

Abstract
The apicomplexan Toxoplasma gondii replicates by endodyogeny, in which replicated organelles assemble into nascent daughter buds within the maternal parasite. The mechanisms governing this complex sequence are not understood. We now report that the kinase inhibitor 3-methlyadenine (3-MA) efficiently blocks T. gondii replication. The inhibition could not be attributed to the effects of 3-MA on mammalian phosphatidylinositol 3-kinase and host cell autophagy. Furthermore, we show that accumulation of host lysosomes around the parasitophorous vacuoles was unaffected. Most 3-MA-treated parasites failed to form daughter buds or replicate DNA, indicating arrest in G1 or early S phase. Some 3-MA-treated parasites displayed abortive cell division, in which nuclear segregation to malformed daughter buds was incomplete or asymmetrical. Electron microscopy revealed the presence of residual body-like structures in many vacuoles, even in the absence of daughter buds. Most treated parasites had otherwise normal morphology and were able to resume replication upon drug removal. 3-MA-treated and control parasites were similar with respect to the extent of Golgi body division and apicoplast elongation; however, treated parasites rarely possessed replicated centrosomes or apicoplasts. These data are suggestive of a generalized blockade of T. gondii cell cycle progression at stages preceding centrosome replication, rather than arrest at a specific checkpoint. We hypothesize that 3-MA treatment triggers a cell cycle pause program that may serve to protect parasites during periods, such as subsequent to egress, when cell cycle progression might be deleterious. Elucidation of the mechanism of 3-MA inhibition may provide insight into the control of parasite growth.
1. Introduction
Toxoplasma gondii, an apicomplexan obligate intracellular parasite, infects about one third of the human population worldwide and causes severe disease in immunocompromised individuals [1]. Following the invasion of host cells and the establishment of a parasitophorous vacuole, Toxoplasma replicates by a mechanism termed endodyogeny, in which two daughter buds form complete cells and subsequently emerge from the mother parasite, the small unused portion of which forms a residual body. During this process, several organelles, including the Golgi apparatus, apicoplast, centrosomes, mitochondrion and nucleus, replicate and segregate into the daughter buds, while others, such as micronemes and rhoptries, form de novo. This sequence of events has recently been elucidated by a series of time-lapse microscopy studies [2–5]. The mechanisms controlling this process, however, are as yet unknown, although the existence of control points is supported by recent studies that use either forward genetic approaches[6] or pharmacologic agents [7] to block cell cycle progression. In addition to signals propagated within the parasite, these mechanisms may also be initiated via interactions with the host cell, which provides a critical source of nutrients. Many host cell organelles, including mitochondria, endoplasmic reticulum, centrosome and endocytic vesicles become closely associated with the parasitophorous vacuole [8,9]. Due to the unique features of endodyogeny, in comparison with the mammalian cell cycle, the elucidation of the mechanisms controlling T. gondii cell division may be of considerable value with respect to the development of novel targets for intervention.
We have previously reported that host cell autophagy contributes to the growth of T. gondii [10]. We now have examined the effect of 3-methyladenine (3-MA), an inhibitor of phosphatidylinositol 3-kinase (PI3K) widely used to suppress autophagy [11], on the parasite. The results of these studies demonstrate that parasite endodyogeny is highly sensitive to 3-MA, independent of effects on host cell autophagy, and suggest that the drug is likely to provide a valuable tool for the elucidation of critical early events in the Toxoplasma cell cycle.
2. Materials and methods
2.1. Parasites and cell culture
RH strain T. gondii and derived strains were maintained in human foreskin fibroblasts (HFF). Green fluorescent protein (GFP)-expressing parasites (GFP-RH) have been described [12]. Yellow fluorescent protein (YFP)-expressing parasites (YFP-RH)[13] were a kind gift of B. Striepen (Univ. of Georgia). RH parasites expressing the apicoplast luminal marker, S+TACP-HcRed (S+T-Red) [14], or additionally expressing the apicoplast membrane protein FtsH1, tagged with V5 and HA epitopes [15], were used for analysis of the apicoplast. A cell line expressing an HA-tagged form of a nucleotide sugar translocator (NST1) was used for analysis of the Golgi apparatus (unpublished results). In some cases the cells also expressed the Golgi marker GRASP55-YFP [16]. Fibroblast monolayers grown on coverslips were infected with the above cell lines. Host cells were cultured in DMEM (Invitrogen) containing 10% fetal bovine serum (Hyclone). Macrophages were obtained by lavage of mice injected 4 days previously with 1 ml of 3% thioglycolate broth (Difco). Cells were cultured for 1 day prior to infection with T. gondii. Multiplicity of infection was either 1 or 4, yielding comparable inhibitor effects. Treatments with 3-MA (Sigma), LY294002 (LC Labs) or wortmannin (EMD) were initiated 3–4 hours post-infection as indicated, to permit completion of invasion and parasitophorous vacuole formation. For plaque assay, infected HFF cultures in multiwell plates were stained with crystal violet after paraformaldehyde fixation and entire wells were photographed. A set of 10 random fields (excluding the outer 20 percent of the well radius) was designed in ImageJ and applied to replicate wells. The value for each well was determined as the mean number of plaques/field. For knockdown of Vps34, HeLa cells were transfected with either nonspecific siRNAs (D-001810-0X, Dharmacon) or predesigned siRNA for hVps34 (GCAAACUCUUCCACAUAGAdTdT, Sigma). Cells were reseeded at 24 hours post-transfection and infected on the following day with YFP-RH at a multiplicity of infection of 4. Infected cells and uninfected controls were harvested for flow cytometry and immunoblotting.
2.2. Flow cytometry
For analysis of intracellular parasite content, cells infected with GFP-RH or YFP-RH parasites were trypsinized, washed with PBS, fixed with 2% buffered paraformaldehyde, washed and analyzed by flow cytometry (FACSCalibur, Becton Dickinson). The data were analyzed with FCS Express (De Novo Software). The number of parasites per infected cell was calculated as the mean fluorescence divided by the fluorescence of single extracellular parasites in the same sample, thereby controlling for differences in GFP expression levels. A sorting analysis verified the linear relationship between fluorescence and parasite number [17].
2.3. Western blotting
Cells were lysed in RIPA buffer (1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS in PBS) supplemented with a protease inhibitor cocktail (Sigma). Protein concentration was determined by the microbicinchoninic acid assay (Pierce). Lysates (20 μg protein) were subjected to SDS-PAGE on 15% gels and transferred to nitrocellulose membranes. After blocking, the membranes were probed with anti-beclin (Novus Biologicals), anti-LC3 (a kind gift of R. Kopito), or anti-β-Actin (Abcam), followed by horseradish peroxidase-conjugated secondary antibody (KPL), and detection with enhanced chemiluminescence (ECL, Amersham). For FtsH1 immunoblots, intracellular V5-FtsH-HA-expressing parasites were treated with 10 mM 3-MA for 20 hours. Lysates from 107 parasites were separated on 7.5% SDS-PAGE gels followed by transfer to nitrocellulose. Blots were blocked and incubated with antibodies as previously described [18]. Bound antibodies were detected using the LI-COR Odyssey.
