first 4 hr of treatment, but was increased by ~3 fold 24 hr after treatment, compared to controls (Figure 6A). These data suggest that while IGF-I-increased β-catenin stability predominately accounts for the protein accumulation during early stages of IGF-I treatment, an increased expression of β-catenin mRNA promoted by IGF-I also contributes to the increase in β-catenin at later times. Pre-treatment with the PI3 kinase inhibitor wortmannin almost completely blunted the IGF-I stimulatory effects on β-catenin mRNA abundance, while the Mek MAP kinase inhibitor PD98095 had little effect (Figure 6B), indicating that PI3-Akt pathway predominantly mediates IGF-I actions to promote β-catenin mRNA expression.
Figure 6.
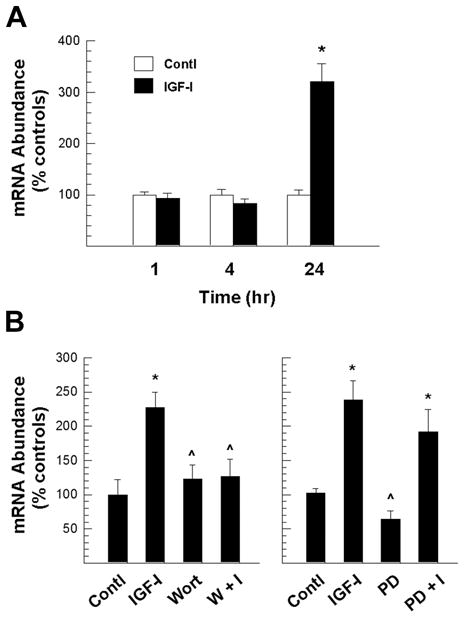
PI3-Akt kinase mediation of IGF-I up-regulated β-catenin mRNA expression in OL-1 cells. Panel A. Time course of IGF-I-stimulated β-catenin mRNA expression. Panel B. Suppression of IGF-I-stimulated β-catenin mRNA expression by inhibition of PI3-Akt (left panel), but not MAP kinase (right panel) pathway. OL-1 cells were pre-treated for 1 hr with either 500 nM of the PI3 kinase inhibitor wortmannin (Wort) or 20 nM of the Erk inhibitor PD98059 (PD) before 24 hr incubation with 100 ng/ml IGF-I. All cells, including controls (Contl), were cultured in the presence of 0.1% DMSO. Values represent mean ± SE of 3–5 samples. *, P < 0.01, compared to control cells. ^, P < 0.05, compared to cells treated with IGF-I only.
It has been shown that β-catenin acts to promote neural cell survival in embryonic mice (Junghans et al., 2005), leading us to believe that β-catenin also may mediate IGF-I promotion of cell survival. To investigate this hypothesis, we suppressed β-catenin expression in IGF-I-treated cultures using β-catenin siRNA, and quantified the number of dead cells in live cultures using propidium iodide (PI), a fluorescent dye that stains the nuclei of dead cells and can not permeate the membranes of viable cells. In OL-1 cultures pre-treated with negative siRNA, ~51% of cells were apoptotic, but only ~11% of cells were apoptotic when IGF-I was added to culture medium (Table 1), a finding that is consistent with published reports that IGF-I enhances the survival of oligodendroglial cells (Barres et al., 1992; Mason et al., 2000; Vemuri and McMorris, 1996; Ye and D’Ercole, 1999). This IGF-I-enhanced cell survival was partially blocked by β-catenin siRNA treatment. Compared to IGF-I-treated cells pre-transfected with control siRNA, the number of PI stained apoptotic cells was increased by 31% and 61% in IGF-I-treated cultures that were pre-treated with 2.5 nM or 5 nM β-catenin siRNAs, respectively (Table 1).
Table 1.
Apoptotic cell number in OL-1 cultures treated with IGF-I in presence or absence of β-catenin siRNAs.
| Negative control siRNA | β-Catenin siRNAs |
||
|---|---|---|---|
| 2.5 nM | 5.0 nM | ||
| Control | 50.67 ± 2.16 | 56.54 ± 2.30 | 55.84 ± 1.06 |
| IGF | 10.63 ± 1.03 | 13.88 ± 1.10 (↑31%) | 17.12 ± 0.90 (↑61%)* |
OL-1 cells were transfected with β-catenin siRNAs or a control siRNA for 24 hr, followed by culture with or without IGF-I (100 ng/ml) for an additional 24 hr. The number of PI-labeled apoptotic cells is expressed as a percentage of total cells. Numbers in parentheses indicate percentage changes over IGF-I treated cells transfected with a control siRNA. Values represent mean ± SE from 3–5 samples.
P <0.05, compared to IGF-I-treated cells pre-transfected with a control siRNA.
In contrast, suppression of the activity of GSK3β, an enzyme that phosphorylates β-catenin protein and promotes its degradation, simulated IGF-I effects on cell survival by increasing the number of surviving cells and decreasing the number of apoptotic cells (Figure 7). When compared to untreated controls or NaCl-treated controls, cultures treated with the GSK3β inhibitor LiCl exhibited an ~71% increase in the number of surviving cells (Figure 7). Consistently, the number of apoptotic cells was reduced by ~64% in LiCl-treated cultures. IGF-I treatment further increased the number of surviving cells and reduced the number of apoptotic cells.
Figure 7.
