

Abstract
Starting with resin-bound glutamic acid, an efficient synthesis of N-substituted pyrrolidinones is described utilizing the Ugi four-component reaction (U-4CR). The same methodology is employed to produce N-substituted pyrrolidinone tethered N-substituted piperidines.
Solid-phase organic synthesis (SPOS) has emerged as a powerful approach for the rapid generation of structurally diverse compounds in the drug discovery community. One major focus of this field is the synthesis of bioactive compounds and their derivatives on the solid phase.1 Multi-component reactions (MCRs) are one of the best tools in modern organic synthesis because of their productivity, simple procedures, convergence and facile execution.2 One of the most important MCRs is the Ugi four-component reaction (U-4CR), in which a carboxylic acid, an amine, a carbonyl compound, and an isocyanide react in a one-step reaction, affording related compounds with variable structures.3 The Ugi four-component reaction is a powerful way to make library scaffolds containing a high number of substitution diversities. Therefore, it has been utilized very efficiently in conjugation with solid-phase organic synthesis to prepare large collections of molecules in a short reaction sequence.4
The N-substituted pyrrolidinone is an attractive drug template because of its extensive biological activities in various drug targets, such as Aspartic Protease,5 Beta-Amyloid Cleaving Enzyme (BACE),6 Progesterone Receptor (PR),7 Human Melanocortin-4 Receptor,8 Factor Xa,9 and HIV-1 Integrase.10 The development of new synthetic protocols for N-substituted pyrrolidinone derivatives are topics of continuous interest.11 Herein, we would like to present our approach toward the solid-phase synthesis of enantiopure N-substituted pyrrolidinones, utilizing glutamic acid as a bifunctional reagent in an Ugi four-center three-component reaction (U-4C-3CR).12 We also used the same methodology to create diversified N-substituted pyrrolidinone tethered N-substituted piperidines.
Polymer supported glutamic acid 2 was readily prepared by the coupling of L-Fmoc-Glu(tBu)-OH to MBHA resin (Scheme 1). The generated resin bound bifunctional compound 2 was optimized for the solid phase Ugi reaction conditions. The initial attempt was carried out with benzyl isocyanide (1.1 equiv.) and cyclohexanone (1.1 equiv.) in acetonitrile/methanol (1:1) for 24 h at 65 °C. Following cleavage of the solid support, the desired product was obtained in 58% yield (Table 1, entry 4a), with traces of starting material. In order to drive the reaction to completion, we used varying stoichiometries of the isocyanide and ketone. The better results were obtained using two fold excess of the reagents with a yield of 83% (Table 1, entry 4c). We also tested the reaction under different reaction conditions by reducing the reaction time (Table 1, entry 4d) or by lowering the temperature (Table 1, entry 4e). In both cases, the reaction was incomplete and lower yields were obtained.
Scheme 1.

Reagents and conditions: (a) 55% TFA/DCM, 1h; (b) 20% piperidine/DMF, 10 min, twice; (c) Benzyl isocyanide/Cyclohexanone (1:1) in Acetonitrile/MeOH (4:1); (d) HF/anisole, 1.5 h, 0 °C.
Table 1.
Screening of reaction conditions for the solid-phase Ugi multi-component reaction
| Entry | Reagents (equiv.) |
time (h) | Temp (°C) | yield (%) a | ||
|---|---|---|---|---|---|---|
| PS Glu | BnNC | Cyclohexanone | ||||
| 4a | 1 | 1.1 | 1.1 | 24 | 65 | 58 |
| 4b | 1 | 1.5 | 1.5 | 24 | 65 | 75 |
| 4c | 1 | 2 | 2 | 24 | 65 | 83 |
| 4d | 1 | 2 | 2 | 12 | 65 | 66 |
| 4e | 1 | 2 | 2 | 24 | 25 | 51 |
Yields based on the weight of purified product and are relative to the initial loading of the resin. The purity of the purified compounds is higher than 95% for all the compounds. The purity was estimated on analytical traces at λ= 214 nm and 254 nm.
Under the optimized conditions (Table 1, entry 4c), the scope of the solid phase Ugi reaction was explored using various isocyanides and ketones (Table 2). Polymer-supported glutamic acid was allowed to react with different isocyanides (2 equiv.) and ketones (2 equiv.) to afford the corresponding products in very good yields. We tried the same reaction conditions starting from resin-bound aspartic acid. The reaction was not successful and very low yields were obtained.
