

Abstract
Objective
Overexpression of the antiapoptotic protein myeloid cell leukemia 1 (Mcl-1) in rheumatoid arthritis (RA) synovial fibroblasts is a major cause of their resistance to tumor necrosis factor α (TNFα)–induced apoptosis. This study was undertaken to evaluate the efficacy of epigallocatechin-3-gallate (EGCG) in down-regulating Mcl-1 expression and its mechanism of RA synovial fibroblast sensitization to TNFα-induced apoptosis.
Methods
EGCG effects on cultured RA synovial fibroblast cell morphology, proliferation, and viability over 72 hours were determined by microscopy and a fluorescent cell enumeration assay. Caspase 3 activity was determined by a colorimetric assay. Western blotting was used to evaluate the apoptosis mediators poly(ADP-ribose) polymerase (PARP), Mcl-1, Bcl-2, Akt, and nuclear translocation of NF-κB.
Results
In RA synovial fibroblasts, EGCG (5–50 μM) inhibited constitutive and TNFα-induced Mcl-1 protein expression in a concentration- and time-dependent manner (P < 0.05). Importantly, EGCG specifically abrogated Mcl-1 expression in RA synovial fibroblasts and affected Mcl-1 expression to a lesser extent in osteoarthritis and normal synovial fibroblasts or endothelial cells. Inhibition of Mcl-1 by EGCG triggered caspase 3 activity in RA synovial fibroblasts, which was mediated via down-regulation of the TNFα-induced Akt and NF-κB pathways. Caspase 3 activation by EGCG also suppressed RA synovial fibroblast growth, and this effect was mimicked by Akt and NF-κB inhibitors. Interestingly, Mcl-1 degradation by EGCG sensitized RA synovial fibroblasts to TNFα-induced PARP cleavage and apoptotic cell death.
Conclusion
Our findings indicate that EGCG itself induces apoptosis and further sensitizes RA synovial fibroblasts to TNFα-induced apoptosis by specifically blocking Mcl-1 expression and, hence, may be of promising adjunct therapeutic value in regulating the invasive growth of synovial fibroblasts in RA.
Apoptosis, or programmed cell death, is a process in which the activation of death receptors or mitochondrial-mediated signaling cascades is initiated in a tightly regulated manner to limit unwanted cell growth (1). In chronic human diseases such as cancer or rheumatoid arthritis (RA), increased expression of antiapoptotic proteins, such as members of the Bcl-2 family, commonly occurs and is associated with disease progression, resistance to therapies, and poor clinical outcome (1,2). Interestingly, among Bcl-2 family members, myeloid cell leukemia 1 (Mcl-1) represents a critical determinant of sensitivity and resistance to apoptosis (2). Cells expressing high levels of Mcl-1 are found to be resistant to a newly developed Bcl-2/Bcl-xL/Bcl-w inhibitor, suggesting the pivotal role of Mcl-1 in suppressing apoptosis (3). This notion was further authenticated by several studies in which knockdown of Mcl-1 dramatically increased drug sensitivity of several tumor cell lines (2).
RA is an inflammatory disorder characterized by hyperplasia of the synovial joint lining, which invades and degrades cartilage and underlying bone, ultimately resulting in joint destruction (4). Studies of the mechanisms of RA pathogenesis have established an essential role for RA synovial fibroblasts in arthritis initiation and progression (5). Although the exact molecular mechanisms regulating synovial hyperplasia are presently unclear, accumulating evidence suggests that RA synovial fibroblasts are resistant to both tumor necrosis factor α (TNFα)– and Fas-mediated apoptosis (6). Recent observations at the cellular level have implicated aberrant expression of Mcl-1 in the resistance of RA synovial fibroblasts to TNFα- and Fas-mediated apoptosis (7,8). Furthermore, constitutive activation of survival protein Akt and transcription factor NF-κB in RA synovial fibroblasts suggests the importance of these molecules in RA synovial fibroblast hyperproliferation and invasion into underlying tissue (9,10). Thus, the purposeful induction of apoptosis in RA synovial fibroblasts, either through suppression of these signaling pathways or through inhibition of Mcl-1 expression, is emerging as a therapeutic strategy for halting deleterious tissue growth (6,11).
Epigallocatechin-3-gallate (EGCG) is a potent antioxidant, antiinflammatory, and antioncogenic compound derived from green tea (12). We and others have shown that EGCG is capable of inducing apoptosis in a variety of cancers both in vitro and in vivo (13,14). However, the use of EGCG in combination with other therapeutic agents in order to enhance the sensitivity or efficacy of the therapy is gaining attention and could serve as an impetus for such mechanisms (2,12). Recent studies have shown that EGCG sensitizes carcinoma cells to trastuzumab and TRAIL-induced apoptosis (14,15). Recently, we found that EGCG blocked interleukin-1β (IL-1β)–induced chemokine production and matrix metalloproteinase 2 (MMP-2) activation in RA synovial fibroblasts (16). In this study, we evaluated whether EGCG inhibits the uncontrolled growth of RA synovial fibroblasts by inducing apoptosis.
MATERIALS AND METHODS
Antibodies and reagents
EGCG, rabbit anti–β-actin, and the colorimetric caspase 3 activity assay kits were purchased from Sigma-Aldrich (St. Louis, MO). Recombinant human TNFα was from R&D Systems (Minneapolis, MN). Rabbit polyclonal antibodies against poly(ADP-ribose) polymerase (PARP), Bcl-2, phospho-Akt, and anti-rabbit and anti-mouse horseradish peroxidase–linked secondary antibodies were purchased from Cell Signaling Technology (Beverly, MA). Rabbit polyclonal antibody against NF-κBp65 and Mcl-1 were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). The signaling inhibitors of NF-κB (pyrrolidine dithiocarbamate [PDTC]), Akt (LY294002), proteasomal degradation (MG132), and caspase 3 were purchased from Calbiochem (La Jolla, CA).
Cell cultures
Fibroblasts were isolated from synovium obtained from de-identified samples of patients with RA or OA who had undergone total joint replacement surgery or synovectomy and fulfilled the American College of Rheumatology (formerly, the American Rheumatism Association) criteria (17). Synovium was obtained according to Institutional Review Board (IRB)–approved protocols, in compliance with the Declaration of Helsinki. Normal synovial tissues were obtained fresh at the time of autopsy or amputation, according to an IRB-approved protocol. Fresh synovial tissues were minced and digested in a solution of Dispase, collagenase, and DNase (16). RA synovial fibroblasts were grown in RPMI 1640 containing 2 mM L-glutamine with 10% fetal bovine serum (FBS), at 37°C, in a humidified atmosphere with 5% CO2. Cells were used between passages 5 and 9.
Preparation of EGCG solution
A stock solution of EGCG (10 mM) was prepared in water, sterile filtered with 0.2-μm syringe filters, and stored at −20°C in aliquots. Fresh EGCG solution was used in each experiment and added directly to the culture medium.
