

Abstract
The Drosophila inhibitor of apoptosis protein DIAP1 ensures cell viability by directly inhibiting caspases. In cells destined to die this IAP-mediated inhibition of caspases is overcome by IAP-antagonists. Genetic evidence indicates that IAP-antagonists are non-equivalent and function synergistically to promote apoptosis. Here we provide biochemical evidence for the non-equivalent mode of action of Reaper, Grim, Hid and Jafrac2. We find that these IAP-antagonists display differential and selective binding to specific DIAP1 BIR domains. Consistently, we show that each DIAP1 BIR region associates with distinct caspases. The differential DIAP1 BIR interaction seen both between initiator and effector caspases and within IAP-antagonist family members suggests that different IAP-antagonists inhibit distinct caspases from interacting with DIAP1. Surprisingly, we also find that the caspase-binding residues of XIAP predicted to be strictly conserved in caspase-binding IAPs, are absent in DIAP1. In contrast to XIAP, residues C-terminal to the DIAP1 BIR1 domain are indispensable for caspase association. Our studies on DIAP1 and caspases expose significant differences between DIAP1 and XIAP suggesting that DIAP1 and XIAP inhibit caspases in different ways.
Keywords: apoptosis/caspase/Drosophila/IAP-antagonist/IAPs
Introduction
Programmed cell death, or apoptosis, is an essential process of animal development and is important for normal morphogenesis and tissue architecture (Meier et al., 2000a). Apoptosis signalling culminates in the activation of a set of highly specific cysteine proteases called caspases. Generally, caspases are ubiquitously expressed as catalytically inactive zymogens consisting of an N-terminal pro-domain, a large (p20) and a small (p10) catalytic subunit. Upon induction of apoptosis the caspase zymogen is proteolytically processed at inter-domain sites generating the catalytically active protease (Shi, 2002). While initiator caspases are activated through an auto-proteolytic process, cleavage and activation of downstream effector caspases require the proteolytic activity of initiator caspases. Once activated, caspases cleave a host of structural and regulatory proteins resulting in the coordinated disassembly of the cell (Shi, 2002).
Activation of the caspase cascade is tightly controlled by members of the evolutionarily conserved Inhibitor of APoptosis (IAP) protein family. IAPs are characterized by the presence of one or more BIR (Baculovirus IAP Repeat) domains (Salvesen and Duckett, 2002). The BIR module is essential for the anti-apoptotic activity of IAPs as it is required for caspase binding. For example, a region containing the BIR2 domain of the mammalian IAP XIAP specifically interacts with the effector caspases-3 and -7, while XIAP’s BIR3 domain binds to the initiator caspase-9. Direct interaction of XIAP to the catalytic pocket of caspase-3 and caspase-7 results in the steric occlusion of these caspases blocking their access to substrates. Residues within a small segment N-terminal to XIAP’s BIR2 domain are essential for the binding of XIAP to activated caspase-3 and -7 (Shi, 2002). Importantly, these residues are highly conserved in human IAPs that potently inhibit caspases. In particular, residue Asp148 of XIAP represents the major anchoring point for IAP–caspase association. Consequently, Asp148 is predicted to be strictly conserved in IAPs capable of binding to caspases (Huang et al., 2001).
In addition to the BIR domain some IAPs also contain a C-terminal RING finger domain that provides these IAPs with E3 ubiquitin–protein ligase activity (Salvesen and Duckett, 2002). Although XIAP has a RING finger domain, this module is dispensable for XIAP’s caspase- neutralizing activity as XIAP mutants lacking the RING finger domain are fully functional in suppressing caspase- mediated cell death. Mere physical association of XIAP with activated caspases seems to be entirely sufficient to suppress caspases. In complete contrast, physical association of the Drosophila IAP DIAP1 with caspases is necessary but not sufficient to inhibit caspases in vivo (Wilson et al., 2002; Ditzel et al., 2003). In addition to caspase binding, DIAP1 requires the E3 ubiquitin protein ligase activity provided by its RING finger domain to effectively neutralize caspases. The RING finger domain of DIAP1 mediates ubiquitylation and inactivation of the Drosophila caspase DRONC (Wilson et al., 2002). In addition to neutralizing DRONC, DIAP1 also potently inhibits the caspases drICE and DCP-1 (Kaiser et al., 1998; Hawkins et al., 1999). While DRONC is an initiator caspase that is most homologous to the mammalian initiator caspase-9, drICE and DCP-1 are effector caspases with sequence and enzymological properties very similar to those of the mammalian effector caspases-3 and -7 (Fraser and Evan, 1997; Song et al., 1997).
In Drosophila, DIAP1 represents the last line of defence against caspase-mediated damage since loss-of-function mutations in diap1 cause spontaneous and unrestrained cell death (Wang et al., 1999; Goyal et al., 2000; Lisi et al., 2000; Rodriguez et al., 2002). Hence, the caspase-neutralizing activity of DIAP1 is essential to maintain cell viability. In cells fated to die, the anti-apoptotic function of DIAP1 is thwarted by a set of specialized IAP-binding proteins called IAP-antagonists. In Drosophila the IAP-antagonists Reaper (Rpr), Grim, Hid, Sickle and Jafrac2 are thought to promote cell death by disrupting DIAP1–caspase association thereby alleviating DIAP1’s inhibition of caspases (White et al., 1994; Grether et al., 1995; Chen et al., 1996; Christich et al., 2002; Srinivasula et al., 2002; Tenev et al., 2002). In mammals, an identical mechanism operates through the IAP-antagonists Smac/DIABLO and HtrA2/Omi (Vaux and Silke, 2003). Common to all IAP-antagonists is the presence of a conserved motif that is critical for IAP binding and is known as IBM (IAP-binding motif). IBMs bear an N-terminal Ala1 that anchors this motif to the BIR surface of IAPs (Huang et al., 2001). The increasing number of Drosophila and mammalian members of the IAP-antagonist protein family invokes the question as to why there are so many distinct IAP-antagonists. Although in Drosophila, over-expression of Rpr, Grim, Hid or Jafrac2 is sufficient to induce apoptosis in a wide variety of cell types, genetic analyses of Rpr, Grim and Hid argue that these IAP-antagonists are not redundant but must act in combination with each other to induce apoptosis (Robinow et al., 1997; Zhou et al., 1997; Wing et al., 1998). Thus, embryos with deletions that remove various combinations of rpr, grim and/or hid indicate that developmental cell death in the embryonic central nervous system (CNS) requires the cooperative actions of Rpr, Grim and Hid. Further, simultaneous ectopic expression of Rpr and Hid in embryonic CNS midline cells induces substantial apoptosis, while expression of two copies of either gene alone has little or no effect on midline cell viability (Zhou et al., 1997). Currently, little is known about the underlying coordinated mode of action through which IAP-antagonists synergistically oppose IAPs.
Here we provide biochemical evidence for the non-redundant mode of action of Rpr, Grim and Hid. We find that Rpr, Grim and Hid display differential and selective binding to specific DIAP1 BIR domains. Further, we show that each BIR domain of DIAP1 associates with distinct caspases. Consistent with the notion that different IAP-antagonists compete with distinct sets of caspases for DIAP1 binding we show that Rpr but not Hid blocks the binding of drICE to DIAP1. We also provide evidence indicating that Rpr, Grim and Hid induce cell death predominantly, if not exclusively, in an IAP-binding-dependent manner. Finally, our biochemical data on the interaction between DIAP1 and caspases expose significant differences between DIAP1 and XIAP. Intriguingly, DIAP1 does not contain sequence homology to the caspase-binding residues of XIAP, which are predicted to be strictly conserved in IAPs capable of binding caspases; yet, DIAP1 specifically interacts with activated caspases such as drICE and DCP-1. Our data indicate that residue Asn117, located immediately C-terminal to the BIR1 domain of DIAP1, is indispensable for caspase association arguing that DIAP1 interacts with activated caspases through a mechanism that is distinct from the one with which XIAP binds to caspase-3 and -7.
