

Abstract
In mammalian cells, the SWI–SNF chromatin-remodeling complex is a regulator of cell proliferation, and overexpression of the catalytic subunit Brm interferes with cell cycle progression. Here, we show that treatment with histone deacetylase (HDAC) inhibitors reduces the inhibitory effect of Brm on the growth of mouse fibroblasts. This observation led to the identification of two carboxy-terminal acetylation sites in the Brm protein. Mutation of these sites into non-acetylatable sequences increased both the growth-inhibitory and the transcriptional activities of Brm. We also show that culture in the presence of HDAC inhibitors facilitates the isolation of clones overexpressing Brm. Removal of the HDAC inhibitors from the growth medium of these clones leads to downregulation of cyclin D1. This downregulation is absent in cell transformed by oncogenic ras.
Keywords: consensus acetylation site/mosaic expression/OV1063/PCAF/Retinoblastoma
Introduction
The SWI–SNF complex is a chromatin-remodeling complex that uses the energy of ATP hydrolysis to alter the accessibility of promoter regions embedded in nucleosomal structures. The complex has been associated with both transcriptional activation and repression, as it may favor recruitment to the DNA of both activators and repressors. In mammals, the SWI–SNF complex appears to play a central role in the control of cell proliferation. The SNF5/INI1 subunit is encoded by a tumor suppressor gene inactivated in malignant rhabdoid tumors, a very aggressive form of pediatric cancer (Versteege et al., 1998). Two other subunits, Brm and Brg1, are also involved in regulation of cell growth. Both these proteins are DNA-dependent ATPases, harboring the catalytic activity of the SWI–SNF complex. Each is associated with different subpopulations of SWI–SNF complexes diverging by one or more subunits and possibly having different cellular functions (for a review see Klochendler-Yeivin et al., 2002). Brm and Brg1 are frequently downregulated, silenced or mutated in malignant cells, including cells derived from several bladder, lung and prostate tumors (Wong et al., 2000; Decristofaro et al., 2001; Reisman et al., 2002, 2003). Brm is also systematically downregulated in rhabdoid tumors lacking SNF5/INI1 (Muchardt and Yaniv, 2001). Inactivation of Brg1 by homologous recombination in mouse is embryonic lethal at a pre-implantation stage but tumors are detected in animals heterozygous for the mutation (Bultman et al., 2000). Mice lacking a functional Brm gene are viable, as inactivation of this gene causes upregulation of the Brg1 gene, possibly compensating for the absence of Brm. However, the mice show increase body weight and a clear deregulation of cellular growth control (Reyes et al., 1998).
The SWI–SNF complex appears to affect the cell cycle control machinery at several levels. First, Brm and Brg1 interact with the Retinoblastoma family of proteins and can cooperate with p105Rb to induce G1-growth arrest in Brm/Brg1-deficient cells (Dunaief et al., 1994; Strober et al., 1996; Strobeck et al., 2000). Brm was also shown to cooperate with p105Rb for repression of E2F-mediated transcription, and several lines of evidence suggest that the SWI–SNF complex is a regulator of cdc 2, cyclin A and cyclin E (Trouche et al., 1997; Zhang et al., 2000). Inversely, the Brg1 growth-arresting activity is controlled by cyclin E by a mechanism apparently independent of Rb (Shanahan et al., 1999). Also, transcriptional regulation by the tumor suppressor p53 requires intact SWI–SNF complex (Lee et al., 2002a). More indirectly, Brg1 has been shown to function as a transcriptional repressor of the AP1 family member c-fos (Murphy et al., 1999).
In earlier studies, we reported that expression of exogenous Brm prevents proliferation of normal mouse fibroblasts. Upon transformation by oncogenic ras, fibroblasts become less sensitive to Brm overexpression, and exogenous Brm does not lead to growth arrest of the transformed cells but rather reverts the transformed phenotype (Muchardt et al., 1998; Bourachot et al., 1999). These observations pinpoint a possible role for the Brm protein in cellular transformation by ras.