Cell survival assays
For cell survival assays, confluent RA synovial fibroblasts in 6- or 96-well plates were cultured for up to 72 hours in RPMI 1640–1% FBS, containing 0, 10, or 50 μM EGCG and/or 20 ng/ml TNFα. For morphologic studies, RA synovial fibroblasts were fixed in 1% glutaraldehyde in RPMI 1640, stained with hematoxylin and eosin, and evaluated microscopically. In survival assays, cell numbers were evaluated using a CyQuant cell enumeration kit (Invitrogen, Carlsbad, CA), with fluorescence measured at 485/528 nm excitation/emission wavelengths. Background from CyQuant-treated wells without cells was subtracted from all values. To evaluate EGCG effects on RA synovial fibroblast growth, RA synovial fibroblasts (1.5 × 103) were plated into 96-well plates in RPMI 1640–1% FBS and were allowed to attach for 16 hours. Medium was replaced with fresh media containing 0–50 μM EGCG, and cell proliferation was measured daily, for up to 72 hours, with CyQuant. In another set of experiments, the effect of the Akt inhibitor LY294002 (20 μM; Calbiochem) or the NF-κB inhibitor PDTC (100 μM; Sigma) on RA synovial fibroblast growth was evaluated (18).
Western immunoblotting
To study the effect of EGCG on antiapoptotic and survival proteins, RA synovial fibroblasts were incubated with 50 μM EGCG alone, with TNFα alone, or with a combination of EGCG and TNFα for up to 72 hours, in RPMI 1640–1% FBS. Cells were lysed in lysis buffer (100 mM Tris [pH 7.4], 100 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1 mM NaF, 20 mM NaP2O4, 2 mM Na3VO4, 1% Triton X-100, 10% glycerol, 0.1% sodium dodecyl sulfate [SDS], 0.5% deoxycholate, and 1 mM phenylmethylsulfonyl fluoride), and protease inhibitors (1 tablet/10 ml; Roche Diagnostics, Indianapolis, IN), and protein was measured using BCA protein assay kits (Pierce, Rockford, IL). Equal amounts of protein (20 μg) were separated by SDS–polyacrylamide gel electrophoresis (PAGE) and transferred onto nitrocellulose membranes (Bio-Rad, Hercules, CA). Blots were probed with rabbit polyclonal antibodies for the specific protein of interest, followed by incubation with the appropriate horseradish peroxidase–conjugated secondary antibody. Immunoreactive protein bands were visualized by enhanced chemiluminescence, and were densitometrically analyzed using Un-Scan-It software (version 5.1; Silk Scientific, Orem, UT). Blots were stripped and reprobed for β-actin to ensure equal protein loading. Data were statistically analyzed using GraphPad Prism software (San Diego, CA).
To study the mechanism of EGCG-mediated inhibition of Mcl-1 protein expression, RA synovial fibroblasts were pretreated with the proteasome inhibitor MG132 (5–20 μM) or caspase 3 inhibitor (10 μM) for 30 minutes, followed by EGCG (50 μM) treatment for 24 hours. DMSO (4 μl) was used as a vehicle control for MG132. Cells were lysed in lysis buffer and processed for evaluating Mcl-1 expression by Western blotting as described above.
Caspase 3 activity assay
RA synovial fibroblasts were treated with EGCG (50 μM) and/or TNFα (20 ng/ml) for 24 hours in RPMI 1640–1% FBS. Cells were washed in ice-cold phosphate buffered saline (PBS), lysed with buffer (50 mM HEPES [pH 7.4], 5 mM CHAPS, and 5 mM dithiothreitol), and centrifuged to collect supernatant in fresh tubes. Protein content of the cell lysates was measured with the BCA assay. Caspase 3 activity in cell lysates was determined using a colorimetric caspase 3 activity assay kit (Sigma-Aldrich). Similar studies were performed using Akt and NF-κB inhibitors alone or in combination with TNFα to evaluate the roles of these signaling pathways in the regulation of caspase 3 activity in RA synovial fibroblasts.
Preparation of nuclear extracts and NF-κBp65 DNA binding activity
To study the effect of EGCG on TNFα-induced NF-κB activation, confluent RA synovial fibroblasts that had been cultured in 60-mm dishes were treated with EGCG (50 μM) and/or TNFα (20 ng/ml) for 30 minutes and 24 hours in RPMI 1640–1% FBS. Upon termination, cells were washed with ice-cold PBS, collected by scraping, and centrifuged at 1,500g for 5 minutes at 4°C. Nuclear and cytoplasmic fractions from different treatment groups were prepared as previously described (16). An equal amount of protein (15 μg) from nuclear and cytoplasmic fractions was evaluated by Western blotting in order to study the level of NF-κBp65 expression. The nuclear extracts (2 μg in each sample) from 30 minutes of stimulation with EGCG and/or TNFα were used to determine NF-κB DNA binding activity using commercially available DNA binding enzyme-linked immunosorbent assay kits (Active Motif, Carlsbad, CA).
Immunocytochemistry
To verify that the presence of EGCG sensitizes RA synovial fibroblasts to TNFα-induced apoptosis and not necrosis, we performed immunocytochemistry on these cells using monoclonal antibody against single-stranded DNA (MAb F7-26; Millipore, Billerica, MA). Briefly, confluent RA synovial fibroblasts (1 × 104/well) in 8-well Labtek chamber slides (BD Falcon, Bedford, MA) were treated with EGCG (50 μM) alone, TNFα (20 ng/ml) alone, or a combination of EGCG and TNFα, or were left untreated, for 24 hours in RPMI 1640 plus 1% FBS. Upon termination, the cells were washed with PBS and fixed with 4% formalin overnight at 4°C. The next day, cells were washed again with PBS followed by incubation with 3% H2O2 for 5 minutes at room temperature to quench endogenous peroxidase. After blocking with 3% nonfat dry milk for 15 minutes at room temperature, slides were probed with mouse anti-human F7-26 (1 μg/ml) or purified nonspecific mouse IgM for 15 minutes at room temperature in 1% nonfat dry milk, followed by PBS washes and incubation with biotinylated goat anti-mouse antibody (10 μg/ml) for 20 minutes at 37°C. After washing, antibody binding was detected using a Vectastain Elite ABC kit (Vector, Burlingame, CA) and the chromogen 3,3′-diaminobenzidine (Vector). The slides were counterstained with Gill’s hematoxylin and then dehydrated, mounted, and coverslipped, and representative photomicrographs were obtained at 40× magnification using an Olympus FV-500 microscope (Olympus America, Melville, NY).
Statistical analysis
Student’s t-tests were performed to calculate statistical differences between the means of the different variables. P values less than 0.05 (2-tailed) were considered significant.
RESULTS
EGCG preferentially down-regulates Mcl-1 expression in RA synovial fibroblasts
We first sought to determine the effect of EGCG incubation on TNFα-induced Mcl-1 expression in RA synovial fibroblasts. RA synovial fibroblasts were treated with EGCG (5–50 μM), TNFα (20 ng/ml), or a combination of EGCG and TNFα for 72 hours. Our initial experiments showed that Mcl-1 protein was constitutively expressed by RA synovial fibroblasts over 72 hours of observation (Figure 1A). Using Western blot analysis, we found that Mcl-1 protein was markedly increased, ~1.8-fold, by TNFα activation (20 ng/ml) (Figure 1B).