Results
The DIAP1 BIR1 region binds to activated effector caspases
To determine the region of DIAP1 that binds to effector caspases, we used wild-type and mutant DIAP1–GST fusion proteins (Figure 1A) as affinity reagents to purify drICE or DCP-1 from cellular extracts. DIAP1–GST was expressed in Drosophila Schneider (S2) cells and affinity purified from cell lysates using glutathione resin. The resin-purified DIAP1–GST was subsequently incubated with total extracts of human 293 cells expressing either drICE or DCP-1. After binding the extracts to the DIAP1–GST proteins, the bound proteins were eluted and analysed by immunoblotting. As expected, DIAP1 exclusively interacted with the processed, activated forms of drICE and DCP-1 but failed to bind to their zymogens (Figure 1B, top right panel, lanes 3 and 7). Significantly, the gain-of-function mutant DIAP16–3s, which suppresses Rpr- and Hid-mediated cell death (Goyal et al., 2000), displayed greatly enhanced binding to activated effector caspases when compared with wild-type DIAP1 (Figure 1B, compare lane 4 with 3, and 8 with 7). The BIR1 region was required for the binding of DIAP1 to activated effector caspases as DIAP1 deletion mutants lacking this region (ΔBIR1) completely failed to associate with drICE and DCP-1. Similarly, DIAP1 fragments consisting of only the DIAP1 RING finger region (RING) also failed to interact with activated caspases. Thus, loss of the BIR1 region abrogated caspase binding indicating that this domain is required for caspase association.




Fig. 1. Co-purification of drICE-V5 or DCP-1-V5 with DIAP1–GST. (A) Schematic representation of the DIAP1 mutants used in this study. (B) The gain-of-function mutant DIAP16–3s binds more efficiently to caspases than wild-type (wt) DIAP1. Top right panel: DIAP1–caspase co-purification; affinity-purified DIAP1–GST was used to precipitate drICE-V5 (lanes 3–6) or DCP-1-V5 (lanes 7–10) from cellular extracts. Top left panel: total extracts of 293 cells expressing the indicated caspases. Caspase expression (top left panel) and caspase–DIAP1 binding (top right panel) were detected by immunoblot analysis using anti-V5 antibody. Bottom right panel: the purification of DIAP1–GST was confirmed by western blot analysis of the eluate using anti-DIAP1 RING antibody. (C and D) The BIR1 region of DIAP1 is necessary and sufficient for caspase binding. (E and F) DIAP111–3e fails to bind to drICE and DCP-1. (C–F) Experiments were conducted as in (B). Top panels: expression (lane 1) and co-purification (lanes 2–end) of drICE-V5 (C and E) and DCP-1-V5 (D and F) with the indicated DIAP1–GST fragments. Bottom panel: purification of the DIAP1–GST fragments was confirmed by immunoblot analysis of the eluate using anti-GST antibody. Note, the purified wild-type and 6–3s mutant DIAP1 BIR1 fragments (E and F; lanes 2 and 3) are cleaved at position 20 by drICE and DCP-1 while BIR111–3e that failed to bind to caspases was only partially processed.
Next, we examined whether the DIAP1 BIR1 region on its own was sufficient for caspase binding. Various DIAP1 deletion mutants (BIR1, Linker1, BIR2 and Linker2; see Figure 1A) were tested for their ability to interact with drICE and DCP-1. drICE exclusively co-purified with the BIR1 fragment (Figure 1C, lane 2). The BIR2, Linker1 or Linker2 regions did not interact with drICE, indicating that the BIR1 region mediates caspase binding. Similarly, activated DCP-1 also preferentially bound to the BIR1 region (Figure 1D, lane 2). However, DCP-1 also interacted with the BIR2 region of DIAP1, albeit only very weakly (lane 3). Taken together, our results indicate that the BIR1 region of DIAP1 is required for efficient binding to activated effector caspases.
A segment immediately C-terminal to the BIR1 domain is indispensable for caspase association
Embryos homozygous for the loss-of-function allele diap111–3e die early during embryogenesis due to unrestrained caspase-dependent cell death (Goyal et al., 2000). Surprisingly, the mutation in diap111–3e was associated with a single amino acid substitution mutation (Asn117Lys) downstream of the DIAP1 BIR1 domain (Table I; Figure 1A). To determine the molecular consequences of the loss-of-function mutation in DIAP111–3e, we compared wild-type and mutant DIAP1 BIR1 regions (1–146) for their ability to bind to activated caspases (Figure 1E and F). Both wild-type and BIR16–3s fragments co-purified with drICE and DCP-1, whereby the BIR16–3s fragment showed enhanced binding to drICE and DCP-1 (compare lanes 2 with 3). In contrast, the BIR111–3e region completely failed to bind to activated drICE and DCP-1 (compare lanes 2 with 4). Thus, amino acid substitution of Asn to Lys at position 117 of DIAP111–3e abrogated DIAP1’s interaction with effector caspases. Although Asn117 in the linker region between the BIR1 and BIR2 domain is critical for caspase binding, the Linker1 segment on its own failed to bind to caspases, indicating that regions preceding Linker1 are also required for caspase association. Our results indicate that diap111–3e is a loss-of-function allele because DIAP111–3e fails to bind to activated effector caspases causing unrestrained caspase- mediated cell death. On the other hand, the 6–3s mutation, which significantly enhances the ability of DIAP to bind activated effector caspases causes a gain-of-function phenotype whereby apoptosis is suppressed.
Table I. Phenotypes and molecular changes associated with the diap1 alleles used in this study.
| diap1 allele | Modifier phenotype |
Recessive phenotype | Mutation | |
|---|---|---|---|---|
| reaper | hid | |||
| 6-3s | suppressor | suppressor | viable | G88S |
| 23-4s | suppressor | suppressor | lethal | G269S |
| 21-4s | enhancer | suppressor | lethal | C406Y |
| th4 | enhancer | enhancer | lethal | H283Y |
| 11-3e | enhancer | enhancer | lethal | N117K |
DIAP1 simultaneously binds to DRONC and activated drICE or DCP-1
Since the BIR1 region of DIAP1 interacts with effector caspases and the BIR2 region binds to initiator caspases (Meier et al., 2000b), we tested whether DIAP1 simultaneously bound to initiator and effector caspases. To this end, we examined whether drICE or DCP-1 interacted with DIAP1 that was pre-bound to the initiator caspase DRONC. DRONC-FLAG was co-expressed with either DIAP1 or controls in S2 cells and anti-FLAG antibody-coupled agarose resin was used to immunoprecipitate DRONC-FLAG from cellular extracts. Resin-purified DRONC-FLAG was subsequently incubated with extracts of 293 cells expressing either drICE-V5 or DCP-1-V5. After binding the extracts to the DRONC-FLAG resin, the bound proteins were eluted and analysed by immunoblotting (Figure 2). In the presence of DIAP1 activated drICE and DCP-1 co-immunoprecipitated with DRONC-FLAG (lanes 1 and 2). In the absence of DIAP1 however, purified DRONC-FLAG on its own failed to interact with either drICE or DCP-1 (lanes 3 and 4). Our results indicate that one molecule of DIAP1 can interact with DRONC and either drICE or DCP-1 at the same time, suggesting that DIAP1 can simultaneously block the activity of both initiator and effector caspases.