Table 2.
Synthesis of substituted pyrrolidinones.
 | |||||
|---|---|---|---|---|---|
| Entry | R1 | Y | X | Obtained MW | Yield (%)a |
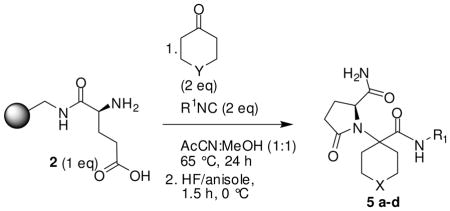
| 5a | 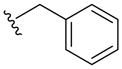 |
-NBoc- | -NH- | 345 (MH+) | 80 |
| 5b | 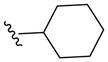 |
-CH2- | -CH2- | 336 (MH+) | 82 |
| 5c | -CH2- | -CH2- | 310 (MH+) | 78 | |
| 5d | 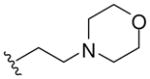 |
-CH2- | -CH2- | 367 (MH+) | 70 |
Yields based on the weight of purified product and are relative to the initial loading of the resin. The purity of the purified compounds is higher than 95% for all the compounds. The purity was estimated on analytical traces at λ= 214 nm and 254 nm.
In order to increase the diversity, we used Boc-piperidone as the carbonyl reagent for the solid-phase Ugi reaction. Following the Ugi condensation of the resin-bound free glutamic acid 2 with isocyanide and Boc-piperidone, the Boc group was deprotected with 55% TFA/DCM and the resin-bound pyrrolidinone tethered piperidine 6 was treated with diversified capping agents such as sulfonyl chlorides, isocyanates, isothiocyanates and carboxylic acids (Scheme 2). In all cases, the reaction proceeded smoothly and the desired products 8 were obtained in high yields (Table 3).
Scheme 2.

Reagents and conditions: (a) Benzyl isocyanide, N-Boc-4-piperidinone, acetonitrile/methanol (4:1), 65 °C, 24 h; (b) 55% TFA/DCM; (c) Acid, DIC, HOBt, DMF, r.t., overnight or Sulfonyl chloride/isocyanate/thioisocyanate, DIEA, DMF, r.t., overnight; (d) HF cleavage, 0 °C, 1.5 h.
Table 3.
Substituted piperidine products obtained
| Entry | R | Obtained MW | Yield (%) a |
|---|---|---|---|
| 8a | 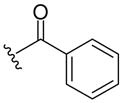 |
449 (MH+) | 76 |
| 8b | 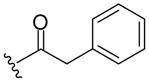 |
463 (MH+) | 72 |
| 8c | 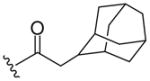 |
521 (MH+) | 68 |
| 8d | 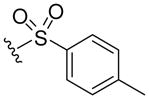 |
499 (MH+) | 82 |
| 8e | 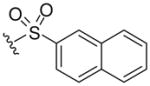 |
535 (MH+) | 85 |
| 8f | 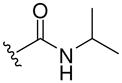 |
430 (MH+) | 74 |
| 8g | 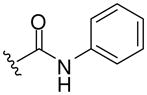 |
464 (MH+) | 79 |
| 8h | 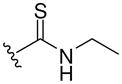 |
432 (MH+) | 72 |
| 8i | 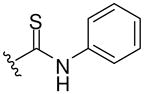 |
480 (MH+) | 80 |
Yields based on the weight of purified product and are relative to the initial loading of the resin. The purity of the purified compounds is higher than 95% for all the compounds. The purity was estimated on analytical traces at λ= 214 nm and 254 nm.
In conclusion, we employed polymer supported glutamic acid as an efficient bifunctional component in Ugi reaction. Using this approach, we performed the parallel solid phase synthesis of enantiopure N-substituted pyrrolidinone derivatives tethered to different biologically relevant heterocycles such as piperidine.