2.4. Immunofluorescence and electron microscopy
For light microscopy, cells were seeded on coverslips in multiwell plates. Cells were stained as described [14] or using the following protocol. Cells were fixed with 4% paraformaldehyde and permeabilized with 0.1% Triton X-100. After blocking with 10% fetal bovine serum in PBS, the samples were incubated with primary antibodies diluted in 1% BSA washed and incubated with Alexa 488-, Alexa 680-, FITC- or Cy5-conjugated secondary antibodies (Invitrogen, Southern Biotechnology) for 1 hour at room temperature. After extensive washing and staining with DAPI, coverslips were mounted with ProLong Gold anti-fade reagent (Invitrogen). The primary antibodies used were anti-V5 (Invitrogen), anti-LAMP1 (BD Bioscience), anti-centrin (a kind gift of J. Salisbury), and anti-IMC1 (a kind gift of G. Ward and C. Beckers) [19]. Mouse anti-HA monoclonal antibody conjugated to Alexa 594 (10 μg/ml; Invitrogen) was used to detect the HA tag. S+TACP-HcRed was detected by intrinsic fluorescence. Images were collected on a fluorescence microscope (Olympus 1X81) or on a DeltaVision deconvolution microscope with an Olympus UPlan/Apo 100X 1.35 NA objective (images were deconvolved using the conservative ratio option). Analysis of DNA content by DAPI intensity was performed in ImageJ. To derive estimated DNA content relative to the 1n haploid value, nuclear intensity, corrected for background, was normalized to the mean of values derived from untreated vacuoles containing closely adjoined parasites and presumed to have recently completed division. For electron microscopy, infected macrophages were harvested, briefly centrifuged into a loose pellet, fixed with glutaraldehyde/paraformaldehyde and processed for microscopy with the assistance of the Analytical Imaging Facility of the Albert Einstein College of Medicine.
3. Results
3.1. 3-MA inhibits the proliferation of T. gondii
We initially investigated the effect of the PI3K inhibitor 3-MA on the growth of T. gondii in HFF. Intracellular parasite content was assessed by flow cytometry using transgenic parasite strains that express either GFP or YFP. The fluorescence intensity of infected cells was compared to that of single extracellular parasites in the same culture to determine the number of parasites per infected cell. As shown in Fig. 1A, overnight infection in the presence of 3-MA resulted in a dose-dependent inhibition of intracellular parasite accumulation. A minimal effect was observed at 1 – 2 mM 3-MA and a maximal effect (70 percent reduction of parasite content) at 10 mM, a concentration previously shown to be required for optimal inhibition of both autophagy and PI3K in mammalian cells[20]. No inhibition of T. gondii proliferation was observed using two other widely used inhibitors of mammalian PI3K, LY294002 and wortmannin, demonstrating that the anti-parasitic action of 3-MA is not the result of inhibition of host cell PI3K. In earlier pilot experiments, we had observed that all three inhibitors partially inhibit parasite invasion (data not shown). Therefore, in this experiment, 3-MA was added four hours after the initiation of infection, in order to specifically examine effects on proliferation. The effect of 3-MA was not host cell-specific, as similar inhibition was observed using either primary macrophages (Fig. 1B), BALB/c-3T3, or HeLa (Fig. 1C) as host cells. Fig. 1B illustrates the use of two time points to quantitate the inhibition of parasite replication by 3-MA. The fold increase in parasites/cell between 7 and 24 hours post-infection was 1.4 ± 0.02 for 3-MA-treated cells compared to 4.8 ± 0.04 for control cultures (71 percent inhibition). Since tachyzoites are partly asynchronous at the time of infection, residual growth during 3-MA treatment may reflect a preferential action of the drug at earlier stages of the cell cycle.
Fig. 1.
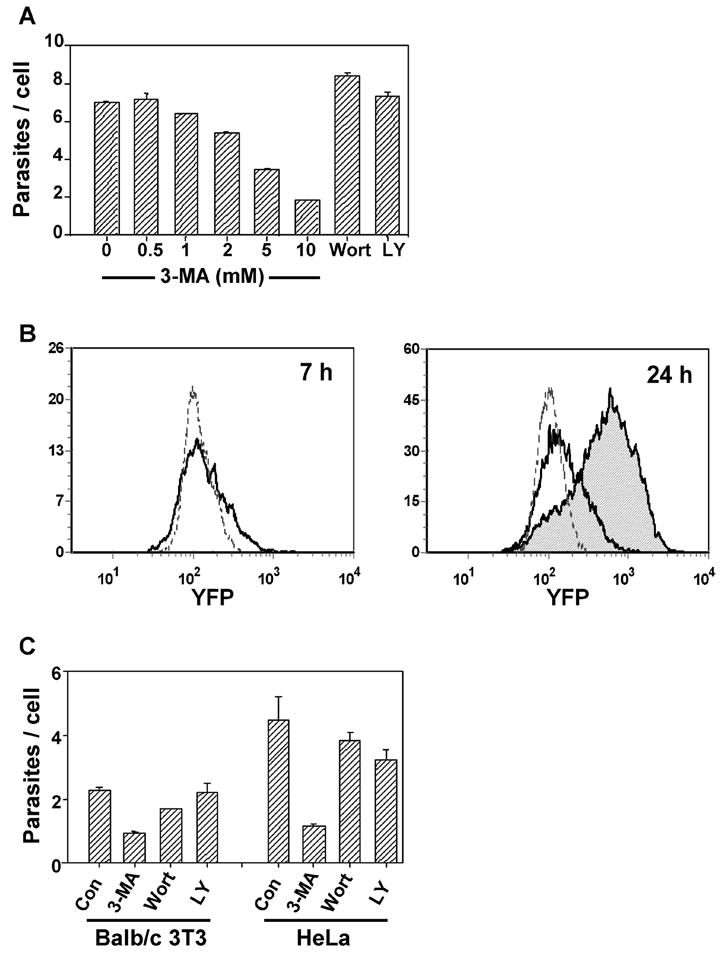
3-MA inhibits T. gondii proliferation. (A) HFF infected with GFP-RH were exposed to the indicated concentrations of 3-MA, or to wortmannin (100 nM) or LY294002 (10 μM). Parasites per infected cell were determined by flow cytometry (mean ± S.E., n = 3). (B) Flow cytometric histograms of parasite content of peritoneal macrophages infected with YFP-RH. After 4 h of infection, cultures were treated with 10 mM 3-MA and analyzed for YFP content at either 7 h or 24 h post-infection. Dotted lines display the intensity of single parasites (extracellular). At 7 h, untreated (solid line) and 3-MA-treated cells (not shown) are similar. At 24 h, 3-MA-treated cells (unshaded) showed reduced proliferation compared to control (shaded). (C) GFP-RH-infected HeLa or BALB/c 3T3 cells were treated for 20 h with 10 mM 3-MA, 100 nM wortmannin, or 10 μM LY 294002.