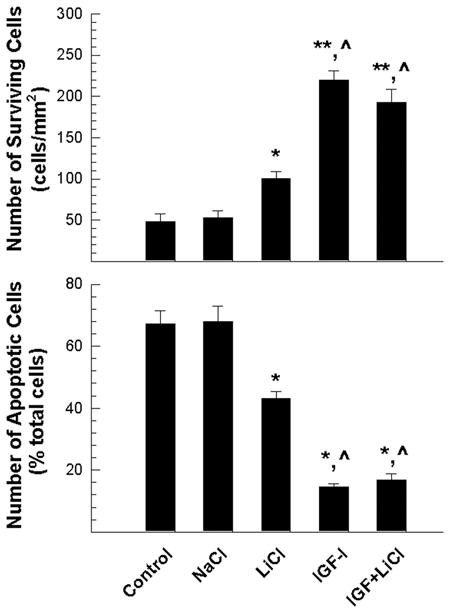
LiCl increase of the number of surviving OL-1 cells. Cells were treated for 24 hr with 20 mM NaCl, 20 mM LiCl, 100 ng/ml IGF-I, or a combination of 20 mM LiCl and 100 ng/ml IGF-I. Values represent mean ± SE of 3–5 samples. *, P < 0.05; **, P<0.01, compared to untreated controls. ^, P < 0.05, compared to cells treated with LiCl alone.
To assess whether IGF-I induced changes in β-catenin expression also occur in primary oligodendrocyte cultures, OPC were treated with IGF-I. Similar to our previous observations in OL-1 cell cultures, treatment of primarily cultured oligodendroglial cells with IGF-I significantly increased the abundance of β-catenin protein (Figure 8 and supplemental Figure S3). Compared to non-treated controls, the abundance of β-catenin protein in cultures treated with IGF-I for 24 hr was more than doubled (Figure 8A and 8B). Consistently, IGF-I also significantly increased the abundance of β-catenin and cyclin D1 mRNA by ~160% and 170%, respectively. Pre-treatment with the PI3 kinase inhibitor wortmannin significantly blunted these IGF-I effects (Figure 8C). In addition, wortmannin significantly suppressed IGF-I-stimulated expression of mRNAs for MBP, PLP and 2′,3′ cyclic nucleotide-3′-phosphodiesterase (CNP), three major oligodendrocyte/myelin-specific proteins (Figure 8).
Figure 8.

IGF-I regulation of the expression of β-catenin protein and its mRNA, and cyclin D1 mRNA in cultured oligodendrocytes. Panel A. Representative Western immunoblot of β-catenin protein. Cells were treated with (IGF-I) or without (Contl) 100 ng/ml IGF-I for 24 hr. Panel B. Quantification of β-catenin protein abundance. Panel C. Expression of mRNA for β-catenin (βCat), cyclin D1 (CycD1), MBP, PLP and CNP. Cells were treated with 100 ng/ml IGF-I or in combination of IGF-I and 250 nM wortmannin (IGF+Wort) for 24 hr. All cells were cultured in the presence of 0.1% DMSO. In both panels B and C, values represent mean ± SE of 3–5 samples. *, P < 0.05; **, P < 0.01, compared to controls. ^, P < 0.05, compared to IGF-I treated cultures.
Consistent with our observations in OL-1 cells, treatment of oligodendrocytes with β-catenin siRNA specifically reduced the expression of β-catenin mRNA by 30–40% in a dose dependent manner, but not that of MBP, PLP, CNP and NG2 mRNA (data not shown), and blocked IGF-I-enhanced expression of β-catenin and cyclin D1 mRNA (Figure 9). In cultures transfected with negative control siRNA, IGF-I treatment increased the abundance of β-catenin and cyclin D1 mRNA by ~60% and ~100%, respectively, as compared to their non-IGF-I treated controls. In cultures transfected with 5 nM β-catenin siRNA, however, IGF-I only increased the abundance of β-catenin and cyclin D1 mRNA by ~7% and ~40%, respectively. Furthermore, β-catenin siRNA treatment also reduced IGF-I-stimulated MBP, CNP and PLP mRNA expression (Figure 9) and IGF-I-promoted cell survival (Figure 10).
Figure 9.

β-catenin siRNA suppression of IGF-I-promoted expression of mRNA for β-catenin, cyclin D1, as well as myelin-specific proteins in primary oligodendrocyte cultures. Rat oligodendrocytes were pre-treated with β-catenin siRNA (βCat siRNA) or a negative control siRNA (neg siRNA) for 24 hr, and then were incubated with or without 100 ng/ml of IGF-I for an additional 24 hr. Values represent mean ± SE of 5–8 samples. *, P < 0.05; **, P < 0.01; ***, P < 0.001, compared to non-IGF-I treated controls. ^, P < 0.05; ^^, P < 0.01, compared to IGF-I treated cells that were pre-transfected with negative control siRNA.
Figure 10.