Fig. 2. DIAP1 simultaneously binds to initiator and effector caspases. DRONC-FLAG was co-expressed with DIAP1 or controls in S2 cells. DRONC-FLAG was purified by immunoprecipitation using anti-FLAG antibody-coupled agarose beads. Resin-bound DRONC-FLAG was subsequently incubated with extracts of 293 cells expressing the indicated caspases. Following incubation of the DRONC-FLAG beads with the extracts, the bound proteins were eluted with FLAG peptides. Co-immunoprecipitation (lanes 1–4) and expression (lanes 5 and 6) of the indicated proteins were determined by immunoblot analysis using the indicated antibodies.
IAP-antagonists display differential binding to specific BIR domains
Next, we assessed whether IAP-antagonists differentially interact with distinct BIR domains of DIAP1 and hence may compete with specific caspases for DIAP1 binding. To identify the region of DIAP1 that is required for the binding to Rpr, Grim, Hid and Jafrac2, we tested various DIAP1 fragments (Figure 1A) for their interaction with IAP-antagonists. S2 cells expressing the baculovirus, pan-caspase inhibitor p35 were used to avoid Rpr-, Hid- and Grim-induced apoptosis and concomitant loss of cellular material. Importantly, Rpr, Grim and Hid specifically co-purified with both the BIR1 as well as the BIR2 regions of DIAP1 (Figure 3). Rpr associated with the BIR1 and BIR2 regions with equal efficiency (Figure 3A, compare lane 2 with 3). Likewise, Grim also bound equally to both BIR regions (Figure 3B, compare lane 2 with 3). However, Hid, Sickle and Jafrac2 interacted preferentially with the BIR2 and associated only weakly with the BIR1 region (Figure 3C and D and Supplementary figure 1 available at The EMBO Journal Online). Under the same conditions, Rpr, Grim, Hid and Jafrac2 did not interact with the Linker regions of DIAP1 or GST alone (lanes 4 and 5). Collectively, our data indicate that the BIR1 and BIR2 regions of DIAP1 are required for caspase binding and that the IAP-antagonists Rpr and Grim target both these BIR domains equally, while Hid, Sickle and Jafrac2 show preferential binding to BIR2.


Fig. 3. IAP-antagonists bind to distinct regions of DIAP1. DIAP1–GST fragments were used to purify Rpr-V5 (A), Grim-TAP (B), Hid-V5 (C) or Jafrac2 (D) from S2/p35 cellular extracts. Top panel: DIAP1–GST deletion mutants were affinity purified from cellular extracts using glutathione–Sepharose beads and associated IAP-antagonists were detected by immunoblotting using anti-V5 (A and C), anti-protein A (B) or anti-Jafrac2 (D) antibodies, respectively. Middle panel: purification of the DIAP1–GST fragments was verified by western blot analysis using anti-GST antibody. Bottom panel: equal expression of the indicated IAP-antagonist was examined by immunoblotting the S2 extracts using the indicated antibodies. The asterisk denotes a cross-reactive band.
To reveal differences in the binding of IAP-antagonists to DIAP1 we tested various DIAP1 mutants with single point mutations for their ability to bind to IAP-antagonists [Table I; Figure 1A; Goyal et al. (2000)]. Since Rpr, Grim and Hid bind to both BIR domains of DIAP1, we used each BIR region in isolation to study the ability of mutant BIR regions to bind to IAP-antagonists. DIAP16–3s with a gain-of-function mutation in the BIR1 domain showed greatly diminished binding to Rpr and Hid (Figure 4A and C, compare lane 1 with 2). Surprisingly, the 6–3s mutation did not abrogate Grim binding as Grim co-purified with wild-type or DIAP16–3s with equal efficiency (Figure 4B, compare lane 1 with 2). The observation that BIR16–3s showed impaired Rpr and Hid but not Grim binding indicates that Grim interacts with DIAP1 in a manner that is distinct from Rpr and Hid. Importantly, the loss-of-function mutation in DIAP111–3e that completely disrupts binding to activated caspases did not impair the association of DIAP1 to Rpr, Grim or Hid, indicating that the 11–3e mutation does not grossly disrupt the structure of the BIR fold. The DIAP1th4 and DIAP123–4s mutants, which carry single amino acid mutations in the BIR2 domains, showed impaired binding to Rpr, Grim and Hid (Figure 4A–C, lanes 4–6). Furthermore, the th4 BIR2 mutation (His283Tyr) completely abolished the interaction with Jafrac2 and Sickle (Figure 4D, compare lane 4 with 5, and see Supplementary figure 1). Interestingly, the same th4 mutation similarly abrogates DRONC binding resulting in a loss-of-function phenotype (Tenev et al., 2002). Thus, physical association between DIAP1 and caspases is essential for DIAP1 function.

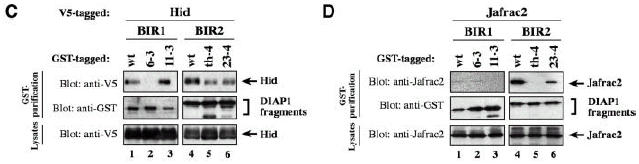
Fig. 4. Rpr, Grim, Hid and Jafrac2 differentially interact with DIAP1. (A–D) Co-purification of Rpr, Grim, Hid and Jafrac2 with wild-type or mutant BIR fragments. Expression and purification of the indicated constructs were determined as in Figure 3. The BIR1 region of DIAP16–3s shows impaired binding to Rpr (A) and Hid (C), while its association to Grim (B) remains unaffected. The 11–3e mutation does not affect the binding of Rpr, Grim or Hid to DIAP1 (A–C, top left panel). (A–D) The th4 and 23–4 BIR2 mutations impair DIAP1’s ability to bind to Rpr (A), Grim (B), Hid (C) and Jafrac2 (D).
IAP-antagonists compete with caspase for DIAP1 binding
The differential interaction between DIAP1 and caspases or IAP-antagonists suggests that different IAP-antagonists may compete with distinct sets of caspases for DIAP1 binding. Therefore, we determined whether different IAP-antagonists displayed contrasting abilities to compete with drICE for DIAP1 binding. As expected, Rpr or Grim, both of which efficiently bind to the BIR1, significantly reduced the ability of DIAP1 to interact with drICE (Figure 5A and data not shown). In parallel experiments, Rpr or Grim also abrogated the association of DIAP1 with DCP-1 (data not shown). In complete contrast however, Hid, that only relatively weakly interacts with the BIR1 region, failed to compete with drICE for DIAP1 binding (Figure 5B). Thus, Rpr and Hid greatly differ in their competence to interfere with DIAP1–drICE interaction. Rpr’s ability to compete with drICE for DIAP1 binding was contingent on Rpr’s IBM. Thus, a Rpr mutant that lacked Ala1 and failed to bind to DIAP1 (see below) also failed to block the binding of DIAP1 to drICE (Figure 5C, compare lane 7 with 8).
