

Summary
Immune memory responses to previously encountered pathogens can sometimes alter the immune response to and the course of infection of an unrelated pathogen by a process known as heterologous immunity. This response can lead to enhanced or diminished protective immunity and altered immunopathology. Here we discuss the nature of T-cell cross-reactivity and describe matrices of epitopes from different viruses eliciting cross-reactive CD8+ T-cell responses. We examine the parameters of heterologous immunity mediated by these cross-reactive T cells during viral infections in mice and humans. We show that heterologous immunity can disrupt T-cell memory pools, alter the complexity of the T-cell repertoire, change patterns of T-cell immunodominance, lead to the selection of viral epitope-escape variants, alter the pathogenesis of viral infections, and, by virtue of the private specificity of T-cell repertoires within individuals, contribute to dramatic variations in viral disease. We propose that heterologous immunity is an important factor in resistance to and variations of human viral infections and that issues of heterologous immunity should be considered in the design of vaccines.
Keywords: heterologous immunity, cross-reactive T cell, private specificity, viral pathogenesis, T-cell memory
Introduction
The term heterologous immunity refers to the immunity that can develop to one pathogen after a host has had exposure to non-identical pathogens (1–3). Heterologous immunity is relatively common within closely related species but can also be seen with unrelated agents. It has been demonstrated within various groups of pathogens, including parasites, protozoa, bacteria, and viruses, and also between highly divergent groups, such as between bacteria and viruses. For example, the tuberculosis vaccine bacterium bacille Calmette-Guérin (BCG) can confer immunity in mice to vaccinia virus (VV)(4). Heterologous immunity can be mediated by specifically cross-reactive T cells or antibodies but may be less specific and mediated, for example, by macrophages activated in infection. Classic experiments by Mackaness (5) showed that macrophages activated by BCG, Mycobacterium tuberculosis, Listeria monocytogenes, and Brucella abortus could protect hosts from infection with the other bacteria. Heterologous immunity, in the case where it is mediated by specific cross-reactive processes, can be very long-lasting, or, in the case mentioned above, where transient activation of macrophages occurring during M. tuberculosis infection may be of short duration. Heterologous immunity is generally not as effective as homologous immunity, where a host immunized against a specific pathogen will usually develop a very strong resistance to re-infection with the same pathogen. Nevertheless, a host may have a much less severe course of infection as a consequence of heterologous immunity (2). Protective immunity, however, is not the only consequence of heterologous immunity. Deviation of the normal immune response by partial but non-protective immunity can lead to altered immunopathology and sometimes higher pathogen loads, morbidity, and mortality (2, 6, 7).
An important question is whether heterologous immunity is the rare event or the usual event. The answer to this question is not yet clear in human studies, as protective heterologous immunity would often go unnoticed, though some vaccine studies with measles vaccine and BCG have yielded unexpected beneficial effects in regards to general morbidity and mortality with diseases unrelated to the vaccine (8–11). We do know, however, from our studies with a variety of pathogens in C57BL/6 mice that heterologous immunity and immunopathology are commonplace (2, 4, 6) (Fig. 1). We find, for example, that infections with BCG, influenza A virus (IAV), lymphocytic choriomeningitis virus (LCMV), murine cytomegalovirus (MCMV), and Pichinde virus (PV) all confer a level of protective immunity against VV (2, 4, 6). LCMV, PV, and MCMV will all cross-protect against each other with different efficiencies. IAV, despite protecting against VV, will render hosts more susceptible to LCMV and MCMV. VV, curiously, does not protect against any of the tested heterologous pathogens (Fig. 1). The considerable overlap in heterologous immunity between these very different agents would either argue that it is a common event or that there is something special about the C57BL/6 mouse. Perhaps one of its major histocompatibility complex (MHC) molecules has an unusual capacity to stimulate cross-reactive T cells. Therefore, we also examined heterologous immunity in BALB/c mice and found that immunity to LCMV partially protected the mice from PV and VV (unpublished). These experimental models would therefore suggest that heterologous immunity is a common and normal feature of immunity, thereby posing a cautionary note about using immunologically naive mice as models for human viral infections.
Fig. 1. Heterologous immunity between viruses in mice.

This figure represents patterns of heterologous immunity between different viruses in C57BL/6 mice. Here, mice immune to one virus, from which the arrow emanates, are challenged with a heterologous virus, toward which the arrow points. The shading of the arrow indicates protective immunity, enhancement of viral titers or no effect. The width of the arrow depicts the relative magnitude of the effect. This figure is based on data from published work (2, 6, 96).
This article focuses on reviewing T-cell-dependent heterologous immunity in viral systems. This type of heterologous immunity may be conferred directly and specifically by T cells cross-reactive between different viruses (12). Alternatively, particularly during smoldering persistent infections, chronically stimulated T cells may secrete cytokines that will be directly antiviral or else activate innate immune system cells such as macrophages to provide resistance to super-infecting pathogens (13, 14). It has also been suggested that memory T cells may be particularly sensitive to non-specific activation by virus-induced cytokines such as IL-12 and IL-18, which in turn may induce the synthesis of interferon-γ (IFNγ) (15, 16). Further, a viral induction of IFNα, β, or γ may upregulate host MHC and self-antigens, which may provide signals that sensitize self-reactive T cells that could theoretically participate in immune responses (17). These possibilities are diagrammed in Fig. 2.
Fig. 2. Potential mechanisms of T-cell-dependent heterologous immunity between viruses.
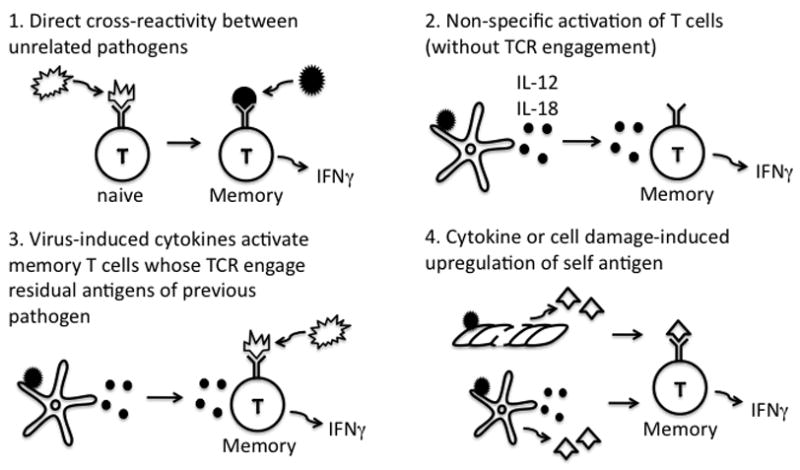
Here the first encountered virus is white and the second encountered virus is black. 1. Direct cross-reactivity between two pathogens, where memory cells specific to the second virus cross-react with antigens of the first virus; 2. Nonspecific activation by cytokines, where the second virus induces cytokines which activate first virus-specific memory cells through cytokines without TCR involvement; 3. Cytokines induced by the second virus plus residual antigen from the persisting original virus trigger the TCR; 4. Activation by self-reactive cellular antigens upregulated by inflammation (IFN) or tissue damage +/− cytokines induced by second virus.
To date the most clear-cut examples of T-cell-mediated heterologous immunity involve T cells cross-reactive between heterologous viruses. This type of cross-reactivity may not only result in enhanced or inhibited clearance of a virus, but may also alter T-cell immunodominance patterns and cause unusual skewing of T-cell repertoires. We begin by discussing these phenomena.
The nature of T-cell cross-reactivity
T cells are characterized by the expression of heterodimeric T-cell receptors (TCRs) generated as a consequence of DNA rearrangement of germ-line genes. T cells expressing the α and β TCR include the CD4+ T cells, which recognize peptides, often about 11 amino acids in length, in association with class II major histocompatibility (MHC) proteins, and the CD8+ T cells, which recognize peptides, often about 9 amino acids in length, in association with class I MHC proteins. The CD4and CD8 co-receptors enhance the avidity of the TCR binding to the peptide-MHC complexes (18). Most but not all crystal structures have shown an orientation of the TCR at a diagonal across the center of the peptide-MHC complex and have revealed that most of the TCR interaction with the peptide-MHC complex is directed against the MHC molecule, rather than the peptide expressed by such a molecule (19). Usually no more than 3 peptide-derived amino acids are actually engaging the TCR, such that it is possible to substitute different amino acids in other positions and still have the TCR engage its target.
T cells are by their nature both specific and highly cross-reactive or ‘polyspecific’ (18–20). They are specific because any given T cell will recognize a small fraction of the universe of MHC-presented peptides, but they are polyspecific because the peptide universe is so large. Considering that there are 20 amino acids and usually 9 residues within a class I MHC-restricted peptide, the potential universe of peptides is 209, or 5 × 1011 peptides for CD8+ T cells and about 400 times higher for 11 amino acid long class II-restricted peptides for CD4+ T cells. This might initially seem manageable without cross-reactivity, given that the theoretical universe of TCR heterodimers in the mouse is about 1015 (and higher in human), but the actual number of T-cell clonotypes present in an individual is much lower than that and has been estimated to be in the range of 106 to 108 for mouse and human, respectively (21–23). Thus, it would seem that to be able to respond to the potential number of antigens present in the peptide universe, the T cells would need to be reactive with more than one epitope (24). This issue becomes increasingly complex when one considers that the T-cell repertoire of the host must not react with host peptides in a negative way and that T cells of high affinity to self-reactive peptides must be clonally deleted or suppressed. It has been estimated that humans encode about 107 unique 9-mers and that probably about 2.5 × 106 9-mers could be processed and presented on available human class I MHC molecules (19). Viral and bacterial pathogens might express 103–106 9-mers, and there are a lot of pathogens, so the host immune system must overcome a number of challenges. It certainly does not need to recognize every 9-mer per pathogen, a subset will do, but there must be enough T cells in the host recognizing enough peptides to be able to mount a rapid immune response to the pathogen, and the specificity of this reaction should be sufficiently high that there is little reactivity against self peptides (18). In the case of sensing pathogens, one should not think that all of this is random, as it is likely that pathogens helped select for T-cell and B-cell variable regions, as well as for MHC molecules, through evolution, perhaps predisposing the immune system to interact with certain classes of pathogens.
The host immune system deals with this problem by the processes of positive and negative selection of T cells in the thymus and by producing a pool of T cells that are polyreactive. The logic of the immune system is to prevent the production of T cells reactive with self without consideration for how many non-self antigens a given T-cell clone will react. Evidence of high levels of cross-reactivity comes from studies in addition to the above mentioned mathematical considerations, which estimate the potential number of peptide-MHC targets for an individual TCR to be in the range of 105 to 106 (18, 20). T cells are positively selected on peptide-presenting MHC in the thymus, but mice engineered to have all the positive T-cell selection mediated by a single peptide produced a T-cell repertoire with a wide range of specificities, indicating widespread TCR cross-reactivity between the positively selected peptides and other antigens; such wide-spread cross-reactivity was facilitated by the poor negative selection found in those mice due to the lack of negative-selecting peptides (25). Mice with greatly reduced TCR diversity as a consequence of transgenic expression of a single TCR β chain or a ‘leaky’ complete αβ transgenic TCR seem well able to generate a T-cell response to a variety of viral infections (26, 27). Further evidence of T-cell polyspecificity comes from the examination of combinatorial peptide libraries and by taking individual T-cell clones and substituting amino acids at different positions (18, 28–30). Such studies predict similar numbers of potential peptide-MHC targets as those derived exclusively from mathematical analyses. These studies together predict that any given T cell may recognize 1 in 104 to 105 9-mers and hence might suggest that T-cell cross-reactivity between pathogens would be of very low frequency. However, a pathogen like VV may encode 40 clearly defined epitopes (31, 32), and epitope-specific T-cell responses may sometimes consist of several hundred T-cell clones (33), thereby dramatically increasing the probability that a T-cell recognizing one of the several epitopes encoded by one virus might recognize one of several epitopes of a heterologous virus. All of these numbers regarding T-cell recognition, of course, imply a certain avidity of binding, and lower avidity interactions might trigger memory cells, which express higher levels of adhesion molecules and whose TCR seem more poised to be fired than those of naive cells (34–36).
Studies with CD4+ and CD8+ T-cell clones using amino acid substitutions in their cognate ligands have shown that CD4+ T cells may be more promiscuous than CD8+ T cells in regards to peptide recognition (18, 28). This may relate to the fact that CD8+ cells seem to have a higher affinity of TCR-pMHC interaction than CD4+ T cells and to the fact that CD4+ T cells tend to see longer peptides. CD4+ T-cell signaling is somewhat different from CD8+ T-cell signaling, in that it involves more pMHC complexes than CD8+ T cells. These potentially promiscuous CD4+ T cells in general seem to be kept more under control than CD8+ T cells in that (i) fewer cells express class II MHC, (ii) the co-receptor CD4 is an absolute requirement for CD4+ T-cell signaling, but CD8 is not an absolute requirement for CD8+ T-cell signaling, (iii) CD4+ T-cell signaling seems more dependent on other costimulatory proteins, and (iv) CD4+ T-cell proliferation is not as great as that of CD8+ T cells after a single contact with a ligand. These distinct properties of the T-cell subsets may actually serve to make it more difficult to detect cross-reactive CD4+ than CD8+ T cells in heterologous viral infections because the clonal burst size of the CD4+ T cells may be smaller than that of the CD8+ T cells. Of note is that most examples of cross-reactive T cells between unrelated viruses have been with CD8+ and not CD4+ T cells (Table 1).
Table 1.