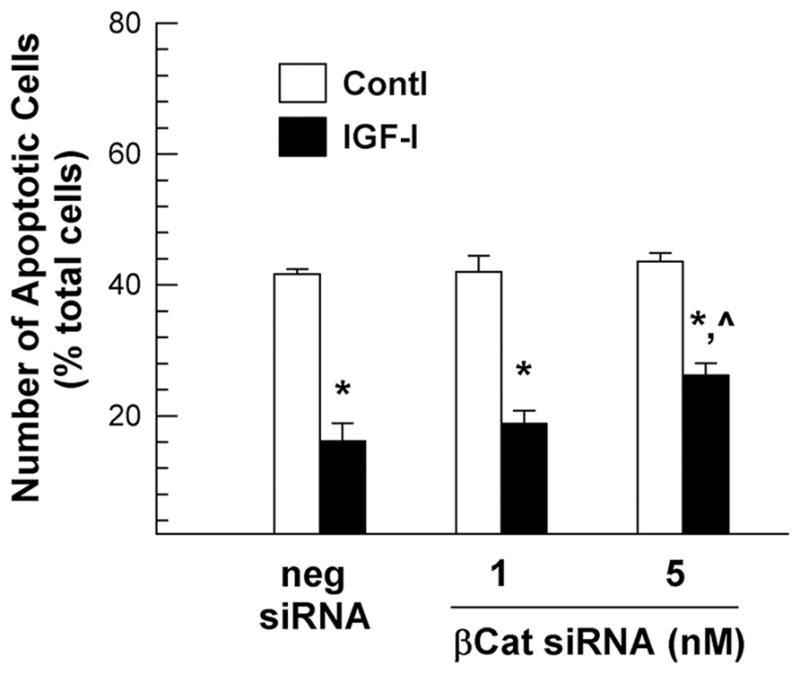
β-catenin siRNA suppression of IGF-I-promoted oligodendrocytes survival. Oligodendrocytes were pre-treated with β-catenin siRNA (βCat) or a negative control siRNA (neg siRNA), as well as 100 ng/ml of IGF-I, as previously described in Figure 9. Values represent mean ± SE of 4 samples. *, P < 0.001, compared to non-IGF-I treated controls. ^, P < 0.01, compared to IGF-I treated cells that were pre-transfected with negative control siRNA.
Similar to LiCl effects on cell survival in OL-1 cultures, GSK3β inhibitor VIII (EMD Chemicals, Gibbstown, NJ) significantly reduced the number of apoptotic oligodendrocytes, simulating IGF-I effects on cell survival. While ~52% of oligodendrocytes were apoptotic in control cultures, ~13% of cells were apoptotic in cultures treated with IGF-I (100 ng/ml; Figure 11). Compared to control cultures, the number of apoptotic cells in oligodendrocyte cultures treated with GSK3β inhibitor VIII (50 or 100 nM) was reduced by ~17 and ~26%, respectively (Figure 11).
Figure 11.
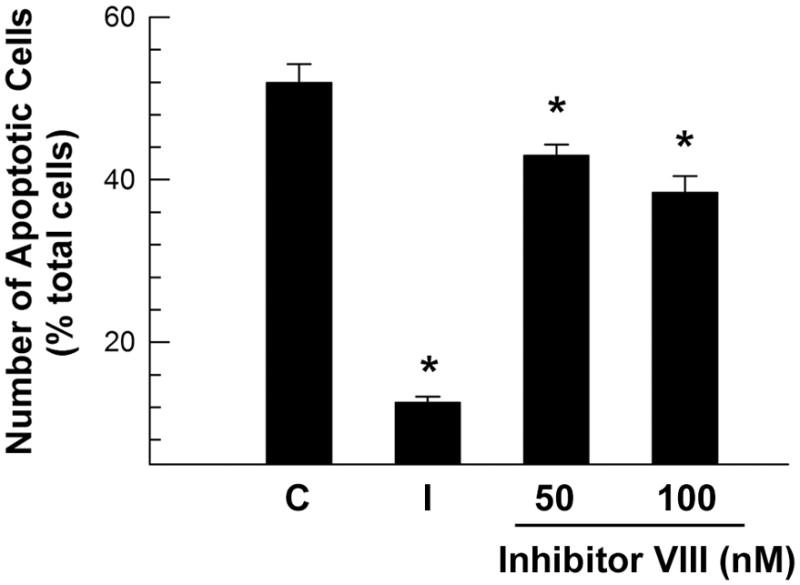
GSK3β inhibitor VIII decrease of apoptotic cell number in oligodendrocyte cultures. Oligodendrocytes were cultured for 24 hr with 50 nM or 100 nM GSK3β inhibitor VIII (inhibitor VIII), or 100 ng/ml IGF-I (I). All cells, including non-treated controls (C), were cultured in the presence of 0.03% DMSO. Values represent mean ± SE of 4 samples. *, P < 0.001, compared to control cells.
DISCUSSION
IGF-I promotion of OPC proliferation, maturation and survival is well documented. While significant evidence shows that the MAP kinase signaling pathway mediates IGF-I actions in the promotion of cell proliferation, accumulating experimental data indicate that PI3-Akt kinase activity also is involved in IGF-I-stimulated proliferation in cultured neural progenitors, including OPC (Cui and Almazan, 2007; Kalluri et al., 2007). Our data from this study strongly support the notion that the PI3-Akt kinase signaling pathway plays a critical role in mediating IGF-I promotion of cell proliferation. Our study reveals that GSK3β and its downstream target β-catenin are signaling molecules in the PI3-Akt kinase pathway and that they mediate IGF-I promotion of oligodendroglial cell proliferation, as well as their survival. This conclusion comes from our data showing that: 1) while IGF-I significantly increases the abundance of β-catenin mRNA and protein, suppression of β-catenin expression blocks cyclin D1 mRNA expression (both under basal and IGF-I stimulated conditions), and the latter is accompanied by blunting of IGF-I-enhanced cell proliferation and survival; 2) IGF-I markedly increases the phosphorylation of Akt and GSK3β, both of which occur prior to IGF-I-stimulated effects on the abundance of β-catenin protein and cyclin D1 mRNA, and inhibition of PI3-Akt activity markedly blunts IGF-I-stimulated increases in the abundance of pGSK3βSer9, β-catenin protein and mRNA, and cyclin D1 mRNA; and 3) suppression of GSK3β activity by LiCl simulates IGF-I actions on β-catenin protein and cell proliferation and survival.