Figure 1.

Inhibition of basal and tumor necrosis factor α (TNFα)–induced myeloid cell leukemia 1 (Mcl-1) expression by epigallocatechin-3-gallate (EGCG) in rheumatoid arthritis (RA) synovial fibroblasts. RA synovial fibroblasts (2 × 105/well) were incubated in RPMI 1640 plus 1% fetal bovine serum. A, Mcl-1 expression in untreated RA synovial fibroblasts after 0, 24, 48, and 72 hours, and in RA synovial fibroblasts treated with TNFα (20 ng/ml) for 72 hours. B, Mcl-1 expression in untreated RA synovial fibroblasts and in RA synovial fibroblasts treated with TNFα alone (20 ng/ml) or with TNFα and EGCG (5–50 μM) for 72 hours. Bars in A and B show the mean and SEM from 3 or more independent experiments using cells from different donors under similar conditions. C, Mcl-1 and Bcl-2 expression in untreated (nonstimulated [NS]) RA synovial fibroblasts and in RA synovial fibroblasts treated with EGCG (50 μM) alone, TNFα (20 ng/ml) alone, or a combination of EGCG and TNFα for 24, 48, and 72 hours. Cells were lysed in extraction buffer containing protease inhibitors and the expression levels of Mcl-1 and Bcl-2 were determined by Western blotting. D, Expression of Mcl-1 in synovial fibroblasts from patients with RA, patients with osteoarthritis (OA), and normal subjects (NL), and in human dermal microvascular endothelial cells (HMVECs) (2 × 105/well). Cells were treated with EGCG (50 μM) alone, TNFα (20 ng/ml) alone, or a combination of EGCG and TNFα for 24 hours. The intensity of the bands was quantified using Un-Scan-It software.
EGCG incubation with TNFα caused a concentration-dependent decrease in the level of Mcl-1 expression. EGCG at 20 μM and at 50 μM caused mean ± SEM inhibition of 45 ± 15% and 80 ± 9%, respectively, of TNFα-induced Mcl-1 expression in RA synovial fibroblasts (Figure 1B) (P < 0.05 versus treatment with TNFα alone). Furthermore, a time-dependent study showed that EGCG (50 μM) was effective in completely blocking the constitutive (un-stimulated) as well as TNFα-induced Mcl-1 expression in RA synovial fibroblasts within 24 hours (Figure 1C). We analyzed the same samples for Bcl-2 expression using Western blotting and found that EGCG required more time (~48 hours) to down-regulate the constitutive or TNFα-induced expression of Bcl-2 (Figure 1C).
We next determined the Mcl-1 specificity of EGCG in synovial fibroblasts obtained from patients with RA, patients with osteoarthritis (OA), or normal subjects, or using human dermal microvascular endothelial cells (HMVECs). These cells were incubated with EGCG (50 μM) alone, with TNFα (20 ng/ml) alone, or with a combination of EGCG and TNFα for 24 hours. Western blot analysis of these samples showed that EGCG had a moderate effect on Mcl-1 expression levels in normal synovial fibroblasts or HMVECs (Figure 1D). However, Mcl-1 expression in RA and OA synovial fibroblasts was almost completely inhibited by EGCG incubation, suggesting its inhibitory role in disease conditions.
Densitometric analysis of these blots obtained from 3 or more experiments performed using synovial fibroblasts from different donors showed that Mcl-1 expression was slightly higher in OA and RA synovial fibroblasts as compared with normal synovial fibroblasts, that TNFα induced Mcl-1 expression in normal synovial fibroblasts by almost 15% of the basal levels, and that EGCG, alone and in combination with TNFα, inhibited Mcl-1 expression in normal, OA, and RA synovial fibroblasts by ~60–70%, ~84–86%, and ~93–99%, respectively. These findings suggest that evaluating the effect of EGCG at lower concentrations on Mcl-1 expression may further clarify the differential effect on normal, OA, and RA synovial fibroblasts.
Role of Akt and NF-κB in EGCG-mediated inhibition of RA synovial fibroblast growth in vitro
Inhibition of either Akt or NF-κB holds promise in regulating abnormal RA synovial fibroblast growth (10,19). We studied the effects of Akt and NF-κB inhibitors (LY294002 and PDTC, respectively) on Mcl-1 expression using concentrations used in prior studies (18). As shown in Figure 2A, TNFα increased constitutive Mcl-1 expression by ~25%. Interestingly, blockade of the Akt pathway using LY294002 (20 μM) caused an ~37% and ~53% decrease in constitutive Mcl-1 expression and in TNFα-induced Mcl-1 expression in RA synovial fibroblasts, respectively (P < 0.05 versus treatment with TNFα alone). Similarly, blockade of the NF-κB pathway using PDTC (100 μM) caused ~51% and 69% inhibition of constitutive Mcl-1 expression and of TNFα-induced Mcl-1 expression, respectively (P < 0.05 versus treatment with TNFα alone). Further analysis of the effects of these pharmacologic inhibitors on RA synovial fibroblast growth showed a significant time-dependent decrease in cell numbers upon NF-κB or Akt inhibition (Figure 2B).
Figure 2.

EGCG regulates Mcl-1 expression by inhibiting the NF-κB and Akt pathways. A, Mcl-1 expression in RA synovial fibroblasts (2 × 105/well) treated with pyrrolidine dithiocarbamate (PDTC; 100 μM) alone, LY294002 (LY; 20 μM) alone, or with a combination of TNFα (20 ng/ml) and PDTC or LY294002 for 72 hours. Bars show the mean and SEM from 3 independent experiments using different donors. B, Decrease in cell numbers upon NF-κB or Akt inhibition. RA synovial fibroblasts (2 × 103/well in 96-well plates) were treated with PDTC or LY294002 over 72 hours, and cell numbers were determined with a CyQuant cell enumeration kit. Values are the mean and SEM from 4 independent experiments using different donors. C, Phosphorylation of Akt and translocation of NF-κBp65 in RA synovial fibroblasts. Fibroblasts were left untreated or were treated with EGCG (50 μM) alone, TNFα (20 ng/ml) alone, or a combination of EGCG and TNFα for 30 minutes or 24 hours. Protein (20 μg) from each sample was used to detect phosphorylation of Akt and activation of NF-κBp65 from cytoplasm (Cyto) to nucleus (Nuc) by Western blotting. D, Quantification of NF-κBp65 DNA binding activity, using equal amounts of nuclear protein (2 μg) from samples treated with EGCG (50 μM) alone, TNFα (20 ng/ml) alone, or a combination of EGCG and TNFα for 30 minutes. TransAM enzyme-linked immunosorbent assay kits were used to quantify NF-κBp65 DNA binding activity. Bars show the mean and SEM from 3 independent experiments using different donors. Mut = mutant NF-κBp65 consensus; WT = wild-type NF-κBp65 consensus (see Figure 1 for other definitions).