Interestingly, in the ras-transformed cells, as well as in cells transformed by other oncogenes such as raf and Polyoma middle T, the levels of endogenous Brm are strongly reduced (Muchardt et al., 1998). Such gene-silencing events are frequently observed in tumor cells and, in many cases, are due to hypo-acetylation of the chromatin embedding the gene (Farias et al., 2002; Guo et al., 2002; Abdollahi et al., 2003). This prompted us to investigate the effect of HDAC inhibitors on Brm expression in ras-transformed cells. We found that expression of Brm is indeed stimulated in the presence of either trichostatin A (TSA) or sodium butyrate (NaBut). However, treatment with HDAC inhibitors was also correlated with a decreased growth-controlling activity of Brm. This observation led to the characterization of an unexpected negative regulation mechanism of Brm activity through acetylation of its carboxy-terminal region.
Results
Expression of Brm is upregulated in ras-transformed mouse fibroblasts in the presence of HDAC inhibitors
We previously showed that DT cells, derived from NIH 3T3 by stable expression of Ki-ras, display strongly decreased levels of Brm compared with the parental mouse fibroblasts. Expression of Brg1, the alternative catalytic subunit of the SWI–SNF complex, is not affected by transformation (Muchardt et al., 1998). To determine whether the reduced expression of Brm was linked to hypo-acetylation, we investigated the effect of HDAC inhibitors on Brm expression in DT cells. Elevated levels of Brm in the presence of HDAC inhibitors have previously been observed in human adrenal adenocarcinoma-derived SW13 cells (Yamamichi-Nishina, 2003). Both trichostatin A (TSA) and sodium butyrate (NaBut) caused increase in the levels of Brm protein, correlated with increased levels of mRNA (Figure 1A, DT, compare lane 1 with lanes 2 and 3, and Figure 1B, compare lanes 2, 4 and 6). HDAC inhibitors had no effect on RNA stability (data not shown) and the expression levels of two other SWI–SNF subunits, Brg1 and BAF155, were unchanged (Figure 1A, DT). We next tested the effect of HDAC inhibitors on a DT-derived clone expressing an HA-tagged version of Brm under the control of the viral MoMuLV LTR (DT21 cells). In this cell line, which has partially lost its transformed phenotype, the total level of Brm is ∼20-fold higher than in the parental DT cells; levels of Brg1 and BAF155 are not significantly different between the two cell lines (Figure 1C). Expression of the Brm transgene, detected via the epitope tag, was, like the endogenous Brm, strongly increased in the presence of TSA/NaBut (Figure 1A, DT21). We also examined two DT-derived cell lines expressing a Brm protein either mutated in its ATP-binding site or deleted in its carboxy-terminal region (DTΔATP and DTΔCter cells, respectively). These two mutants were previously shown to be defective in reversing the transformed phenotype of DT cells. Expression of these two mutant proteins was not increased in the presence of HDAC inhibitors (Figure 1A, DTΔCter and DTΔATP).
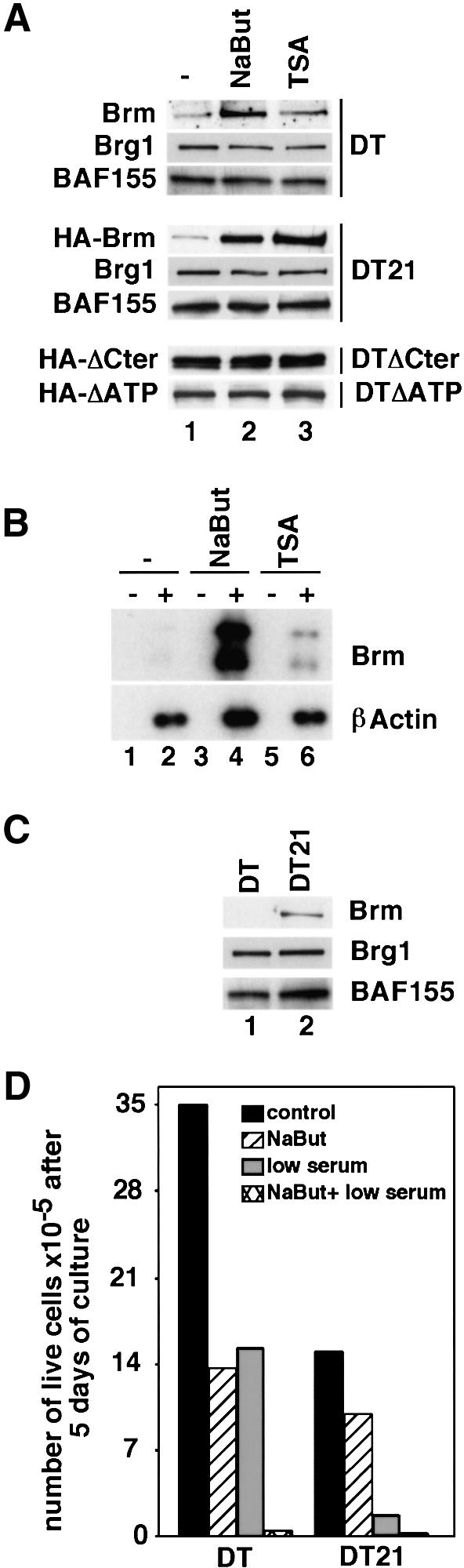