Our studies also indicate a role for the Mek-Erk MAP kinase pathway in mediating IGF-I stimulation of cyclin D1 mRNA expression, which appears to be independent of the Akt/SK3β/β-catenin pathway. We found that IGF-I rapidly increased Erk phosphorylation and that blockade of the Mek-Erk pathway with PD98059 reduced both basal and IGF-I stimulated cyclin D1 expression, but not β-catenin expression. Our results are consistent with published data showing IGF-I activates Mek-Erk MAP kinase pathway in cultured oligodendrocytes (Cui and Almazan, 2007; Pang et al., 2007) and activation of Mek-Erk MAP kinase pathway up-regulates cycle D1 expression in neuroendocrine tumor cells (von Wichert et al., 2005). In addition, Frederick et al (2007) reported that in cultured oligodendrocytes IGF-I at low concentrations synergized FGF stimulatory effects on cyclin D1 expression, suggesting that IGF-I actions on Erk MAP kinase pathway and cyclin D1 expression are likely dependent on experimental conditions.
Previously, we showed that in Tg mice IGF-I overexpression stimulated proliferation of neural precursors, including OPC (Hodge et al., 2004; Popken et al., 2004; Ye et al., 1995, 1996). In embryonic brains that overexpress IGF-I in nestin-expressing cells, IGF-I exerts its proliferating actions by shortening the length of the cell cycle and by increasing re-entry of proliferating cells. The shorter cell cycle observed with in vivo IGF-I overexpression is due entirely to a shortened duration of the G1 phase (Hodge et al., 2004). The latter finding is in line with earlier studies showing that IGF-I promotes cell cycle progression through G1 phase or G0/G1 transition in cultured fibroblasts (Olashaw et al., 1987; Russell et al., 1984; Stiles et al., 1979) and skeletal muscle satellite cells (Chakravarthy et al., 2000). Consistent with these data, our current studies demonstrate that IGF-I markedly increases the abundance of mRNA for cyclin D1, a cyclin that is critical for progression through G1 phase of the cell cycle, and the proliferation of oligodendroglial cells.
β-catenin is abundantly expressed during CNS development and plays a key role as a “limiting” factor in the Wnt canonical signaling pathway (Aberle et al., 1997; Liu et al., 2002; Salic et al., 2000; Schwarz-Romond et al., 2002). Modulation of β-catenin abundance by phosphorylation, and its resultant degradation, effectively controls the transduction of Wnt canonical signaling. More specifically, β-catenin phosphorylation effected by GSK3β and casein kinase I promotes its degradation through the ubiquitin-proteasome pathway, and thus, the reduction in GSK3β activity results in an increase in β-catenin abundance, which in turn promotes the expression of multiple genes (including cyclin D1) through interaction with the Tcf/Lef (T-cell factor/lymphoid enhancer factor) family transcription factors (see review: Patapoutian and Reichardt, 2000). Consistent with previous data (Romanelli et al., 2007), our study demonstrates that in cultured oligodendroglial cells stimulation of the IGF1R-PI3-Akt pathway significantly promotes phosphorylation of GSK3βSer9. Furthermore, our data show that the PI3-Akt pathway mediates IGF-I-stimulated increases in β-catenin mRNA and protein, which in turn promotes cell proliferation and survival. These data indicate that a portion of IGF-I and Wnt signaling converge by regulating GSK3βSer9 phosphorylation and β-catenin abundance.
Studies of IGF-I and IGF1R mutant mice and cultured oligodendrocytes, including our studies, have convincingly demonstrated that IGF-I-IGF1R signaling plays a key role in promoting the development of oligodendrocyte lineage cells (Carson et al., 1993; Hsieh et al., 2004; Mozell and McMorris, 1991; Palacios et al., 2007; Ye and D’Ercole, 1999; Ye et al., 1995, 2000, 2002; Zeger et al., 2007). While the exact mechanisms and intracellular signaling pathways that mediate IGF-I actions remain to be completely defined, evidence from our previous studies indicate that promotion of oligodendrocyte/myelin-specific protein mRNA expression by IGF-I-IGF1R signaling is one of the mechanisms (Ye and D’Ercole, 1999; Ye et al., 1995, 2000, 2002). In the current studies, we observed that the PI3 kinase inhibitor wortmannin significantly blunts IGF-I stimulation of MBP and CNP mRNA expression. The latter findings strongly indicate a critical role for the PI3-Akt pathway in mediating the IGF-I actions to promote the expression of myelin-specific protein mRNA. Consistent with our data, Bibollet-Bahena and Almazan (2009) have reported that in cultured oligodendrocytes new mRNA transcription is required for IGF-I-stimulated protein synthesis during the early stage of treatment, and that IGF-I-stimulated protein synthesis is mediated by the PI3-Akt pathway. The PI3-Akt actions to promote the synthesis of myelin proteins are likely mediated partially by the mammalian target of rapamycin pathway (Bibollet-Bahena and Almazan, 2009; Narayanan et al., 2009). When β-catenin expression is suppressed, IGF-I-enhanced expression of PLP and MBP mRNA, however, is blunted. Our studies, therefore, also suggest that β-catenin, acting as a downstream target of the PI3-Akt pathway, in part mediates IGF-I actions to promote the expression of myelin-specific protein genes and cell maturation. These results indicate that in addition to its function in promotion of cell proliferation and survival, the PI3-Akt pathway plays a key role in IGF-I signaling that promotes the production of myelin proteins at both the mRNA and protein levels.