To study the early and prolonged effect of EGCG on these signaling pathways, we treated RA synovial fibroblasts with EGCG (50 μM) and/or TNFα for 30 minutes or 24 hours. The phosphorylation of Akt was studied in total cell lysates, while nuclear and cytoplasmic fractions were prepared to study the nuclear translocation of NF-κBp65 in RA synovial fibroblasts (Figures 2C and D). EGCG was capable of suppressing phosphorylation of Akt by 24 hours, but not at 30 minutes (Figure 2C). However, EGCG was effective in inhibiting constitutive and TNFα-induced nuclear translocation of NF-κBp65 both at 30 minutes and 24 hours (Figure 2C), suggesting its efficacy in suppressing NF-κB–regulated genes.
Since EGCG was capable of inhibiting nuclear translocation of NF-κBp65 at both time points studied, we tested whether EGCG also inhibits NF-κBp65 DNA binding activity in these cells. Using the nuclear extracts obtained from similar treatment for 30 minutes, we observed that TNFα increased NF-κBp65 DNA binding activity by ~35-fold as compared with untreated samples (P < 0.05) (Figure 2D). Interestingly, the presence of EGCG (50 μM) significantly inhibited the TNFα-induced NF-κBp65 DNA binding activity, by >50% when compared with samples treated with TNFα alone (P < 0.05), providing direct evidence of the ability of EGCG to inhibit the NF-κB pathway by reducing the pool of NF-κBp65 translocating to the nucleus and concomitantly inhibiting its DNA binding activity in RA synovial fibroblasts.
Mcl-1 abrogation by EGCG enhances RA synovial fibroblast caspase 3 activity
We studied the mechanism by which EGCG inhibits Mcl-1 expression in RA synovial fibroblasts. EGCG (50 μM) almost completely inhibited the expression of Mcl-1 in RA synovial fibroblasts (P < 0.05 versus unstimulated fibroblasts) (Figure 3A). Interestingly, preincubation with the proteasome inhibitor (MG132) at varying concentrations completely blocked the ability of EGCG to inhibit Mcl-1 expression, implicating the role of EGCG in degrading Mcl-1 protein in RA synovial fibroblasts to make them susceptible to apoptosis (P < 0.05 for unstimulated fibroblasts versus fibroblasts treated with EGCG, and for fibroblasts treated with EGCG versus fibroblasts treated with EGCG plus MG132) (Figure 3A). However, preincubation of RA synovial fibroblasts with a caspase 3 inhibitor (10 μM) failed to reverse ECGG-inhibited Mcl-1 expression, suggesting that abrogation of Mcl-1 protein by EGCG is an upstream event that leads to the activation of caspase 3 (Figure 3B).
Figure 3.
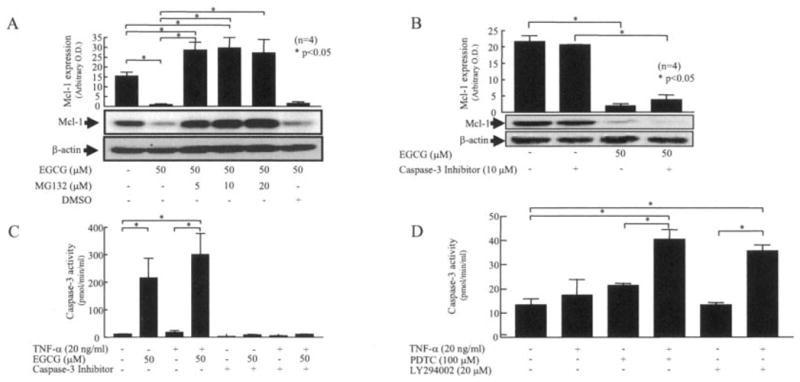
Mechanism of EGCG activation of caspase 3 in RA synovial fibroblasts. A and B, RA synovial fibroblasts (2 × 105/well) were preincubated with MG132 (5–20 μM) or DMSO (A) or with caspase 3 inhibitor (10 μM) (B) for 30 minutes, and then exposed to EGCG (50 μM) for 24 hours. Cells were lysed, and the expression level of Mcl-1 was determined. C, For caspase 3 activity assays, RA synovial fibroblasts (2 × 105/well) were incubated with EGCG (50 μM) alone, TNFα (20 ng/ml) alone, or a combination of EGCG and TNFα for 24 hours in RPMI 1640 plus 1% fetal bovine serum. Cells were lysed in lysis buffer, and 10 μl of lysate was used to estimate caspase 3 activity using commercially available colorimetric assay kits. Results were normalized to protein content in each sample. D, RA synovial fibroblasts were treated with pyrrolidine dithiocarbamate (PDTC; 100 μM) alone, LY294002 (LY; 20 μM) alone, or with a combination of TNFα (20 ng/ml) and PDTC or LY294002 for 24 hours. Cells were lysed with lysis buffer and used to estimate caspase 3 activity. Bars show the mean and SEM from 3 or 4 independent experiments using cells from different donors under similar conditions. * = P < 0.05. OD = optical density (see Figure 1 for other definitions).
Further experiments showed that EGCG (50 μM) significantly induced caspase 3 activity, by 18-fold, in RA synovial fibroblasts as compared with the activity in unstimulated cells (P < 0.05) (Figure 3C). TNFα (20 ng/ml) alone caused no modulation of caspase 3 activity in RA synovial fibroblasts. Interestingly, the combination of EGCG and TNFα further enhanced caspase 3 activation by ~25% more than that induced by EGCG alone, suggesting a possible increase in the susceptibility of RA synovial fibroblasts to TNFα in the presence of EGCG (Figure 3C).
To study the possible role of the Akt and NF-κB pathways in regulating TNFα-induced caspase 3 activity, RA synovial fibroblasts were incubated with the Akt inhibitor (LY294002) alone, the NF-κB inhibitor (PDTC) alone, or a combination of TNFα and each inhibitor for 24 hours. The samples were analyzed for levels of caspase 3 activity (Figure 3D). The results showed that TNFα, PDTC, or LY294002 alone had no significant effect on caspase 3 activation in RA synovial fibroblasts after 24 hours of treatment (Figure 3D). However, coincubation of PDTC and TNFα enhanced caspase 3 activity by ~1.9-fold compared with PDTC alone (P < 0.05), whereas LY294002 and TNFα in combination enhanced caspase 3 activity by ~2.6 fold compared with LY294002 alone (P < 0.05). These results provide evidence that EGCG sensitizes RA synovial fibroblasts to TNFα-induced apoptosis, possibly by blocking these signaling pathways. These results are consistent with the results of previous studies showing Akt and NF-κB involvement in RA synovial fibroblast proliferation and suggest that EGCG-mediated Mcl-1 suppression via the blockade of either or both of these pathways may be the mechanism by which apoptosis is induced (10,19).