3.2. The inhibitory effect of 3-MA is independent of host cell autophagy
3-MA is a well-established inhibitor of macroautophagy in mammalian cells [11], and we have recently demonstrated that host cell macroautophagy can be induced by T. gondii and enhances parasite replication [10]. Therefore we considered whether the inhibitory effect of 3-MA on Toxoplasma proliferation might result from its effect on host cell autophagy. Macroautophagy requires the presence of Atg5, which functions as part of a ubiquitin-like conjugation system that results in the conversion of LC3 (Atg8) to a lipidated form (LC3-II) that associates with the developing autophagosome [21]. The extent of LC3 conversion is a widely used indicator of autophagy, although this conversion can occur even during 3-MA blockade of autophagy [22]. We examined the effect of 3-MA on T. gondii proliferation in the presence or absence of host Atg5. As expected, Atg5−/− cells lacked LC3-II (Fig. 2A). However, Atg5 status had no effect on 3-MA inhibition of parasite growth, demonstrating independence of this inhibition from host cell autophagy (Fig. 2B). Similarly, we observed no alteration of 3-MA inhibition upon siRNA-mediated knockdown of Vps34, another essential component of the autophagic pathway [23] (Fig. 2C, D).
Fig. 2.

The inhibitory effect of 3-MA is independent of host cell autophagy. (A, B) Wildtype or Atg5−/− embryonic fibroblasts were infected with GFP-RH for 4 h and then treated for 20 h with 10 mM 3-MA. (C, D). HeLa cells were transfected with nonspecific (NS) or Vps34 RNAi for 24 h, infected for 4 h with GFP-RH and then treated with 3-MA for 20 h. (A, C) Total cell lysates were subjected to Western blot analysis with the indicated probes. ‘Toxo’, lysate prepared from extracellular tachyzoites. (B, D) Parasite content was determined by flow cytometry (mean ± S.E., n = 3).
3.3. 3-MA does not affect the sequestration of host cell lysosomes by the parasitophorous vacuole
PI3Ks play an important role in endosomal trafficking in mammalian cells [24]. In T. gondii-infected cells, host endolysosomes become closely associated with the parasitophorous vacuole, and acquisition of these vesicles by the vacuole may play an important nutritive function for the parasite [25]. Since, in comparison to LY294002 and wortmannin, 3-MA has additional effects on the endolysosomal system [26], it was possible that the effect of 3-MA on T. gondii proliferation was due to an inhibition of host lysosome trafficking to the vacuole. However, in both HFF and HeLa cells, we observed that the association of LAMP1-bearing vesicles with the parasitophorous vacuole was unaffected by 3-MA, even though parasite proliferation was efficiently inhibited (Fig. 3).
Fig. 3.

3-MA does not alter host cell lysosome distribution. HFF (A) or HeLa cells (B) were infected with T. gondii for 4 h and then incubated with or without 10 mM 3-MA for 20 h. Cells were fixed and labeled with anti-LAMP1 and Alexa488-conjugated anti-mouse IgG. Samples in (A) are stained with DAPI (blue). Insets in (B) display phase contrast images of the same field. Scale bar = 2 μm.
3.4. Morphology of 3-MA-treated parasites
The majority of T. gondii exposed to 3-MA retain normal size and shape by phase contrast microscopy (Fig. 3). To more definitively determine the structural integrity of 3-MA-treated parasites, we assessed electron micrographs of macrophages infected overnight in the presence or absence of the drug. 3-MA-treated vacuoles typically contained only a single parasite, which displayed a normal organization of organelles (Fig. 4A). Host mitochondria surrounding the vacuole were considerably enlarged. Notably, many vacuoles were observed to contain large round bodies (asterisks) containing what appeared to be parasite-derived cytoplasm and mitochondria. Nuclei were not observed in these bodies, which were delimited by a simple plasmalemma, in contrast to the three-layered pellicle (plasma membrane + inner membrane complex (IMC)) surrounding the tachyzoite. These features are reminiscent of the residual bodies that form during endodyogeny from mother cell components not incorporated into the emerging daughter buds. Images of transverse sections (Fig. 4C, D) revealed that these bodies were often in continuity with the tachyzoite, implying that they were not simply products of parasite demise. Similar structures were also apparent in light microscope fluorescent images (Fig. 4E).
Fig. 4.

Morphology of 3-MA-treated T. gondii. (A – D) Electron microscopy of thioglycolate-elicited peritoneal macrophages infected overnight with T. gondii in the presence (A, C, D) or absence (B) of 10 mM 3-MA. Samples were prepared as cell pellets in order to view parasites in both longitudinal (A, B) and transverse section (C, D). (A) Representative 3-MA-treated parasite displaying normal morphology, except for the presence of a large residual body-like structure (asterisk) and the absence of any evidence of replication. N, nucleus; G, Golgi body; PV, parasitophorous vacuole; Mt, mitochondrion; A, apicoplast; R, rhoptry; M, microneme; C, conoid; Host Mt, host cell mitochondrion (note characteristic enlargement in 3-MA-treated samples). (B) Normal morphology and replication in control cells. (C, D) Examples of the continuity of the residual body-like structure (asterisks) with the tachyzoite. Arrowheads indicate the points of transition between the pellicle that surrounds the tachyzoite and the single-membrane plasmalemma that surrounds the residual body-like structure. Scale bars = 0.5 um (A, B, D), 0.2 um (C). (E) GFP-RH-infected HFF were incubated with 10 mM 3-MA. The image displays GFP signal. The scale bar is 2 μm.
3.5. Inhibition by 3-MA is reversible
The largely normal appearance of 3-MA-treated parasites suggested that they may retain viability. To determine the reversibility of inhibition, infected HFF cells were subjected to treatment with 3-MA for 20 hours, followed by a 24 hour washout period. As shown in Fig. 5A, vigorous parasite proliferation resumed during the washout period. This proliferation resulted in a 7.4-fold increase in intracellular parasite content, comparable to the 6.8-fold increase observed in untreated cells during the first day of culture. The viability of 3-MA-treated parasites was confirmed by plaque assay (Fig. 5B). Parasitophorous vacuoles displayed a normal rosette structure following inhibitor washout (Fig. 5C), further indicating that for most vacuoles a complete reversal of inhibition was obtained.
Fig. 5.