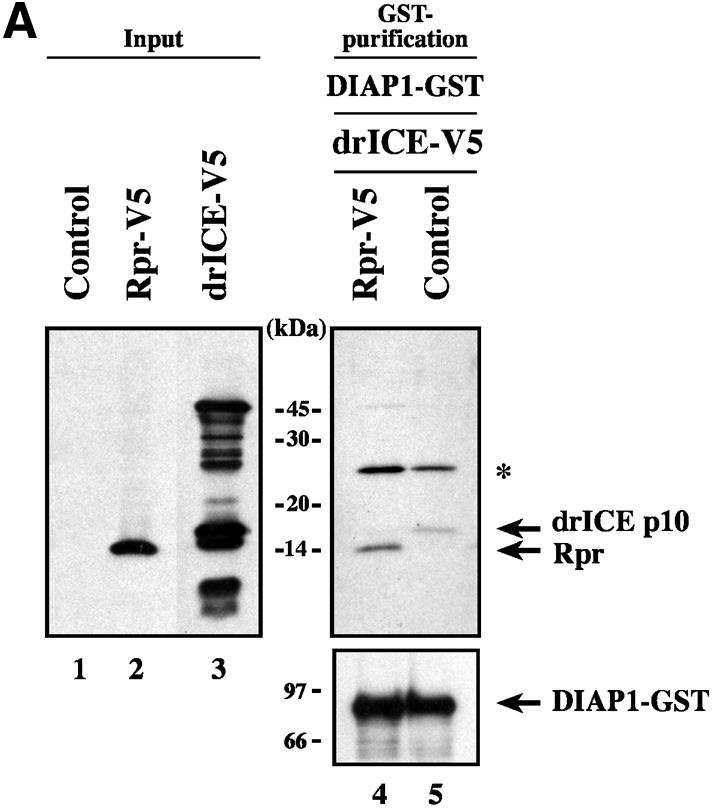
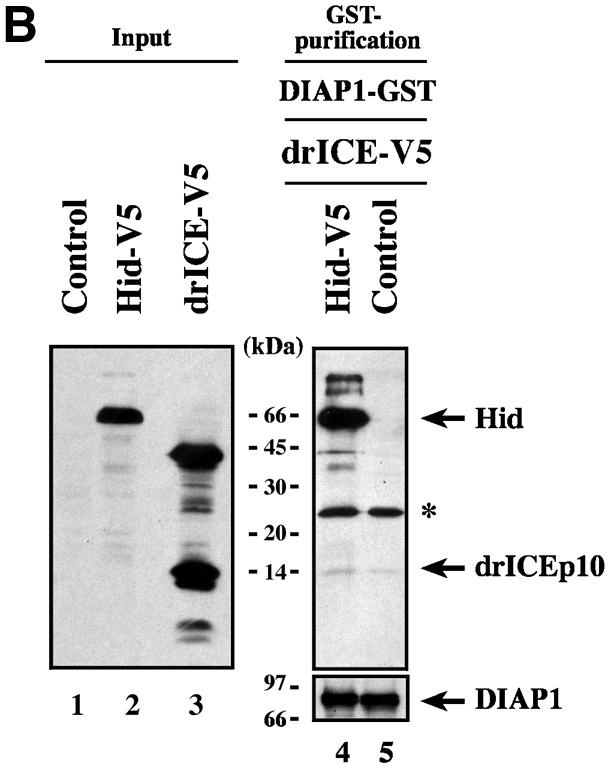

Fig. 5. Rpr but not Hid directly competes with drICE for DIAP1 binding. (A and B) Co-purification of drICE with DIAP1–GST in the presence or absence of Rpr (A) or Hid (B). DIAP–GST was affinity purified from S2 cellular extracts. Resin-bound DIAP1 was subsequently incubated with a mixture of cellular extracts containing drICE-V5 and Rpr-V5 (A) or drICE-V5 and Hid-V5 (B). Bound proteins were eluted and analysed by immunoblotting using anti-V5 antibody (top right panel, lanes 4 and 5). Left panel: total extracts of cells expressing the indicated constructs; western blot analysis using anti-V5 antibody. Bottom right panel: effective DIAP1 purification was confirmed by immunoblotting the eluate with anti-DIAP1 antibody. (C) Rpr’s ability to compete with drICE for DIAP1 binding is contingent on its association with the BIR1 domain. Co-purification of drICE with the indicated DIAP1–GST proteins in the presence of wild-type or mutant Rpr or Jafrac2. Top right panel: DIAP1–GST co-purification. Top left panel: total extracts expressing the indicated constructs; immunoblot analysis using anti-V5 antibody. Bottom right panel: purification of DIAP1–GST was confirmed by western blot analysis of the eluate using anti-DIAP1 RING antibody. The asterisk denotes a cross-reactive band.
Since Rpr binds to both BIR domains of DIAP1 we next examined whether Rpr needed to bind to the BIR1 domain in order to successfully compete with drICE for DIAP1 association. To this end, DIAP16–3s was used because the 6–3s mutation selectively impairs BIR1–Rpr binding while the interaction of Rpr to the BIR2 domain remains unaffected (Figure 5C, lane 9 and data not shown). Intriguingly, unlike wild-type DIAP1, DIAP16–3s maintained drICE binding in the presence of Rpr. In contrast, the IAP-antagonist Jafrac2, which predominantly targets the BIR2 domain, eliminated drICE–DIAP1 association. Together these results indicate that Rpr must interact with the first BIR domain to thwart drICE binding. Conversely, Jafrac2 appears to antagonize DIAP1–drICE association from a BIR2 position.
Rpr, Grim and Hid promote apoptosis exclusively in an IAP-binding-dependent manner
Recent evidence suggest that some IAP-antagonists such as Rpr and Grim promote apoptosis not only through a mechanism that relies on IAP binding but also by a mechanism that is entirely independent of IAP association (Claveria et al., 1998; Holley et al., 2002; Yoo et al., 2002). To examine the contribution of the IBM in Rpr, Grim and Hid for IAP binding and induction of apoptosis, we tested wild-type and mutant IAP-antagonists, lacking Ala1, for their ability to bind to DIAP1 and induce cell death. Wild-type Rpr, Grim, Hid and their respective Ala1 mutants were expressed in S2/p35 cells through the ubiquitin (Ub) fusion technique in which a Ub reporter is fused to the test protein. Ubiquitin-specific proteases cleave these fusion proteins at the C-terminus of the Ub moiety, yielding the test proteins (Varshavsky, 2000). Wild-type AVA-Rpr, AIA-Grim and AVP-Hid were efficiently co-purified by DIAP1 (Figure 6A). Conversely, the Ala1 mutants VA-Rpr, IA-Grim and VP-Hid displayed greatly diminished binding to DIAP1. In parallel experiments neither Rpr nor Grim or Hid bound to GST alone (data not shown). Since the Ala1 mutant Rpr and Grim proteins still bound to DIAP1, albeit weakly, we examined whether the residual IBM of Rpr and Grim was responsible for the weak interaction. As shown in Figure 6B, Rpr and Grim lacking the entire IBM completely failed to interact with DIAP1.