Cross-reactive class 1–restricted viral T-cell epitopesa
| Virus/epitope | Sequence | Reference |
|---|---|---|
| Mouse | ||
| LCMV NP205 (Kb): | Y T V K Y P N L | (50, 51) |
| PV NP205 (Kb): | Y T V K F P N M | |
| VVa11r198 (Kb) | A I V N Y A N L | (12, 156) |
| LCMV NP205 (Kb): | Y T V K Y P N L | |
| PV NP205 (Kb): | Y T V K F P N M | |
| LCMV GP118(Kb): | I S H N F C N L | |
| LCMV GP34(Kb): | A V Y N F A T C | |
| VV e7r130 (Kb) | S T L N F N N L | |
| IAV PA224 (Db) | S S L E N F R A Y V | Wlodarczyk & Selinb |
| LCMV GP276 (Db) | S G V E N P G G Y C L | |
| IAV PB1703(Kb) | S S Y R R P V G I | Wlodarczyk & Selinb |
| LCMV GP34(Kb) | A V Y N F A T C | |
| IAV NP147 (Kd): | T Y Q R T R A L V R T G | (42) |
| IAV PB2146 (Kd): | V F P N E V G A R I L T S E | |
| IAV PB2185 (Kd): | E R E L V R K T R | |
| RSV M282 (Kd): | S Y I G S I N N I | (157) |
| RSV M271 (Kd): | E Y A L G V V G V | |
| IAV HA515 (Kd): | ILAIYSTVASSL | (158) |
| IAV NS150 (Kd): | LGLDIETATRAGKQIVERI | |
| IAV NP366 (Db): | A S N E N M D A M | (55) |
| IAV NP366 variant (Db): | A S N E N M E T M | |
| Dengue 2 NS3298 (Kd): | G Y I S T R V E M | (159) |
| Dengue 3 NS3299 (Kd): | G Y I S T R V G M | |
| Dengue 1 E331 (Ld) | A P C K I P F S S | (160) |
| Dengue 2 E331 (Ld) | S P C K I P F E I | |
| Dengue 3 E331(Ld) | A P C K I P F S T | |
| Dengue 4 E331(Ld) | A P C K V P I E I | |
| Sin nombre virus N331(Kb): | F A I L Q D M R N T I | (161) |
| Puumala virus N331(Kb) | F S I L Q D M R N T I | |
| Hanta virus N331(Kb) | F S I L Q D M R N T I | |
| VV-WR F2L26 (Ld) | S P Y A A G Y D L | (162) |
| MVA F2L26 (Ld) | S P G A A G Y D L | |
| Ectomelia (ECTV) F2L26 (Ld) | S N H A A G Y D L | |
| Human | ||
| EBV BMLF1280 (A2): | G L C T LV A M L | (58) |
| IAV A M158 (A2): | G I L G F V F T L | |
| EBV BRLF1 (A2): | Y V L D H L I V V | cornberg& selinb |
| EBV LMP2329(A2): | L L W T L V V L L | |
| IAV NP85(A2): | K L G E F Y N Q M M | |
| IAV NA231 (A2): | C V N G S C F T V | (68, 119) |
| HCV NS31073 (A2): | C V N G V C W T V | |
| IAV M158 (A2): | G I L G F V F T L | (163) |
| HIV-1P17 GAG77(A2): | S L Y N T I A V L | |
| HPV16 E711 (A2): | Y M L D L Q P E T | (164) |
| Coronavirus NS252 (A2): | T M L D I Q P E D | |
| HIVenvgp120 (A2): | V P T D P N P P E V | (164) |
| M. tuberculosis 19 kDa (A2): | V L T D G N P P E V | |
| Rotavirus VP486(A2): | CPTNQQVVLEGTNKTD | (165) |
| IAV M155 (A2): | LTKGILGFVFTLTVPSERG | |
| Dengue 2 NS371 (B62) | D V K K D L I S Y | (166) |
| Dengue 3 NS371 (B62) | S V K K D L I S Y | |
| Dengue 1 NS3133 (A11) | G T S G S P I V N R | (49) |
| Dengue 2 NS3133 (A11) | G T S G S P I I D K | |
| Dengue 3/4 NS3133 (A11) | G T S G S P I I N R | |
| Dengue 1 NS4b111 (A2) | V L M L V A H Y A | (167) |
| Dengue 2 NS4b111 (A2) | F L L V A H Y A I | |
| Dengue 3 NS4b111 (A2) | V L L L V T H Y A | |
| Dengue 3 NS4b111 (A2) | L V M L L V H Y A | |
| Dengue 1 NS4b181 (A2) | I L L M R T T W A | (167) |
| Dengue 2 NS4b181 (A2) | V L L M R T T W A | |
| Dengue 3 NS4b181 (A2) | L L L M R T S W A | |
| Dengue 4 NS4b181 (A2) | L L L M R T T W A | |
| Dengue 1 NS4a56 (A2) | M L L A L I A V L | (167) |
| Dengue 2 NS4a56 (A2) | L L L T L L A T V | |
| Dengue 3 NS4a56 (A2) | L L L G L M I L L | |
| Dengue 4 NS4a56 (A2) | M L V A L L G A M | |
| Dengue 1 E211 (A2) | F L D L P L P W T | (167) |
| Dengue 2 E211 (A2) | F L D L P L P W L | |
| Dengue 3 E211 (A2) | F F D L P L P W T | |
| Dengue 4 E211 (A2) | F F D L P L P W L | |
| Hantavirus Sin Nombre | I S N Q E P L K L | (168) |
| N421 (A1): | ||
| Hantavirus Seoul virus N421 (A1): | I S N Q E P M K L | |
This table lists the virus, viral protein, epitope number, and class I MHC-restricted element for mouse (top) and human (bottom) cross-reactive CD8 T cell epitopes. With paired epitopes the amino acids in common are underlined. With clusters of epitopes the amino acid in common with the top bolded epitope are underlined. Within clusters of epitopes there may also be T cell cross-reactivity within subsets of the epitopes.
Unpublished epitope data from L.K. Selin and coworkers.
T cells can show high levels of cross-reactivity, because the T cell first scans the pMHC complex by binding to the MHC, and then it settles into a higher affinity interaction as it molds itself around the peptide. The complementarity dependent region 3 (CDR3) undergoes a conformational adjustment to accommodate the pMHC, and this conformational wobble may allow for recognition of different pMHC complexes (37, 38). Many CDR3 regions are rich in glycines, which allow for enhanced flexibility; alanines and serines also can confer such flexibility. By this property, some T-cell clones may be inherently more cross-reactive than others. It is noteworthy that the TCR α chains of human influenza A virus (IAV) HLA-A2-restricted M158-66 epitope-specific CD8+ T cells have extensive glycine runs and that the M1-58 IAV-specific memory T-cell populations are enriched in poly-glycine sequences compared to the T-cell repertoire coming out of the thymus (39). This implies that flexibility through glycine runs may be advantageous for creating a memory repertoire. Some TCRs may be more cross-reactive than others in part because of their relative dependence on the MHC vs. the peptide in their binding and by their ability to conformationally adjust to the pMHC complex. The TCR mostly binds to the MHC and usually contacts only 2–4 amino acids in the peptide, so the total energy of the TCR-pMHC interaction is heavily influenced by the MHC rather than the peptide 2 (18, 19). This could allow for dramatic variations in the peptide sequences without inhibiting the pMHC-TCR interaction. Amino acid substitution experiments on epitopes recognized by T-cell clones have revealed that many amino acids can be replaced by biochemically similar amino acids and preserve reactivity with the TCR (40). What this indicates is that simply looking for identical amino acids between peptides to predict cross-reactivity is likely to extremely underestimate potential cross-reactivities. One study analyzed several defined cross-reactive but amino acid-divergent epitopes between viruses and found that their cross-reactivity could be predicted by an analysis of biochemical similarities of the amino acids in discrete positions of the peptide (40).
It could be argued that some T cells may be more intrinsically cross-reactive than others as a consequence of the orientation of their TCR on the pMHC complex. Crystal structures of several polyspecific autoimmune TCRs have revealed an unusual orientation of the TCR toward the end rather than diagonal across the middle of the pMHC (19). Further, it is possible that some MHC molecules may be intrinsically more capable at cross-reactively signaling T cells. Our studies with several heterologous viral infections in C57BL/6 mice, which have two class I MHC molecules, Db and Kb, have shown more cross-reactivity with Kb-restricted epitopes than with Db-restricted epitopes (Table 1). This observation might reflect the fact that Kb presents an 8 amino acid peptide epitope instead of the more usual 9 amino acid peptide presented by Db, and the shorter the peptide, the more likely the cross-reactivity. It might also reflect the fact that the Kb molecule has a deep pocket and that a relatively small amount of the peptide projects out to engage the TCR. If it is true that some MHC molecules are more promiscuous than others, then it is likely that some individuals may more likely be influenced by heterologous immunity than others, depending on the MHC they express. The nature of the peptide epitope might also dictate the breadth of T-cell responses directed against it and, as a consequence, its potential cross-reactivity with T cells specific to other epitopes. Structural studies have indicated that the IAV PA224-233 epitope generates a far more diverse TCR repertoire than the IAV NP366-374 epitope (41). The PA224 epitope has an arginine emanating at position 4 for the TCR to see, whereas the NP366 amino acids are more buried within the groove of the MHC. Genetically engineering the PA224 epitope to be ‘flattened’ by alanine substitutions led to the induction of a much more restricted repertoire.
Table 1 presents a partial list of cross-reactive T cells between pathogens that have thus far been defined, and cross-reactivity may be more common than mathematically predicted. It should be noted that screens for T-cell epitopes in viruses have found cross-reactivity between two distinct epitopes on two different proteins within the same virus (42), between epitopes from related viruses, such as within the arena-, flavi-, hanta-, and orthomyxovirus groups, and between epitopes from such widely diverse viruses as LCMV and VV in the mouse and IAV and hepatitis C virus (HCV) in the human (3, 43). Further, when screening for immunodominant epitopes on a protein, it is not unusual to get signals from more than one sequence, and the strongest sequence is arbitrarily considered representative of the ‘true’ epitope. This review discusses the significance of these cross-reactivities.
T-cell immunodominance and repertoire skewing
Studies are currently estimating the number of epitope-specific T cells in immunologically naive mice. A recent in vivo limiting dilution assay designed to quantify the number of CD8+ T cells that can grow out in response to viral infections in vivo has estimated frequencies of CD8+ T-cell precursors to LCMV (1 in 2958) and VV (1 in 1444), or a total of about 7000 and 14000 per mouse, respectively (44). MHC tetramer binding studies on T cells from naive mice have now estimated precursors to a variety of previously defined epitopes to be at 15–500 per immunodominant epitope, as previously reviewed (44). Extensive sequencing reactions have estimated that the number of clonotypes specific for certain epitopes can be quite high, such as about 500 calculated for a mouse hepatitis virus-encoded epitope (33). Thus, there is a relatively large number of T cells available to interact with a variety of epitopes from different viruses. During viral infections, there is a generation of T cells reactive with a subset of the peptides encoded by a pathogen, but as epitope-detecting methods have been getting more sophisticated, increasing numbers of such epitopes are now being found. In the C57BL/6 mice there have now been defined over 25 class 1 and 2 epitopes for LCMV and over 40 for VV (31, 32, 45, 46). T-cell responses to a viral pathogen in an infected immunologically naive mouse tend to fall into an epitope immunodominance hierarchy, where T-cell responses to some epitopes are high and to others are low (47). This hierarchy is achieved by the efficiency of processing viral proteins and presenting peptides to the MHC, particularly early during viral infections, and by the availability of T cells with high enough affinity to the epitope to expand during infection. A given T-cell clone may undergo 12 to 15 or so divisions during an infection, thereby making the progeny from individual clones rather easy to detect (44). T-cell clones also compete with each other at the level of the antigen-presenting cells. Because of high affinity TCR-peptide-MHC (pMHC) interactions or because of a high frequency of a presented epitope a clone of one affinity or specificity may ‘immunodominate’ a clone of lower affinity or of a different specificity (47).
The immunodominance hierarchy of epitopes can change under conditions of heterologous immunity, when an infecting pathogen encodes an epitope cross-reactive with a pathogen that has previously infected the host. This is sometimes referred to as ‘original antigenic sin’ in T-cell responses (48, 49). LCMV and Pichinde virus (PV) are arenaviruses that share cross-reactive epitopes, both called NP205-212, which have 6 of 8 amino acids in common and are presented by the Kb class I MHC molecule (50). With either of these infections this NP205 response is subdominant, accounting for 1–3% of the CD8+ T cells, and other epitopes are immunodominant. However, in mice immune to LCMV and challenged with PV or vice versa, the NP205 response becomes dominant, accounting for 15 to 30% of the CD8+ T cells, and the normally dominant epitopes become subdominant. In this case the available repertoire of T cells specific for NP205 is so high in an immune mouse that it overwhelms other factors contributing to immunodominance. In addition, memory T cells seem easier to activate that naive T cells (34–36) Thus, immunodominance can be dictated by one’s prior history of infections.
Our analysis of the TCR repertoire generated in response to either LCMV or PV infection revealed a highly diverse repertoire consisting of many distinct T-cell clonotypes (51). Re-challenge of LCMV-immune mice with LCMV led to a modest narrowing of the repertoire, as T cells competed with each other for expansion in response to the small amount of antigen generated on homologous re-challenge. Most T cells generated in response to either LCMV or PV could recognize the NP205 peptide encoded by either virus, though there were subtle differences in the ability of epitope-charged tetramers to bind the T cells, suggesting different affinities of interaction. Examination of the evolution of the TCR repertoire under conditions of heterologous immunity between LCMV and PV revealed a striking narrowing of the repertoire-much more so than that on homologous challenge, perhaps in part because of the presence of more antigen to drive antigen-specific expansion. This response was markedly oligoclonal, meaning that a small number, not many, distinct clonotypes could be found (51). Narrow T-cell responses to viral epitopes have been seen in certain human viral infections, such as with HIV (52), HCV (53), and CMV (54), but there have been no insights on why such narrow repertoires evolved. One explanation could be heterologous immunity, where only a subset of the T cells in a memory pool specific to an epitope would grow out in response to a different but cross-reactive epitope (51, 55). In human infections and in primate models, narrow oligoclonal responses have been associated with bad prognosis and the generation of epitope escape viral variants (52, 53, 56, 57). We therefore asked whether a narrow TCR repertoire created by conditions of heterologous immunity would give rise to escape mutants in our tractable LCMV/PV system. PV-immune mice were challenged with a high, persistent infection-generating dose of LCMV, which would induce a strong but narrow oligoclonal NP205 response, and an NP205-escape variant was indeed generated (58).
The concept of private specificity
Genetically identical individuals can have genetically different immune systems, because their T and B-cell repertoires are generated by random DNA rearrangements, followed by positive and negative selection. T-cell repertoires that are similar among individuals are referred to as the ‘public’ repertoire, whereas as those that are different among individuals are referred to as the ‘private’ repertoire (12, 59–62). The personalities of the T-cell populations in genetically identical immunologically naive individuals will be similar because they have been selected in the presence of similar antigens and because there are so many T-cell clones. Hence, if naive mice are infected with LCMV, their T cells will respond with similar immunodominant epitope hierarchies, e.g. NP396>GP276>NP205, and their epitope-specific T cells will preferentially employ TCR with similar CDR1 and CDR2, encoded by discrete variable (V) region genes (51, 62, 63). These similarities among individuals are referred to as the public response. Nevertheless, a closer examination of TCR clonotypes reveals that individual hosts generate different TCRs that can be particularly divergent in their CDR3, which comformationally contort to bind the epitope in the context of the MHC (51, 61). These variations in TCR sequences create the private repertoire. All is not random, however, as CDR3 from different individuals may still maintain certain amino acid motifs to accommodate appropriate binding to the peptide. For example, extensive analyses of CDR3 of Vβ19 human HLA-A2-restricted T cells binding a conserved IAV-encoded M158-66 immunodominant epitope reveal a centrally located xRSx motif (64), and analysis of LCMV-encoded Vβ16 responses to NP205 reveal a GGN or GGA motif.
In the LCMV system, both viral clearance and meningoencephalitis (on intracranial infection) are mediated exclusively by CD8+ T cells (65, 66), and in our experience, virus is cleared at the same rate and the time of death due to central nervous system (CNS) involvement is also the same in genetically identical naive mice. Hence, the impact of the private specificity of the T-cell repertoire in regards to immunodominance, Vβ usage, viral clearance, and immunopathology in this virus infection of immunologically naive mice seems minimal. However, studies have shown that the impact of private specificity is much greater under conditions of heterologous immunity, where a pathogen may allow a greatly expanded cross-reactive memory T-cell pool to dictate the personality of its immune response (67). Memory T-cell pools include greatly (~100-fold) expanded populations of epitope-specific T cells, whereas epitope-specific T cells in naive T-cell pools, while being diverse, are present at low frequencies (44, 46). It is the high frequency of memory pools that can change immunodominance and affect heterologous immunity. The private specificities of these greatly expanded T-cell populations can introduce marked variation in regards to whether they will cross-reactively recognize a different epitope (12, 68). A variation in a CDR3 amino acid may not matter for interaction with the original inducing epitope, but it may decrease or enhance the ability of the T cell to interact with another epitope. This presents the possibility that subsets of a T-cell repertoire generated against an epitope in one individual may react with a putative cross-reactive peptide, whereas the repertoire generated in another individual would not. This is clearly demonstrated in both human and mouse systems and will be discussed with specific examples below (Fig. 3). It should be clear that heterologous immunity can not only be responsible for the narrow expansions of oligoclonal TCR repertoires but also for marked variations in T-cell responses between genetically identical individuals.
Fig. 3. Cross-reactivity matrices of CD8+ T-cell epitopes between viruses defined in our laboratories.

(A). Cross-reactivity matrix of Kb-restricted epitopes encoded by LCMV, PV, and VV in C57BL/6 mice. (B). Cross-reactivity matrix of HLA-A2-restricted epitopes between IAV and EBV in humans. Numbers in parentheses represent the numbers of subjects having this cross-reactivity. Width of the lines represents the magnitude of the cross-reactivity when present.