Our results suggest that nMyc is not a downstream signaling molecule in the IGF-I-Akt-GSK3β pathway in oligodendroglial cells. nMyc, an oncogene protein, is capable of promoting cell proliferation in certain cell types, such as cerebellar neuronal precursors (Kenney et al., 2003; Kenney et al., 2004). The latter studies suggest that inactivation of GSK3β activity suppresses nMyc degradation, and in turn results in cell proliferation. Our Western immunoblot analyses, however, demonstrate that IGF-I treatment does not affect the abundance of either total or phosphorylated nMyc, indicating that nMyc is not involved in signaling IGF-I actions in oligodendroglial cells.
Taken together with the studies by others, our current experimental data strongly support the concept that the PI3-Akt kinase pathway is required for a full mitogenic response to IGF-I. Our studies also indicate that while β-catenin is a regulatory molecule downstream of PI3-Akt kinases and GSK3β, more than one pathway mediates IGF-I actions on cyclin D1 mRNA expression, cell survival, proliferation and maturation. Clearly, more studies are needed to precisely define IGF-I signaling pathways and their interaction with Wnt signaling pathways.
Supplementary Material
Acknowledgments
This work was supported by NIH grants RO1 NS038891, RO1 HD008299, and RO1 NS048868.
REFERENCE LIST
- Aberle H, Bauer A, Stappert J, Kispert A, Kemler R. beta-catenin is a target for the ubiquitin-proteasome pathway. EMBO J. 1997;16:3797–3804. doi: 10.1093/emboj/16.13.3797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adachi K, Mirzadeh Z, Sakaguchi M, Yamashita T, Nikolcheva T, Gotoh Y, Peltz G, Gong L, Kawase T, varez-Buylla A, Okano H, Sawamoto K. Beta-catenin signaling promotes proliferation of progenitor cells in the adult mouse subventricular zone. Stem Cells. 2007;25:2827–2836. doi: 10.1634/stemcells.2007-0177. [DOI] [PubMed] [Google Scholar]
- Baker J, Liu J-P, Robertson EJ, Efstratiadis A. Role of Insulin-like Growth Factors in embryonic and postnatal growth. Cell. 1993;75:73–82. [PubMed] [Google Scholar]
- Barres BA, Hart IK, Coles HS, Burne JF, Voyvodic JT, Richardson WD, Raff MC. Cell death and control of cell survival in the oligodendrocyte lineage. Cell. 1992;70:31–46. doi: 10.1016/0092-8674(92)90531-g. [DOI] [PubMed] [Google Scholar]
- Beck KD, Powell-Braxton L, Widmer HR, Valverde J, Hefti F. Igf1 gene disruption results in reduced brain size, CNS hypomyelination, and loss of hippocampal granule and striatal parvalbumin-containing neurons. Neuron. 1995;14:717–730. doi: 10.1016/0896-6273(95)90216-3. [DOI] [PubMed] [Google Scholar]
- Bibollet-Bahena O, Almazan G. IGF-1-stimulated protein synthesis in oligodendrocyte progenitors requires PI3K/mTOR/Akt and MEK/ERK pathways. J Neurochem. 2009;109:1440–1451. doi: 10.1111/j.1471-4159.2009.06071.x. [DOI] [PubMed] [Google Scholar]
- Carson MJ, Behringer RR, Brinster RL, McMorris FA. Insulin-like growth factor I increases brain growth and central nervous system myelination in transgenic mice. Neuron. 1993;10:729–740. doi: 10.1016/0896-6273(93)90173-o. [DOI] [PubMed] [Google Scholar]
- Chakravarthy MV, Abraha TW, Schwartz RJ, Fiorotto ML, Booth FW. Insulin-like growth factor-I extends in vitro replicative life span of skeletal muscle satellite cells by enhancing G1/S cell cycle progression via the activation of phosphatidylinositol 3′-kinase/Akt signaling pathway. J Biol Chem. 2000;275:35942–35952. doi: 10.1074/jbc.M005832200. [DOI] [PubMed] [Google Scholar]
- Chenn A, Walsh CA. Regulation of cerebral cortical size by control of cell cycle exit in neural precursors. Science. 2002;297:365–369. doi: 10.1126/science.1074192. [DOI] [PubMed] [Google Scholar]
- Chenn A, Walsh CA. Increased neuronal production, enlarged forebrains and cytoarchitectural distortions in beta-catenin overexpressing transgenic mice. Cereb Cortex. 2003;13:599–606. doi: 10.1093/cercor/13.6.599. [DOI] [PubMed] [Google Scholar]
- Chrysis D, Calikoglu AS, Ye P, D’Ercole AJ. Insulin-like growth factor-I overexpression attenuates cerebellar apoptosis by altering the expression of Bcl family proteins in a developmentally specific manner. J Neurosci. 2001;21:1481–1489. doi: 10.1523/JNEUROSCI.21-05-01481.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui QL, Almazan G. IGF-I-induced oligodendrocyte progenitor proliferation requires PI3K/Akt, MEK/ERK, and Src-like tyrosine kinases. J Neurochem. 2007;100:1480–1493. doi: 10.1111/j.1471-4159.2006.04329.x. [DOI] [PubMed] [Google Scholar]