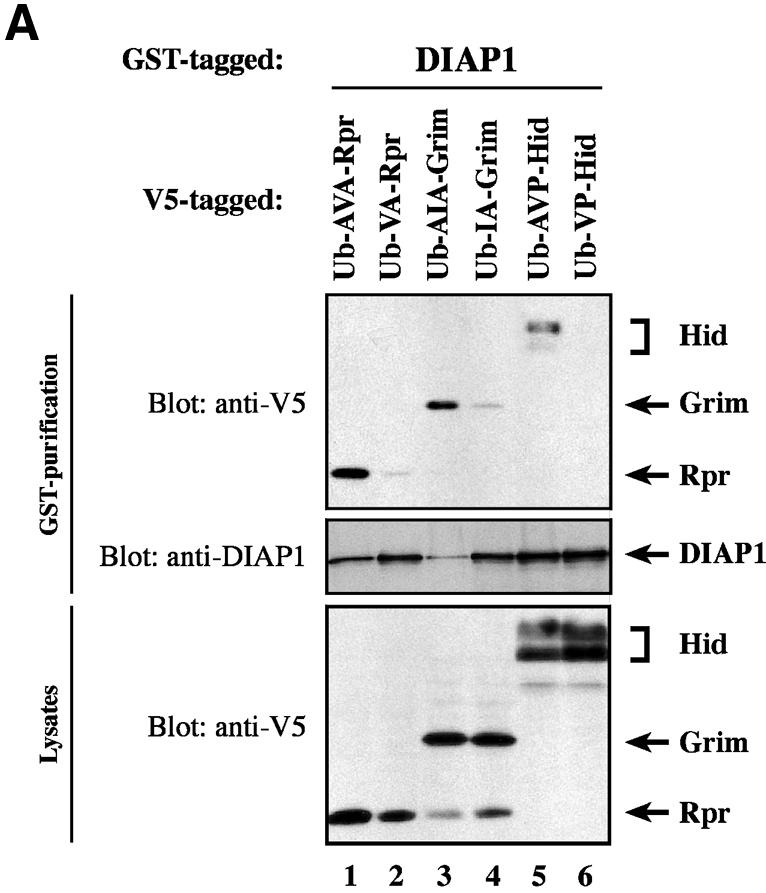

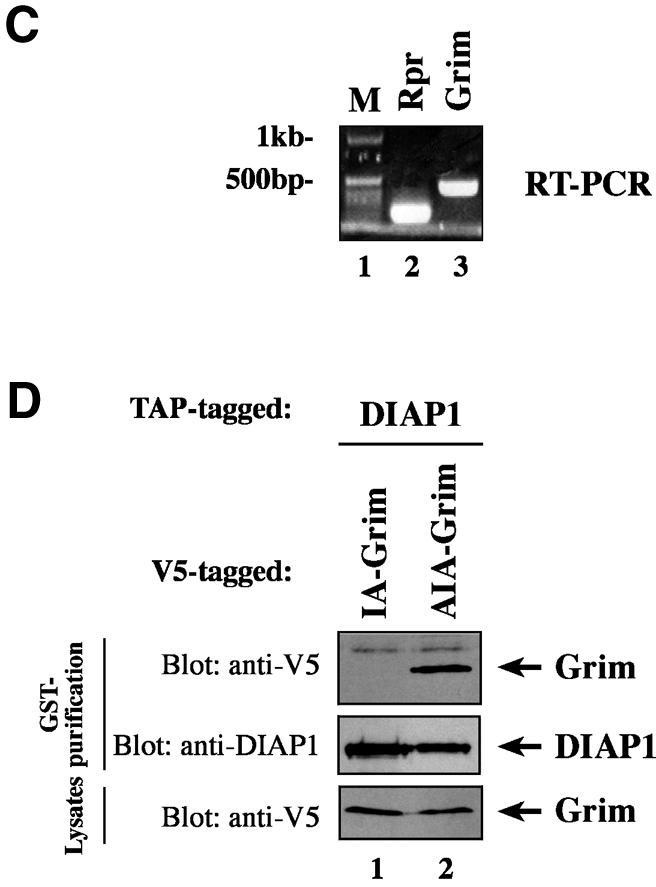
Fig. 6. Ala1 of the IBM of Rpr, Grim and Hid is indispensable for their binding to DIAP1. (A) Co-purification of Rpr-V5, Grim-V5 or Hid-V5 with DIAP1–GST from cellular extracts. Wild-type but not mutant Rpr, Grim or Hid lacking Ala1 efficiently interacted with DIAP1. Rpr, Grim and Hid were expressed using the ubiquitin fusion technique. Protein expression (bottom panel) and co-purification (top panel) was examined by immunoblot analysis with the indicated antibodies. Purification of DIAP1–GST was verified by western blot analysis of the eluate using anti-DIAP1 RING antibody (middle panel). (B) Rpr and Grim proteins that lack their entire IBM completely failed to bind to DIAP1. Note, while the difference in mobility between Grim and ΔIBM-Grim is readily detectable, Rpr and ΔIBM-Rpr appear to migrate at the same level due to the SDS–acrylamide gel used. (C) Detection of rpr and grim mRNAs by RT–PCR from total RNA of healthy S2 cells. (D) No residual binding of IA-Grim to DIAP1 is detectable in 293 cells suggesting that the residual binding shown in (A) may be due to the association of the Ala1-mutants with endogenous Rpr or Grim proteins.
Crystal structure analysis of Smac/DIABLO and HtrA2/Omi indicate that IAP-antagonists function as multimers (Chai et al., 2000; Li et al., 2002). Therefore, we examined the possibility that the residual binding of VA-Rpr or IA-Grim to DIAP1 was simply due to their association with endogenous, wild-type Rpr or Grim proteins. To this end, we first tested whether rpr or grim was expressed in S2 cells. Using RT–PCR on total RNA from S2 cells we found that rpr and grim mRNAs were indeed present in healthy S2 cells (Figure 6C). To test the importance of Ala1 for DIAP1 binding in the absence of endogenous Rpr and Grim proteins, we repeated the above binding study in mammalian 293 cells. Under these conditions, Ala1 was essential for the binding of Grim to DIAP1 as no residual interaction of IA-Grim to DIAP1 was detectable in 293 cells (Figure 6D). Together, these results suggest that the residual interaction between DIAP1 and VA-Rpr or IA-Grim in Drosophila S2 cells is likely to be due to the association of the Ala1 mutants with endogenous Rpr or Grim proteins.
We next examined the contribution of the IBM for the overall pro-apoptotic activity of Rpr, Grim and Hid. To assess the mechanism by which Rpr, Grim and Hid induce apoptosis, we performed cell death assays in S2 cells using wild-type and Ala1 mutants of Rpr, Grim and Hid. As expected, induced expression of Ub–AVA-Rpr, Ub–AIA-Grim or Ub–AVP-Hid caused rapid and efficient cell death (Figure 7A–O). In complete contrast, Ub–VA-Rpr, Ub–IA-Grim and Ub–VP-Hid that lacked the N-terminal Ala1 and failed to efficiently bind to DIAP1, also failed to notably induce apoptosis. Thus, apoptosis of S2 cells induced by expression of Rpr, Grim or Hid is contingent upon IBM-mediated DIAP1 binding.

Fig. 7. IAP-antagonists induce apoptosis exclusively in an IAP-binding-dependent manner. Induced expression of AVA-Rpr, AIA-Grim and AVP-Hid promotes apoptosis, while expression of VA-Rpr, IA-Grim or VP-Hid fail to trigger cell death. The indicated Ub fusion constructs were co-transfected with lacZ reporter plasmids, and 24 h post-transfection cells from each well were divided into two dishes to avoid variations in transfection efficiencies. Expression of the constructs was induced by copper sulfate and cells were examined for β-galactosidase activity. (A–D) and (I–L), untreated cells; (E–H) and (M–O), cells treated with copper sulfate (induced state).
Discussion
In Drosophila, the IAP-antagonists Rpr, Grim, Hid, Sickle and Jafrac2 promote apoptosis by binding to DIAP1 thereby obstructing caspase–IAP association. The interaction between IAP-antagonists and DIAP1 is mediated through an evolutionarily conserved IBM present in the extreme N-terminal portion of IAP-antagonists. IBMs require an unblocked, N-terminal Ala1 to anchor this motif to the BIR surface of IAPs (Wu et al., 2000). Although genetic evidence indicate that Rpr, Grim and Hid are non-equivalent and promote apoptosis in a coordinated mode of action (Robinow et al., 1997; Zhou et al., 1997; Wing et al., 1998), the published biochemical data suggest that IAP-antagonists indistinguishably bind to one and the same BIR2 domain of DIAP1 liberating BIR2-bound caspases. However, despite the high degree of sequence conservation among IBMs of IAP-antagonists, particularly among Rpr, Grim and Hid, we find that these IAP-antagonists display differential and selective binding to specific DIAP1 BIR domains (Figure 8) providing a biochemical explanation for the non-equivalent function of these IAP-antagonists in vivo. While Rpr and Grim show the same binding preference for both BIR1 and BIR2 domains, Hid, Sickle and Jafrac2 preferentially interact with the BIR2 domain. Furthermore, despite Rpr and Grim displaying the same BIR-binding preference, they interact with DIAP1 in distinct modes. This is evident because the 6–3s mutation in the DIAP1 BIR1 domain selectively impairs Rpr binding while the DIAP1–Grim association is unaffected. The ability of Rpr to bind to both BIR domains resides in the first 10 amino acids of the Rpr protein. A chimeric Rpr mutant, in which the first 10 residues of Rpr’s IBM were replaced with the ones of the IBM of Jafrac2, displayed the same DIAP1-binding preference as mature Jafrac2 and interacted preferentially with the BIR2 domain (unpublished data).