Experimental Section
Procedure for the synthesis of polymer supported glutamic acid 2
A 100 mg sample of p-methylbenzhydrylamine hydrochloride salt (MBHA) resin (1.15 mequiv/g, 100–200 mesh) was contained within a sealed polypropylene mesh packet. 13 Reactions were carried out in polyethylene bottles. Following neutralization of the resin with 5% diisopropylethylamine (DIEA) in dichloromethane (DCM), L Fmoc-Glu(tBu)-OH (6eq) was coupled using the conventional reagents hydroxybenzotriazole (HOBt, 6 equiv) and diisopropylcarbodiimide (DIC, 6 equiv.) in anhydrous DMF for 2 h. Completion of the coupling was monitored by the ninhydrin test. 14 Following removal of t-Butyl group with 55% TFA/DCM and washing with DCM (3 ×), the Fmoc group was deprotected with 20% piperidine in DMF (2×10min). The resin was then washed with DMF (3×), iPrOH (3×), DCM (3×).
General procedure for the solid-phase Ugi reaction: Synthesis of compounds 4 and 5
Resin 2 was put in a solution of ketone (2 equiv.) in acetonitrile/methanol (4:1) for 1 h at 65 °C, and then isocyanide (2 equiv.) was added. After allowing the mixture to react at 65 °C for 24 h, the resin was washed with MeOH (3×), DMF (3×), DCM (3×) and dried. The completeness of the reaction was verified by cleavage and analysis with LC-MS. 4: 1H NMR (DMSO d6): δ 1.30–1.86 (m, 10H, H on cyclohexane ring); 2.12–2.29 (m, 4H, H on pyrrolidine ring); 4.24 (m, 2H, H on benzyl group); 4.56 (m, 1H, H on pyrrolidine ring); 7.08–7.18 (m, 5H, H on benzyl group); 7.49 (s, 1H, H on amide group); 7.98 (s, 1H, H on amide group); 9.00 (t, 1H, J= 5.8 Hz, H on amide group). 13 C NMR (DMSO d6): δ 21.69 (C on piperidine ring), 22.12 (C on piperidine ring), 24.75 (C on pyrrolidine ring), 25.20 (C on piperidine ring), 29.64 (C on pyrrolidine ring), 31.00 (C on piperidine ring), 32.44 (C on piperidine ring), 42.18 (C on benzyl group), 58.24 (C on piperidine ring), 61.79 (C on pyrrolidine ring), 126.40 (C on benzyl group), 126.80 (2C, C on benzyl group), 128.03 (2C, C on benzyl group), 139.66 (C on benzyl group), 173.12 (C on amide group), 174.64 (C on amide group), 176.81 (C on amide group) ESI-MS: 343 (MH+).
5a: 1H NMR (DMSO d6): δ 1.71–1.89 (m, 4H, H on piperidine ring); 2.15–2.34 (m, 3H, H on piperidine ring); 2.76–2.89 (m, 2H, H on piperidine ring); 3.02–3.23 (m, 4H, m, 4H, H on pyrrolidine ring); 4.28 (m, 2H, H on benzyl group); 4.52 (m, 1H, H on pyrrolidine ring); 7.19–7.30 (m, 5H, H on benzyl group); 7.53 (s, 1H, H on amide group); 8.06 (s, 1H, H on amide group); 9.18 (t, 1H, J= 5.7 Hz, H on amide group)