The inhibitory effect of 3-MA is reversible. (A) HFF cells were infected with YFP-RH for 4 h (multiplicity of infection = 4) and then either harvested for analysis (4h) or incubated in the presence or absence of 10 mM 3-MA for 20 h (Day 1). Cells were then either harvested for analysis or else washed and incubated for a further 24 h in the presence or absence of 3-MA (Day 2). Parasites per cell were determined by flow cytometry (mean ± S.E., n = 3). (B) HFF were infected for 4 h (multiplicity of infection = 0.01) and incubated in the presence or absence of 10 mM 3-MA for 24 h. Culture was continued in the absence of 3-MA for a further 4 d (Control) or 5 days (3-MA), followed by fixation to determine plaque density (mean ± S.E. for replicate wells, n = 3). (C) YFP or phase contrast images of infected cells from the experiment shown in panel A. The scale bar is 5 μm.
3.6. 3-MA inhibits progression through S-phase and daughter bud formation
To locate the effect of 3-MA within the parasite cell cycle, we assessed daughter bud formation in 3-MA treated cells. To avoid interference from secondary effects arising from prolonged drug treatment, the duration of treatment was limited to six hours. Buds were readily detected in control cells by the presence of nascent IMC (Fig. 6A, B). In contrast, the frequency of budding was reduced by 95 percent in 3-MA-treated parasites (Fig. 6A, B) and few buds were observed even after 20 hours of treatment (Fig. 6C). DAPI staining of 3-MA-treated cells revealed an absence of nuclear growth or division in treated cells, suggesting an arrest either prior to or close to the onset of S-phase (data not shown). This was confirmed by quantitative analysis. In parasites treated with 3-MA for six hours, the distribution of DAPI intensity was markedly restricted compared with control cells, consistent with an inability of treated parasites to progress through S phase (Fig. 6D).
Fig. 6.

3-MA-treated parasites are inhibited in bud formation and S phase progression. (A) HFFs were infected with T. gondii for 3 h followed by 6 h of incubation with or without 10 mM 3-MA. Cells were fixed and labeled with anti-IMC1. Representative vacuoles are displayed. (B) The percentage of vacuoles displaying daughter buds. Data from two independent experiments were pooled. A total of 198 control and 178 3-MA-treated vacuoles were scored. (C) IMC1 staining in cell treated with 10 mM 3-MA for 24 h. The scale bar is 2 μm. (D) Inhibition of S phase progression. HFF were infected for 3 h, treated with or without 10 mM 3-MA for 6 h, and then fixed and stained with DAPI. Nuclear DAPI intensity was measured in 52 treated and 39 untreated parasites (pool of two independent experiments). Only the values from vacuoles containing single parasites are displayed. The p value by Student’s t test is < 10−10. (E) Examples of abnormal bud formation and nuclear segregation in HFF infected for 3 h followed by 6 h of incubation in 3-MA.
While most 3-MA-treated parasites were blocked near or prior to S-phase entry, a few parasites (< 10 percent) displayed an abnormal progression to a bud-forming stage (Fig. 6E). The buds in these parasites were characteristically asymmetrical and irregular: at least one of the buds typically had a pinched or otherwise deformed appearance. In some instances, only a single daughter bud was evident. Nuclear segregation was also highly irregular in these cells, and was characterized by asymmetric partitioning between the two buds, as well as the retention of considerable nuclear material by the posterior portion of the mother cell (residual body).
3.7. Early replicative events in 3-MA-inhibited T. gondii
During the T. gondii cell cycle, a number of parasite organelles, including the Golgi apparatus, the centrosome and the apicoplast, initiate replication in a series of clearly delineated events that precede the formation of daughter buds [2,3,5]. We therefore investigated the effect of 3-MA on these early events using transgenic parasite strains expressing tagged Golgi or apicoplast proteins. Division of the Golgi body is one of the earliest organellar duplication events in T. gondii, occurring prior to division of the apicoplast [5]. To investigate the effects of 3-MA on early events during replication, transgenic parasites expressing a Golgi marker, nucleotide-sugar transporter 1 (NST1), were examined. This marker overlaps with GRASP55-YFP (unpublished results), which in higher eukaryotes is a peripheral protein marker for the medial Golgi [27] and is a Golgi marker in T. gondii [16]. In two experiments we examined the effect of treatment with 10 mM 3-MA for 20 hours. As seen in Fig 7A, Golgi body length was broadly distributed in both control and treated cells, although the Golgi body tended to be more extended in the treated cells, where the average length was about 23% longer (1.25 vs. 1.00 μm in experiment 1 and 1.28 vs. 1.06 μm in experiment 2). These data are suggestive of a generalized inhibition of the progression of the Golgi body division cycle by the drug, rather than a block at a specific stage. As already seen with IMC staining, there were indications that occasional progression to late stages in the presence of 3-MA was associated with aberrant morphology: in experiment 1, of the 20 cells displaying a divided Golgi body, four showed abnormal localization of one of the Golgi bodies, and in experiment 2, four of the 14 cells displaying a divided Golgi body showed abnormal localization (compare Figs. 7B and 7C).
Fig. 7.

Golgi body replication in 3-MA-treated parasites. (A) Parasites expressing the Golgi marker NST1-HA were grown with or without 10 mM 3-MA for 20 h in two separate experiments. The length of Golgi body was measured (μm). Each diamond represents a single cell. Cells with divided Golgi bodies are indicated at top. (B, C) Immunofluorescence analysis of Golgi body division in 3-MA-treated parasites. Examples of normal (B) and abnormal (C) Golgi body positioning are shown. NST1-HA was detected using Alexa 594-conjugated anti-HA (red) and cells were counterstained with DAPI (blue)
To examine duplication and division of other organelles, a division cycle analysis was performed. This analysis was based on the assessment of the formation of the inner membrane complex, the shape and location of the apicoplast (as visualized by the luminal marker S+T-Red), and the division of the nucleus [14] as described by Striepen and coworkers [2]. The apicoplast is oval or round in stage 1 and in stage 2 starts to elongate and move closer to the nucleus. By the stage 3 it is further elongated and sits on top of the nucleus. In stage 4, the apicoplast has assumed a “V” shape and daughter buds are detected by the presence of internal IMC. (see Fig. 8A for typical patterns). Later stages include apicoplast and nuclear division, and complete formation of the daughter cells. Cells expressing the apicoplast membrane marker V5-FtsH-HA [15] and the apicoplast luminal marker S+T-Red were treated with 3-MA as above, and full staging of 72 vacuoles from the treated cells and 95 from untreated was performed. As shown in Fig 8B, stages 4 through 6 were absent in the 3-MA treated sample, consistent with the previously observed absence of bud formation. Mistargeting of the luminal marker S+T-Red was not observed. In a second, independent experiment, we screened 250 additional 3-MA-treated vacuoles expressing S+T-Red and found no normal stage 4–6 cells (data not shown). At the same time, consistent with the initial IMC data (Fig. 6), we observed an increase in aberrant forms. About 15 percent of cells displayed an abnormal division or segregation of the detected organelles, a much higher percentage than normally seen (Fig. 8B). Some of these abnormal forms displayed bud formation, but either nuclear or apicoplast division was deficient (Fig. 8D). In other cases the cells contained two daughter IMC complexes and two apicoplasts but only one DAPI-stained nucleus, indicating that the apicoplast was able to divide but not the nucleus (data not shown). These results could suggest that cell cycle progression is blocked by 3-MA at a stage prior to stage 4. Alternatively, since the distribution of apicoplast stages prior to stage 4 did not appear to be altered by 3-MA (Fig. 8B), the results are also consistent with a generalized 3-MA-induced pause at stages 1–3, and in this respect are similar to our observations of Golgi body replication.