EGCG sensitization of RA synovial fibroblasts to TNFα-induced apoptosis
Therapeutic strategies aimed at inducing apoptosis in RA synovial fibroblasts are an emerging option for controlling their expansion into the adjacent tissues in RA joints (4,5). To study whether RA synovial fibroblasts that are stimulated with TNFα in the presence of EGCG (50 μM) indeed undergo apoptosis, we incubated these cells with EGCG (50 μM) alone, TNFα (20 ng/ml) alone, or a combination of EGCG and TNFα for 24, 48, and 72 hours. Western blot analysis of cell lysates from the treated RA synovial fibroblasts showed extremely low levels of constitutively cleaved PARP (Figure 4A). EGCG (50 μM), however, significantly induced PARP cleavage by 4-fold in RA synovial fibroblasts within 24 hours of incubation (P < 0.05), and these levels remained elevated up to 72 hours.
Figure 4.
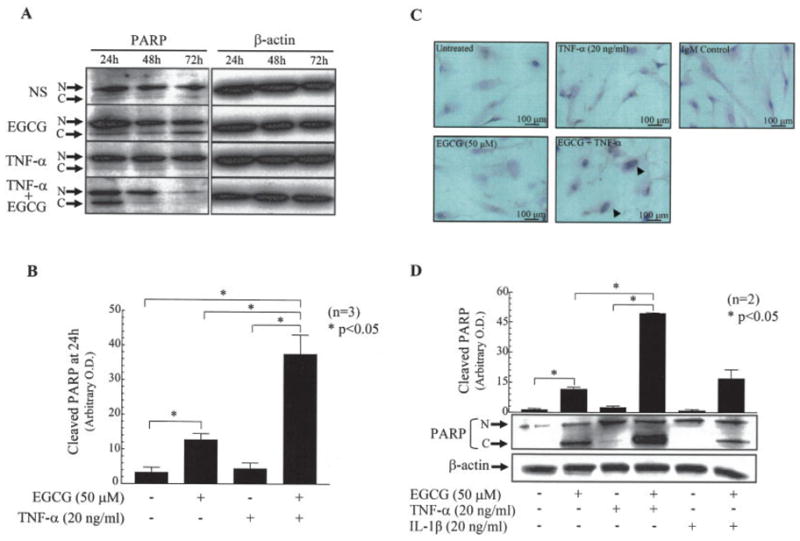
EGCG sensitization of RA synovial fibroblasts to TNFα-induced apoptosis. A, Poly(ADP-ribose) polymerase (PARP) cleavage in cell lysates, as determined by Western blotting, in RA synovial fibroblasts (2 × 105/well) incubated with EGCG (50 μM) alone, TNFα (20 ng/ml) alone, or a combination of EGCG and TNFα for 24, 48, or 72 hours in RPMI 1640 plus 1% fetal bovine serum (FBS). N = native; C = cleaved. B, Intensity of the cleaved PARP bands at 24 hours, as quantified using Un-Scan-It software. C, Staining for F7-26 protein (arrowheads) in RA synovial fibroblasts (1 × 104/well) that were left untreated or were treated with EGCG (50 μM) alone, TNFα (20 ng/ml) alone, or a combination of EGCG and TNFα as for 24 hours in RPMI 1640 plus 1% FBS. Results are representative of 1 experiment using cells from 3 different donors. D, PARP cleavage, as determined by Western blotting, in RA synovial fibroblasts (2 × 105/well) incubated with EGCG (50 μM) alone, TNFα (20 ng/ml) alone, interleukin-1β (IL-1β; 10 ng/ml) alone, or a combination of EGCG and TNFα or IL-1β for 24 hours in RPMI 1640 plus 1% FBS. Results were normalized to protein content in each sample. Bars in B and D show the mean and SEM from 3 independent experiments using cells from different donors under similar conditions. OD = optical density (see Figure 1 for other definitions).
Interestingly, although TNFα alone did not induce PARP cleavage in RA synovial fibroblasts at any time point studied, the combination of TNFα and EGCG resulted in significantly enhanced PARP cleavage, by ~9-fold, within 24 hours of incubation compared with treatment with TNFα alone (P < 0.05) (Figure 4B). To confirm this phenomenon, similarly treated RA synovial fibroblasts were stained with monoclonal antibody for single-stranded DNA (F7-26) (Figure 4C). The results of the cell immunostaining confirmed the previous observation that PARP cleavage was enhanced in fibroblasts treated with both EGCG and TNFα, as evident from markedly enhanced F7-26 nuclear staining (Figure 4C). Furthermore, to test whether synergistic induction of PARP cleavage by EGCG was TNFα specific, we performed similar experiments using IL-1β and a combination of EGCG plus IL-1β. Western blot analysis of PARP cleavage showed that IL-1β in combination with EGCG (50 μM) did not enhance PARP cleavage as compared with EGCG alone, thus implicating the relative specificity of EGCG for TNFα sensitization of RA synovial fibroblasts (Figure 4D).
EGCG enhances TNFα-induced RA synovial fibroblast cytotoxicity
To study the effect of EGCG and TNFα on RA synovial fibroblast viability, we treated confluent RA synovial fibroblasts with EGCG (10 and 50 μM) alone and in combination with TNFα (20 ng/ml) for up to 72 hours. After glutaraldehyde fixation, morphology was determined by hematoxylin and eosin staining of RA synovial fibroblasts, and photomicrographs were obtained. As shown in Figure 5A, EGCG (10 μM) was nontoxic to RA synovial fibroblasts when given alone or in combination with TNFα. However, moderate morphologic changes and apoptosis were visible at 50 μM EGCG. TNFα treatment alone for up to 72 hours was nontoxic to RA synovial fibroblasts. In the presence of EGCG, however, TNFα-induced apoptosis-like changes were accelerated and enhanced in RA synovial fibroblasts (Figure 5A).
Figure 5.

Synergistic effect of EGCG and TNFα in inducing RA synovial fibroblast cytotoxicity. A, RA synovial fibroblasts were incubated with EGCG (10 and 50 μM) alone or with a combination of EGCG and TNFα (20 ng/ml) for 72 hours in RPMI 1640 plus 1% fetal bovine serum (FBS). Fibroblasts were stained with hematoxylin and eosin, and photomicrographs were obtained. Results are representative of experiments conducted using RA synovial fibroblasts from 3 different donors (original magnification × 200). B, Cell survival was determined using quantitative cell enumeration assays in RA synovial fibroblasts treated with EGCG (10 and 50 μM) alone, TNFα (20 ng/ml) alone, or a combination of EGCG and TNFα for 72 hours in RPMI 1640 plus 1% FBS. Bars show the mean and SEM from 3 independent experiments using RA synovial fibroblasts from 3 different donors, conducted in 4 wells per condition in each experiment. See Figure 1 for other definitions.
In quantitative cell survival assays (Figure 5B), EGCG at 10 μM showed no synergistic effect on cytotoxicity with TNFα treatment. However, EGCG at 50 μM caused a significant reduction in the RA synovial fibroblast population, resulting in >40% of cells being killed by 72 hours (P < 0.05). Interestingly, TNFα treatment alone resulted in a modest yet significant increase of 13% in the RA synovial fibroblast population after 72 hours, as compared with unstimulated cells. When incubated with EGCG (50 μM) and TNFα (20 ng/ml), cell survival was reduced to ~25% by 72 hours (P < 0.05) (Figure 5B).