Fig. 1. HDAC inhibitors cause re-expression of Brm in transformed mouse fibroblasts. (A) DT, as well as cell lines derived from DT and expressing HA–Brm constructs either wild-type (DT21), mutated in the ATP-binding site (DTΔATP) or lacking amino acids 1342–1586 (DTΔCter), were cultured in the absence (lane 1) or presence of either NaBut (lane 2) or TSA (lane 3). DT cells: 60 µg of total extract was analyzed by western blot using anti-Brm, anti-Brg1 or anti-BAF155 antibodies. DT21, DTΔATP and DTΔCter cells: 20 µg of total extract was analyzed by western blot using anti-HA, anti-Brg1 or anti-BAF155 antibodies. It should be noted that, under these conditions, detection of Brm in DT cells required ∼10-fold longer exposure times compared with DT21 cells. (B) Total RNA extracted from DT cells grown in either the absence (lanes 1 and 2) or the presence of NaBut (lanes 3 and 4) or trichostatin A (lanes 5 and 6) was used to prepare cDNA in either the absence (– lanes) or the presence (+ lanes) of reverse transcriptase. cDNAs were then amplified in semiquantitative PCR using primers specific for either Brm or β-actin as indicated. PCR products were detected by Southern blot. (C) Extracts from either ras-transformed mouse fibroblasts (DT, lane 1) or ras-transformed mouse fibroblasts expressing an HA–Brm transgene (DT21, lane 2) were analyzed by western blot with anti-Brm, anti-Brg1 or anti-BAF155 antibodies as indicated. (D) Cells (105) were inoculated in medium containing 7% serum (control), 7% serum and 1 mM NaBut (NaBut), 0.25% serum and no NaBut (low serum) or 0.25% serum and 1 mM NaBut (NaBut + low serum). Live cells were counted after 5 days. Indicated measures are averages from two independent experiments.
Owing to the presence of oncogenic ras, growth of the DT cells is only moderately slowed when the cells are cultured in medium containing reduced concentrations of serum (∼2-fold reduction, see Figure 1D). In contrast, growth of the derived DT21 cells expressing exogenous wild-type Brm is severely impaired at low serum concentrations (10-fold reduction). Culture of the DT and DT21 cells in the presence of NaBut also reduced the growth rate. However, we noted that the effect was more pronounced in the DT cells that express very low levels of Brm than in the DT21 cells that express very high levels of the protein under these conditions. Surprisingly, this suggested that the Brm protein accumulating in the DT21 cells upon treatment with HDAC inhibitors was poorly effective in reducing cell growth.
Taken together, our observations show that HDAC inhibitors can at least partially relieve repression of Brm expression in ras-transformed cells. However, the mechanism leading to increased levels of Brm is not promoter specific and only affects expression of wild-type active Brm, but not mutants that fail to affect cell growth. Finally, the pool of Brm protein expressed in the presence of HDAC inhibitors seems to show reduced growth-inhibitory activity. These observations favor a model in which HDAC inhibitors do not directly control Brm expression, but rather increase the cellular tolerance for augmented levels of partially inactive Brm.