- D’Ercole AJ, Ye P. Expanding The Mind: IGF-I and Brain Development. Endocrinology. 2008 doi: 10.1210/en.2008-0920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Ercole AJ, Ye P, Calikoglu AS, Gutierrez-Ospina G. The role of the insulin-like growth factors in the central nervous system. Mol Neurobiol. 1996;13:227–255. doi: 10.1007/BF02740625. [DOI] [PubMed] [Google Scholar]
- Dentremont KD, Ye P, D’Ercole AJ, O’Kusky JR. Increased insulin-like growth factor-I (IGF-I) expression during early postnatal development differentially increases neuron number and growth in medullary nuclei of the mouse. Brain Res Dev Brain Res. 1999;114:135–141. doi: 10.1016/s0165-3806(99)00024-3. [DOI] [PubMed] [Google Scholar]
- Efstratiadis A. Genetics of mouse growth. Int J Dev Biol. 1998;42:955–976. [PubMed] [Google Scholar]
- Frederick TJ, Min J, Altieri SC, Mitchell NE, Wood TL. Synergistic induction of cyclin D1 in oligodendrocyte progenitor cells by IGF-I and FGF-2 requires differential stimulation of multiple signaling pathways. Glia. 2007;55:1011–1022. doi: 10.1002/glia.20520. [DOI] [PubMed] [Google Scholar]
- Gehmert S, Sadat S, Song YH, Yan Y, Alt E. The anti-apoptotic effect of IGF-1 on tissue resident stem cells is mediated via PI3-kinase dependent secreted frizzled related protein 2 (Sfrp2) release. Biochem Biophys Res Commun. 2008;371:752–755. doi: 10.1016/j.bbrc.2008.04.151. [DOI] [PubMed] [Google Scholar]
- Hsieh J, Aimone JB, Kaspar BK, Kuwabara T, Nakashima K, Gage FH. IGF-I instructs multipotent adult neural progenitor cells to become oligodendrocytes. J Cell Biol. 2004;164:111–122. doi: 10.1083/jcb.200308101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodge RD, D’Ercole AJ, O’Kusky JR. Insulin-like growth factor-I accelerates the cell cycle by decreasing G1 phase length and increases cell cycle reentry in the embryonic cerebral cortex. J Neurosci. 2004;24:10201–10210. doi: 10.1523/JNEUROSCI.3246-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodge RD, D’Ercole AJ, O’Kusky JR. Increased expression of insulin-like growth factor-I (IGF-I) during embryonic development produces neocortical overgrowth with differentially greater effects on specific cytoarchitectonic areas and cortical layers. Brain Res Dev Brain Res. 2005;154:227–237. doi: 10.1016/j.devbrainres.2004.10.016. [DOI] [PubMed] [Google Scholar]
- Hodge RD, D’Ercole AJ, O’Kusky JR. Insulin-like growth factor-I (IGF-I) inhibits neuronal apoptosis in the developing cerebral cortex in vivo. Int J Dev Neurosci. 2007;25:233–241. doi: 10.1016/j.ijdevneu.2007.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughson E, Dowler S, Geall K, Johnson G, Rumsby M. Rat oligodendrocyte O-2A precursor cells and the CG-4 oligodendrocyte precursor cell line express cadherins, beta-catenin and the neural cell adhesion molecule, NCAM. Neurosci Lett. 1998;251:157–160. doi: 10.1016/s0304-3940(98)00523-0. [DOI] [PubMed] [Google Scholar]
- Junghans D, Hack I, Frotscher M, Taylor V, Kemler R. Beta-catenin-mediated cell-adhesion is vital for embryonic forebrain development. Dev Dyn. 2005;233:528–539. doi: 10.1002/dvdy.20365. [DOI] [PubMed] [Google Scholar]
- Kalluri HS, Vemuganti R, Dempsey RJ. Mechanism of insulin-like growth factor I-mediated proliferation of adult neural progenitor cells: role of Akt. Eur J Neurosci. 2007;25:1041–1048. doi: 10.1111/j.1460-9568.2007.05336.x. [DOI] [PubMed] [Google Scholar]
- Kenney AM, Cole MD, Rowitch DH. Nmyc upregulation by sonic hedgehog signaling promotes proliferation in developing cerebellar granule neuron precursors. Development. 2003;130:15–28. doi: 10.1242/dev.00182. [DOI] [PubMed] [Google Scholar]
- Kenney AM, Widlund HR, Rowitch DH. Hedgehog and PI-3 kinase signaling converge on Nmyc1 to promote cell cycle progression in cerebellar neuronal precursors. Development. 2004;131:217–228. doi: 10.1242/dev.00891. [DOI] [PubMed] [Google Scholar]