Fig. 8.

Apicoplast replication in 3-MA-treated parasites. Parasites expressing the apicoplast membrane marker V5-FtsH1-HA and the apicoplast luminal marker, S+T-Red were treated with 10 mM 3-MA for 20 h. Vacuoles were stained with anti-V5 (green), anti-IMC1 (red) and DAPI (blue) and staged for cell cycle analysis. (A) Representative cells at stages 3 and 4. (B) Vacuole stage distribution in untreated and 3-MA-treated cells (95 and 72 vacuoles, respectively). (C) Immunoblot analysis of cells expressing V5-FtsH-HA. Full-length protein (FL) as well as N-terminally processed (NP) and C-terminally processed (CP) cleavage products were detected. (D) Representative abnormal 3-MA-treated parasites, showing defective duplication of apicoplasts.
In the 3-MA-treated population, we observed some staining of the ER with the apicoplast membrane marker in about ten percent of the cells (data not shown), suggesting that localization of apicoplast membrane proteins might be impeded. V5-FtsH1-HA undergoes distinct processing events during trafficking, with the N-terminus being (non-quantitatively) processed in the endoplasmic reticulum and the C-terminus being processed at or near the apicoplast [18]. Immunoblot analysis of treated cells showed that both cleavage events occurred, and the steady state levels of the processed proteins were not distinguishable from the untreated cells (Fig. 8C). Taken together, the data indicate that the effect of 3-MA on trafficking of proteins to the apicoplast is minimal.
We next examined centrosome replication. This event has been associated with apicoplast elongation, since it occurs at approximately the same time and the replicated centrosomes are found at the ends of elongated apicoplast [2]. However, a recent study suggests that at least initial phases of apicoplast elongation may take place before centrosomes become associated with these ends [5]. Remarkably, we observed that centrosome replication was largely absent in parasites treated for 6 hours with 3-MA, at which time centrosomes had already replicated in the majority of control cells (Fig. 9). These results suggest that the apicoplast can initiate lateral extension in the absence of replicated centrosomes. More importantly, the data imply that 3-MA inhibition of T. gondii proliferation occurs, at least in part, through alteration of events upstream of centrosome duplication in the T. gondii cell cycle.
Fig. 9.
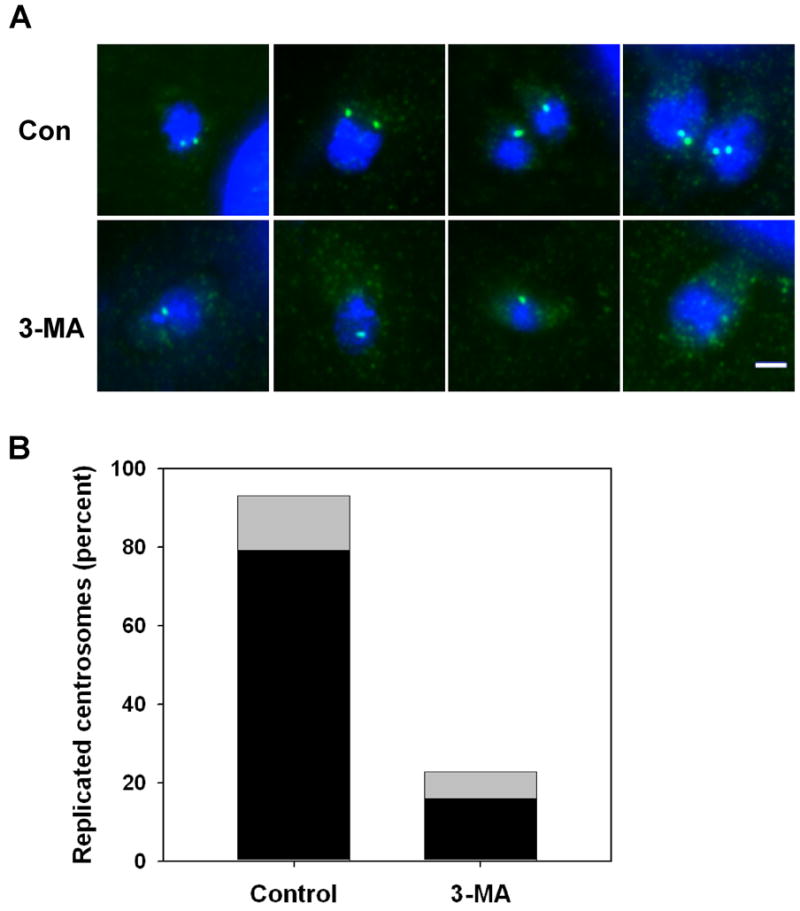
3-MA inhibits centrosome replication in T. gondii. HFF were infected for 3 h and then incubated with or without 10 mM 3-MA for 6 h. Cells were fixed and labeled with anti-centrin (green) and DAPI (blue). (A) Representative images of control and 3-MA-treated vacuoles. (B) Quantitation of centrosome replication in control and 3-MA-treated parasites. The data represent the percentage of parasites in which centrosome replication had occurred since invasion, including both single parasites displaying two centrosomes (black) and divided parasites displaying one centrosome each (gray). A total of 72 control and 44 3-MA-treated parasites were examined. The data are representative of two independent experiments.
4. Discussion
Endodyogeny in Toxoplasma is characterized by a highly choreographed sequence of events in which multiple organelles relocalize and divide, or else form de novo, and assemble into nascent daughter buds within the mother cell. This intricate sequence suggests the existence of regulatory mechanisms, as yet not elucidated, that order and coordinate these events [28]. A forward genetic approach to this problem has shown promise, leading to the identification of several novel loci by the complementation of temperature-sensitive growth mutants [6]. A second approach is pharmacologic, with the goal of targeting specific events or processes that control replication. In this study, we demonstrate that early stages of endodyogeny are effectively targeted by treatment of infected cells with 3-MA.