Fig. 8. Caspases, Rpr, Grim, Hid and Jafrac2 display differential and selective binding to specific DIAP1 BIR domains. The differential interaction seen both between DIAP1 and caspases and IAP-antagonists indicate that different IAP-antagonists compete with distinct caspases for DIAP1 binding. The relative affinity of individual IAP-antagonists to specific BIR domains is indicated by +.
Although caspases, Rpr, Grim, Hid, Sickle and Jafrac2 all bind to DIAP1, these proteins interact with individual BIR domains in very distinct manners. In agreement with the notion that different IAP-antagonists compete with distinct sets of caspases for DIAP1 binding we find that Rpr and Grim but not Hid effectively interfere with drICE–DIAP1 association. Thus, Rpr and Grim that efficiently bind to DIAP1’s BIR1 domain block the binding of DIAP1 to drICE. In complete contrast, Hid which associates preferentially with the BIR2 and binds only relatively weakly to the BIR1 domain is unable to prevent DIAP1–drICE association. Surprisingly, Jafrac2, which also preferentially binds to the BIR2 domain, is capable of inhibiting DIAP1 from drICE binding. Thus, Jafrac2 may function similarly to Smac/DIABLO, which displays an enhanced affinity to the XIAP BIR domains when simultaneously bound to two BIR domains (Huang et al., 2003). While the Smac–BIR2 interaction does not sterically prevent caspase-7 from binding to a fragment of XIAP containing the BIR2 domain, Smac efficiently blocks the binding of caspase-7 to XIAP fragments containing both the BIR2 and BIR3 domains (Huang et al., 2003). Rpr’s ability to interfere with DIAP1–drICE/DCP-1 interaction is contingent upon BIR1 binding. Thus, Rpr that only interacts with the BIR2 domain fails to displace BIR1-bound caspases (Figure 4F). Thus, the differential DIAP1–BIR interaction seen both between initiator and effector caspases and within IAP-antagonist family members is consistent with the notion that different IAP-antagonists displace distinct sets of caspases from DIAP1 complexes. The non-equivalent mode of DIAP1 binding and caspase liberation displayed by Rpr and Hid may hence provide a biochemical explanation for the observed genetic differences of these IAP-antagonists (Robinow et al., 1997; Zhou et al., 1997; Wing et al., 1998).
While the association of Rpr, Grim and Hid with the DIAP1 BIR2 domain is well documented, currently little biochemical data is available implicating the first BIR domain in Rpr, Grim or Hid binding. Several technical issues may have helped to detect the interaction between IAP-antagonists and the BIR1 domain. Firstly, in contrast to earlier studies (Goyal et al., 2000), we have used IAP-antagonists with unblocked IBMs in our binding assays. The presence of an unblocked Ala1 is crucial for efficient association of Rpr, Grim and Hid with DIAP1 as Rpr, Grim and Hid mutants that lack Ala1 failed to establish stable interactions with DIAP1 (Figure 6). Secondly, we have used the caspase inhibitor p35 to avoid Rpr-, Grim- and Hid-induced apoptosis and concomitant loss of cellular material. Importantly, p35 also prevents caspase-mediated N-terminal DIAP1 cleavage and subsequent N-end rule-mediated DIAP1 degradation (Ditzel et al., 2003). Thus, p35-mediated stabilization of DIAP1 protein levels may have helped to detect the interaction between the DIAP1 BIR1 region and IAP-antagonists.
Mature forms of Jafrac2, Rpr, Grim and Hid efficiently induce cell death in various Drosophila tissues and cultured cells. While these IAP-antagonists can induce apoptosis in a DIAP1-binding-dependent manner, it has been suggested that Rpr and Grim also promote cell death by a mechanism that does not rely on IAP association (Claveria et al., 1998; Holley et al., 2002; Yoo et al., 2002). In an in vitro assay it was found that Rpr and Grim repress global protein synthesis in an IAP-binding-independent manner. Using the ubiquitin fusion technique we have found that in vivo the pro-apoptotic activities of Rpr, Grim and Hid are contingent upon IAP binding. Rpr, Grim or Hid mutants that lack Ala1 and fail to efficiently bind to DIAP1, completely fail to promote apoptosis. The observation that IAP-antagonists induce apoptosis mainly, if not exclusively, in an IAP-binding dependent manner is further supported by in vivo studies expressing wild-type and mutant Rpr or Jafrac2 proteins in the developing Drosophila eye. Expression of wild-type but not Ala1-mutant Rpr or Jafrac2 proteins caused prominent mutant eye phenotypes due to ectopic activation of cell death (Tenev et al., 2002).
DIAP1 is essential for cell survival and is a potent inhibitor of the caspases DRONC, drICE and DCP-1. Several lines of evidence indicate that DIAP1 associates with these initiator and effector caspases in distinct manners. First, we find that two separate domains in DIAP1 are required for caspase binding. While the BIR1 region of DIAP1 is essential for its binding to the effector caspases drICE and DCP-1, the BIR2 region directly associates with the initiator caspase DRONC (Meier et al., 2000b). Secondly, the requirement for caspase cleavage and DIAP1 binding also differs greatly depending on the caspase involved. Proteolytic cleavage of drICE/DCP-1 is essential for DIAP1 binding, however, cleavage of DRONC is not required for DIAP1 association. The DRONC zymogen readily binds to the BIR2 region of DIAP1 whereby the CARD-containing pro-domain of DRONC mediates DIAP1 association (Meier et al., 2000b).
Importantly, the mechanism through which DIAP1 binds to Drosophila caspases appears to differ significantly from the one with which XIAP interacts with mammalian caspases. In mammals, XIAP binds exclusively to activated, processed forms of caspase-3, -7 or -9 and does not interact with their zymogenic forms. DIAP1, on the other hand, interacts with both activated drICE/DCP-1 and the zymogenic form of DRONC. Crystal structure analysis of active caspase-3 and -7 in complex with the BIR2 region of XIAP shows that the linker segment preceding the BIR2 domain of XIAP is critical for caspase inhibition (Shi, 2002). This linker segment completely occupies the catalytically active site of these caspases in a reverse orientation resulting in a blockade of substrate entry. Asp148 and Val146 in the N-terminal linker sequence of the XIAP BIR2 domain (166–231) are major anchoring points for the interaction of XIAP with caspase-3 and -7. Based on the specific hydrogen bonds formed between the side chain of Asp148 and activated caspases, it was predicted that Asp148 must be strictly conserved in IAPs that bind to activated effector caspases (Huang et al., 2001) as is the case for c-IAP1, c-IAP2, NAIP and the Drosophila IAPs DIAP2 and dBruce. Surprisingly these sites are not conserved in the flanking sequences of the corresponding BIR1 or BIR2 domains of DIAP1. Of note, the BIR1 domain of DIAP1 is most homologous to XIAP’s BIR2 domain. The absence of sequence conservation of Asp148 and Val146 between XIAP and DIAP1 is highly unexpected because IAPs lacking Asp148 and Val146 are predicted not to interact with activated caspases. The fact that DIAP1 interacts with activated caspases despite lacking the corresponding Asp148 residue, may suggest that DIAP1 and XIAP interact with activated caspases in distinct modes.