T-cell stability and loss of memory
Both CD8+ and CD4+ T cells undergo programmed proliferation after engaging their cognate ligands. Although in the context of viral infections, the CD8+ T cells tend to expand more, and the CD8+ to CD4+ T-cell ratio often shifts from 1:2 to 3:1 (69–72). After clearance of virus, the T cells undergo an apoptosis-dependent programmed contraction (73), which can be reduced in part by combined mutations in the pro-apoptotic Bim and Fas proteins (74). Thereafter, the host maintains a population of memory T cells, whose numbers are sustained by low level homeostatic proliferation (75–78). A characteristic of type 1 IFN-inducing viral infections is that they will cause an attrition of previously existing memory T-cell populations early in infection, in association with the IFN response (79–84). This attrition involves apoptosis, as shown by Annexin V staining and TUNEL assays, and it preferentially affects memory T cells. T-cell death can certainly be caused by a variety of other mechanisms during viral infection, such as in the case of direct HIV infection of CD4+ T cells, but in the LCMV system it seems to be mostly mediated by IFN 1, as the apoptosis and memory loss do not occur in IFN1 receptor (R) knockout (KO) mice (81, 83). Recovery of these depleted memory cell populations is impaired because they poorly compete with the new virus-specific T-cell response, and they also tend to compete poorly with naive T cells undergoing homeostatic proliferation in a lymphopenic environment (85). Homeostatic proliferation of naive T cells requires interaction with self MHC, and perhaps those T cells with highest affinity for self MHC are less likely to be present in a foreign antigen-specific memory pool. The impact of this early apoptosis of memory cells is that it can be long lasting, in that a series of infections can cause a substantial reduction in memory to previously encountered pathogens and also result in loss of protective immunity against tumor antigens (79, 80, 86). This paradigm has been challenged in a recent publication, but that study used agents and regimens that probably did not induce enough IFN to cause the memory cell loss (87, 88). Table 2 shows new data from our laboratory to address this point, by providing data showing that an LCMV infection causes a subsequent long term reduction in VV-specific CD8+ T cells and that an MCMV infection causes a subsequent long-term reduction in LCMV-specific CD8+ T cells.
Table 2.
Loss of memory (A) CD8+ T-cell number (× 104) and (B) percentage in spleen after heterologous viral infection
| (A) | VV B8R 20-27-specific | LCMV GP 33/34-41-specific | LCMV NP 396-404-specific | |||
|---|---|---|---|---|---|---|
| VV | VV+LCMV | LCMV | LCMV+MCMV | LCMV | LCMV+MCMV | |
| 27 | 16 | 23 | 7.4 | 13 | 4.5 | |
| 32 | 10 | 19 | 9.1 | 17 | 6.8 | |
| 8.4 | 12 | 19 | 9.5 | 11 | 8.6 | |
| 24 | 3.3 | 12 | 17 | 5.8 | 7.8 | |
| 35 | 3.8 | 33 | 9.1 | 19 | 6.3 | |
| mean | 25 | 9.0 | 21 | 10 | 13 | 6.8 |
| ↓64% (p = 0.01) | ↓51% (p = 0.02) | ↓ 48% (p = 0.03) | ||||
| (B) | VV B8R 20-27-specific | LCMV GP33/34-specific | LCMV NP 396-404-specific | |||
|---|---|---|---|---|---|---|
| VV | VV+LCMV | LCMV | LCMV+MCMV | LCMV | LCMV+MCMV | |
| 2.2% | 1.2% | 3.1% | 0.84% | 1.9% | 0.51% | |
| 3.0% | 0.87% | 2.7% | 0.73% | 2.3% | 0.55% | |
| 0.96% | 0.94% | 1.9% | 1.3% | 1.1% | 1.2% | |
| 2.3% | 0.42% | 1.9% | 2.4% | 0.95% | 1.1% | |
| 2.7% | 0.38% | 3.0% | 0.92% | 1.7% | 0.64% | |
| mean | 2.2% | 0.76% | 2.5% | 1.2% | 1.6% | 0.80% |
| ↓66% (p = 0.005) | ↓51% (p = 0.01) | ↓50% (p = 0.03) | ||||
C57BL/6 mice were infected with sublethal doses of VV, strain WR, or LCMV, strain Armstrong, and allowed to clear virus and enter a resting memory state. The VV-immune mice were then challenged with LCMV or vehicle, and the LCMV-immune mice were challenged with MCMV or vehicle. The numbers represent the total numbers (top) or frequencies per CD8 T cell (bottom) of some of their immundominant epitopes per spleen. GP33/34 represents the combined response of overlapping Db- and Kb-restricted epitopes. Values given are for 5 individual mice per group and their means and p values calculated by Student’s t-test.
It has been shown in several systems that reducing lymphocyte numbers by irradiation or cytotoxic drug treatment can actually enhance the development of new T-cell responses (89, 90), so IFN-induced memory T-cell attrition early in infection might actually enhance antiviral T-cell responses. Both memory T-cell attrition and the induction of LCMV-specific CD8+ T cells are reduced in aged mice, consistent with this concept (91, 92). An important question in the context of heterologous immunity is whether antigen engagement would protect a memory cell from the IFN1-induced apoptosis. Experiments addressing whether the presence of cognate ligand in vivo would inhibit memory loss showed that TCR engagement with peptide did not prevent the loss, but the surviving memory cells that did not die could then rapidly proliferate in the presence of their ligand (83). Our computer modeling of this system using the IMMSIM (www.immsim.org) virtual mouse program predicted that, under conditions of heterologous immunity, if T cells specific for the cross-reactive epitope did not undergo apoptosis, they would more dramatically dominate the T-cell response (83). Hence, this attrition of memory cells dampens the effect of heterologous immunity and actually can contribute to more diversified T-cell responses, which are thought to be important for appropriate control of viral infections.
Mouse models of heterologous immunity with defined T cell cross-reactivity
Above we introduced the concept of heterologous immunity and how it is influenced by T-cell repertoires and population dynamics. In summary, heterologous immunity can arise as a consequence of T-cell cross-reactivity, and it can manifest itself in terms of enhanced protective immunity, reduced protective immunity, and altered immunopathology. It can lead to changes in T-cell immunodominance hierarchies and skewed oligoclonal T-cell responses that likely enable the formation of epitope-escape viral variants. Dramatic variations in these responses can be a consequence of the private specificities of the T-cell repertoires unique to the individual. Further, while cross-reactive T cells may enter and dominate a memory pool after a heterologous viral infection, there will simultaneously be a reduction of memory to previously encountered pathogens, due to the IFN1-induced apoptosis of memory cells. Below we address several systems that support these concepts.
LCMV and PV
LCMV and PV encode cross-reactive nucleoprotein epitopes at positions 205–212 that have 6 of 8 amino acids in common and differing in amino acids only in their anchoring sites to the Kb MHC (50). This represents relatively conserved sequences on the nucleoproteins of both Old World and New World arenaviruses, but LCMV, an Old World arenavirus, has a valine in position 3, as do the New World arenaviruses, whereas most other Old World viruses, such as the LCMV-related Lassa fever virus, have an alanine at that position. PV is a typical New World virus with a valine in the third position. Thus, these PV and LCMV epitopes should be similarly recognized by T cells. Indeed, most T cells generated in response to one virus will recognize the alternative peptide epitope (50), but they seem to do so with different affinities, as judged by tetramer binding patterns. Notably, the PV-induced repertoire seems to include more Vβ5-specific T cells, whereas the LCMV-induced repertoire is almost exclusively oriented to Vβ16 and oriented toward different CDR3 motifs than the PV-specific Vβ16 T cells (51). Sequential infections with these viruses led to the generation of highly skewed narrow oligoclonal TCR repertoires that varied with the host, reflecting the private specificity of the T-cell response. It is clear that these variations in repertoire reflected private specificity patterns rather than random stochastic patterns that could be produced, for instance, by a programmed expansion of the first randomly encountered cross-reactive T-cell clone that engaged its cognate ligand. This was shown by transferring splenocyte populations from one immune mouse into three recipient mice, which were then infected with the heterologous virus. All three recipients from a single immune donor generated similar T-cell repertoires, but recipients from other donors all generated a different set of repertoires (51). This indicates that the repertoire available for cross-reactive expansions was relatively unique for each immune mouse.
This two virus system was used to demonstrate other basic concepts of heterologous immunity, including the observation of changes in epitope immunodominance hierarchies in hosts sequentially infected with viruses containing a cross-reactive epitope (50). It was uncertain whether T-cell responses to a cross-reactive epitope would be selectively retained in memory or selectively depleted, perhaps due to activation-induced cell death. Although one could imagine different scenarios depending on the antigen load, in this system the cross-reactive cells were retained, thereby dramatically skewing the memory pool.
PV grows to titers about 10-fold lower in LCMV-infected mice than in naive mice (2, 50), and our assumption is that is due to the cross-reactive T-cell response, in part because T cells from LCMV-immune mice can be transferred into naive mice and partially protect them from PV infection. However, LCMV-immune mice are relatively resistant to infections with VV and MCMV, and it could be argued that nonspecific activation of memory cells might have mediated this protective immunity and that some non-specific mechanisms were at work. We thus tested the NP205 epitope-escape variant of LCMV and showed that it lost its ability to protect mice against PV (A. Chen and R.M. welsh, unpublished data). As a further indicator that heterologous immunity can be mediated by MHC-restricted cellular mechanisms, we tested immunity between these viruses in mice with different MHC alleles, H2b (C57BL/6, B10), H2d (BALB/c, B10.D2), and H2k (C3H, B10.BR). We found evidence of T-cell cross-reactivity between epitopes and heterologous protective immunity in the H2b and H2d systems but not in the H2k system (M. A. Brehm, K. A. Daniels, and R.M. Welsh, unpublished data). This correlation of protective immunity and cross-reactivity adds to the argument that the protective immunity is not a non-specific phenomenon but is instead a consequence of MHC-restricted T-cell cross-reactivity.
LCMV and VV
Our first demonstration of heterologous immunity between viruses was with LCMV-immune mice challenged with VV. To our surprise at that time, the immune mice infected with VV generated a pathology that differed from naïve mice infected with VV, in that there was severe panniculitis, in the form of inflammation and necrosis of visceral fat tissue, bearing resemblance to the human syndromes Weber-Christian disease and erythema nodosum (2, 93). We next found that VV infection activated LCMV-specific memory T-cell populations, and we sought to clarify the nature of the cross-reactivity (94). We reasoned that if VV were cross-reactive with an LCMV epitope, then a VV infection of an LCMV-immune mouse should expand LCMV-specific T cells with the cross-reactive specificity (95). We found that VV sometimes but not all the time expanded populations of LCMV NP205-specific T cells (96), so we searched the VV proteome for potentially cross-reactive epitopes based on sequence similarity to NP205 and found two, E7R and A11R, to be recognized by VV-specific T cells (43). E7R was an immunodominant epitope, and E7R-specific CD8+ T-cell lines proliferated in vivo and protected mice from VV infection (97). Further, immunization of mice with a minigene encoding E7R also protected mice from VV infection, arguing that CD8+ T-cell immunity can be important for that virus. There was no evidence, however, despite its sequence similarity, that the E7R epitope was cross-reactive with LCMV.
Adoptive transfers of CFSE-labeled LCMV-immune splenocyte populations into naive mice, which were then infected with VV showed that about half the time the VV infection expanded populations of NP205-specific T cells, but it sometimes expanded populations of LCMV GP34-specific T cells and other times populations of LCMV GP118-specific T cells (12). This variability in responses was not due to random stochastic events but instead reflected the private specificity of the LCMV-immune T-cell repertoire in individual mice. This was shown by adoptively transferring splenocytes from one immune mouse into three recipient mice. All recipients from the same donor generated the same specificity of outgrowth of LCMV epitope-specific T cells, but recipients from a different donor sometimes stimulated a different specificity. This means that the private specificity of the immune host dictated the specificity of the cross-reactive response to VV. This is conceptually the same as the results found with repertoire skewing and variation in LCMV-immune mice infected with PV (51). There the private specificities dictated TCR usage within an epitope-specific T-cell population. In the LCMV + VV system, the private specificities dictated which epitope would be recognized. The VV-encoded A11R epitope was Kb-restricted, and T cells specific to A11R could be cross-reactive with either LCMV-encoded NP205, GP34, and GP118, all of which are Kb-restricted, though no T cell recognized all epitopes (12, M. Cornberg and L. Selin, unpublished data). In fact, we found a whole matrix of cross-reactivity of epitopes encoded by VV, LCMV, and PV (Fig. 3A). Even though the E7R-specific T-cell population was not cross-reactive with any of the LCMV-encoded epitopes, a part of the E7R T-cell population was cross-reactive with A11R, which engaged some T cells specific to each of the three LCMV epitopes or to the PV-encoded NP205 epitope. These experiments indicate that the epitope specificity of a T-cell response in genetically identical individuals with the same histories of infection can be influenced by the private specificity of the individual. Further, in VV-infected naïve mice the T-cell response to E7R is always greater than that to A11R, but, because of the cross-reactive nature of the A11R epitope, a prior history of an LCMV infection can change the immunodominance pattern to A11R>E7R, but that only happens about 1/3 of the time, presumably due to the private specificity of potentially A11R-reactive T cells in LCMV-immune mice (Cornberg and Selin, unpublished data).
The aberrant immune pathology in LCMV-immune mice challenged with VV is in the form of panniculitis dependent on CD8+ and CD4+ T cells from LCMV-immune mice (2, 93). Further, there is variation in immunopathology in VV-infected LCMV-immune recipients, and this variation is a product of the private specificity of the T cells in the LCMV-immune hosts. Paired naive recipient mice adoptively reconstituted with splenocytes from LCMV-immune mice developed, on VV infection, various degrees of immunopathology that depended on the individual LCMV-immune donor (S. Nie, S. Lin, R.M. Welsh, and L.K. Selin, unpublished data). Thus, private specificities can not only influence T-cell repertoires but also T-cell-dependent immune pathology.
A history of an LCMV infection, as well as infections with BCG, PV, IAV, and MCMV, all provide a level of protective immunity to VV, and VV titers at day 3–4 after infection of LCMV-immune mice are generally 1–2 logs lower than those in naive mice (2, 4, 6, 96) (Fig. 1). The combination of CD8+ and CD4+ spleen T cells from LCMV-immune mice can provide protective immunity to VV (2), as can cross-reactive T-cell lines generated by stimulating LCMV-immune T-cell populations with the VV-encoded and cross-reactive A11R epitope (Cornberg and Selin, unpublished data). Both protective immunity and the induction of immunopathology in this system seem to be heavily dependent on IFNγ, as neither protective immunity nor panniculitis occur in mice treated with antibody to IFNγ or in mice lacking IFNγ receptors (2, 96). The panniculitis but not the protective immunity was also dependent on tumor necrosis factor (TNF), as shown in TNF-deficient mice and by blocking TNF with soluble TNF receptors (Enbrel) (Nie and Selin, unpublished data). In fact, whereas the control of VV infection was partially dependent on TNF in naive mice, the presence of heterologous protective immunity made resistance to VV less dependent on TNF. Perhaps heterologous immunity may help explain why TNF blockers such as Enbrel and Reminade (anti-TNF antibody) used in the treatment of human autoimmune diseases such as rheumatoid arthritis are tolerated with relatively few infectious complications (98).