DISCUSSION
The present study provides novel insights into the mechanism of Mcl-1 abrogation through which EGCG sensitizes RA synovial fibroblasts to TNFα-induced apoptosis. A schematic representation of this mechanism is presented in Figure 6. Interestingly, EGCG-mediated blockade of Mcl-1 expression was observed in RA and OA synovial fibroblasts, but was moderate or absent in normal synovial fibroblasts and HMVECs. This finding is important, especially given the present state of RA therapies, in which treatment options do not always halt the joint destruction in RA, and adjunct therapy is needed to overcome drug resistance.
Figure 6.

Schematic representation of the mechanism of EGCG sensitization of RA synovial fibroblasts to TNFα-induced apoptosis. The findings of the present study suggested the down-regulation of Mcl-1 by EGCG as a key mechanism in RA synovial fibroblast sensitization to TNFα-induced apoptosis, which was mediated via inhibition of Akt and NF-κB activation. PARP = poly(ADP-ribose) polymerase (see Figure 1 for other definitions).
The Bcl-2 family of proteins, which includes antiapoptotic members (e.g., Mcl-1, Bcl-2, and Bcl-xL), plays a critical role in regulating apoptosis induced by a wide variety of stimuli (20,21). Mcl-1 is a caspase-sensitive antiapoptotic Bcl-2 family member that has been shown to act independently of Bcl-2 and Bcl-xL (22). This notion is supported by studies showing that induction of apoptosis by ultraviolet irradiation or adenoviral infection requires proteasomal degradation of Mcl-1, but not Bcl-2 or Bcl-xL, and that specific cleavage of Mcl-1, but not Bcl-2 or Bcl-xL, by caspase 3 is essential for spontaneous apoptosis (23). RA synovial fibroblasts constitutively express high levels of Mcl-1, which is further induced by cytokines such as TNFα, IL-6, or IL-1β (24). This suggests that Mcl-1 overexpression in RA synovial fibroblasts may contribute to apoptosis resistance, thereby enhancing the participation of RA synovial fibroblasts in RA pathogenesis.
In our study, RA synovial fibroblasts constitutively expressed Mcl-1, and its level was further elevated upon TNFα activation. Importantly, the inhibitory effect of EGCG on constitutive Mcl-1 expression and TNFα-induced Mcl-1 expression in RA synovial fibroblasts was much higher when compared with Bcl-2 expression. EGCG induced apoptosis in leukemia cells by inhibiting down-regulation of Mcl-1 and X-linked inhibitor of apoptosis proteins (25). Our results further strengthen the hypothesis that EGCG induces apoptosis via proteasomal degradation of Mcl-1, resulting in the activation of caspase 3 activity to break down RA synovial fibroblast resistance to TNFα-induced apoptosis. This notion is further supported by recent studies showing that inhibition of Mcl-1 resulted in increased efficacy of ABT-737, a drug designed to specifically target the Bcl-2 member of the antiapoptotic family, in inducing apoptosis in cancer cells (26).
Caspases are crucial mediators of apoptosis and catalyze the cleavage of key structural proteins and important signaling, homeostatic, and repair enzymes (27). Among these, caspase 3 is a main downstream activator of death proteases. PARP is an intracellular enzyme that is essential for DNA repair and replication, and is a prime target of caspase 3 (28). Cleavage of PARP results in the subsequent DNA fragmentation that is characteristic of apoptosis (29,30). In the present study, incubation of RA synovial fibroblasts with EGCG induced caspase 3 activity, which was synergistically elevated by costimulation of these cells with TNFα. Furthermore, we observed a similar synergistic effect of EGCG and TNFα on activation of PARP cleavage and enhanced nuclear staining for fragmented DNA, resulting in the induction of apoptosis in an accelerated manner. We and others have shown that EGCG induces apoptosis in cancer cells, and that this apoptosis is mediated via activation of caspases and PARP-mediated DNA fragmentation (12,13,31). Given that RA synovial fibroblasts are known to be resistant to TNFα-induced apoptosis, the salient finding of this study is that EGCG reverses this insensitivity and renders RA synovial fibroblasts susceptible to TNFα-induced apoptosis. Several studies of the pathologic role of RA synovial fibroblasts suggest induction of apoptosis as a promising and effective therapeutic strategy in regulating unwanted synovial pannus growth in RA (5,11,32).
Synovial hyperplasia is a hallmark of RA, which is at least in part mediated by an important survival protein, Akt, and a transcription factor, NF-κB (9,10,19). Akt is a serine/threonine protein kinase regulated by phosphatidylinositol 3,4,5-trisphosphate and has been implicated in signaling of survival in a wide variety of cells, including RA synovial fibroblasts (33). NF-κB is a transcription factor composed of dimers of the NF-κB/Rel family of proteins (34). Constitutive activation of Akt and NF-κB in RA synovial fibroblasts, but not OA or normal synovial fibroblasts (10), suggests their role in RA synovial fibroblast survival and proliferation. Additionally, EGCG has been shown to be an effective inhibitor of phosphatidylinositol 3-kinase/Akt and NF-κB pathways in neoplastic cells (12).
At the molecular level, TNFα up-regulated both phosphorylation of Akt and nuclear translocation of NF-κBp65 in RA synovial fibroblasts within 30 minutes to 24 hours of stimulation in the present study. EGCG markedly inhibited the accumulation of p-Akt and the nuclear translocation of NF-κBp65 in TNFα-stimulated RA synovial fibroblasts. Additionally, the presence of EGCG also markedly suppressed TNFα-induced NF-κBp65 DNA binding activity in RA synovial fibroblasts. Importantly, pharmacologic inhibitors of both Akt and NF-κB, while having no significant effect by themselves, synergistically increased caspase 3 activation in the presence of TNFα in RA synovial fibroblasts within 24 hours, to levels ~3-fold greater than constitutive activity. Taken together, our findings and the results of previous studies support the notion that EGCG-mediated down-regulation of uncontrolled growth and induction of apoptosis in RA synovial fibroblasts is, at least in part, achieved by the inhibition of Akt and NF-κB signaling pathways.
TNFα-targeted therapeutic approaches have demonstrated promising potential for treating RA (35). However, even in patients with a good response to TNFα therapy, the synthesis of TNFα is not completely abrogated (36). This suggests that future optimization of anti-TNFα therapies will require the use of complementary approaches to achieve the desired efficacy in halting joint destruction in RA. We have previously shown that EGCG is a potent inhibitor of cytokine-induced chondrocyte MMP-1 and MMP-13 activation, and RA synovial fibroblast chemokine production and MMP-2 activation (16,37). Recently, we found that EGCG possesses the ability to inhibit IL-6 synthesis in human RA synovial fibroblasts and rat adjuvant-induced arthritis (38). This finding suggests that EGCG, besides inducing apoptosis, can down-regulate the possible inflammatory or reactive proteins produced by IL-6 in RA to suppress local or systemic inflammation and joint destruction. The cooperation of ECGC in TNFα-induced RA synovial fibroblast apoptosis suggests that EGCG holds potential for use as an adjunct or combination option with anti-TNFα therapies to inhibit synovial hyperproliferation in the RA joint. Additionally, EGCG appears to have an acceptable safety profile in humans, and plasma levels at the micromolar concentration are achievable and could be maintained by oral consumption (39).