- Lagarde WH, Benjamin R, Heerens AT, Ye P, Cohen RI, Moats-Staats BM, D’Ercole AJ. A non-transformed oligodendrocyte precursor cell line, OL-1, facilitates studies of insulin-like growth factor-I signaling during oligodendrocyte development. Int J Dev Neurosci. 2007;25:95–105. doi: 10.1016/j.ijdevneu.2006.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Li Y, Semenov M, Han C, Baeg GH, Tan Y, Zhang Z, Lin X, He X. Control of beta-catenin phosphorylation/degradation by a dual-kinase mechanism. Cell. 2002;108:837–847. doi: 10.1016/s0092-8674(02)00685-2. [DOI] [PubMed] [Google Scholar]
- Liu H, Hu Q, D’Ercole AJ, Ye P. Histone deacetylase 11 regulates oligodendrocyte-specific gene expression and cell development in OL-1 oligodendroglial cells. Glia. 2009;57:1–12. doi: 10.1002/glia.20729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J-P, Baker J, Perkins AS, Robertson EJ, Efstratiadis A. Mice carrying null mutations of the genes encoding Insulin-like Growth Factor I (Igf-1) and type 1 IGF receptor. Cell. 1993;75:59–72. [PubMed] [Google Scholar]
- Liu W, Ye P, O’Kusky JR, D’Ercole AJ. Type 1 insulin-like growth factor receptor signaling is essential for the development of the hippocampal formation and dentate gyrus. J Neurosci Res. 2009;87:2821–2832. doi: 10.1002/jnr.22129. [DOI] [PubMed] [Google Scholar]
- Machon O, van den Bout CJ, Backman M, Kemler R, Krauss S. Role of beta-catenin in the developing cortical and hippocampal neuroepithelium. Neurosci. 2003;122:129–143. doi: 10.1016/s0306-4522(03)00519-0. [DOI] [PubMed] [Google Scholar]
- Mason JL, Ye P, Suzuki K, D’Ercole AJ, Matsushima GK. Insulin-like growth factor-1 inhibits mature oligodendrocyte apoptosis during primary demyelination. J Neurosci. 2000;20:5703–5708. doi: 10.1523/JNEUROSCI.20-15-05703.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMorris FA, Dubois-Dalcq M. Insulin-like growth factor I promotes cell proliferation and oligodendroglial commitment in rat glial progenitor cells developing in vitro. J Neurosci Res. 1988;21:199–209. doi: 10.1002/jnr.490210212. [DOI] [PubMed] [Google Scholar]
- Mozell RL, McMorris FA. Insulin-like growth factor I stimulates oligodendrocyte development and myelination in rat brain aggregate cultures. J Neurosci Res. 1991;30:382–390. doi: 10.1002/jnr.490300214. [DOI] [PubMed] [Google Scholar]
- Narayanan SP, Flores AI, Wang F, Macklin WB. Akt signals through the mammalian target of rapamycin pathway to regulate CNS myelination. J Neurosci. 2009;29:6860–6870. doi: 10.1523/JNEUROSCI.0232-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni W, Rajkumar K, Nagy JI, Murphy LJ. Inpaired brain development and reduced astrocyte response to injury in transgenic mice expressing IGF binding protein-1. Brain Res. 1997;769:97–107. doi: 10.1016/s0006-8993(97)00676-8. [DOI] [PubMed] [Google Scholar]
- Olashaw NE, Van Wyk JJ, Pledger WJ. Control of late G0/G1 progression and protein modification by SmC/IGF I. Am J Physiol. 1987;253:C575–C579. doi: 10.1152/ajpcell.1987.253.4.C575. [DOI] [PubMed] [Google Scholar]
- Palacios N, Sanchez-Franco F, Fernandez M, Sanchez I, Villuendas G, Cacicedo L. Opposite effects of two PKA inhibitors on cAMP inhibition of IGF-I-induced oligodendrocyte development: a problem of unspecificity? Brain Res. 2007;1178:1–11. doi: 10.1016/j.brainres.2007.07.018. [DOI] [PubMed] [Google Scholar]
- Pang Y, Zheng B, Fan LW, Rhodes PG, Cai Z. IGF-1 protects oligodendrocyte progenitors against TNFalpha-induced damage by activation of PI3K/Akt and interruption of the mitochondrial apoptotic pathway. Glia. 2007;55:1099–1107. doi: 10.1002/glia.20530. [DOI] [PubMed] [Google Scholar]
- Patapoutian A, Reichardt LF. Roles of Wnt proteins in neural development and maintenance. Curr Opin Neurobiol. 2000;10:392–399. doi: 10.1016/s0959-4388(00)00100-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popken GJ, Hodge RD, Ye P, Zhang J, Ng W, O’Kusky JR, D’Ercole AJ. In vivo effects of insulin-like growth factor-I (IGF-I) on prenatal and early postnatal development of the central nervous system. Eur J Neurosci. 2004;19:2056–2068. doi: 10.1111/j.0953-816X.2004.03320.x. [DOI] [PubMed] [Google Scholar]
- Richards RG, Klotz DM, Bush MR, Walmer DK, DiAugustine RP. E2-induced degradation of uterine insulin receptor substrate-2: Requirement for an IGF-I-stimulated, proteasome-dependent pathway. Endocrinology. 2001:3842–3849. doi: 10.1210/endo.142.9.8370. [DOI] [PubMed] [Google Scholar]