A VV infection will not confer any protection of mice to LCMV, PV, or MCMV (2). This finding indicates that heterologous immunity is not necessarily reciprocal. This lack of reciprocity is also seen at the level of T-cell proliferation. When CFSE-labeled LCMV-immune cells are transferred into mice which then get infected with VV, there is a substantial proliferation (CFSE-loss) of LCMV-specific T cells. However, when VV-immune cells are transferred into mice that are then infected with LCMV, there is little to no T-cell expansion (99). The reason for this is unclear, but there are far more (>10-fold) potentially cross-reactive NP205-, GP34-, and GP118-specific T cells in the LCMV-immune memory pool than there are A11R-specific cells in the VV-immune memory pool, and those frequencies could influence reciprocity. We also doubt that we have resolved all patterns of cross-reactivity between these two viruses, and it may be that one of the many thousand of potential epitopes encoded by the very large VV, which encodes >200 proteins, could engage a sufficient number of memory cells to have a biologically meaningful effect. In contrast, LCMV encodes only four proteins, which might reduce the likelihood that they could successfully engage the VV-specific memory pool. Large viruses like herpes simplex virus (HSV) and CMV encode many proteins that interfere with the class 1 antigen presentation system (100). Perhaps they do so to escape the protective effects of heterologous immunity. There also remains the possibility that VV-specific memory cells may be qualitatively different than LCMV-specific memory cells. For one thing, different cytokines play a role in the maturation of their T-cell responses, where VV-specific T cells seem dependent on IL-12 but not IFN1 and LCMV-specific T cells do not require IL-12 but are dependent on IFN1 (101, 102). The long term influence of these cytokines on the responsiveness of these memory populations is uncertain. What is clear, however, is that VV infection will readily expand populations of VV-specific memory cells, so there is no reason to think that they are incapable of a recall response.
Heterologous immunity between LCMV and VV has also been studied in the lung after intranasal infection, which is probably the most common route of entry of all of these viruses in nature (6, 96). Again, there was protective immunity and aberrant immunopathology. Under the prescribed conditions, the acute VV infection of naive mice was characterized by high virus loads, necrotizing bronchiolitis, acute inflammatory neutrophilic infiltrates, pulmonary edema, and severe respiratory distress of the host. In contrast, in LCMV-immune mice, the VV infection was characterized by reduced virus loads, little edema, areas of massive chronic lymphocytic infiltration, and enhanced bronchus-associated lymphoid tissue (BALT). Some mice presented with bronchiolitis obliterans, which is an obstruction of the airways with plugs of fibrin and inflammatory cells. This is a pathology with a poorly defined etiology sometimes seen in humans undergoing viral infections and is strongly associated with lung transplant rejection. Despite these pathologies, the LCMV-immune VV-infected mice experienced less respiratory distress, due to areas of the lung being reasonably intact rather than edematous. LCMV- immune mice were also able to survive doses of VV that were lethal to naive mice. Thus, models of heterologous immunity can reveal new diseases with pathologies resembling poorly defined syndromes in humans.
Another interesting feature of heterologous immunity between LCMV and VV in the lung model is that there is a profound deviation in virus-induced cytokine production. The acute VV infection of the naïve mouse is associated with high levels of the proinflammatory cytokine IL-6 and low levels of the immune response cytokine IFNγ. However, presumably because of the presence of IFNγ-producing memory T cells, the cytokine levels in VV-infected LCMV-immune mice are high in IFNγ and low in IL-6 (6, 96). This is consistent with the concept that heterologous immunity is influenced by the cytokine producing capacity of memory cells and that memory cells skewed in one cytokine direction or another might have the capacity to influence Th1 versus Th2 cell deviation during infection. This issue is discussed later in the section on respiratory syncytial virus (RSV).
There is always a question of how much of heterologous immunity is mediated exclusively by cross-reactive mechanisms and how much by non-specific bystander mechanisms, and this remains an issue even when cross-reactive epitopes can be found (Fig. 2). Several studies have concluded that memory T cells can be subjected to bystander activation by cytokines independent of TCR signaling and that these non-specifically activated cells may contribute to heterologous immunity (15, 103–105). It has also been reported that IL-12 and IL-18 can non-specifically stimulate the production of IFNγ by memory T cells (16), and VV infection will induce the synthesis of IL-12 in mice (6). This observation leads to the question of whether the IFNγ-dependent protection against VV is entirely a consequence of T-cell cross-reactivity or is in part due to a non-specific liberation of IFNγ from memory cells. In our hands, LCMV-specific memory cells required TCR engagement to produce elevated IFNγ levels in the presence of IL-12, but this does not rule out a non-specific effect under certain conditions. Further, it is possible that the upregulation of host MHC by IFNs and other cytokines may provide some low level TCR signaling to memory cells, making them more receptive to activation by cytokines without engagement of viral peptides (17, 106). This area is in need of more study, but it is noteworthy that BCG immunization can provide protective immunity to VV by a CD4+ T-cell-dependent mechanism (4). We have examined the IFNγ production by BCG-induced or LCMV-induced peritoneal and fat memory cell populations in vivo after i.p. VV infection by injecting mice with Brefeldin A, which traps the cytokine in the cytoplasm, allowing analysis by flow cytometry (107). Although both primary infections resulted in memory phenotype CD4+ and CD8+ T-cell populations in the peritoneal cavity and visceral fat pads, VV infection preferentially induced IFNγ from CD4+ T cells in BCG-immune mice but from CD8+ T cells in LCMV-immune mice (4). This T-cell subtype bias would argue against a non-specific stimulation of cytokines from memory cells under conditions of the natural infection in vivo.
IAV and LCMV
A history of an intranasal IAV infection of mice can generate a level of protective immunity against VV, possibly through a similar mechanism as that between LCMV and VV described above. However, a history of IAV infection can lead to enhanced titers of LCMV and MCMV, with altered immunopathology (6). It has long been known that IAV infection can break down structural barriers to bacterial infection in the lung, and bacterial invasion of the lung is often responsible for the mortality in human influenza patients. Here, however, mice are receiving LCMV and MCMV long after the IAV infection has resolved and when the lung no longer shows signs of structural damage. This setting suggests that dysregulated protective immunity can occur as a consequence of heterologous immunity.
Heterologous immunity between IAV and LCMV is quite complicated. IAV-immune mice developed enhanced replication of LCMV and enhanced immune pathology in the lung, characterized by massive consolidating mononuclear pneumonia with brochiolization instead of the mild lymphocytic pneumonitis observed during LCMV infection of naive mice (6). Memory T-cell depletion studies indicated that the enhanced LCMV titers seem to be dependent on IAV-specific memory CD4+ T cells (perhaps by regulating CD8+ T cells), and the enhanced immune pathology is caused by cross-reactive IAV-specific CD8+ T-cell responses (M. Wlodarczik and L. K. Selin, unpublished data). In this system, T-regulatory (Treg) cells stimulated by IAV infection might be influencing the response to LCMV. Enhanced numbers of FoxP3+CD25+ Treg phenotype CD4+ cells are found in the lung and draining lymph nodes after IAV infection, and depletion of these cells with monoclonal antibody to CD25 decreases the lung pathology induced by LCMV infection (A. Kraft and L. K. Selin, unpublished data). Cross-reactive epitopes for IAV and LCMV have been defined in these studies in C57BL/6 mice (Table 1). The importance of these epitopes in these patterns of heterologous immunity was shown by using variants of IAV and LCMV with deletions in these epitopes, and those deletions inhibited the immunopathology associated with the heterologous immunity. In addition, epitope-specific T cells were deleted or functionally inactivated by intravenous infusion of the epitope into mice. This selectively depleted the epitope-specific memory T cells and decreased lung pathology induced by the heterologous immunity.
Mouse models of heterologous immunity with undefined T-cell cross-reactivity
Some models of heterologous immunity are not mediated by T cells and are out of the scope of this review, but others have some less defined T-cell involvement.
Persistent viral infections
Mice infected with certain herpes group viruses, such as MCMV and murine γ–herpesvirus, have resistance to intracellular bacterial pathogens such as L. monocytogenes and Yersinia pestis, leading to speculation that there may under certain conditions be a survival benefit to harboring herpesviruses, which tend to be host species-specific and which have evolved over a long time with their host (13).The mechanism of resistance here seems related to enhanced IFNγ production, which would put macrophages in a higher state of activation, leading to their enhanced digestion of intracellular bacteria in a manner similar to the heterologous immunity between intracellular bacteria reported by Mackaness in the 1960s. This does, however, appear to be a T-cell-dependent heterologous immunity, as T cells responding to low levels of persisting viral antigen seem to be the source of the IFNγ. This resistance, therefore, is a T-cell-dependent heterologous immunity that clearly is not a consequence of cross-reactivity.
Resistance to super-infections of persistently infected hosts can also be conferred by other means, including type 1 IFN, which is induced during infections and which can confer resistance to many viruses. It has long been known that mice congenitally infected with LCMV have a life-long persistent infection and that such relatively asymptomatic persistent infections are relatively common in wild mice. These immunologically tolerant mice produce low levels of type 1 IFN and are relatively refractory to infections with other viruses (14). Thus, like in the persistent herpesvirus infections, there may be some benefit to the host to be harbor a persistent viral infection. In this persistent LCMV infection, heterologous immunity seems unrelated to T-cell responses, though that aspect should probably be more carefully studied.
BCG and VV
BCG is a strain of Mycobacterium bovis that can act as a strong adjuvant for innate and adaptive immune response, and it also is used in many parts of the world as a vaccine against M. tuberculosis, due to the high sequence similarity and cross-reactivity between these two bacterial species (108). This vaccination, in effect, is an exercise in heterologous immunity, much as is the original ‘vaccination’ with VV against the smallpox virus, variola. Epidemiological studies, however, have shown unexpected reductions in morbidity and mortality among recipients of BCG immunizations in third world countries, suggesting that BCG may have some role in heterologous immunity to other pathogens (10, 11). Indeed, immunization of C57BL/6 mice with BCG can result in a level of protective immunity against some viruses, such as VV, but not other viruses, such as LCMV (4). Here the protective immunity seems to be mediated by IFNγ produced by CD4+ T cells, as depletion of IFNγ or CD4+ T cells but not CD8+ T cells negates the heterologous immunity. No cross-reactive CD4+ T-cell epitopes have been defined in this system, and there is only a very modest selective proliferation of CD4+ T cells when BCG-immune T-cell populations are transferred into VV-infected mice. This proliferation is much less than the CD8+ T-cell proliferation that occurs in LCMV-immune populations transferred into VV-infected mice. This again leads to the question of whether this BCG-induced heterologous immunity is regulated by cross-reactive T cells or by some other non-specific process, as argued above. An additional argument in favor of a non-specific mechanism is that BCG immunization provides heterologous immunity to VV in other strains of mice.
The arguments that BCG-induced heterologous immunity to VV is due to T-cell cross-reactivity are several but are not yet completely convincing without the demonstration of cross-reactive epitopes (4). The first is that there is selectivity in the protective immunity, as BCG protects against VV but not LCMV or MCMV. However, this pattern of protection could occur if VV were more effective than the other viruses at inducing cytokines like IL-12 or IL-18 to non-specifically activate the memory T cells (Fig. 2). Alternatively, VV might be more susceptible to the antiviral effects of IFNγ than are LCMV and MCMV, though both have some sensitivity to this cytokine. IFNγ seems particularly adept at controlling VV in the mouse, perhaps because the VV-encoded IFNγ decoy receptor has very low affinity to mouse IFNγ, rendering it an ineffective viral immune evasion protein in this system (109). It could well be that the role of IFNγ in human pox virus vaccinations or infections is of reduced importance due to the viral scavenging of IFNγ with decoy receptors of high affinity to human IFNγ. As noted in the discussion above on heterologous immunity between LCMV and VV, we questioned whether memory T cells in the BCG-immune mice showed a pattern of IFNγ secretion in vivo that appeared to be nonspecific. Instead, we found that many more BCG-induced memory phenotype CD4+ T cells than CD8+ T cells produced IFNγ in vivo after VV infection, whereas the VV-induced IFNγ production from LCMV-immune memory populations was predominantly in the CD8+ T cells. These different patterns of IFNγ production are more consistent with a selective, antigen-specific activation. Additionally, the in vivo IFNγ production is blocked by treatment of mice with cyclosporine A, which blocks T-cell signaling. This would argue that the IFNγ production by the memory T cells is not completely due to a non-specific process, but it does not discriminate between signaling of BCG-immune CD4+ T cells with VV antigens, residual BCG antigens (though mice had been treated with antibiotics to clear the infection), or self-antigens, that might be upregulated by IFN, as depicted in Fig. 2.
There are few examples of cross-reactive CD4+ T cells in models of heterologous immunity, even though CD4+ T cells, due to their low affinities and longer peptide targets, should be more cross-reactive than CD8+ T cells. A problem may lie in the possibility that there may be broad-based CD4+ T-cell cross-reactivity between the two pathogens, coupled with a rather low proliferative index that would prevent a CD8+ T-cell-like expansion that can lead to the identification of the cross-reactive epitope. As it stands, this system is in need of further investigation, though it clearly shows T-cell-dependent heterologous immunity between different classes of pathogens.
Immune deviation and heterologous immunity
It seems likely that, if memory T cell pools are skewed in a Th1, Th2, or Th17 direction, infection with a cross-reactive pathogen could activate and induce cytokine secretion by some of those T cells, which could then alter the differentiation of the immune response against the pathogen. There have not been studies to unambiguously show this, though there are hints with infection of BALB/c mice with RSV. RSV causes severe respiratory infections in children and adults, and inactive vaccines against RSV in the past have resulted in increased morbidity and mortality in children when the children became naturally exposed to the virus (110). These children presented with severe lung pathology associated with intense eosinophilia, presumably brought above by a type 2 cytokine response involving IL-5 and other Th2 cell-produced cytokines. This phenomenon can be mimicked in mice immunized with RSV G protein and later challenged with live RSV (111, 112). These mice develop a narrowly focused Vβ14-expressing CD4 Th2 response associated with high levels of IL-5 production and severe eosinophilia. These T cells are directed against a single G-encoded epitope and are rather similar in CDR3 sequences between mice (111). Priming with RSV G can be done in the context of a VV-G recombinant virus, but if a host first has an infection with IAV, VV-G will no longer prime for a Th2 response (7). IAV provides a modest level of heterologous immunity to VV, but, importantly, there is a different cytokine milieu in VV-infected IAV-immune mice in comparison to VV-infected naive mice (6). This milieu is deviated in a type 1 cytokine, IFNγ-oriented direction. Thus, it is possible that the IAV infection caused an immune deviation of the VV-G immunization, such that on infection with the third virus, RSV, severe Th2-dependent pathology did not develop.
Human examples of heterologous immunity
Heterologous immunity is, of course, more difficult to study in human systems, but there is evidence that it occurs. Some studies have been done on what has been called ‘heterotypic’ immunity in the IAV system, where many viral strains and variants are found. T cells cross-reactive between these variants are easy to detect, and an unresolved question is how important these cross-reactive T cells are in immunity or immune pathology associated with human IAV infections (113, 114). Two epidemiological studies have indicated that exposure to one strain of IAV (H1N1) apparently provided some level of protection against another strain of IAV (H2N2) under conditions of very little serological cross-reactivity among the IAV targets of neutralizing antibody(115, 116). Different IAV strains can also encode some extremely conserved epitopes, such as the HLA-A2-restricted M1 epitope, which generates an immunodominant CD8+ T-cell response (117, 118). Whether that specific response provides any resistance to infection is unclear.