In summary, the present study provides evidence that EGCG efficiently induces apoptosis and concomitantly enhances the susceptibility of RA synovial fibroblasts to TNFα-induced apoptosis. Taken together, these findings suggest that EGCG could be the basis of an efficacious adjunct therapy for preventing or treating RA.
Acknowledgments
The authors thank Phillip L. Campbell and Pamela J. Mansfield for technical assistance.
Dr. Ahmed’s work was supported by the NIH (grants AT-003633 and AR-055741). Dr. Silverman’s work was supported by an American Heart Association Postdoctoral Fellowship (0425749Z). Dr. Marotte’s work was supported by the French Society of Rheumatology, Lavoisier, and the Philippe Foundation. Dr. Koch’s work was supported by the NIH (grants AI-40987 and AR-48267), the Frederick G. L. Huetwell and William D. Robinson, MD, Professorship in Rheumatology, and the Office of Research and Development, Medical Research Service, Department of Veterans Affairs.
Footnotes
AUTHOR CONTRIBUTIONS
Dr. Ahmed had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Study design. Ahmed, Silverman, Koch.
Acquisition of data. Ahmed, Silverman, Marotte, Kwan, Matuszczak.
Analysis and interpretation of data. Ahmed, Silverman, Marotte, Kwan, Koch.
Manuscript preparation. Ahmed, Koch.
Statistical analysis. Ahmed, Silverman, Marotte, Kwan.
References
- 1.Thompson CB. Apoptosis in the pathogenesis and treatment of disease. Science. 1995;267:1456–62. doi: 10.1126/science.7878464. [DOI] [PubMed] [Google Scholar]
- 2.Dai Y, Grant S. Targeting multiple arms of the apoptotic regulatory machinery. Cancer Res. 2007;67:2908–11. doi: 10.1158/0008-5472.CAN-07-0082. [DOI] [PubMed] [Google Scholar]
- 3.Certo M, Del Gaizo Moore V, Nishino M, Wei G, Korsmeyer S, Armstrong SA, et al. Mitochondria primed by death signals determine cellular addiction to antiapoptotic BCL-2 family members. Cancer Cell. 2006;9:351–65. doi: 10.1016/j.ccr.2006.03.027. [DOI] [PubMed] [Google Scholar]
- 4.Huber LC, Distler O, Tarner I, Gay RE, Gay S, Pap T. Synovial fibroblasts: key players in rheumatoid arthritis. Rheumatology (Oxford) 2006;45:669–75. doi: 10.1093/rheumatology/kel065. [DOI] [PubMed] [Google Scholar]
- 5.Meinecke I, Rutkauskaite E, Gay S, Pap T. The role of synovial fibroblasts in mediating joint destruction in rheumatoid arthritis. Curr Pharm Des. 2005;11:563–8. doi: 10.2174/1381612053381945. [DOI] [PubMed] [Google Scholar]
- 6.Liu H, Pope RM. Apoptosis in rheumatoid arthritis: friend or foe. Rheum Dis Clin North Am. 2004;30:603–25. doi: 10.1016/j.rdc.2004.04.010. [DOI] [PubMed] [Google Scholar]
- 7.Liu H, Eksarko P, Temkin V, Haines GK, 3rd, Perlman H, Koch AE, et al. Mcl-1 is essential for the survival of synovial fibroblasts in rheumatoid arthritis. J Immunol. 2005;175:8337–45. doi: 10.4049/jimmunol.175.12.8337. [DOI] [PubMed] [Google Scholar]
- 8.Liu H, Pope RM. The role of apoptosis in rheumatoid arthritis. Curr Opin Pharmacol. 2003;3:317–22. doi: 10.1016/s1471-4892(03)00037-7. [DOI] [PubMed] [Google Scholar]
- 9.Zhang HG, Huang N, Liu D, Bilbao L, Zhang X, Yang P, et al. Gene therapy that inhibits nuclear translocation of nuclear factor κB results in tumor necrosis factor α–induced apoptosis of human synovial fibroblasts. Arthritis Rheum. 2000;43:1094–105. doi: 10.1002/1529-0131(200005)43:5<1094::AID-ANR20>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 10.Zhang HG, Wang Y, Xie JF, Liang X, Liu D, Yang P, et al. Regulation of tumor necrosis factor α–mediated apoptosis of rheumatoid arthritis synovial fibroblasts by the protein kinase Akt. Arthritis Rheum. 2001;44:1555–67. doi: 10.1002/1529-0131(200107)44:7<1555::AID-ART279>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 11.Pope RM. Apoptosis as a therapeutic tool in rheumatoid arthritis. Nat Rev Immunol. 2002;2:527–35. doi: 10.1038/nri846. [DOI] [PubMed] [Google Scholar]
- 12.Khan N, Afaq F, Saleem M, Ahmad N, Mukhtar H. Targeting multiple signaling pathways by green tea polyphenol (−)-epigallocatechin-3-gallate. Cancer Res. 2006;66:2500–5. doi: 10.1158/0008-5472.CAN-05-3636. [DOI] [PubMed] [Google Scholar]
- 13.Hafeez BB, Ahmed S, Wang N, Gupta S, Zhang A, Haqqi TM. Green tea polyphenols-induced apoptosis in human osteosarcoma SAOS-2 cells involves a caspase-dependent mechanism with down-regulation of nuclear factor-κB. Toxicol Appl Pharmacol. 2006;216:11–9. doi: 10.1016/j.taap.2006.03.013. [DOI] [PubMed] [Google Scholar]
- 14.Siddiqui IA, Malik A, Adhami VM, Asim M, Hafeez BB, Sarfaraz S, et al. Green tea polyphenol EGCG sensitizes human prostate carcinoma LNCaP cells to TRAIL-mediated apoptosis and synergistically inhibits biomarkers associated with angiogenesis and metastasis. Oncogene. 2008;27:2055–63. doi: 10.1038/sj.onc.1210840. [DOI] [PubMed] [Google Scholar]
- 15.Eddy SF, Kane SE, Sonenshein GE. Trastuzumab-resistant HER2-driven breast cancer cells are sensitive to epigallocatechin-3 gallate. Cancer Res. 2007;67:9018–23. doi: 10.1158/0008-5472.CAN-07-1691. [DOI] [PubMed] [Google Scholar]
- 16.Ahmed S, Pakozdi A, Koch AE. Regulation of interleukin-1β–induced chemokine production and matrix metalloproteinase 2 activation by epigallocatechin-3-gallate in rheumatoid arthritis synovial fibroblasts. Arthritis Rheum. 2006;54:2393–401. doi: 10.1002/art.22023. [DOI] [PubMed] [Google Scholar]
- 17.Arnett FC, Edworthy SM, Bloch DA, McShane DJ, Fries JF, Cooper NS, et al. The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum. 1988;31:315–24. doi: 10.1002/art.1780310302. [DOI] [PubMed] [Google Scholar]