The view that DIAP1 and XIAP bind to activated caspases differently is further supported by our molecular and genetic studies on the loss-of-function mutant DIAP111–3e in which Asn117 is replaced with Lys. The Asn117Lys substitution mutation completely abrogates the binding of DIAP111–3e to drICE and DCP-1 indicating that residues in the linker segment downstream of the DIAP1 BIR1 domain are essential for DIAP1–caspase association. Importantly, Asn117Lys selectively impairs caspase–DIAP1 association and does not generally disrupt DIAP1 binding. We find that DIAP111–3e fails to bind to drICE/DCP-1, while its interaction with Rpr, Grim or Hid is unaffected. Hence, the 11–3e mutation is unlikely to disrupt the overall structure of the BIR fold. Instead, the selective disruption of caspase binding may suggest that Asn117 makes direct contact with activated caspases. Although the linker region between the BIR1 and BIR2 is necessary for drICE and DCP-1 binding, it is insufficient to mediate DIAP1 binding on its own. The linker segment in isolation fused to GST failed to interact with activated effector caspases suggesting that the linker region requires the adjacent BIR1 domain for caspase binding. This observation is in contrast to the linker segment of XIAP, which when fused to GST, readily binds to caspase-3 (Sun et al., 1999). While the caspase-binding residues of XIAP are located N-terminal to the BIR domain, the DIAP1 residue critical for effector caspase binding (Asn117) is positioned C-terminal to the DIAP1 BIR1 domain. DIAP1’s Asn117 appears to be conserved in the BIR1 region of the IAPs from Anopheles gambiae and Aedes triseriatus, however, it is absent from the corresponding BIR regions of DIAP2 or the mammalian caspase inhibitors XIAP, cIAP-1 and cIAP-2 (data not shown).
Our biochemical analyses of various diap1 alleles provide a molecular explanation of the genetic phenotypes associated with these gain- and loss-of-function diap1 mutations. Thus, mutations (i.e. 6–3s) that enhance DIAP1’s ability to associate with activated effector caspases and abrogate IAP-antagonist binding result in a gain-of-function phenotype. On the other hand, mutations (i.e. 11–3e) that impair caspase binding cause a loss-of-function phenotype leading to aberrant caspase-mediated cell death. Hence, regulation of DIAP1–caspase association and liberation represents a pivotal step in the regulation of the apoptotic caspase cascade. The identification of DIAP1 loss- and gain-of-function mutations that promote or thwart apoptosis suggests that analogous mutations in mammalian IAPs may similarly alter the regulation of apoptosis and thus may contribute to human pathologies such as cancer, autoimmune diseases or neuro-degenerative disorders (Thompson, 1995).
Materials and methods
Constructs
Constructs encoding wild-type and mutant DIAP1 were generated by PCR using Expand High Fidelity (Roche, Germany) and cloned into the CuSO4-inducible expression vector pMTIZ–GST. DIAP1 mutants were generated by PCR-mediated mutagenesis. cDNAs encoding Rpr, Grim, Hid and the catalytic mutant Dronc C318A were cloned into pMTIZ (Invitrogen, The Netherlands) in frame with the V5 tag. grim and hid cDNAs were cloned into pMTIZ in frame with the TAP tag. ΔIBM-Rpr lacks the first 14 amino acids, whereas ΔIBM-Grim lacks the first 15 amino acids. All tags were expressed at the C-terminus of the proteins. pcDNA3.1-based constructs encoding drICE-V5 and DCP-1-V5 were generated by PCR. Ub fusion constructs were generated by PCR and cloned into the pMTIZ in frame with the V5 tag. All constructs were verified by DNA sequencing.
Tissue culture, transfections and cell death assays
S2 and S2/p35 stable cell lines were cultured in Drosophila Schneider medium (Gibco-BRL, UK) in the presence of 10% fetal calf serum. hEK 293 cells were cultured as previously described (Wilson et al., 2002). Cells were transfected with calcium phosphate (Clontech, Palo Alto, CA) or Effectene (Qiagen, Germany) according to the manufacturers’ instructions. The cell death assay was carried out as described previously (Tenev et al., 2002). Briefly, S2 cells were co-transfected with pAC5.1/LacZ (Invitrogen) and pMT-based Ub fusion constructs in a ratio of 1:10. Twenty-four hours post-transfection, cells from each well were divided into two wells and expression of pMT-based constructs was induced in one of the two wells using CuSO4. Cells were then scored for β-gal activity. Cell viability was determined based on the morphology and the reduction of β-gal positive cells of induced versus non-induced constructs.
Protein extracts and immunoprecipitations
Preparation of S2 cell extracts and immunoprecipitations were conducted as previously described (Tenev et al., 2002). For the caspase-binding assay, expression constructs encoding either drICE-V5 or DCP-1-V5 were transfected into 293 cells. Sixteen hours post-transfection the cells were lysed and cell extracts prepared. Wild-type and mutant DIAP1–GST proteins were expressed in S2 cells and affinity purified from cellular extracts using glutathione–Sepharose resin. Resin-purifed DIAP1–GST proteins were subsequently incubated with extracts containing either drICE or DCP-1. Following 1 h incubation at 4°C samples were washed, eluted with reduced glutathione and examined by immunoblot analysis. For the competition experiment described in Figure 5 DIAP1–GST was generated and purified as described above. Resin-bound DIAP1–GST was incubated with a mixture of cell lysates containing drICE-V5 and either Rpr-V5, Grim-V5, Hid-V5 or Jafrac2-V5. drICE-V5-containing lysates were generated from 293 as described above, while lysates containing Rpr-V5, Grim-V5, Hid-V5 or Jafrac2-V5 were generated from S2/p35 cells. After 1 h incubation at 4°C, resin-bound DIAP–GST was washed and assayed for DIAP1-associated proteins.
Supplementary data
Supplementary data are available at The EMBO Journal Online.
Acknowledgments
Acknowledgements
We thank B.Seraphin for the TAP and A.Varshavsky for the ubiquitin fusion construct. We especially thank Mark Ditzel, John Silke, Shila Schneider, Rebecca Wilson and Emma Deas for helpful discussions and critical reading of the manuscript. We apologize to the scientists whose work we could not cite because of space limitations.