ESI-MS: 345 (MH+). 5b: 1H NMR (DMSO d6): δ 1.11–1.86 (m, 20H, H on tow piperidine rings); 2.08–2.28 (m, 4H, H on pyrrolidine ring); 3.48 (m, 1H, H on piperidine ring); 4.53 (m, 1H, H on pyrrolidine ring); 7.52 (s, 1H, H on amide group); 7.93 (s, 1H, H on amide group); 8.47 (m, 1H, H on amide group). ESI-MS: 336 (MH+). 5c: 1H NMR (DMSO d6): δ 0.84 (t, 3H, J= 7.3 Hz, H on the n-butyl group); 1.23–1.86 (m, 14H, H on the n-butyl group and cyclohexane ring); 2.09–2.28 (m, 4H, H on pyrrolidine ring); 2.99 (q, 2H, J= 6.5 Hz, H on the n-butyl group); 4.53 (m, 1H, H on pyrrolidine ring); 7.50 (s, 1H, H on amide group); 7.96 (s, 1H, H on amide group); 8.48 (m, 1H, H on amide group). ESI-MS: 310 (MH+). 5d: 1H NMR (DMSO d6): δ 1.28–1.86 (m, 10H, H on cyclohexane ring); 2.07–2.31 (m, 10H, H on 2-morpholinethyl group and pyrrolidine ring); 3.11 (m, 2H, H on 2-morpholinethyl group); 3.53 (t, 4H, J= 4.6 Hz, H on 2-morpholinethyl group); 4.51 (m, 1H, H on pyrrolidine ring); 7.44 (m, 1H, H on amide group); 7.92 (m, 1H, H on amide group); 8.47 (t, 1H, J= 5.5 Hz, H on amide group). ESI-MS: 367 (MH+)
General procedure for the synthesis of compounds 8a-8c
Resin 5a was shaken in 55% TFA/DCM for 30 min to remove the Boc group. Following neutralization with 5% DIEA/DCM, the resin was coupled with a carboxylic acid (6 equiv.) using conventional reagents HOBt (6 equiv.) and DIC (6 equiv.) in anhydrous DMF overnight. The resin was then washed with DMF (3×), DCM (3×), dried and cleaved with HF for 1.5 h at 0 °C. All samples were purified by preparative HPLC and characterized by LC-MS and 1H-NMR. 8a: 1H NMR (DMSO d6): δ 1.75–2.32 (m, 8H, H on piperidine ring); 3.46 (m, 4H, H on pyrrolidine ring); 4.28 (m, 2H, H on benzyl group); 4.57 (m, 1H, H on pyrrolidine ring); 7.20–7.28 (m, 5H, H on benzyl group); 7.37–7.39 (m, 2H, H on benzoyl group); 7.42–7.45 (m, 3H, H on benzoyl group); 7.53 (m, 1H, H on amide group); 7.96 (m, 1H, H on amide group); 9.11 (m, 1H, H on amide group). ESI-MS: 449 (MH+). 8b: 1H NMR (DMSO d6): δ 1.70–2.37 (m, 8H, H on piperidine ring); 3.40 (m, 4H, H on pyrrolidine ring); 3.67 (m, 2H, H on 2-phenylacetyl group); 4.26 (m, 2H, H on benzyl group); 4.48 (m, 1H, H on pyrrolidine ring); 7.19–7.23 (m, 6H, H on benzyl group and 2-phenylacetyl group); 7.26–7.32 (m, 4H, H on 2-phenylacetyl group); 7.52 (m, 1H, H on amide group); 7.95 (m, 1H, H on amide group); 9.12 (t, 1H, J= 5.7 Hz, H on amide group). ESI-MS: 463 (MH+). 8c: 1H NMR (DMSO d6): δ 1.58–2.31 (m, 25H, H on piperidine ring and adamaneacetyl group); 3.40 (m, 4H, H on pyrrolidine ring); 4.27 (m, 2H, H on benzyl group); 4.51 (m, 1H, H on pyrrolidine ring); 7.19–7.30 (m, 5H, H on benzyl group); 7.50 (m, 1H, H on amide group); 7.94 (s, 1H, H on amide group); 9.07–9.13 (m, 1H, H on amide group). ESI-MS: 521 (MH+)
General procedure for the synthesis of compounds 8d-8i
Resin 5a was shaken in 55% TFA/DCM for 30 min to remove the Boc group. Following neutralization with 5% DIEA/DCM, the resin was treated with DIEA (10 equiv.) and corresponding reagents, such as sulfonyl chloride, isocyanate or thioisocyanate (10 equiv.) in anhydrous DMF overnight. The resin was then washed with DMF (3×), DCM (3×), dried and cleaved by HF for 1.5 h at 0 °C. All samples were purified by preparative HPLC and characterized. 