Many genetic and pharmacologic interventions in the Toxoplasma cell cycle produce lethal effects characterized by the loss of proper coordination between the parallel processes of the normal cycle. For example, when parasite tubulin is targeted with high concentrations of colchicine or dinitroanilines, normal spindle formation is prevented, most organelles do not divide, and daughter buds cannot form. Nevertheless, centrosome duplication and DNA replication continue, leading to abnormal, inviable forms [29]. The failure of parasites to arrest in a viable state in such experiments raised a question as to whether the T. gondii cell cycle includes control points (checkpoints) analogous to those described in other eukaryotes. Several recent studies, however, have demonstrated reversible arrest at early stages of the cell cycle, consistent with the existence of checkpoints. Forward genetic analysis has yielded two temperature-sensitive mutants (63H4 and 31F1) that reversibly arrest with 1n DNA content [6]. Another study from this group has shown similar reversible arrest upon pharmacologic intervention with pyrrolidone dithiocarbamate (PDTC) [7]. The state of organelle division in these new systems has not been reported, except that centrosomes remain unduplicated. Reversible arrest by thymidine block in early S phase (with centrosomes largely duplicated) has also been described; this strategy requires overexpression of heterologous thymidine kinase in the parasite [30]. Finally, arginine deprivation has been shown to generate stable arrest of T. gondii [31].
The majority of our findings appear similarly consistent with a checkpoint model, since 3-MA reversibly blocks T. gondii replication prior to DNA synthesis, centrosome division and budding. However, this model predicts that 3-MA-treated parasites should accumulate at a single stage in the cell cycle. We did not observe such an accumulation, with respect to either apicoplast progression or Golgi body replication, even after 20 hours of 3-MA treatment. While we cannot rule out that a partial accumulation might be revealed by further analysis, the simplest interpretation of the results is that 3-MA induces a uniform pausing of cell cycle progression in stage 1–3 parasites. In the case of the Golgi apparatus, a slow progression may still take place, as indicated by the increased mean lateral extension of this organelle in drug-treated cells. It will be of interest to compare this phenotype to that of the reversible arrest obtained with PDTC or the 63H4 and 31F1 mutants. Arrest with PDTC was reported to result in parasite synchronization upon release, implying that the blockade occurred within a narrow cell cycle window [7].
How might uniform pausing occur in stages 1–3? Several of the replicative events examined, including apicoplast progression and centrosome division, involve organellar motility (the centrosome is reported to migrate caudally prior to division [5]). Therefore it is possible that a disruption by 3-MA of signaling required for such motility could affect multiple early events. Effects on organellar localization might be related to previously observed effects of 3-MA on vesicle trafficking [26]. Alternatively, 3-MA might act by interfering with parasite acquisition of energy or nutrients. An energy-poor state of the 3-MA-treated parasite, perhaps the result of defective acquisition of energy-rich nutrients, is suggested by the prominence of enlarged host mitochondria adjoining the parasitophorous vacuole. Enlargement of mitochondria may represent a compensatory mechanism responding to local ATP depletion [32,33].
Although the concept of uniform pausing, perhaps linked to nutrient deprivation, appears to be at variance with the notion of a specific parasite-determined checkpoint, a reconciliation of these ideas may be possible. This is due to a unique feature of the Toxoplasma cell cycle: the continual insertion, at random points in the cycle, of brief intervals of extracellularity that punctuate intracellular proliferation. These extracellular intervals are characterized by a sudden alteration in parasite access to host nutrients critical to parasite growth, as well as a need to expend energy on motility. The maintenance of an energy-intensive replicative program during these intervals might well be deleterious to the parasite. Extracellular intervals occur with considerable randomness throughout much of the cell cycle, as indicated by the observation that egress of viable parasites can be rapidly and efficiently induced by a variety of agents in cultures of infected cells [34–37]. Furthermore, we have recently demonstrated that highly efficient, rapid inducible egress occurs physiologically in infected mice [17]. It is therefore likely that parasites are adapted to sudden, temporary abrogations of the replication-supporting environment that occur in a non-specific manner with respect to cell cycle stage. It is plausible that a broad-window pause program, which can be accessed throughout early stages of endodyogeny, provides the needed adaptation, and it is conceivable that a checkpoint program of this kind, responding to nutrient level or other signals, could be triggered by 3-MA treatment and result in reversible arrest.
A small number of the 3-MA-treated cells were able to progress to daughter cell bud formation. These parasites have a highly abnormal morphology, characterized by asymmetric nuclear partition, with nuclear material often remaining in the residual body. Daughter buds with reduced nuclei have a pinched, irregular appearance and sometimes only one bud forms. These features are suggestive of defective spindle function. Toxoplasma cell cycle mutants with abnormal nuclear replication or segregation often show spindle defects and incomplete bud formation [6], and direct targeting of spindle microtubules with dintroanalines leads to aberrant nuclear division [29]. Our data are therefore consistent with the notion that duplicated T. gondii centrosomes play a critical role in spindle formation or function. For example, centrosomes may act to maintain spindle orientation or anchoring during the progressive curvature of this structure that accompanies apicoplast and nuclear scission.
Proliferating cells require a mechanism to maintain correct cell size, and a variety of such mechanisms have been demonstrated or proposed in yeast and animal cells [38–40]. In T. gondii, this function may be subsumed by the formation, near the end of the cell cycle, of a residual body containing unused portions of the mother cell. However, it has been reported that various forms of stress during endodyogeny can lead to expansion of this compartment [5]. Remarkably, 3-MA-treated parasites, even in the absence of any bud formation, often display a large appendage with properties of a residual body. A possible explanation of this finding is that 3-MA treatment, while effectively inhibiting bud formation, centrosome duplication and other replicative events, may not completely block growth of the parasite. The parasite may continue to acquire biomass at a reduced rate during drug treatment, and then jettison this excess material by activating the pathway that generates the residual body. The observation that PDTC-treated parasites generate a large amount of ‘cytoplasmic discard’ shortly after release from the block [7] may reflect a similar phenomenon of volume regulation. This notion is compatible with the hypothesis of a ‘host nutrient’ checkpoint advanced earlier; under this hypothesis, the parasite pauses organelle replication under unfavorable conditions, but may not actively arrest general growth processes, which will diminish naturally in the energy-poor extracellular milieu. It will be of interest to examine the modulation of parasite anabolic function in the context of treatments or mutations that lead to cell cycle arrest.
In mammalian cells, 3-MA has been characterized as an inhibitor of PI3K, acting via the competitive inhibition of ATP binding [20], and as a negative regulator of autophagy. We consider it unlikely that these effects account for the inhibition of parasite replication, since the blockade is not replicated with other known PI3K inhibitors, is independent of the state of autophagy in the host cell and is not correlated with any alteration of host endolysosome localization to the PV. It is possible that 3-MA affects another host process, or a parasite-encoded kinase, such as a PI3K homolog. The family of PI3K-related kinases includes target-of-rapamycin (Tor), a central regulator of cell growth that has been shown to be sensitive to PI3K inhibitors, such as LY294002, that act (like 3-MA) as competitive inhibitors of the ATP binding site [41,42]. Proteins related to both PI3K and mTOR are predicted from the T. gondii genome sequence. Inhibition of a PI3K-like kinase might lead to alterations in parasite vesicular trafficking, as observed in 3-MA-treated mammalian cells [26]. Finally, the drug may target a kinase that participates in centrosome duplication. Evidence from other organisms suggests the involvement of multiple kinases in centrosome duplication and separation [43].