References
- Chai J., Du,C., Wu,J.W., Kyin,S., Wang,X. and Shi,Y. (2000) Structural and biochemical basis of apoptotic activation by Smac/DIABLO. Nature, 406, 855–862. [DOI] [PubMed] [Google Scholar]
- Chen P., Nordstrom,W., Gish,B. and Abrams,J.M. (1996) grim, a novel cell death gene in Drosophila. Genes Dev., 10, 1773–1782. [DOI] [PubMed] [Google Scholar]
- Christich A., Kauppila,S., Chen,P., Sogame,N., Ho,S.I. and Abrams,J.M. (2002) The damage-responsive Drosophila gene sickle encodes a novel IAP binding protein similar to but distinct from reaper, grim and hid. Curr. Biol., 12, 137–140. [DOI] [PubMed] [Google Scholar]
- Claveria C., Albar,J.P., Serrano,A., Buesa,J.M., Barbero,J.L., Martinez,A.C. and Torres,M. (1998) Drosophila grim induces apoptosis in mammalian cells. EMBO J., 17, 7199–7208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ditzel M., Wilson,R., Tenev,T., Zachariou,A., Paul,A., Deas,E. and Meier,P. (2003) Degradation of DIAP1 by the N-end rule pathway is essential for regulating apoptosis. Nat. Cell Biol., 5, 467–473. [DOI] [PubMed] [Google Scholar]
- Fraser A.G. and Evan,G.I. (1997) Identification of a Drosophila melanogaster ICE/CED-3-related protease, drICE. EMBO J., 16, 2805–2813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goyal L., McCall,K., Agapite,J., Hartwieg,E. and Steller,H. (2000) Induction of apoptosis by Drosophila reaper, hid and grim through inhibition of IAP function. EMBO J., 19, 589–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grether M.E., Abrams,J.M., Agapite,J., White,K. and Steller,H. (1995) The head involution defective gene of Drosophila melanogaster functions in programmed cell death. Genes Dev., 9, 1694–1708. [DOI] [PubMed] [Google Scholar]
- Hawkins C.J., Wang,S.L. and Hay,B.A. (1999) A cloning method to identify caspases and their regulators in yeast: identification of Drosophila IAP1 as an inhibitor of the Drosophila caspase DCP-1. Proc. Natl Acad. Sci. USA, 96, 2885–2890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holley C.L., Olson,M.R., Colon-Ramos,D.A. and Kornbluth,S. (2002) Reaper eliminates IAP protein through stimulated IAP degradation and generalized translational inhibition. Nat. Cell Biol., 4, 439–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y., Park,Y.C., Rich,R.L., Segal,D., Myszka,D.G. and Wu,H. (2001) Structural basis of caspase inhibition by XIAP: differential roles of the linker versus the BIR domain. Cell, 104, 781–790. [PubMed] [Google Scholar]
- Huang Y., Rich,R.L., Myszka,D.G. and Wu,H. (2003) Requirement of both the BIR2 and BIR3 domains for the relief of XIAP-mediated caspase inhibition by Smac. J. Biol. Chem., in press. [DOI] [PubMed] [Google Scholar]
- Kaiser W.J., Vucic,D. and Miller,L.K. (1998) The Drosophila inhibitor of apoptosis D-IAP1 suppresses cell death induced by the caspase drICE. FEBS Lett., 440, 243–248. [DOI] [PubMed] [Google Scholar]
- Li W., Srinivasula,S.M., Chai,J., Li,P., Wu,J.W., Zhang,Z., Alnemri,E.S. and Shi,Y. (2002) Structural insights into the pro-apoptotic function of mitochondrial serine protease HtrA2/Omi. Nat. Struct. Biol., 9, 436–441. [DOI] [PubMed] [Google Scholar]
- Lisi S., Mazzon,I. and White,K. (2000) Diverse domains of THREAD/DIAP1 are required to inhibit apoptosis induced by REAPER and HID in Drosophila. Genetics, 154, 669–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier P., Finch,A. and Evan,G. (2000a) Apoptosis in development. Nature, 407, 796–801. [DOI] [PubMed] [Google Scholar]
- Meier P., Silke,J., Leevers,S.J. and Evan,G.I. (2000b) The Drosophila caspase DRONC is regulated by DIAP1. EMBO J., 19, 598–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinow S., Draizen,T.A. and Truman,J.W. (1997) Genes that induce apoptosis: transcriptional regulation in identified, doomed neurons of the Drosophila CNS. Dev. Biol., 190, 206–213. [DOI] [PubMed] [Google Scholar]
- Rodriguez A., Chen,P., Oliver,H. and Abrams,J.M. (2002) Unrestrained caspase-dependent cell death caused by loss of Diap1 function requires the Drosophila Apaf-1 homolog, Dark. EMBO J., 21, 2189–2197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvesen G.S. and Duckett,C.S. (2002) Apoptosis: IAP proteins: blocking the road to death’s door. Nat. Rev. Mol. Cell Biol., 3, 401–410. [DOI] [PubMed] [Google Scholar]
- Shi Y. (2002) Mechanisms of caspase activation and inhibition during apoptosis. Mol. Cell, 9, 459–470. [DOI] [PubMed] [Google Scholar]
- Song Z., McCall,K. and Steller,H. (1997) DCP-1, a Drosophila cell death protease essential for development [published erratum appears in Science, 277, 167]. Science, 275, 536–540. [DOI] [PubMed] [Google Scholar]
- Srinivasula S.M. et al. (2002) sickle, a novel Drosophila death gene in the reaper/hid/grim region, encodes an IAP-inhibitory protein. Curr. Biol., 12, 125–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun C. et al. (1999) NMR structure and mutagenesis of the inhibitor-of-apoptosis protein XIAP. Nature, 401, 818–822. [DOI] [PubMed] [Google Scholar]
- Tenev T., Zachariou,A., Wilson,R., Paul,A. and Meier,P. (2002) Jafrac2 is an IAP antagonist that promotes cell death by liberating Dronc from DIAP1. EMBO J., 21, 5118–5129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson C.B. (1995) Apoptosis in the pathogenesis and treatment of disease. Science, 267, 1456–1462. [DOI] [PubMed] [Google Scholar]
- Varshavsky A. (2000) Ubiquitin fusion technique and its descendants. Methods Enzymol., 327, 578–593. [DOI] [PubMed] [Google Scholar]
- Vaux D.L. and Silke,J. (2003) Mammalian mitochondrial IAP binding proteins. Biochem. Biophys. Res. Commun., 304, 499–504. [DOI] [PubMed] [Google Scholar]
- Wang S.L., Hawkins,C.J., Yoo,S.J., Muller,H.A. and Hay,B.A. (1999) The Drosophila caspase inhibitor DIAP1 is essential for cell survival and is negatively regulated by HID. Cell, 98, 453–463. [DOI] [PubMed] [Google Scholar]
- White K., Grether,M.E., Abrams,J.M., Young,L., Farrell,K. and Steller,H. (1994) Genetic control of programmed cell death in Drosophila. Science, 264, 677–683. [DOI] [PubMed] [Google Scholar]
- Wilson R., Goyal,L., Ditzel,M., Zachariou,A., Baker,D.A., Agapite,J., Steller,H. and Meier,P. (2002) The DIAP1 RING finger mediates ubiquitination of Dronc and is indispensable for regulating apoptosis. Nat. Cell Biol., 4, 445–450. [DOI] [PubMed] [Google Scholar]
- Wing J.P., Zhou,L., Schwartz,L.M. and Nambu,J.R. (1998) Distinct cell killing properties of the Drosophila reaper, head involution defective and grim genes. Cell Death Differ., 5, 930–939. [DOI] [PubMed] [Google Scholar]
- Wu G., Chai,J., Suber,T.L., Wu,J.W., Du,C., Wang,X. and Shi,Y. (2000) Structural basis of IAP recognition by Smac/DIABLO. Nature, 408, 1008–1012. [DOI] [PubMed] [Google Scholar]
- Yoo S.J. et al. (2002) Apoptosis inducers Hid, Rpr and Grim negatively regulate levels of the caspase inhibitor DIAP1 by distinct mechanisms. Nat. Cell Biol., 4, 416–424. [DOI] [PubMed] [Google Scholar]
- Zhou L., Schnitzler,A., Agapite,J., Schwartz,L.M., Steller,H. and Nambu,J.R. (1997) Cooperative functions of the reaper and head involution defective genes in the programmed cell death of Drosophila central nervous system midline cells. Proc. Natl Acad. Sci. USA, 94, 5131–5136. [DOI] [PMC free article] [PubMed] [Google Scholar]