IAV and HCV
A more concrete example of heterologous immunity between unrelated human viruses occurs between IAV and HCV. A T-cell response to a defined HCV-encoded HLA-A2-restricted epitope NS31073-1081 was found to stimulate a T-cell response in non-HCV-immune individuals and ended up being strongly cross-reactive and sharing 7 of 9 amino acids with an IAV-encoded NA231-239 epitope (119). In a remarkable study, the breadth of immune responses to the HCV proteome was addressed in a number of HCV-infected patients by enzyme-linked immunospot assay (ELISPOT) analyses of their T cells stimulated against HCV-encoded peptides. Most patients presented with a broadly reactive response with signals seen among a large number of the HCV peptides. Two patients, however, had an extremely focused response against a peptide spanning the HCV and IAV cross-reactive epitopes. These two patients had an unusual presentation for HCV infection, with severe fulminant necrotizing hepatitis (68). Hence, many of the parameters of heterologous immunity addressed above in mouse models are in play in this study, including a (i) narrowly focused cross-reactive response, (ii) associated with severe pathology, and (iii) reflective of private specificity, as all of the patients had likely been exposed to the IAV strains encoding the epitope, but only a subset mounted this immunodominant and narrow response.
IAV and EBV
An unresolved phenomenon of viral infections in humans is that infections with a number of viruses, such as Epstein-Barr virus (EBV), varicella-zoster virus, measles, or mumps are more severe in young adults than in children (120, 121). Usually there is more immune pathology associated with the young adult infections, and we propose that one reason for that phenomenon may be heterologous immunity. As the memory T-cell repertoire of children becomes more diversified due to the acquisition of a sequence of infections, the probability of having memory cells cross-reactive with another pathogen increases, and these may not be the best cells to rapidly clear the virus. Acute infectious mononucleosis (AIM) associated with EBV infections is one of the best examples of such a phenomenon. Children usually have subclinical infections, but teenagers of college age and young adults can have a much more severe and longer lasting infection. The characteristic pathology of AIM is the appearance of ‘atypical lymphocytes’, which are, in fact, cytotoxic granule-containing activated CD8+ T cells responding to EBV-infected B cells and epithelial tissue. There is no evidence that the EBV load is any higher in AIM patients than in the subclinically infected (122). The major pathological feature of AIM, then, is that of an overzealous CD8+ T-cell response that is not very effective at controlling the virus. Many HLA-A2+ AIM patients have an increase in the frequency of their IAV-M158 –specific CD8+ T-cell responses. We have demonstrated human T-cell cross-reactivity between the major immunodominant HLA-A2-restricted epitopes of IAV and EBV and find that a substantial part of the acute EBV-specific CD8+ T-cell response during AIM is mediated by T cells cross-reactive with IAV (58).
Our continued analyses of the CD8+ T-cell response to EBV in AIM patients and immune controls has revealed a whole matrix of HLA-A2-restricted cross-reactivities between EBV-encoded and IAV-encoded epitopes (Fig. 3B), much like the Kb-restricted matrix of cross-reactivities discussed between LCMV-, VV-, and PV- encoded epitopes (Fig. 3A). These cross-reactive T cells were found in some subjects but not others, likely revealing a strong role for private specificity in this process.
Dengue viruses
Dengue viruses are closely related viruses that are found in four serotypes (123, 124). Other than by neutralization assay, these viruses serologically cross-react and encode cross-reactive T-cell epitopes. The most severe manifestation of dengue disease is dengue hemorrhagic fever and shock syndrome, and this most commonly presents when individuals immune to one strain (serotype) of dengue virus become infected with another strain. This has long been thought due to cross-reactive non-neutralizing antibody that binds to viruses without inactivating them and instead enhances the infection of cells like macrophages that bear Fc receptors, in a process known as immune enhancement (124). However, this may be only part of the mechanism, as extensive T-cell cross-reactivity occurs between these viruses (125) (Table 1). One report shows that a substantial part of the T-cell response to the second dengue virus infection consisted of CD8+ T cells having higher affinity to the previously encountered dengue virus than to the one causing the current infection (49). Thus, a combination of enhanced viral load due to antibody-dependent immune enhancement plus cross-reactive low affinity T-cell responses may help contribute to the severity of the disease.
Heterologous immunity and allograft rejection
A characteristic of the T-cell repertoire that emerges from the thymus is the ability of many of the cells to recognize uninfected allogeneic cells (3, 126, 127). There is enough similarity of allogeneic MHC to self-MHC that many allospecific T cells are positively selected in the thymus and not extinguished by negative selection. The frequencies of allospecific T cells then can be quite high in the periphery. A recent assay developed to directly assess the frequencies of naive allospecific T cells utilizes the ability of naive T cells to synthesize TNF on contact with a target. This assay, which can be used in both mouse and human studies, showed that about 1% of the CD8+ T cells in a C57BL/6 (H2b) mouse reacted with BALB/c (H2d) mouse targets (128, 129). This frequency is in the range previously predicted by more cumbersome estimates. Because these allospecific T cells are positively selected by self-MHC, many of them should specifically recognize self-MHC presenting foreign peptide epitopes. It was noticed that viral infections will induce the activation of allospecific cytotoxic T lymphocytes (130), and experiments designed to determine whether this was due to a non-specific polyclonal bystander activation or due to cross-reactivity clearly indicated a cross-reactive mechanism with little evidence of bystander stimulation (27, 131). We developed an assay to directly visualize cross-reactive T cells during an LCMV infection in C57BL/6 mice by first stimulating virus-induced T cells with allogeneic targets in vitro in a standard intracellular cytokine assay to produce IFNγ and then by staining the cells for intracellular IFNγ and LCMV-peptide-charged MHC tetramers. These studies showed that the allo-cross-reactivity was very broad based and of high frequency (132). For example, 1/8 of the LCMV NP396-specific CD8+ T cells cross-reacted with H2d-expressing allogeneic targets. The proportions of epitope-specific T cells reacting with alloantigen varied with the epitope, and these proportions varied with the alloantigen. Each of four tested LCMV epitope-specific T cells contained populations cross-reactive with H2d targets, and two of the four had populations reactive with H2k targets. Variations in the abilities of different strains of mice to generate LCMV-induced allospecific T cells genetically mapped to the MHC locus (131). Allospecific T cells are also induced during human EBV infections (133, 134), and some of these have been shown to be cross-reactive with EBV antigens (135). In this model, human CD8+ T cells recognizing an EBV-epitope (FLRGRAYGL) presented by HLA-B8 cross-reacted with HLA molecules B14, B44, and B35. Structural studies on how the same T-cell clone interacts with self pMHC versus allogeneic pMHC are limited, but one study comparing the 2C TCR cross-recognition of a syngenic Kb pMHC with an allogeneic Ld MHC complex revealed engagement of different parts of the TCR with the complexes (136).
Most allospecific T cells in a young immunologically naive mouse are of a naive (CD44lo, CD11alo) phenotype, but memory phenotype (CD45RO) allospecific T cells are not uncommon in humans. Experiments in the mouse have shown that the cross-reactive naive allospecific T cells activated during a viral infection will be deposited in the memory pool after resolution of the infection, and presumably the same thing happens after human infections (132, 137). Hence, memory phenotype allospecific T cells may well be cross-reactive with pathogens or other antigens the host has previously encountered. Memory allospecific T cells cross-reactive with viral antigens can be quite active in vivo. Fig. 4 shows an experiment where CFSE-labeled spleen leukocytes from an LCMV-immune C57BL/6 mouse were transferred into another C57BL/6 mouse, which also received an intraperitoneal inoculation of allogeneic (H2d) P-815 cells, which form tumors in the peritoneal cavity. These donor spleen cell populations containing cross-reactive T cells expanded in both the spleen (Fig. 4A) and the peritoneal cavity (Fig. 4B) and that they made IFNγ after being stimulated with alloantigen in vitro. These same expanded cell populations made IFNγ in response to certain LCMV encoded epitopes (Fig. 4C,D), thereby indicating that virus-specific T cells will proliferate in response to and migrate into regions of cells displaying allo-antigens in vivo. The qualitative and quantitative nature of the allospecific T-cell pool will change during sequences of viral infections. LCMV induces a higher level of anti-H2k-allospecific T cells if mice had previously had a PV infection (138). This result may relate to the fact that the cross-reactive NP205-specific T cells, which are enriched as a consequence of heterologous immunity, tend to cross-react with H2k alloantigens.
Fig. 4. Implantation of an allogeneic cell line stimulates LCMV-specific memory CD8+ T cells to proliferate in vivo.

CFSE-labeled splenocytes (3 × 107) from LCMV-immune C57BL6 mice were adoptively transferred into naïve congenic recipient mice, which were inoculated with allogeneic (H2d) P815 cells 1 day later. Thirteen days after immunization, donor CD8+ T cells (CD45.2) from the spleens (A) and the peritoneum (B) of recipient mice were evaluated for division by dilution of CFSE, and the values represent the percentage of CD8+ T cells that are CFSElo. Recovered splenocytes (A) and peritoneal exudate cells (B) were incubated with either syngeneic (C57BL/6) or allogeneic (BALB/c) splenocytes for 5 h and then evaluated for the production of IFN-γ. Alternatively, recovered splenocytes (C) and peritoneal exudate cells (D) were incubated with the indicated LCMV-encoded peptides for 5 h and then evaluated for the production of IFN-γ. For the intracellular cytokine assays, samples were gated on CD8+ cells, and the values shown represent the percentage of either CFSElo or CFSEhi CD8+ T cells staining positive for IFN-γ.
The immune system does not seem to care about how many targets its T cells can recognize, as long as they do not have high affinity recognition of self. Although mouse and human MHC molecules may have evolved from primordial genes that allowed clustering cells in an organism to exclude non-alike cells, there is little need for allospecific T cells in a mammalian host. Pregnancy is an issue whereby alloreactivity needs to be controlled, as the mother’s T cells might respond to paternal MHC antigens of the fetus, but a number of structural barriers and suppressive factors reduce the impact of that reaction. The study of physiologically relevant alloreactive T cells for an immunologist primarily relates to their role in the rejection of transplanted allografts. Attempts are usually made to reduce MHC mismatches, but transplant patients are still usually treated with immunosuppressive drugs. These drugs suppress allospecific T-cell responses but at the same time suppress virus-specific T-cell responses that control graft-threatening viruses such as CMV, EBV, and polyoma (139–142). For this reason, considerable effort has gone into development of protocols to tolerize the host to alloantigens prior to transplant and to prevent the need for immunosuppressive drugs. Most of these protocols interfere with T-cell costimulation and block CD40/CD40L interactions or B7-1 and B7-2/CD28 or CTLA4 interactions. For example, transfer of BALB/c (H2d) splenocytes into C57BL/6 (H2b) mice in the presence of monoclonal antibody to CD40L (otherwise known as CD154) tolerizes host anti-H2d alloreactive T cells and allows for the successful transplant of skin or pancreatic islet H2d allografts (143).
Anti-CD154-dependent costimulation blockade works very well in immunologically naive mice but not well in mice that had previously experienced a viral infection (132, 144). This difference may be because the virus infection has created a population of viral epitope-cross-reactive, allospecific memory T cells. Memory cells tend to be less dependent on costimulation and are therefore more difficult to tolerize than naive cells. This property poses problems in the clinic, as humans have memory allospecific T-cell populations.
Acute viral infections will also interfere with the initiation of the anti-CD40L tolerization process, in part through their ability to induce type 1 IFN (145–147). Some but not all viruses may also induce rejection of the allograft after the tolerization process has already occurred (146). There is some allospecific CTL activity induced by LCMV in tolerized mice. We suggest that low affinity allospecific T cells may escape the tolerization process and then get activated at high affinity by a cross-reactive viral epitope. Once activated, they will acquire additional adhesion molecules and enhanced signaling properties that will enable them to attack an allograft, even though they are of low affinity. This mechanism, however, still needs to be proven.
Heterologous immunity and autoimmunity
Viral infections have been shown to induce autoimmunity in animal models and are thought to precipitate such human autoimmune diseases as multiple sclerosis and type 1 diabetes (148). One potential mechanism for this is molecular mimicry, whereby a virus may encode a sequence of amino acids similar to that in a host protein and somehow break through the barrier of tolerance and stimulate the generation of cross-reactive T cells that attack self-tissue (149). Several experimental models of autoimmunity and human multiple sclerosis are diseases of the central nervous system (CNS), for which there may be less tolerance due to the brain being an immunologically privileged site, protected by the blood/brain barrier. Multiple sclerosis (MS) is a disease characterized by exacerbations and remissions, and it has been speculated that these exacerbations might be precipitated by viral infections that induce autoreactive T cells by virtue of T-cell cross-reactivity or as a consequence of a bystander activation event. Since the target antigen for autoreactive T cells is present, a virus infection might only need to induce helper cytokines to boost the autoreactive T-cell response (150). Some interesting work in the MS system also would suggest a cross-reactive heterologous immune mechanism for MS. Some CD4+ T-cell clones found in the CNS of MS patients react with a polyarginine sequence that is found on the CNS α-1B adrenergic receptor, and very similar sequences are encoded by several DNA viruses (151). In a mouse model of MS, mice first infected with a recombinant VV encoding myelin proteolipid protein normally found in the CNS cleared the infection without incident. If these mice were later challenged with MCMV or M. tuberculosis, they developed white matter lesions in the CNS (150, 152).
Transgenic models of autoimmunity have also illustrated the potential for heterologous immunity to be an augmenter of autoimmunity. Mice harboring a transgenic LCMV protein in the brain developed a transient encephalitis when infected with LCMV, but a second wave of CNS inflammation was seen if these LCMV-immune mice were later challenged with PV or VV, both of which are cross-reactive with LCMV (153). When LCMV NP was transgenically expressed in the pancreatic islets, a PV infection after the LCMV infection pushed the host into full-blown diabetes, and the T cells infiltrating the islets were predominantly the cross-reactive NP205-specific T cells, like those discussed above in this review (154). In both brain and islet models, it would appear that the first infection probably overcame tolerance and generated a memory pool of T cells that could then be further stimulated by the heterologous viral infection, which caused an exacerbation of disease.
New frontiers in heterologous immunity
We have reviewed here work that shows that heterologous immunity can disrupt T-cell memory pools, alter the complexity of the T-cell repertoire, change patterns of T-cell immunodominance, lead to the selection of viral epitope-escape variants, alter the pathogenesis of viral infections, and, by virtue of the private specificity phenomenon, contribute to dramatic variations in viral disease. Further, as shown in the LCMV plus VV system, it may change the requirements for certain cytokines such as TNF to control infections. It is now clear that in addition to the widely acknowledged contributors to one’s response to viral infections, such as host genetics and physiological state and viral dose and infection route, one’s history of infections with putatively unrelated pathogens can also influence the course of disease. At this moment we can only point to three or four human diseases where heterologous immunity may be manifest, but, given the ease of demonstrating heterologous immunity against unrelated viruses in mouse models, we suspect that it should be a common trait in humans. We would suggest that the effects of heterologous immunity in humans may occur in the following situations: (i) with infections which present with more severe pathology in young adults than in children, such as those caused by EBV, measles, mumps, and varicella-zoster viruses; (ii) with diseases characterized by high variations in pathology among individuals, such as hepatitis and tuberculosis; (iii) with infections with viruses that are in groups containing genetically related human pathogens, such as within flavi (dengue), papilloma (wart viruses), and picorna viral families; (iv) with persistent viral infections associated with the generation of epitope-escape mutants, such as with HCV and HIV; and (v) with diseases in the elderly, who often have compromised responses to infections due to their decreasing frequencies of naive T cells and increased dependence on de novo responses from memory T cells specific to previously encountered pathogens. Preliminary data in our laboratories suggest that the effects of heterologous immunity may be more profound in aged than in young mice.