- Romanelli RJ, LeBeau AP, Fulmer CG, Lazzarino DA, Hochberg A, Wood TL. Insulin-like growth factor type-I receptor internalization and recycling mediate the sustained phosphorylation of Akt. J Biol Chem. 2007;282:22513–22524. doi: 10.1074/jbc.M704309200. [DOI] [PubMed] [Google Scholar]
- Russell WE, Van Wyk JJ, Pledger WJ. Inhibition of the mitogenic effects of plasma by a monoclonal antibody to somatomedin C. Proc Natl Acad Sci. 1984;81:2389–2392. doi: 10.1073/pnas.81.8.2389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salic A, Lee E, Mayer L, Kirschner MW. Control of beta-catenin stability: reconstitution of the cytoplasmic steps of the wnt pathway in Xenopus egg extracts. Mol Cell. 2000;5:523–532. doi: 10.1016/s1097-2765(00)80446-3. [DOI] [PubMed] [Google Scholar]
- Schuller U, Rowitch DH. Beta-catenin function is required for cerebellar morphogenesis. Brain Res. 2007;1140:161–169. doi: 10.1016/j.brainres.2006.05.105. [DOI] [PubMed] [Google Scholar]
- Schwarz-Romond T, Asbrand C, Bakkers J, Kuhl M, Schaeffer HJ, Huelsken J, Behrens J, Hammerschmidt M, Birchmeier W. The ankyrin repeat protein Diversin recruits Casein kinase Iepsilon to the beta-catenin degradation complex and acts in both canonical Wnt and Wnt/JNK signaling. Genes Dev. 2002;16:2073–2084. doi: 10.1101/gad.230402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stiles CD, Capone GT, Scher CD, Antoniades HN, Van Wyk JJ, Pledger WJ. Dual control of cell growth by somatomedins and platelet-derived growth factor. Proc Natl Acad Sci. 1979;76:1279–1283. doi: 10.1073/pnas.76.3.1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vemuri GS, McMorris FA. Oligodendrocytes and their precursors require phosphatidylinositol 3-kinase signaling for survival. Development. 1996;122:2529–2537. doi: 10.1242/dev.122.8.2529. [DOI] [PubMed] [Google Scholar]
- von Wichert T, Haeussler U, Greten FR, Kliche S, Dralle H, Bohm BO, Adler G, Seufferlein T. Regulation of cyclin D1 expression by autocrine IGF-I in human BON neuroendocrine tumour cells. Oncogene. 2005;24:1284–1289. doi: 10.1038/sj.onc.1208264. [DOI] [PubMed] [Google Scholar]
- Ye P, Carson J, D’Ercole AJ. In vivo actions of insulin-like growth factor-I (IGF-I) on brain myelination: studies of IGF-I and IGF binding protein-1 (IGFBP-1) transgenic mice. J Neurosci. 1995;15:7344–7356. doi: 10.1523/JNEUROSCI.15-11-07344.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye P, D’Ercole AJ. Insulin-like growth factor I protects oligodendrocytes from tumor necrosis factor-alpha-induced injury. Endocrinology. 1999;140:3063–3072. doi: 10.1210/endo.140.7.6754. [DOI] [PubMed] [Google Scholar]
- Ye P, D’Ercole AJ. Insulin-like growth factor actions during development of neural stem cells and progenitors in the central nervous system. J Neurosci Res. 2006;83:1–6. doi: 10.1002/jnr.20688. [DOI] [PubMed] [Google Scholar]
- Ye P, Lee KH, D’Ercole AJ. Insulin-like growth factor-I (IGF-I) protects myelination from under nutritional insult: studies of transgenic mice overexpressing IGF-I in brain. J Neurosci Res. 2000;62:700–708. doi: 10.1002/1097-4547(20001201)62:5<700::AID-JNR9>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- Ye P, Li L, Richards RG, DiAugustine RP, D’Ercole AJ. Myelination is altered in insulin-like growth factor-I null mutant mice. J Neurosci. 2002;22:6041–6051. doi: 10.1523/JNEUROSCI.22-14-06041.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye P, Popken GJ, Kemper A, McCarthy K, Popko B, D’Ercole AJ. Astrocyte-specific overexpression of insulin-like growth factor-I promotes brain overgrowth and glial fibrillary acidic protein expression. J Neurosci Res. 2004;78:472–484. doi: 10.1002/jnr.20288. [DOI] [PubMed] [Google Scholar]
- Ye P, Xing Y, Dai Z, D’Ercole AJ. In vivo actions of insulin-like growth factor-I (IGF-I) on cerebellum development in transgenic mice: evidence that IGF-I increases proliferation of granule cell progenitors. Brain Res Dev Brain Res. 1996;95:44–54. doi: 10.1016/0165-3806(96)00492-0. [DOI] [PubMed] [Google Scholar]
- Zeger M, Popken G, Zhang J, Xuan S, Lu QR, Schwab MH, Nave KA, Rowitch D, D’Ercole AJ, Ye P. Insulin-like growth factor type 1 receptor signaling in the cells of oligodendrocyte lineage is required for normal in vivo oligodendrocyte development and myelination. Glia. 2007;55:400–411. doi: 10.1002/glia.20469. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.