- 18.Morel J, Audo R, Hahne M, Combe B. Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) induces rheumatoid arthritis synovial fibroblast proliferation through mitogen-activated protein kinases and phosphatidylinositol 3-kinase/Akt. J Biol Chem. 2005;280:15709–18. doi: 10.1074/jbc.M414469200. [DOI] [PubMed] [Google Scholar]
- 19.Bai S, Liu H, Chen KH, Eksarko P, Perlman H, Moore TL, et al. NF-κB–regulated expression of cellular FLIP protects rheumatoid arthritis synovial fibroblasts from tumor necrosis factor α–mediated apoptosis. Arthritis Rheum. 2004;50:3844–55. doi: 10.1002/art.20680. [DOI] [PubMed] [Google Scholar]
- 20.Adams JM, Cory S. The Bcl-2 protein family: arbiters of cell survival. Science. 1998;281:1322–6. doi: 10.1126/science.281.5381.1322. [DOI] [PubMed] [Google Scholar]
- 21.Perlman H, Georganas C, Pagliari LJ, Koch AE, Haines K, 3rd, Pope RM. Bcl-2 expression in synovial fibroblasts is essential for maintaining mitochondrial homeostasis and cell viability. J Immunol. 2000;164:5227–35. doi: 10.4049/jimmunol.164.10.5227. [DOI] [PubMed] [Google Scholar]
- 22.Weng C, Li Y, Xu D, Shi Y, Tang H. Specific cleavage of Mcl-1 by caspase-3 in tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-induced apoptosis in Jurkat leukemia T cells. J Biol Chem. 2005;280:10491–500. doi: 10.1074/jbc.M412819200. [DOI] [PubMed] [Google Scholar]
- 23.Nijhawan D, Fang M, Traer E, Zhong Q, Gao W, Du F, et al. Elimination of Mcl-1 is required for the initiation of apoptosis following ultraviolet irradiation. Genes Dev. 2003;17:1475–86. doi: 10.1101/gad.1093903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Michels J, Johnson PW, Packham G. Mcl-1. Int J Biochem Cell Biol. 2005;37:267–71. doi: 10.1016/j.biocel.2004.04.007. [DOI] [PubMed] [Google Scholar]
- 25.Lee YK, Shanafelt TD, Bone ND, Strege AK, Jelinek DF, Kay NE. VEGF receptors on chronic lymphocytic leukemia (CLL) B cells interact with STAT 1 and 3: implication for apoptosis resistance. Leukemia. 2005;19:513–23. doi: 10.1038/sj.leu.2403667. [DOI] [PubMed] [Google Scholar]
- 26.Van Delft MF, Wei AH, Mason KD, Vandenberg CJ, Chen L, Czabotar PE, et al. The BH3 mimetic ABT-737 targets selective Bcl-2 proteins and efficiently induces apoptosis via Bak/Bax if Mcl-1 is neutralized. Cancer Cell. 2006;10:389–99. doi: 10.1016/j.ccr.2006.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sellers WR, Fisher DE. Apoptosis and cancer drug targeting. J Clin Invest. 1999;104:1655–61. doi: 10.1172/JCI9053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Soldani C, Scovassi AI. Poly(ADP-ribose) polymerase-1 cleavage during apoptosis: an update. Apoptosis. 2002;7:321–8. doi: 10.1023/a:1016119328968. [DOI] [PubMed] [Google Scholar]
- 29.Janicke RU, Sprengart ML, Wati MR, Porter AG. Caspase-3 is required for DNA fragmentation and morphological changes associated with apoptosis. J Biol Chem. 1998;273:9357–60. doi: 10.1074/jbc.273.16.9357. [DOI] [PubMed] [Google Scholar]
- 30.Porter AG, Janicke RU. Emerging roles of caspase-3 in apoptosis. Cell Death Differ. 1999;6:99–104. doi: 10.1038/sj.cdd.4400476. [DOI] [PubMed] [Google Scholar]
- 31.Shimizu M, Weinstein IB. Modulation of signal transduction by tea catechins and related phytochemicals. Mutat Res. 2005;591:147–60. doi: 10.1016/j.mrfmmm.2005.04.010. [DOI] [PubMed] [Google Scholar]
- 32.Davis LS. A question of transformation: the synovial fibroblast in rheumatoid arthritis. Am J Pathol. 2003;162:1399–402. doi: 10.1016/S0002-9440(10)64272-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tas SW, Remans PH, Reedquist KA, Tak PP. Signal transduction pathways and transcription factors as therapeutic targets in inflammatory disease: towards innovative antirheumatic therapy. Curr Pharm Des. 2005;11:581–611. doi: 10.2174/1381612053381918. [DOI] [PubMed] [Google Scholar]
- 34.Firestein GS. NF-κB: holy grail for rheumatoid arthritis? [editorial] Arthritis Rheum. 2004;50:2381–6. doi: 10.1002/art.20468. [DOI] [PubMed] [Google Scholar]
- 35.Lorenz HM, Kalden JR. Perspectives for TNF-α-targeting therapies. Arthritis Res. 2002;4 (Suppl 3):S17–24. doi: 10.1186/ar564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ulfgren AK, Andersson U, Engstrom M, Klareskog L, Maini RN, Taylor PC. Systemic anti-tumor necrosis factor α therapy in rheumatoid arthritis down-regulates synovial tumor necrosis factor α synthesis. Arthritis Rheum. 2000;43:2391–6. doi: 10.1002/1529-0131(200011)43:11<2391::AID-ANR3>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 37.Ahmed S, Wang N, Lalonde M, Goldberg VM, Haqqi TM. Green tea polyphenol epigallocatechin-3-gallate (EGCG) differentially inhibits interleukin-1 β-induced expression of matrix metalloproteinase-1 and -13 in human chondrocytes. J Pharmacol Exp Ther. 2004;308:767–73. doi: 10.1124/jpet.103.059220. [DOI] [PubMed] [Google Scholar]
- 38.Ahmed S, Marotte H, Kwan K, Ruth JH, Campbell PL, Rabquer BJ, et al. Epigallocatechin-3-gallate inhibits IL-6 synthesis and suppresses transsignaling by enhancing soluble gp130 production. Proc Natl Acad Sci U S A. 2008;105:14692–7. doi: 10.1073/pnas.0802675105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ullmann U, Haller J, Decourt JP, Girault N, Girault J, Richard-Caudron AS, et al. A single ascending dose study of epigallocatechin gallate in healthy volunteers. J Int Med Res. 2003;31:88–101. doi: 10.1177/147323000303100205. [DOI] [PubMed] [Google Scholar]