In summary, this study identifies 3-methyladenine as a new pharmacologic tool for the reversible blockade of T. gondii replication. The findings are suggestive of a novel pause mechanism affecting multiple early stages of the cell cycle. The elucidation of the mechanism of the 3-MA blockade should provide insight into pathways governing the parasite cell cycle and identify new targets for intervention in tachyzoite expansion.
Acknowledgments
We are grateful to Drs. Michael Lenardo (NIH), Noboru Mizushima (Tokyo Medical and Dental University), Jeffrey Salisbury (Mayo Clinic), Gary Ward (University of Vermont), and Con Beckers (University of North Carolina) for the generous gifts of reagents. We appreciate the discussion with Drs. Ana Maria Cuervo and Jonathan Backer at the Albert Einstein College of Medicine. We also thank Ms. Yanfen Ma for her technical assistance. This study was financially supported by the National Institutes of Health grants (AI-55358 to AO and R01 AI50506 to MP. The authors are solely responsible for the content.
Abbreviations
- 3-MA
3-methyladenine
- GFP
green fluorescent protein
- HFF
human foreskin fibroblasts
- IMC
inner membrane complex
- NST
nuclear sugar transporter
- PI3K
phosphatidylinositol 3-kinase
- PDTC
pyrrolidone dithiocarbamate
- S+T-Red
S+TACP-HcRed
- YFP
yellow fluorescent protein
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hall S, Ryan KA, Buxton D. The epidemiology of Toxoplasma infection. In: Joynson DH, Wreghitt TJ, editors. Toxoplasmosis: A Comprehensive Clinical Guide. Cambridge: Cambridge University Press; 2001. pp. 58–124. [Google Scholar]
- 2.Striepen B, Crawford MJ, Shaw MK, et al. The plastid of Toxoplasma gondii is divided by association with the centrosomes. J Cell Biol. 2000;151:1423–34. doi: 10.1083/jcb.151.7.1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hartmann J, Hu K, He CY, et al. Golgi and centrosome cycles in Toxoplasma gondii. Mol Biochem Parasit. 2006;145:125–7. doi: 10.1016/j.molbiopara.2005.09.015. [DOI] [PubMed] [Google Scholar]
- 4.Hu K, Johnson J, Florens L, et al. Cytoskeletal components of an invasion machine - The apical complex of Toxoplasma gondii. PLoS Pathog. 2006;2:121–38. doi: 10.1371/journal.ppat.0020013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nishi M, Hu K, Murray JM, et al. Organellar dynamics during the cell cycle of Toxoplasma gondii. J Cell Sci. 2008;121:1559–68. doi: 10.1242/jcs.021089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gubbels MJ, Lehmann M, Muthalagi M, et al. Forward genetic analysis of the apicomplexan cell division cycle in Toxoplasma gondii. Plos Pathog. 2008:4. doi: 10.1371/journal.ppat.0040036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.de Felipe MMC, Lehmann MM, Jerome ME, et al. Inhibition of Toxoplasma gondii growth by pyrrolidine dithiocarbamate is cell cycle specific and leads to population synchronization. Mol Biocheml Parasit. 2008;157:22–31. doi: 10.1016/j.molbiopara.2007.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Coppens I, Dunn JD, Romano JD, et al. Toxoplasma gondii sequesters lysosomes from mammalian hosts in the vacuolar space. Cell. 2006;125:261–74. doi: 10.1016/j.cell.2006.01.056. [DOI] [PubMed] [Google Scholar]
- 9.Sinai AP, Webster P, Joiner KA. Association of host cell endoplasmic reticulum and mitochondria with the Toxoplasma gondii parasitophorous vacuole membrane: a high affinity interaction. J Cell Sci. 1997;110:2117–28. doi: 10.1242/jcs.110.17.2117. [DOI] [PubMed] [Google Scholar]
- 10.Wang YB, Weiss LM, Orlofsky A. Host Cell Autophagy Is Induced by Toxoplasma gondii and Contributes to Parasite Growth. J Biol Chem. 2009;284:1694–701. doi: 10.1074/jbc.M807890200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Seglen PO, Gordon PB. 3-Methyladenine: specific inhibitor of autophagic/lysosomal protein degradation in isolated rat hepatocytes. Proc Natl Acad Sci U S A. 1982;79:1889–92. doi: 10.1073/pnas.79.6.1889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ma YF, Zhang YW, Kim K, et al. Identification and characterisation of a regulatory region in the Toxoplasma gondii hsp70 genomic locus. Int J Parasitol. 2004;34:333–46. doi: 10.1016/j.ijpara.2003.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gubbels MJ, Li C, Striepen B. High-throughput growth assay for Toxoplasma gondii using yellow fluorescent protein. Antimicrob Agents Ch. 2003;47:309–16. doi: 10.1128/AAC.47.1.309-316.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Karnataki A, Derocher A, Coppens I, et al. Cell cycle-regulated vesicular trafficking of Toxoplasma APT1, a protein localized to multiple apicoplast membranes. Mol Microbiol. 2007;63:1653–68. doi: 10.1111/j.1365-2958.2007.05619.x. [DOI] [PubMed] [Google Scholar]
- 15.Karnataki A, Derocher AE, Coppens I, et al. A membrane protease is targeted to the relict plastid of Toxoplasma via an internal signal sequence. Traffic. 2007;8:1543–53. doi: 10.1111/j.1600-0854.2007.00637.x. [DOI] [PubMed] [Google Scholar]
- 16.Pelletier L, Stern CA, Pypaert M, et al. Golgi biogenesis in Toxoplasma gondii. Nature. 2002;418:548–52. doi: 10.1038/nature00946. [DOI] [PubMed] [Google Scholar]
- 17.Tomita T, Yamada T, Weiss LM, et al. Externally Triggered Egress Is the Major Fate of Toxoplasma gondii during Acute Infection. J Immunol. 2009;183:6667–6680. doi: 10.4049/jimmunol.0900516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Karnataki A, Derocher AE, Feagin JE, et al. Sequential processing of the Toxoplasma apicoplast membrane protein FtsH1 in topologically distinct domains during intracellular trafficking. Mol Biochem Parasit. 2009;166:126–33. doi: 10.1016/j.molbiopara.2009.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mann T, Beckers C. Characterization of the subpellicular network, a filamentous membrane skeletal component in the parasite Toxoplasma gondii. Mol Biochem Parasit. 2001;115:257–68. doi: 10.1016/s0166-6851(01)00289-4. [DOI] [PubMed] [Google Scholar]