It could be argued that heterologous immunity is too complex to study in humans, due to the variations in human MHC molecules, the uncertain infection history of any individual, and the variations caused by the private specificity of immune responses. However, considering that evidence of heterologous immunity has already appeared with several human viruses, we would argue that it is not too complex to study. Nevertheless, some extensive epidemiological studies and systematic identification of CD4+ and CD8+ T-cell epitopes for the most common human viral infections may be needed to start making predictions along the lines of ‘a person with a given MHC and a history of infection with a given virus is likely to respond in a given way to an infection with another given virus.’ It could be argued that variations in disease caused by private specificity patterns add too much complexity to the system to be resolvable. However, our studies on the private specificity of T-cell cross-reactivity have shown that while it is variable, it is not random. We can predict, for example, that a certain percentage of the time (~50%) a VV infection will expand the LCMV NP205 response in LCMV-infected C57BL/6 mice. We can predict that about 30% of the time, an HLA-A2+ IAV-immune individual will have T-cell cross-reactivity between the IAV M1 epitope and the EBV BMLF1 epitope. We contend that this type of variability is manageable and understanding that this type of variability can exist will help in interpreting human genetic studies on resistance to infections. New strategies are being developed to selectively deplete or inactivate epitope-specific T cells in vivo, so it may in the future be possible to prevent certain disease manifestations by depleting a cross-reactive T cell.
The potential detrimental effects of heterologous immunity should be considered in the rational design of new vaccines (148). For example, using recombinant adenovirus or vaccinia vectors to immunize against a protein encoded by another virus may give strong immune responses because of the reactivation of vector-specific CD4+ T cells, but we suggest that the pronounced helper effect of such vaccines might allow for the generation of lower affinity CD8+ T-cell responses against the target protein. We suggest that immunization with live IAV strains that encode new heterotypic strain-specific proteins but still express immunodominant epitopes in common with all IAV strains, such as the HLA-A2-restricted M1 epitope, may boost responses to the common epitope that will immunodominate and suppress the desired responses to the heterologous epitopes. Perhaps vaccines should be engineered to delete epitopes that might interfere with the desired target response (148). As another example, given the observed epitope-dependent heterologous immunity reported and discussed above between IAV and HCV (68, 155), would it not be wise to delete the cross-reactive epitope from any live HCV vaccine? The damaging effects of heterologous immunity must, of course, be balanced with its positive effects. Mouse model studies have indicated that most of the time heterologous immunity is a good thing for the host (Fig. 1), in that it usually leads to more rapid clearance of the virus and, in fact, can lower mortality when the host is exposed to normally lethal doses of virus (96). With the selection of the correct cross-reactive epitopes, there perhaps could be manufactured a vaccine that would give good protective immunity but not immunopathology against a wide variety of pathogens.
Acknowledgments
This work was supported by United States National Institutes of Health (NIH) Research Grants AI017672, AI081675, AI073871, AI46692, AI046578, AI049320, AI054455, and AI073540. The opinions expressed are those of the authors and not necessarily those of the NIH. We thank Keith Daniels for help with the artwork and acknowledge the unpublished research contributions of Alex Chen, Shalyn Clute, Markus Cornberg, Keith Daniels, Anke Kraft, Siwei Nie, Levi Watkin, and Myriam Wlodarczyk.
Reference List
- 1.Clark IA. Heterologous immunity revisited. Parasitology. 2001;122 (Suppl):S51–S59. [PubMed] [Google Scholar]
- 2.Selin LK, Varga SM, Wong IC, Welsh RM. Protective heterologous antiviral immunity and enhanced immunopathogenesis mediated by memory T cell populations. J Exp Med. 1998;188:1705–1715. doi: 10.1084/jem.188.9.1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Welsh RM, Selin LK. No one is naive: the significance of heterologous T-cell immunity. Nat Rev Immunol. 2002;2:417–426. doi: 10.1038/nri820. [DOI] [PubMed] [Google Scholar]
- 4.Mathurin KS, Martens GW, Kornfeld H, Welsh RM. CD4 T-cell-mediated heterologous immunity between mycobacteria and poxviruses. J Virol. 2009;83:3528–3539. doi: 10.1128/JVI.02393-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mackaness GB. The immunological basis of acquired cellular resistance. J Exp Med. 1964;120:105–120. [PubMed] [Google Scholar]
- 6.Chen HD, Fraire AE, Joris I, Welsh RM, Selin LK. Specific history of heterologous virus infections determines antiviral immunity and immunopathology in the lung. Am J Pathol. 2003;163:1341–1355. doi: 10.1016/S0002-9440(10)63493-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Walzl G, Tafuro S, Moss P, Openshaw PJ, Hussell T. Influenza virus lung infection protects from respiratory syncitial virus-induced immunopathology. J Exp Med. 2000;192:1317–1326. doi: 10.1084/jem.192.9.1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aaby P, Samb B, Simondon F, Seck AM, Knudsen K, Whittle H. Non-specific beneficial effect of measles immunisation:analysis of mortality studies from developing countries. Brit Med J. 1995;311:481–485. doi: 10.1136/bmj.311.7003.481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kristensen I, Aaby P, Jensen H. Routine vaccinations and child survival: follow up study in Guinea-Bissau, West Africa. BMJ. 2000;321:1435–1438. doi: 10.1136/bmj.321.7274.1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Roth A, et al. BCG vaccination scar associated with better childhood survival in Guinea-Bissau. Int J Epidemiol. 2005;34:540–547. doi: 10.1093/ije/dyh392. [DOI] [PubMed] [Google Scholar]
- 11.Roth AE, Stensballe LG, Garly ML, Aaby P. Beneficial non-targeted effects of BCG--ethical implications for the coming introduction of new TB vaccines. Tuberculosis. 2006;86:397–403. doi: 10.1016/j.tube.2006.02.001. [DOI] [PubMed] [Google Scholar]
- 12.Kim SK, Cornberg M, Wang XZ, Chen HD, Selin LK, Welsh RM. Private specificities of CD8 T cell responses control patterns of heterologous immunity. J Exp Med. 2005;201:523–533. doi: 10.1084/jem.20041337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Barton ES, et al. Herpesvirus latency confers symbiotic protection from bacterial infection. Nature. 2007;447:326–329. doi: 10.1038/nature05762. [DOI] [PubMed] [Google Scholar]
- 14.Bukowski JF, Biron CA, Welsh RM. Elevated natural killer cell-mediated cytotoxicity, plasma interferon, and tumor cell rejection in mice persistently infected with lymphocytic choriomeningitis virus. J Immunol. 1983;131:991–996. [PubMed] [Google Scholar]
- 15.Gilbertson B, Germano S, Steele P, Turner S, Fazekas de St GB, Cheers C. Bystander activation of CD8+ T lymphocytes during experimental mycobacterial infection. Infect Immun. 2004;72:6884–6891. doi: 10.1128/IAI.72.12.6884-6891.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Raue HP, Brien JD, Hammarlund E, Slifka MK. Activation of virus-specific CD8+ T cells by lipopolysaccharide-induced IL-12 and IL-18. J Immunol. 2004;173:6873–6881. doi: 10.4049/jimmunol.173.11.6873. [DOI] [PubMed] [Google Scholar]
- 17.Bukowski JF, Welsh RM. Enhanced susceptibility to cytotoxic T lymphocytes of target cells isolated from virus-infected or interferon-treated mice. J Virol. 1986;59:735–739. doi: 10.1128/jvi.59.3.735-739.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wilson DB, et al. Specificity and degeneracy of T cells. Mol Immunol. 2004;40:1047–1055. doi: 10.1016/j.molimm.2003.11.022. [DOI] [PubMed] [Google Scholar]
- 19.Wucherpfennig KW, et al. Polyspecificity of T cell and B cell receptor recognition. Semin Immunol. 2007;19:216–224. doi: 10.1016/j.smim.2007.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mason D. A very high level of crossreactivity is an essential feature of the T cell repertoire. Immunol Today. 1998;19:395–404. doi: 10.1016/s0167-5699(98)01299-7. [DOI] [PubMed] [Google Scholar]
- 21.Casrouge A, Beaudoing E, Dalle S, Pannetier C, Kanellopoulos J, Kourilsky P. Size estimate of the alpha beta TCR repertoire of naive mouse splenocytes. J Immunol. 2000;164:5782–5787. doi: 10.4049/jimmunol.164.11.5782. [DOI] [PubMed] [Google Scholar]
- 22.Nikolich-Zugich J, Slifka MK, Messaoudi I. The many important facets of T-cell repertoire diversity. Nat Rev Immunol. 2004;4:123–132. doi: 10.1038/nri1292. [DOI] [PubMed] [Google Scholar]
- 23.Arstila TP, Casrouge A, Baron V, Even J, Kanellopoulos J, Kourilsky P. A direct estimate of the human alphabeta T cell receptor diversity. Science. 1999 Oct 29;286:958–961. doi: 10.1126/science.286.5441.958. [DOI] [PubMed] [Google Scholar]
- 24.Borghans JA, De Boer RJ. Crossreactivity of the T-cell receptor. Immunol Today. 1998;19:428–429. doi: 10.1016/s0167-5699(98)01317-6. [DOI] [PubMed] [Google Scholar]
- 25.Ignatowicz L, Kappler J, Marrack P. The repertoire of T cells shaped by a single MHC/peptide ligand. Cell. 1996;84:521–529. doi: 10.1016/s0092-8674(00)81028-4. [DOI] [PubMed] [Google Scholar]
- 26.Ewing C, et al. Virus-specific CD8+ T-cell responses in mice transgenic for a T-cell receptor beta chain selected at random. J Virol. 1994;68:3065–3070. doi: 10.1128/jvi.68.5.3065-3070.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zarozinski CC, Welsh RM. Minimal bystander activation of CD8 T cells during the virus- induced polyclonal T cell response. J Exp Med. 1997;185:1629–1639. doi: 10.1084/jem.185.9.1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hemmer B, Vergelli M, Pinilla C, Houghten R, Martin R. Probing degeneracy in T-cell recognition using peptide combinatorial libraries. Immunol Today. 1998;19:163–168. doi: 10.1016/s0167-5699(97)01217-6. [DOI] [PubMed] [Google Scholar]
- 29.Frankild S, De Boer RJ, Lund O, Nielsen M, Kesmir C. Amino acid similarity accounts for T cell cross-reactivity and for “holes” in the T cell repertoire. PLoS ONE. 2008;3:e1831. doi: 10.1371/journal.pone.0001831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee JK, et al. T cell cross-reactivity and conformational changes during TCR engagement. J Exp Med. 2004;200:1455–1466. doi: 10.1084/jem.20041251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Moutaftsi M, et al. A consensus epitope prediction approach identifies the breadth of murine T(CD8+)-cell responses to vaccinia virus. Nat Biotechnol. 2006;24:817–819. doi: 10.1038/nbt1215. [DOI] [PubMed] [Google Scholar]
- 32.Moutaftsi M, Bui HH, Peters B, Sidney J, Salek-Ardakani S, Oseroff C, et al. Vaccinia virus-specific CD4+ T cell responses target a set of antigens largely distinct from those targeted by CD8+ T cell responses. J Immunol. 2007;178:6814–6820. doi: 10.4049/jimmunol.178.11.6814. [DOI] [PubMed] [Google Scholar]
- 33.Pewe LL, Netland JM, Heard SB, Perlman S. Very diverse CD8 T cell clonotypic responses after virus infections. J Immunol. 2004;172:3151–3156. doi: 10.4049/jimmunol.172.5.3151. [DOI] [PubMed] [Google Scholar]
- 34.Pihlgren M, Dubois PM, Tomkowiak M, Sjogren T, Marvel J. Resting memory CD8+ T cells are hyperactive to antigenic challenge in vitro. J Exp Med. 1996;184:2141–2151. doi: 10.1084/jem.184.6.2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Slifka MK, Whitton JL. Functional avidity maturation of CD8(+) T cells without selection of higher affinity TCR. Nat Immunol. 2001;2:711–717. doi: 10.1038/90650. [DOI] [PubMed] [Google Scholar]
- 36.Curtsinger JM, Lins DC, Mescher MF. CD8+ memory T cells (CD44high, Ly-6C+) are more sensitive than naive cells (CD44low, Ly6C-) to TCR/CD8 signaling in response to antigen. J Immunol. 1998;160:3236–3243. [PubMed] [Google Scholar]
- 37.Wu LC, Tuot DS, Lyons DS, Garcia KC, Davis MM. Two-step binding mechanism for T-cell receptor recognition of peptide MHC. Nature. 2002;418:552–556. doi: 10.1038/nature00920. [DOI] [PubMed] [Google Scholar]
- 38.Lee JK, et al. T cell cross-reactivity and conformational changes during TCR engagement. J Exp Med. 2004;200:1455–1466. doi: 10.1084/jem.20041251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Naumov YN, Naumova EN, Yassai MB, Kota K, Welsh RM, Selin LK. Multiple glycines in TCR alpha-chains determine clonally diverse nature of human T cell memory to influenza A virus. J Immunol. 2008;181:7407–7419. doi: 10.4049/jimmunol.181.10.7407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Frankild S, de Boer RJ, Lund O, Nielsen M, Kesmir C. Amino acid similarity accounts for T cell cross-reactivity and for “holes” in the T cell repertoire. PLoS One. 2008;3:e1831. doi: 10.1371/journal.pone.0001831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Turner SJ, et al. Lack of prominent peptide-major histocompatibility complex features limits repertoire diversity in virus-specific CD8+ T cell populations. Nat Immunol. 2005;6:382–389. doi: 10.1038/ni1175. [DOI] [PubMed] [Google Scholar]
- 42.Anderson RW, Bennick JR, Yewdell JW, Maloy WL, Coligan JE. Influenza basic polymerase 2 peptides are recognized by influenza nucleoprotein-specific cytotoxic T lymphocytes. Mol Immunol. 1992;29:1089–1096. doi: 10.1016/0161-5890(92)90041-u. [DOI] [PubMed] [Google Scholar]
- 43.Welsh RM, Selin LK, Szomolanyi-Tsuda E. Immunological memory to viral infections. Annu Rev Immunol. 2004;22:711–743. doi: 10.1146/annurev.immunol.22.012703.104527. [DOI] [PubMed] [Google Scholar]
- 44.Seedhom MO, Jellison ER, Daniels KA, Welsh RM. High frequencies of virus-specific CD8+ T cell precursors. J Virol. 2009;83:12907–12916. doi: 10.1128/JVI.01722-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kotturi MF, et al. The CD8+ T-cell response to lymphocytic choriomeningitis virus involves the L antigen: uncovering new tricks for an old virus. J Virol. 2007;81:4928–4940. doi: 10.1128/JVI.02632-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kotturi MF, et al. Naive precursor frequencies and MHC binding rather than the degree of epitope diversity shape CD8+ T cell immunodominance. J Immunol. 2008;181:2124–2133. doi: 10.4049/jimmunol.181.3.2124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yewdell JW, Bennink JR. Immunodominance in major histocompatibility complex class I-restricted T lymphocyte responses. Annu Rev Immunol. 1999;17:51–88. doi: 10.1146/annurev.immunol.17.1.51. [DOI] [PubMed] [Google Scholar]