HDAC inhibitors reduce the ability of Brm to affect cell growth
To address more directly the effect of HDAC inhibitors on Brm activity, we used the non-transformed NIH 3T3 mouse fibroblasts. Unlike the ras-transformed DT cells, NIH 3T3 display a poor tolerance for overexpression of Brm, and generation of clones expressing exogenous Brm is very inefficient. Therefore these cells provide a good model for evaluation of the effect of HDAC inhibitors on the growth-suppressing properties of Brm. Cells were transfected with either a plasmid expressing HA–Brm or an empty plasmid. In both cases, the constructs included a selective marker (neomycin resistance). Selection was carried out for 2 weeks in either the absence or the presence of NaBut. Although less specific, NaBut was chosen over TSA as it appeared less deleterious for cell growth during long-term treatment. Results are summarized in Table I. In the absence of NaBut, ∼6-fold fewer neomycin-resistant colonies were detected in the presence of HA–Brm. Western blot analysis showed that out of 18 colonies tested, only one expressed the HA–Brm protein, and only at very low levels. In the presence of NaBut, approximately the same number of neomycin-resistant colonies was detected but in this case more than one third of these expressed the HA–Brm protein. These observations clearly indicate that in non-transformed cells, HDAC inhibitors increase the tolerance to overexpressed Brm.
Table I. Mutation of Brm acetylation sites is incompatible with colony formation.
| Transfected plasmids | Number of clones |
|
|---|---|---|
| Without NaBut | With NaBut | |
| Selection of neomycin-resistant clonesa | ||
| neor | 269 (100%) | 181 (67.3%) |
| neor + HA–Brm wt | 48 (16%) | 44 (24%) |
| neor + HA–Brm mutK | 9 (3%) | 14 (7%) |
| Brm expression in neomycin-resistant clonesb | ||
| neor + HA–Brm wt | 1/18 (5.5%) | 13/35 (37.1%) |
| neor + HA–Brm mutK | 0/9 (0%) | 0/14 (0%) |
aWT HA–Brm and HA–Brm mutK constructs were inserted in an expression vector harboring a neomycin resistance gene. NIH 3T3 cells were transfected with either vector without insert or the indicated Brm constructs. After 14 days of selection in the presence of neomycin and in the absence or in the presence of 1 mM NaBut, colonies were counted. The experiment was repeated three times. The table shows a representative experiment
bSeveral neomycin-resistant clones were isolated and assayed by western blot for expression of the transgene.
Brm is a substrate for acetylation
The decreased growth-inhibitory activity of Brm in the presence of HDAC inhibitors led us to investigate whether the Brm protein may itself be a substrate for factor acetyltransferases (FATs). Brm was immunopreciptated from NIH 3T3 extracts, resolved by SDS–PAGE and blotted on membrane. The immunoprecipitated protein could be detected with antibodies specific for acetylated lysines (anti-AcK antibodies), indicating that Brm is acetylated in vivo (Figure 2A). Transfection of several deletion mutants, followed by immunoprecipitation and detection with the anti-AcK antibodies, showed that acetylation sites were distributed throughout the protein (Figure 2B). As a control, we observed that the anti-AcK antibodies did not detect a large non-modified carboxy-terminal fragment of Brm expressed in Escherichia coli as a GST fusion (Figure 2B, lanes 5–7; see also a schematic of GST–Cter, Figure 2C). To determine whether Brm could associate with FATs in a cellular context, we prepared extracts from cells cotransfected with expression vectors for HA–Brm and the PCAF acetyltransferase. Using these extracts, we found that PCAF was co-immunoprecipitated with HA–Brm but not with HA-USF used as a control (Figure 2D).