- 20.Blommaart EFC, Krause U, Schellens JPM, et al. The phosphatidylinositol 3-kinase inhibitors wortmannin and LY294002 inhibit autophagy in isolated rat hepatocytes. Eur J Biochem. 1997;243:240–6. doi: 10.1111/j.1432-1033.1997.0240a.x. [DOI] [PubMed] [Google Scholar]
- 21.Hanada T, Noda NN, Satomi Y, et al. The Atg12-Atg5 conjugate has a novel E3-like activity for protein lipidation in autophagy. J Biol Chem. 2007;282:37298–302. doi: 10.1074/jbc.C700195200. [DOI] [PubMed] [Google Scholar]
- 22.Gimenez-Xavier P, Francisco R, Platini F, et al. LC3-I conversion to LC3-II does not necessarily result in complete autophagy. Int J Mol Med. 2008;22:781–5. [PubMed] [Google Scholar]
- 23.Backer JM. The regulation and function of Class III PI3Ks: novel roles for Vps34. Biochem J. 2008;410:1–17. doi: 10.1042/BJ20071427. [DOI] [PubMed] [Google Scholar]
- 24.Lindmo K, Stenmark H. Regulation of membrane traffic by phosphoinositide 3-kinases. J Cell Sci. 2006;119:605–14. doi: 10.1242/jcs.02855. [DOI] [PubMed] [Google Scholar]
- 25.Coppens I, Dunn JD, Romano JD, et al. Toxoplasma gondii sequesters lysosomes from mammalian hosts in the vacuolar space. Cell. 2006;125(2):261–74. doi: 10.1016/j.cell.2006.01.056. [DOI] [PubMed] [Google Scholar]
- 26.Hirosako K, Imasato H, Hirota Y, et al. 3-Methyladenine specifically inhibits retrograde transport of cation-independent mannose 6-phosphate/insulin-like growth factor II receptor from the early endosome to the TGN. Biochem Biophys Res Comm. 2004;316:845–52. doi: 10.1016/j.bbrc.2004.02.119. [DOI] [PubMed] [Google Scholar]
- 27.Shorter J, Watson R, Giannakou ME, et al. GRASP55, a second mammalian GRASP protein involved in the stacking of Golgi cisternae in a cell-free system. EMBO J. 1999;18:4949–60. doi: 10.1093/emboj/18.18.4949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gubbels MJ, White M, Szatanek T. The cell cycle and Toxoplasma gondii cell division: Tightly knit or loosely stitched? Int J Parasitol. 2008;38:1343–58. doi: 10.1016/j.ijpara.2008.06.004. [DOI] [PubMed] [Google Scholar]
- 29.Morrissette NS, Sibley LD. Disruption of microtubules uncouples budding and nuclear division in Toxoplasma gondii. J Cell Sci. 2002;115:1017–25. doi: 10.1242/jcs.115.5.1017. [DOI] [PubMed] [Google Scholar]
- 30.Radke JR, White MW. A cell cycle model for the tachyzoite of Toxoplasma gondii using the Herpes simplex virus thymidine kinase. Mol Biochem Parasit. 1998;94:237–47. doi: 10.1016/s0166-6851(98)00074-7. [DOI] [PubMed] [Google Scholar]
- 31.Fox BA, Gigley JP, Bzik DJ. Toxoplasma gondii lacks the enzymes required for de novo arginine biosynthesis and arginine starvation triggers cyst formation. Int J Parasitol. 2004;34:323–31. doi: 10.1016/j.ijpara.2003.12.001. [DOI] [PubMed] [Google Scholar]
- 32.Bertoni-Freddari C, Mocchegiani E, Malavolta M, et al. Synaptic and mitochondrial physiopathologic changes in the aging nervous system and the role of zinc ion homeostasis. Mech Ageing Dev. 2006;127:590–6. doi: 10.1016/j.mad.2006.01.019. [DOI] [PubMed] [Google Scholar]
- 33.Bertonifreddari C, Fattoretti P, Casoli T, et al. Morphological Plasticity of Synaptic Mitochondria During Aging. Brain Res. 1993;628:193–200. doi: 10.1016/0006-8993(93)90955-m. [DOI] [PubMed] [Google Scholar]
- 34.Stommel EW, Ely KH, Schwartzman JD, et al. Toxoplasma gondii: Dithiol-induced Ca2+ flux causes egress of parasites from the parasitophorous vacuole. Exp Parasitol. 1997;87:88–97. doi: 10.1006/expr.1997.4187. [DOI] [PubMed] [Google Scholar]
- 35.Persson EK, Agnarson AM, Lambert H, et al. Death receptor ligation or exposure to perforin trigger rapid egress of the intracellular parasite Toxoplasma gondii. J Immunol. 2007;179:8357–65. doi: 10.4049/jimmunol.179.12.8357. [DOI] [PubMed] [Google Scholar]
- 36.Moudy R, Manning TJ, Beckers CJ. The loss of cytoplasmic potassium upon host cell breakdown triggers egress of Toxoplasma gondii. J Biol Chem. 2001;276:41492–501. doi: 10.1074/jbc.M106154200. [DOI] [PubMed] [Google Scholar]
- 37.Arrizabalaga G, Ruiz F, Moreno S, et al. Ionophore-resistant mutant of Toxoplasma gondii reveals involvement of a sodium/hydrogen exchanger in calcium regulation. J Cell Biol. 2004;165:653–62. doi: 10.1083/jcb.200309097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Umen JG. The elusive sizer. Curr Opin Cell Biol. 2005;17:435–41. doi: 10.1016/j.ceb.2005.06.001. [DOI] [PubMed] [Google Scholar]
- 39.Kellogg DR. Wee1-dependent mechanisms required for coordination of cell growth and cell division. J Cell Sci. 2003;116:4883–90. doi: 10.1242/jcs.00908. [DOI] [PubMed] [Google Scholar]
- 40.Grebien F, Dolznig H, Beug H, et al. Cell size control - New evidence for a general mechanism. Cell Cycle. 2005;4:418–21. doi: 10.4161/cc.4.3.1523. [DOI] [PubMed] [Google Scholar]
- 41.Vlahos CJ, Matter WF, Brown RF. A Specific Inhibitor of Phosphatidylinositol 3-Kinase. J Cell Biochem. 1994:274. [PubMed] [Google Scholar]
- 42.Walker EH, Pacold ME, Perisic O, et al. Structural determinants of phosphoinositide 3-kinase inhibition by wortmannin, LY294002, quercetin, myricetin, and staurosporine. Molec Cell. 2000;6:909–19. doi: 10.1016/s1097-2765(05)00089-4. [DOI] [PubMed] [Google Scholar]
- 43.Azimzadeh J, Bornens M. Structure and duplication of the centrosome. J Cell Sci. 2007;120:2139–42. doi: 10.1242/jcs.005231. [DOI] [PubMed] [Google Scholar]