- 48.Klenerman P, Zinkernagel RM. Original antigenic sin impairs cytotoxic T lymphocyte responses to viruses bearing variant epitopes. Nature. 1998;394:421–422. doi: 10.1038/28860. [DOI] [PubMed] [Google Scholar]
- 49.Mongkolsapaya J, Dejnirattisai W, Xu XN, Vasanawathana S, Tangthawornchaikul N, Chairunsri A, et al. Original antigenic sin and apoptosis in the pathogenesis of dengue hemorrhagic fever. Nat Med. 2003;9:921–927. doi: 10.1038/nm887. [DOI] [PubMed] [Google Scholar]
- 50.Brehm MA, Pinto AK, Daniels KA, Schneck JP, Welsh RM, Selin LK. T cell immunodominance and maintenance of memory regulated by unexpectedly cross-reactive pathogens. Nat Immunol. 2002;3:627–634. doi: 10.1038/ni806. [DOI] [PubMed] [Google Scholar]
- 51.Cornberg M, et al. Narrowed TCR repertoire and viral escape as a consequence of heterologous immunity. J Clin Invest. 2006 May;116:1443–1456. doi: 10.1172/JCI27804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Borrow P, et al. Antiviral pressure exerted by HIV-1-specific cytotoxic T lymphocytes (CTLs) during primary infection demonstrated by rapid selection of CTL escape virus. Nat Med. 1997;3:205–211. doi: 10.1038/nm0297-205. [DOI] [PubMed] [Google Scholar]
- 53.Meyer-Olson D, et al. Limited T cell receptor diversity of HCV-specific T cell responses is associated with CTL escape. J Exp Med. 2004;200:307–319. doi: 10.1084/jem.20040638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bitmansour AD, et al. Clonotypic structure of the human CD4+ memory T cell response to cytomegalovirus. J Immunol. 2001;167:1151–1163. doi: 10.4049/jimmunol.167.3.1151. [DOI] [PubMed] [Google Scholar]
- 55.Haanan JB, Wolkers MC, Kruisbeek AM, Schumacher TN. Selective expansion of cross-reactive CD8(+) memory T cells by viral variants. J Exp Med. 1999;190:1319–1328. doi: 10.1084/jem.190.9.1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Manfras BJ, et al. Oligoclonal CD8+ T-cell expansion in patients with chronic hepatitis C is associated with liver pathology and poor response to interferon-alpha therapy. J Clin Immunol. 2004;24:258–271. doi: 10.1023/B:JOCI.0000025447.23473.ab. [DOI] [PubMed] [Google Scholar]
- 57.Price DA, et al. T cell receptor recognition motifs govern immune escape patterns in acute SIV infection. Immunity. 2004;21:793–803. doi: 10.1016/j.immuni.2004.10.010. [DOI] [PubMed] [Google Scholar]
- 58.Clute SC, et al. Cross-reactive influenza virus-specific CD8+ T cells contribute to lymphoproliferation in Epstein-Barr virus-associated infectious mononucleosis. J Clin Invest. 2005;115:3602–3612. doi: 10.1172/JCI25078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Maryanski JL, Jongeneel CV, Bucher P, Casanova JL, Walker PR. Single-cell PCR analysis of TCR repertoires selected by antigen in vivo: a high magnitude CD8 response is composed of very few clones. Immunity. 1996;4:47–55. doi: 10.1016/s1074-7613(00)80297-6. [DOI] [PubMed] [Google Scholar]
- 60.Maryanski JL, et al. Individuality of Ag-selected and preimmune TCR repertoires. Immunol Res. 2001;23:75–84. doi: 10.1385/IR:23:1:75. [DOI] [PubMed] [Google Scholar]
- 61.Lin MY, Welsh RM. Stability and diversity of T cell receptor (TCR) repertoire usage during lymphocytic choriomeningitis virus infection of mice. J Exp Med. 1998;188:1993–2005. doi: 10.1084/jem.188.11.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Welsh RM, Kim SK, Cornberg M, Clute SC, Selin LK, Naumov YN. The privacy of T cell memory to viruses. Curr Top Microbiol Immunol. 2006;311:117–153. doi: 10.1007/3-540-32636-7_5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Blattman JN, Sourdive DJ, Murali-Krishna K, Ahmed R, Altman JD. Evolution of the T cell repertoire during primary, memory, and recall responses to viral infection. J Immunol. 2000;165:6081–6090. doi: 10.4049/jimmunol.165.11.6081. [DOI] [PubMed] [Google Scholar]
- 64.Naumov YN, Naumova EN, Hogan KT, Selin LK, Gorski J. A fractal clonotype distribution in the CD8+ memory T cell repertoire could optimize potential for immune responses. J Immunol. 2003;170:3994–4001. doi: 10.4049/jimmunol.170.8.3994. [DOI] [PubMed] [Google Scholar]
- 65.Zinkernagel RM, Doherty PC. MHC-restricted cytotoxic T cells: studies on the biological role of polymorphic major transplantation antigens determining T-cell restriction-specificity, function, and responsiveness. Adv Immunol. 1979;27:51–177. doi: 10.1016/s0065-2776(08)60262-x. [DOI] [PubMed] [Google Scholar]
- 66.Baenziger J, Hengartner H, Zinkernagel RM, Cole GA. Induction or prevention of immunopathological disease by cloned cytotoxic T cell lines specific for lymphocytic choriomeningitis virus. Eur J Immunol. 1986;16:387–393. doi: 10.1002/eji.1830160413. [DOI] [PubMed] [Google Scholar]
- 67.Welsh RM. Private specificities of heterologous immunity. Curr Opin Immunol. 2006;18:331–337. doi: 10.1016/j.coi.2006.03.002. [DOI] [PubMed] [Google Scholar]
- 68.Urbani S, et al. Heterologous T cell immunity in severe hepatitis C virus infection. J Exp Med. 2005;201:675–680. doi: 10.1084/jem.20041058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kaech SM, Ahmed R. Memory CD8+ T cell differentiation: initial antigen encounter triggers a developmental program in naive cells. Nat Immunol. 2001;2:415–422. doi: 10.1038/87720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.van Stipdonk MJ, Lemmens EE, Schoenberger SP. Naive CTLs require a single brief period of antigenic stimulation for clonal expansion and differentiation. Nat Immunol. 2001;2:423–429. doi: 10.1038/87730. [DOI] [PubMed] [Google Scholar]
- 71.Mercado R, Vijh S, Allen SE, Kerksiek K, Pilip IM, Pamer EG. Early programming of T cell populations responding to bacterial infection. J Immunol. 2000;165:6833–6839. doi: 10.4049/jimmunol.165.12.6833. [DOI] [PubMed] [Google Scholar]
- 72.Foulds KE, Zenewicz LA, Shedlock DJ, Jiang J, Troy AE, Shen H. Cutting edge: CD4 and CD8 T cells are intrinsically different in their proliferative responses. J Immunol. 2002;168:1528–1532. doi: 10.4049/jimmunol.168.4.1528. [DOI] [PubMed] [Google Scholar]
- 73.Razvi ES, Jiang Z, Woda BA, Welsh RM. Lymphocyte apoptosis during the silencing of the immune response to acute viral infections in normal, lpr and Bcl-2-transgenic mice. Am J Pathol. 1995;147:79–91. [PMC free article] [PubMed] [Google Scholar]
- 74.Weant AE, Michalek RD, Khan IU, Holbrook BC, Willingham MC, Grayson JM. Apoptosis regulators Bim and Fas function concurrently to control autoimmunity and CD8+ T cell contraction. Immunity. 2008;28:218–230. doi: 10.1016/j.immuni.2007.12.014. [DOI] [PubMed] [Google Scholar]
- 75.Selin LK, Welsh RM. Cytolytically active memory CTL present in lymphocytic choriomeningitis virus (LCMV)-immune mice after clearance of virus infection. J Immunol. 1997;158:5366–5373. [PubMed] [Google Scholar]
- 76.Razvi ES, Welsh RM, McFarland HI. In vivo state of antiviral CTL precursors: characterization of a cycling population containing CTL precursors in immune mice. J Immunol. 1995;154:620–632. [PubMed] [Google Scholar]
- 77.Tough DF, Sprent J. Turnover of naive- and memory-phenotype T cells. J Exp Med. 1994;179:1127–1135. doi: 10.1084/jem.179.4.1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zimmermann C, Brduscha-Riem K, Blaser C, Zinkernagel RM, Pircher H. Visualization, characterization, and turnover of CD8+ memory T cells in virus-infected hosts. J Exp Med. 1996;183:1367–1375. doi: 10.1084/jem.183.4.1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Selin LK, Vergilis K, Welsh RM, Nahill SR. Reduction of otherwise remarkably stable virus-specific cytotoxic T lymphocyte memory by heterologous viral infections. J Exp Med. 1996;183:2489–2499. doi: 10.1084/jem.183.6.2489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Selin LK, et al. Attrition of T cell memory:selective loss of lymphocytic choriomeningitis virus (LCMV) epitope-specific memory CD8 T cells following infections with heterologous viruses. Immunity. 1999;11:733–742. doi: 10.1016/s1074-7613(00)80147-8. [DOI] [PubMed] [Google Scholar]
- 81.McNally JM, Zarozinski CC, Lin MY, Brehm MA, Chen HD, Welsh RM. Attrition of bystander CD8 T cells during virus-induced T cell and interferon responses. J Virol. 2001;75:5965–5976. doi: 10.1128/JVI.75.13.5965-5976.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kim SK, Welsh RM. Comprehensive early and lasting loss of memory CD8 T cells and functional memory during acute and persistent viral infections. J Immunol. 2004;172:3139–3150. doi: 10.4049/jimmunol.172.5.3139. [DOI] [PubMed] [Google Scholar]
- 83.Bahl K, et al. Interferon-induced attrition of CD8 T cells in the presence or absence of cognate antigen during the early stages of viral infections. J Immunol. 2006;176:4284–4295. doi: 10.4049/jimmunol.176.7.4284. [DOI] [PubMed] [Google Scholar]
- 84.Jiang J, Gross D, Nogusa S, Elbaum P, Murasko DM. Depletion of T cells by type I interferon: differences between young and aged mice. J Immunol. 2005;175:1820–1826. doi: 10.4049/jimmunol.175.3.1820. [DOI] [PubMed] [Google Scholar]
- 85.Peacock CD, Kim S-K, Welsh RM. Attrition of virus-specific memory CD8(+) T cells during reconstitution of lymphopenic environments. Journal of Immunology. 2003;171:655–663. doi: 10.4049/jimmunol.171.2.655. [DOI] [PubMed] [Google Scholar]
- 86.Smith DK, Dudani R, Pedras-Vasconcelos JA, Chapdelaine Y, van Faassen H, Sad S. Cross-reactive antigen is required to prevent erosion of established T cell memory and tumor immunity: a heterologous bacterial model of attrition. J Immunol. 2002;169:1197–1206. doi: 10.4049/jimmunol.169.3.1197. [DOI] [PubMed] [Google Scholar]
- 87.Vezys V, et al. Memory CD8 T-cell compartment grows in size with immunological experience. Nature. 2009;457:196–199. doi: 10.1038/nature07486. [DOI] [PubMed] [Google Scholar]
- 88.Welsh RM, Selin LK. Attrition of memory CD8 T cells. Nature. 2009;459:E3–E4. doi: 10.1038/nature08091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Pfizenmaier K, Jung H, Starzinski-Powitz A, Rollinghoff M, Wagner H. The role of T cells in anti-herpes simplex virus immunity. I. Induction of antigen-specific cytotoxic T lymphocytes. J Immunol. 1977;119:939–944. [PubMed] [Google Scholar]
- 90.Dummer W, et al. T cell homeostatic proliferation elicits effective antitumor autoimmunity. J Clin Invest. 2002;110:185–192. doi: 10.1172/JCI15175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Jiang J, Anaraki F, Blank KJ, Murasko DM. Cuttine edge: T cells from aged mice are resistant to depletion early during virus infection. J Immunol. 2003;171:3353–3357. doi: 10.4049/jimmunol.171.7.3353. [DOI] [PubMed] [Google Scholar]
- 92.Kapasi ZF, Murali-Krishna K, McRae ML, Ahmed R. Defective generation but normal maintenance of memory T cells in old mice. Eur J Immunol. 2002;32:1567–1573. doi: 10.1002/1521-4141(200206)32:6<1567::AID-IMMU1567>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 93.Yang H, Joris I, Majno G, Welsh RM. Necrosis of adipose tissue induced by sequential infections with unrelated viruses. Am J Pathol. 1985;120:173–177. [PMC free article] [PubMed] [Google Scholar]
- 94.Yang H, Dundon PL, Nahill SR, Welsh RM. Virus-induced polyclonal cytotoxic T lymphocyte stimulation. J Immunol. 1989;142:1710–1718. [PubMed] [Google Scholar]
- 95.Selin LK, Nahill SR, Welsh RM. Cross-reactivities in memory cytotoxic T lymphocyte recognition of heterologous viruses. J Exp Med. 1994;179:1933–1943. doi: 10.1084/jem.179.6.1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Chen HD, Fraire AE, Joris I, Brehm MA, Welsh RM, Selin LK. Memory CD8+ T cells in heterologous antiviral immunity and immunopathology in the lung. Nat Immunol. 2001;2:1067–1076. doi: 10.1038/ni727. [DOI] [PubMed] [Google Scholar]
- 97.Cornberg M, Sheridan BS, Saccoccio FM, Brehm MA, Selin LK. Protection against vaccinia virus challenge by CD8 memory T cells resolved by molecular mimicry. J Virol. 2007;81:934–944. doi: 10.1128/JVI.01280-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Nie S, Cornberg M, Selin LK. Resistance to vaccinia virus is less dependent on TNF under conditions of heterologous immunity. J Immunol. 2009;183:6554–6560. doi: 10.4049/jimmunol.0902156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kim SK, Brehm MA, Welsh RM, Selin LK. Dynamics of memory T cell proliferation under conditions of heterologous immunity and bystander stimulation. J Immunol. 2002;169:90–98. doi: 10.4049/jimmunol.169.1.90. [DOI] [PubMed] [Google Scholar]
- 100.Ploegh HL. Viral strategies of immune evasion. Science. 1998;280:248–253. doi: 10.1126/science.280.5361.248. [DOI] [PubMed] [Google Scholar]
- 101.Xiao Z, Casey KA, Jameson SC, Curtsinger JM, Mescher MF. Programming for CD8 T cell memory development requires IL-12 or type I IFN. J Immunol. 2009;182:2786–2794. doi: 10.4049/jimmunol.0803484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kolumam GA, Thomas S, Thompson LJ, Sprent J, Murali-Krishna K. Type I interferons act directly on CD8 T cells to allow clonal expansion and memory formation in response to viral infection. J Exp Med. 2005;202:637–650. doi: 10.1084/jem.20050821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Berg RE, Crossley E, Murray S, Forman J. Memory CD8+ T cells provide innate immune protection against Listeria monocytogenes in the absence of cognate antigen. J Exp Med. 2003;198:1583–1593. doi: 10.1084/jem.20031051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Bangs SC, Baban D, Cattan HJ, Li CK, McMichael AJ, Xu XN. Human CD4+ memory T cells are preferential targets for bystander activation and apoptosis. J Immunol. 2009;182:1962–1971. doi: 10.4049/jimmunol.0802596. [DOI] [PubMed] [Google Scholar]
- 105.Polley R, Sanos SL, Prickett S, Haque A, Kaye PM. Chronic Leishmania donovani infection promotes bystander CD8+-T-cell expansion and heterologous immunity. Infect Immun. 2005;73:7996–8001. doi: 10.1128/IAI.73.12.7996-8001.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Bukowski JF, Welsh RM. Interferon enhances the susceptibility of virus-infected fibroblasts to cytotoxic T cells. J Exp Med. 1985;161:257–262. doi: 10.1084/jem.161.1.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Liu F, Whitton JL. Cutting edge: re-evaluating the in vivo cytokine responses of CD8+ T cells during primary and secondary viral infections. J Immunol. 2005;174:5936–5940. doi: 10.4049/jimmunol.174.10.5936. [DOI] [PubMed] [Google Scholar]
- 108.Oettinger T, Jorgensen M, Ladefoged A, Haslov K, Andersen P. Development of the Mycobacterium bovis BCG vaccine: review of the historical and biochemical evidence for a genealogical tree. Tuber Lung Dis. 1999;79:243–250. doi: 10.1054/tuld.1999.0206. [DOI] [PubMed] [Google Scholar]