8d: 1H NMR (DMSO d6): δ 1.64–2.40 (m, 11H, H on piperidine ring and tosyl group); 3.41 (m, 4H, H on pyrrolidine ring); 4.15 (m, 2H, H on benzyl group); 4.49 (m, 1H, H on pyrrolidine ring); 7.13–7.26 (m, 5H, H on benzyl group); 7.44 (d, 2H, J= 8.3 Hz, H on tosyl group); 7.50 (s, 1H, H on amide group); 7.59 (d, 2H, J= 8.3 Hz, H on tosyl group); 7.94 (s, 1H, H on amide group); 8.98 (t, 1H, J= 5.7 Hz, H on amide group). ESI-MS: 499 (MH+). 8e: 1H NMR (DMSO d6): δ 1.66–2.28 (m, 8H, H on piperidine ring); 3.49 (m, 4H, H on pyrrolidine ring); 4.09 (m, 2H, H on benzyl group); 4.49 (m, 1H, H on pyrrolidine ring); 7.07–7.20 (m, 5 H, H on benzyl group); 7.50 (s, 1H, H on amide group); 7.69–7.76 (m, 3H, H on naphthalene-2-ylsulfonyl); 7.94 (s, 1H, H on amide group); 8.08 (d, 1H, J= 8.0 Hz, H on naphthalene-2-ylsulfonyl); 8.17 (d, 1H J= 8.8 Hz, H on naphthalene-2-ylsulfonyl); 8.21 (d, 1H, J= 8.2 Hz, H on naphthalene-2-ylsulfonyl); 8.41 (s, 1H, H on naphthalene-2-ylsulfonyl); 8.95 (t, 1H, J= 5.6 Hz, H on amide group). ESI-MS: 535 (MH+). 8f: 1H NMR (DMSO d6): δ 1.04 (d, 6H, J= 6.5, H on the isopropyl group); 1.58–2.37 (m, 8H, H on piperidine ring); 3.44 (m, 4H, H on pyrrolidine ring); 3.73 (m, 1H, H on the isopropyl group); 4.27 (m, 2H, H on benzyl group); 4.51 (m, 1H, H on pyrrolidine ring); 6.86 (s, 1H, H on urea); 7.19–7.30 (m, 5H, H on benzyl group); 7.52 (s, 1H, H on amide group); 7.96 (s, 1H, H on amide group); 9.09 (t, 1H, J= 5.7Hz, H on amide group). ESI-MS: 430 (MH+). 8g: 1H NMR (DMSO d6): δ 1.24–2.34 (m, 8H, H on piperidine ring); 3.58 (m, 4H, H on pyrrolidine ring); 4.29 (m, 2H, H on benzyl group); 4.54 (m, 1H, H on pyrrolidine ring); 7.20–7.30 (m, 7H, H on benzyl group and phenyl group); 7.44 (m, 2H, H on phenyl group); 7.53 (s, 1H, H on amide group); 7.96 (s, 1H, H on amide group); 8.47 (s, 1H, H on urea); 9.13 (t, 1H, J= 5.8 Hz, H on amide group). ESI-MS: 464 (MH+). 8h: 1H NMR (DMSO d6): δ 1.08 (t, 3H, J= 7.2 Hz, H on ethyl group); 1.23–2.38 (m, 8H, H on piperidine ring); 3.41–3.48 (m, 6H, H on pyrrolidine ring and ethyl group); 4.28 (m, 2H, H on benzyl group); 4.50 (m, 1H, H on pyrrolidine ring); 6.87 (s, 1H, H on thiourea); 7.19–7.30 (m, 5H, H on benzyl group); 7.54 (s, 1H, H on amide group); 7.97 (s, 1H, H on amide group); 9.15 (t, 1H, J= 5.7 Hz, H on amide group). ESI-MS: 432 (MH+). 8i: 1H NMR (DMSO d6): δ 1.23–2.38 (m, 8H, H on piperidine ring); 3.50–3.89 (m, 4H, H on pyrrolidine ring); 4.30 (m, 2H, H on benzyl group); 4.56 (m, 1H, H on pyrrolidine ring); 7.09 (m, 1H, H on benzyl group); 7.28 (m, 9H, H on benzyl group and phenyl group); 7.56 (s, 1H, H on amide group); 8.00 (s, 1H, H on amide group); 9.19 (t, 1H, J= 5.7 Hz, H on amide group); 9.28 (s, 1H, H on thiourea). ESI-MS: 480 (MH+).
Supplementary Material
Acknowledgments
The authors would like to thank the State of Florida Funding, NIH (1R03DA025850-01A1, Nefzi), NIH (5P41GM081261-03, Houghten) and NIH (3P41GM079590-03S1, Houghten) for their financial support.
Footnotes
Supporting Information Available:
1H-NMR and LC-MS of all the compounds. This information is available free of charge via the Internet at http://pubs.acs.org/.