- 109.Alcami A, Smith GL. Vaccinia, cowpox, and camelpox viruses encode soluble gamma interferon receptors with novel broad species specificity. J Virol. 1995;69:4633–4639. doi: 10.1128/jvi.69.8.4633-4639.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Kapikian AZ, Mitchell RH, Chanock RM, Shvedoff RA, Stewart CE. An epidemiological study of altered clinical reactivity to respiratory syncitial (RS) virus infection in children previously vaccinated with an inactivated RS virus vaccine. Am J Epidemiol. 1969;89:405–421. doi: 10.1093/oxfordjournals.aje.a120954. [DOI] [PubMed] [Google Scholar]
- 111.Varga SM, Wang X, Welsh RM, Braciale TJ. Immunopathology in RSV infection is mediated by a discrete oligoclonal subset of antigen-specific CD4+ T cells. Immunity. 2001;15:637–646. doi: 10.1016/s1074-7613(01)00209-6. [DOI] [PubMed] [Google Scholar]
- 112.Johnson TR, Graham BS. Secreted respiratory syncytial virus G glcoprotein induces interleukin-5 (IL-5), IL-13, and eosinophilia by an IL-4-dependent mechanism. J Virol. 1999;73:8485–8495. doi: 10.1128/jvi.73.10.8485-8495.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Effros RB, Doherty PC, Gerhard W, Bennink J. Generation of both cross-reactive and virus-specific T-cell populations after immunization with serologically distinct influenza A viruses. J Exp Med. 1977;145:557–568. doi: 10.1084/jem.145.3.557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Lee LY, et al. Memory T cells established by seasonal human influenza A infection cross-react with avian influenza A (H5N1) in healthy individuals. J Clin Invest. 2008;118:3478–3490. doi: 10.1172/JCI32460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Epstein SL. Prior H1N1 influenza infection and susceptibility of Cleveland Family Study participants during the H2N2 pandemic of 1957: an experiment of nature. J Infect Dis. 2006;193:49–53. doi: 10.1086/498980. [DOI] [PubMed] [Google Scholar]
- 116.Slepushkin AN. The effect of a previous attack of A1 influenza on susceptibility to A2 virus during the 1957 outbreak. Bull World Health Organ. 1959;20:297–301. [PMC free article] [PubMed] [Google Scholar]
- 117.Moss PAH, et al. Extensive conservation of a and b chains of the human T-cell antigen receptor recognizing HLA-A2 and influenza A matrix peptide. Proc Natl Acad Sci. 1991;88:8987–8990. doi: 10.1073/pnas.88.20.8987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Naumov YN, Naumova EN, Yassai MB, Kota K, Welsh RM, Selin LK. Multiple glycines in TCR alpha-chains determine clonally diverse nature of human T cell memory to influenza A virus. J Immunol. 2008;181:7407–7419. doi: 10.4049/jimmunol.181.10.7407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Wedemeyer H, Mizukoshi E, Davis AR, Bennink JR, Rehermann B. Cross-reactivity between hepatitis C virus and influenza A virus determinant-specific cytotoxic T cells. J Virol. 2001;75:11392–11400. doi: 10.1128/JVI.75.23.11392-11400.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Weinstein L, Meade RH. Respiratory manifestations of chickenpox. Arch Intern Med. 1956;98:91–99. doi: 10.1001/archinte.1956.00250250097013. [DOI] [PubMed] [Google Scholar]
- 121.Rickinson AB, Kieff E. Epstein-Barr virus. In: Fields BN, et al., editors. Virology. Vol. 2. Philadelphia: Lippincott-Raven Publishers; 1996. pp. 2397–2446. [Google Scholar]
- 122.Silins SL, et al. Asymptomatic primary Epstein-Barr virus infection occurs in the absence of blood T-cell repertoire perturbations despite high levels of systemic viral load. Blood. 2001;98:3739–3744. doi: 10.1182/blood.v98.13.3739. [DOI] [PubMed] [Google Scholar]
- 123.Morens DM. Antibody-dependent enhancement of infection and the pathogenesis of viral disease. Clin Infect Dis. 1994;19:500–512. doi: 10.1093/clinids/19.3.500. [DOI] [PubMed] [Google Scholar]
- 124.Halstead SB. Antibody, macrophages, dengue virus infection, shock and hemorrhage: a pathogenetic cascade. Rev Infect Dis. 1989;11:S830–S839. doi: 10.1093/clinids/11.supplement_4.s830. [DOI] [PubMed] [Google Scholar]
- 125.Matthew A, et al. Predominance of HLA-restricted cytotoxic T-lymphocyte responses to serotype-cross-reactive epitopes on nonstructural proteins following natural secondary dengue virus infection. J Virol. 1998;72:3999–4004. doi: 10.1128/jvi.72.5.3999-4004.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Allen PM, Kersh GJ. Essential flexibility in the T-cell recognition of antigen. Nature. 1996;380:495–498. doi: 10.1038/380495a0. [DOI] [PubMed] [Google Scholar]
- 127.Daniel C, Horvath S, Allen PM. A basis for alloreactivity: MHC helical residues broaden peptide recognition by the TCR. Immunity. 1998;8:543–552. doi: 10.1016/s1074-7613(00)80559-2. [DOI] [PubMed] [Google Scholar]
- 128.Brehm MA, Daniels KA, Welsh RM. Rapid production of TNF-alpha following TCR engagement of naive CD8 T cells. J Immunol. 2005;175:5043–5049. doi: 10.4049/jimmunol.175.8.5043. [DOI] [PubMed] [Google Scholar]
- 129.Brehm MA, et al. Rapid quantification of naive alloreactive T cells by TNF-alpha production and correlation with allograft rejection in mice. Blood. 2007;109:819–826. doi: 10.1182/blood-2006-03-008219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Yang H, Welsh RM. Induction of alloreactive cytotoxic T cells by acute virus infection of mice. J Immunol. 1986;136:1186–1193. [PubMed] [Google Scholar]
- 131.Nahill SR, Welsh RM. High frequency of cross-reactive cytotoxic T lymphocytes elicited during the virus-induced polyclonal cytotoxic T lymphocyte response. J Exp Med. 1993;177:317–327. doi: 10.1084/jem.177.2.317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Brehm MA, Markees TG, Daniels KA, Greiner DL, Rossini AA, Welsh RM. Direct visualization of cross-reactive effector and memory allo-specific CD8 T cells generated in response to viral infections. J Immunol. 2003;170:4077–4086. doi: 10.4049/jimmunol.170.8.4077. [DOI] [PubMed] [Google Scholar]
- 133.Tomkinson BE, Maziarz R, Sullivan JL. Characterization of the T cell-mediated cellular cytotoxicity during infectious mononucleosis. J Immunol. 1989;143:660–670. [PubMed] [Google Scholar]
- 134.Strang G, Rickinson AB. Multiple HLA class I-dependent cytotoxicities constitute the “non-HLA-restricted” response in infectious mononucleosis. Eur J Immunol. 1987;17:1007–1013. doi: 10.1002/eji.1830170717. [DOI] [PubMed] [Google Scholar]
- 135.Burrows SR, et al. Cross-reactive memory T cells for Epstein-Barr virus augment the alloresponse to common human leukocyte antigens: degenerate recognition of major histocompatibility complex-bound peptide by T cells and its role in alloreactivity. Eur J Immunol. 1997;27:1726–1736. doi: 10.1002/eji.1830270720. [DOI] [PubMed] [Google Scholar]
- 136.Speir JA, Garcia C, Brunmark A, Degano M, Peterson PA, Teyton L, et al. Structural basis of 2C TCR allorecognition of H-2Ld peptide complexes. Immunity. 1998;8:553–562. doi: 10.1016/s1074-7613(00)80560-9. [DOI] [PubMed] [Google Scholar]
- 137.Burrows SR, Khanna R, Silins SL, Moss DJ. The influence of antiviral T-cell responses on the alloreactive repertoire. Immunol Today. 1999;20:203–207. doi: 10.1016/s0167-5699(98)01429-7. [DOI] [PubMed] [Google Scholar]
- 138.Brehm MA, Markees TG, Daniels KA, Greiner DL, Rossini AA, Welsh RM. Direct visualization of cross-reactive effector and memory allo-specific CD8 T cells generated in response to viral infections. J Immunol. 2003;170:4077–4086. doi: 10.4049/jimmunol.170.8.4077. [DOI] [PubMed] [Google Scholar]
- 139.Briggs JD, Timbury MC, Paton AM, Bell PR. Viral infection and renal transplant rejection. Br Med J. 1972;4:520–522. doi: 10.1136/bmj.4.5839.520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Caldas C, Ambinder R. Epstein-Barr virus and bone marrow transplantation. Curr Opin Oncology. 1995;7:626–629. doi: 10.1097/00001622-199503000-00002. [DOI] [PubMed] [Google Scholar]
- 141.Kadakia MP, et al. Human herpes virus 6:infection and disease following autologous and allogeneic bone marrow transplantation. Blood. 1996;87:5341–5354. [PubMed] [Google Scholar]
- 142.May AG, Betts RF, Freeman RB, Andrus CH. An analysis of cytomegalovirus infection and HLA antigen matching on the outcome of renal transplantation. Ann Surg. 1978;187:110–117. doi: 10.1097/00000658-197802000-00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Parker DC, et al. Survival of mouse pancreatic islet allografts in recipients treated with allogeneic small lymphocytes and antibody to CD40 ligand. Proc Natl Acad Sci USA. 1995;92:9560–9564. doi: 10.1073/pnas.92.21.9560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Adams AB, et al. Heterologous immunity provides a potent barrier to transplantation tolerance. J Clin Invest. 2003;111:1887–1895. doi: 10.1172/JCI17477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Williams MA, et al. Cutting edge: persistent viral infection prevents tolerance induction and escapes immune control following CD28/CD40 blockade-based regimen. J Immunol. 2002;169:5387–5391. doi: 10.4049/jimmunol.169.10.5387. [DOI] [PubMed] [Google Scholar]
- 146.Welsh RM, et al. Virus-induced abrogation of transplantation tolerance induced by donor-specific transfusion and anti-CD154 antibody. J Virol. 2000;74:2210–2218. doi: 10.1128/jvi.74.5.2210-2218.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Thornley TB, et al. Type 1 IFN mediates cross-talk between innate and adaptive immunity that abrogates transplantation tolerance. J Immunol. 2007;179:6620–6629. doi: 10.4049/jimmunol.179.10.6620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Welsh RM, Fujinami RS. Pathogenic epitopes, heterologous immunity and vaccine design. Nat Rev Microbiol. 2007;5:555–563. doi: 10.1038/nrmicro1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Barnett LA, Fujinami RS. Molecular mimicry: a mechanism for autoimmune injury. FASEB J. 1992;6:840–844. doi: 10.1096/fasebj.6.3.1740233. [DOI] [PubMed] [Google Scholar]
- 150.McCoy L, Tsunoda I, Fujinami RS. Multiple sclerosis and virus induced immune responses: autoimmunity can be primed by molecular mimicry and augmented by bystander activation. Autoimmunity. 2006;39:9–19. doi: 10.1080/08916930500484799. [DOI] [PubMed] [Google Scholar]
- 151.Sospedra M, et al. Recognition of conserved amino acid motifs of common viruses and its role in autoimmunity. PLoS Pathog. 2005;1:e41. doi: 10.1371/journal.ppat.0010041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Theil DJ, Tsunoda I, Rodriguez F, Whitton JL, Fujinami RS. Viruses can silently prime for and trigger central nervous system autoimmune disease. J Neurovirol. 2001;7:220–227. doi: 10.1080/13550280152403263. [DOI] [PubMed] [Google Scholar]
- 153.Evans CF, Horwitz MS, Hobbs MV, Oldstone MBA. Viral infection of transgenic mice expressing a viral protein in oligodendrocytes leads to chronic central nervous system autoimmune disease. J Exp Med. 1996;184:2371–2384. doi: 10.1084/jem.184.6.2371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Christen U, et al. A viral epitope that mimics a self antigen can accelerate but not initiate autoimmune diabetes. J Clin Invest. 2004;114:1290–1298. doi: 10.1172/JCI22557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.He X-S, et al. Quantitative analysis of hepatitis C virus-specific CD8+ T cells in peripheral blood and liver using peptide-MHC tetramers. Proc Natl Acad Sci USA. 1999;96:5692–5697. doi: 10.1073/pnas.96.10.5692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Selin LK, et al. Memory of mice and men: CD8+ T-cell cross-reactivity and heterologous immunity. Immunol Rev. 2006;211:164–181. doi: 10.1111/j.0105-2896.2006.00394.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Kulkarni AB, Morse HC, III, Bennink JR, Yewdell JW, Murphy BR. Immunization of mice with vaccinia virus-M2 recombinant induces epitope-specific and cross-reactive Kd-restricted CD8+ cytotoxic T cells. J Virol. 1993;67:4086–4092. doi: 10.1128/jvi.67.7.4086-4092.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Kuwano K, Reyes RE, Humphreys RE, Ennis FA. Recognition of disparate HA and NS1 peptides by an H-2kd- restricted, influenza specific CTL clone. Mol Immunol. 1991;28:1–7. doi: 10.1016/0161-5890(91)90080-4. [DOI] [PubMed] [Google Scholar]
- 159.Spaulding AC, Kurane I, Ennis FA, Rothman AL. Analysis of murine CD8(+) T-cell clones specific for the Dengue virus NS3 protein: flavivirus cross-reactivity and influence of infecting serotype. J Virol. 1999;73:398–403. doi: 10.1128/jvi.73.1.398-403.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Beaumier CM, Mathew A, Bashyam HS, Rothman AL. Cross-reactive memory CD8(+) T cells alter the immune response to heterologous secondary dengue virus infections in mice in a sequence-specific manner. J Infect Dis. 2008;197:608–617. doi: 10.1086/526790. [DOI] [PubMed] [Google Scholar]
- 161.Maeda K, West K, Toyosaki-Maeda T, Rothman AL, Ennis FA, Terajima M. Identification and analysis for cross-reactivity among hantaviruses of H-2b-restricted cytotoxic T-lymphocyte epitopes in Sin Nombre virus nucleocapsid protein. J Gen Virol. 2004;85:1909–1919. doi: 10.1099/vir.0.79945-0. [DOI] [PubMed] [Google Scholar]
- 162.Tscharke DC, et al. Poxvirus CD8+ T-cell determinants and cross-reactivity in BALB/c mice. J Virol. 2006;80:6318–6323. doi: 10.1128/JVI.00427-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Acierno PM, Newton DA, Brown EA, Maes LA, Baatz JE, Gattoni-Celli S. Cross-reactivity between HLA-A2-restricted FLU-M1:58–66 and HIV p17 GAG:77–85 epitopes in HIV-infected and uninfected individuals. J Transl Med. 2003;1:3. doi: 10.1186/1479-5876-1-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Nilges K, et al. Human papillomavirus type 16 E7 peptide-directed CD8+ T cells from patients with cervical cancer are cross-reactive with the coronavirus NS2 protein. J Virol. 2003;77:5464–5474. doi: 10.1128/JVI.77.9.5464-5474.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Shimojo N, Maloy WL, Anderson RW, Biddison WE, Coligan JE. Specificity of peptide binding by the HLA-A2.1 molecule. J Immunol. 1989;143:2939–2947. [PubMed] [Google Scholar]
- 166.Zivny J, et al. Partial agonist effect influences the CTL response to a heterologous dengue virus serotype. J Immunol. 1999;163:2754–2760. [PubMed] [Google Scholar]
- 167.Bashyam HS, Green S, Rothman AL. Dengue virus-reactive CD8+ T cells display quantitative and qualitative differences in their response to variant epitopes of heterologous viral serotypes. J Immunol. 2006;176:2817–2824. doi: 10.4049/jimmunol.176.5.2817. [DOI] [PubMed] [Google Scholar]
- 168.Van Epps HL, Schmaljohn CS, Ennis FA. Human memory cytotoxic T-lymphocyte (CTL) responses to Hantaan virus infection: identification of virus-specific and cross-reactive CD8(+) CTL epitopes on nucleocapsid protein. J Virol. 1999;73:5301–5308. doi: 10.1128/jvi.73.7.5301-5308.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]