References and Notes
- 1.(a) Ziegert RE, Toräng J, Knepper K, Bräse S. J Comb Chem. 2005;7:147–169. doi: 10.1021/cc049879v. [DOI] [PubMed] [Google Scholar]; (b) Kundu B. Curr Opin Drug Discovery Dev. 2003;6:815–826. [PubMed] [Google Scholar]; (c) Feliu L, Vera-Luque P, Albericio F, Alvarez M. J Comb Chem. 2007;9:521–565. doi: 10.1021/cc070019z. [DOI] [PubMed] [Google Scholar]; (d) Gil C, Bräse S. J Comb Chem. 2009;11:175–197. doi: 10.1021/cc800102t. [DOI] [PubMed] [Google Scholar]
- 2.(a) Zhu J, Bienaymé H, editors. Multicomponent Reactions. Wiley-VCH; Weinheim: 2005. and references therein. [Google Scholar]; (b) Simila STM, Martin SF. Tet Lett. 2008;49:450. doi: 10.1016/j.tetlet.2008.05.073. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) El Kaim L, Grimaud L. Tetrahedron. 2009;65:2153. [Google Scholar]; (d) Hulme C, Dietrich J. Molecular Diversity. 2009;13:195. doi: 10.1007/s11030-009-9111-6. [DOI] [PubMed] [Google Scholar]; (e) Domling A. Chem Rev. 2006;106:17. doi: 10.1021/cr0505728. [DOI] [PubMed] [Google Scholar]
- 3.(a) Dömling A. Chem Rev. 2006;106:17–89. doi: 10.1021/cr0505728. [DOI] [PubMed] [Google Scholar]; (b) Marcaccini S, Torroba T. Nat Protoc. 2007;2:632–639. doi: 10.1038/nprot.2007.71. [DOI] [PubMed] [Google Scholar]; (c) Dömling A, Ugi I. Angew Chem, Int Ed. 2000;39:3168–3210. doi: 10.1002/1521-3773(20000915)39:18<3168::aid-anie3168>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]; (d) Ugi I, Werner B, Dömling A. Molecules. 2003;8:53–56. [Google Scholar]; (e) Main M, Snaith JS, Meloni MM, Jauregui M, Sykes D, Faulkner S, Kenwright AM. Chem Commun. 2008:5212–5214. doi: 10.1039/b810083g. [DOI] [PubMed] [Google Scholar]
- 4.(a) Pulici M, Quartieri F, Felder ER. J Comb Chem. 2005;7:463–473. doi: 10.1021/cc049831h. [DOI] [PubMed] [Google Scholar]; (b) Hoel AML, Nielson J. Tetrahedron Lett. 1999;40:3941–3944. [Google Scholar]; (c) Tempest RA, Brown SD, Armstrong RW. Angewandte Chemie, International Edition in English. 1996;35:640. [Google Scholar]; (d) Lin Q, O’Neill JC, Blackwell HE. Organic Letters. 2005;7:4455. doi: 10.1021/ol051684o. [DOI] [PubMed] [Google Scholar]; (e) Stocker AM, Keating TA, Tempest PA, Armstrong RW. Tetrahedron Letters. 1996. p. 1149.; (f) Golebiowski A, Jozwik J, Klopfenstein SR, Colson A-O, Grieb AL, Russell AF, Rastogi VL, Diven CF, Portlock DE, Chen JJ. Journal of Combinatorial Chemistry. 2002;4:584. doi: 10.1021/cc020029u. [DOI] [PubMed] [Google Scholar]; (g) Golebiowski A, Jozwik J, Klopfenstein SR, Colson A-O, Grieb AL, Russell AF, Rastogi VL, Diven CF, Portlock DE, Chen JJ. Journal of Combinatorial Chemistry. 2002;4:584–590. doi: 10.1021/cc020029u. [DOI] [PubMed] [Google Scholar]; (e) Suda A, Ohta A, Sudoh M, Tsukuda T, Shimma N. Heterocycles. 2001;55:1023. [Google Scholar]; (f) Sutherlin DP, Stark TM, Hughes R, Armstrong RW. Journal of Organic Chemistry. 1996;61:8350. doi: 10.1021/jo960119j. [DOI] [PubMed] [Google Scholar]; (h) Stocker AM, Keating TA, Tempest PA, Armstrong RW. Tetrahedron Letters. 1996;37:1149. [Google Scholar]
- 5.Hanessian S, Yun H, Hou Y, Tintelnot-Blomley M. J Org Chem. 2005;70:6746–6756. doi: 10.1021/jo050740w. [DOI] [PubMed] [Google Scholar]
- 6.Iserloh U, Pan J, Stamford AW, Kennedy ME, Zhang Q, Zhang L, Parker EM, McHugh NA, Favreau L, Strickland C, Voit J. Bioorg Med Chem Lett. 2008;18:418–422. doi: 10.1016/j.bmcl.2007.10.053. [DOI] [PubMed] [Google Scholar]
- 7.Washburn DG, Hoang TH, Frazee JS, Johnson L, Hammond M, Manns S, Madauss KP, Williams SP, Duraiswami C, Tran TB, Stewart EL, Grygielko ET, Glace LE, Trizna W, Nagilla R, Bray JD, Thompson SK. Bioorg Med Chem Lett. 2009;19:4664–4668. doi: 10.1016/j.bmcl.2009.06.081. [DOI] [PubMed] [Google Scholar]
- 8.Jiang W, Tucci FC, Tran JA, Fleck BA, Wen J, Markison S, Marinkovic D, Chen CW, Arellano M, Hoare SR, Johns M, Foster AC, Saunders J, Chen C. Bioorg Med Chem Lett. 2007;17:5610–5613. doi: 10.1016/j.bmcl.2007.07.097. [DOI] [PubMed] [Google Scholar]
- 9.(a) Gong Y, Becker M, Choi-Sledeski YM, Davis RS, Salvono JM, Chu V, Brown KD, Pauls HW. Bioorg Med Chem Lett. 2000;10:1033–1036. doi: 10.1016/s0960-894x(00)00151-7. [DOI] [PubMed] [Google Scholar]; (b) Choi-Sledeski YM, Mcgarry DG, Green DM, Mason HJ, Becker MR, Davis RS, Ewing WR, Dankulich WP, manetta VE, Morris RL, Spada AP, Cheney DL, Brown KD, Colussi DJ, Chu V, Heran CL, Morgan SR, Bentley RG, Leadley RJ, Maigan S, Guilloteau JP, Dunwiddie CT, Pauls HW. J Med Chem. 1999;42:3572–3587. doi: 10.1021/jm990041+. [DOI] [PubMed] [Google Scholar]
- 10.Melamed JY, Egbertson MS, Varga S, Vacca JP, Moyer G, Gabryelski L, Felock PJ, Stillmock KA, Witmer MV, Schleif W, Hazuda DJ, Leonard Y, Jin L, Ellis JD, young SD. Bioorg Med Chem Lett. 2008;18:5307–5310. doi: 10.1016/j.bmcl.2008.08.038. [DOI] [PubMed] [Google Scholar]
- 11.(a) Pelletier SMC, Ray PC, Dixon DJ. Org Lett. 2009;11:4512–4515. doi: 10.1021/ol901640v. [DOI] [PubMed] [Google Scholar]; (b) Donohoe TJ, Chiu JYK, Thomas RE. Org Lett. 2007;9:421–424. doi: 10.1021/ol062705x. [DOI] [PubMed] [Google Scholar]; (c) Tellitu I, Serna S, Herrero MT, Moreno I, Dominguez E, SanMartin R. J Org Chem. 2007;72:1526–1529. doi: 10.1021/jo062320s. [DOI] [PubMed] [Google Scholar]; (d) Medina JR, Blackledge CW, Erhard KF, Axten JM, Miller WH. J Org Chem. 2008;73:3946–3949. doi: 10.1021/jo7027163. [DOI] [PubMed] [Google Scholar]; (e) Wardrop DJ, Burge MS. J Org Chem. 2005;70:10271–10284. doi: 10.1021/jo051252r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gedey S, Van der Eychen J, Fulop F. Org Lett. 2002;4:1967–1969. doi: 10.1021/ol025986r. [DOI] [PubMed] [Google Scholar]
- 13.Houghten RA. Proc Natl Acad Sci USA. 1985;82:5131. doi: 10.1073/pnas.82.15.5131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kaiser E, Colescott RL, Bossinger CD, Cook PI. Analytical Biochemistry. 1970;34:595. doi: 10.1016/0003-2697(70)90146-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.