Fig. 2. The Brm protein is acetylated in vivo. (A) Brm was immunoprecipitated from NIH 3T3 cell extracts and analyzed by western blot using either anti-Brm antibodies (lane 1) or antibodies specific for acetylated lysines (lane 2). (B) C33A cells were transfected with the indicated constructs. Brm proteins were immunoprecipitated using anti-HA antibodies and then analyzed by western blot using either anti-Brm or anti-acetyl lysine antibodies as indicated. In lane 3, the Brm protein is indicated by an asterisk. Specificity of the anti-acetyl lysine antibodies was tested on the indicated amounts of bacterially produced non-modified GST–Cter fusion protein (lanes 5 and 6). (C) Upper panel: schematic representation of the Brm constructs. The black box represents the HA epitope tag, and the shaded box represents GST. Lower panel: carboxy-terminal region of either wild-type Brm (Brm) or Brm mutated in consensus acetylation sequences (mutK). Consensus acetylation sequences are boxed. (D) C33A cells were transfected with a Flag-PCAF expression vector and expression vectors for either HA–Brm (lanes 1 and 3) or HA–USF (lanes 2 and 4). Immunoprecipitations were performed using anti-HA antibodies. The presence of PCAF in the extracts (lanes 1 and 2) and in the immunoprecipitate (lanes 3 and 4) was analyzed by western blot using anti-flag antibodies. (E) Carboxy-terminal GST fusion protein spanning amino acids 1189–1569 (GST–Cter) or purified histone H4 from calf thymus was incubated in the presence of the catalytic domain of PCAF produced in E.coli and radioactively labeled acetyl coenzyme A. Reactions were then resolved by SDS–PAGE and protein acetylation was detected by autoradiography. (F) GST–Cter either WT or mutated in the consensus acetylation sequences shown in panel C (GST–mutK) was incubated as above in the presence of the catalytic domain of PCAF. Reactions were then resolved by SDS–PAGE and protein acetylation was detected by autoradiography (top). The protein content of the reactions was visualized by Coomassie blue staining (bottom).
Examining the Brm amino acid sequence, we identified several G/SK motifs throughout the protein, corresponding to putative acetylation sites (Bannister et al., 2000). Two of these sites matched the extended recognition motif GKXXP (Kouzarides, 2000; Roth et al., 2001). These two sites were clustered in the carboxy-terminal region, immediately downstream of the Bromo-domain (see diagram in Figure 2C). This region has previously been shown to be essential for reversion of the ras-induced transformed phenotype in mouse fibroblasts by Brm (Bourachot et al., 1999). To determine whether the two sites could actually be acetylated, we used the GST–Cter fusion protein as a substrate for a GST–PCAF construct containing the catalytic domain of the acetyltransferase. The GST–Cter protein was found to be acetylated in vitro, the reaction being ∼50-fold less efficient than with histone H4 (Figure 2E). This level of acetylation is comparable with that previously reported for p53 (Liu et al., 2000). We then mutated the lysine residues present in the two putative acetylation sites into arginines, conserving the overall charge but rendering the sites non-acetylatable (GST–Brm mutK construct). This construct was not acetylated by GST–PCAF (Figure 2F, lane 2).
To determine whether the carboxy-terminal consensus acetylation sites were acetylated in vivo, we raised polyclonal rabbit antibodies directed against a peptide spanning amino acids 1531–1546 and acetylated on lysine residues 1532, 1534, 1535, 1541 and 1543. These antibodies (anti-BrmAcK) detected only the acetylated peptide and not a non-modified peptide with the same sequence (Figure 3A). We then transfected 293T cells with expression vectors for full-length WT or mutK Brm in either the absence or the presence of NaBut. Using the anti-BrmAcK antibodies, we detected WT Brm upon treatment with NaBut. Under the same conditions, the mutK construct was not detected (Figure 3B). These experiments show that treatment with the HDAC inhibitor leads to increased acetylation of the carboxy-terminal consensus acetylation sites. To address acetylation of endogenous Brm, we used the OV1063 cell line derived from an ovarian carcinoma. This cell line fails to express endogenous Brg1 but contains high levels of Brm. We compared it with the adrenal cortex carcinoma cell line SW13 that expresses neither Brg1 nor Brm (Figure 3C). Using the anti-BrmAcK, we observed a strong increase in the nuclear signal detected when OV1063 but not SW13 cells were treated with NaBut (compare Figure 3D and E with Figure 3F and G). Consistent with mitotic degradation of Brm in human cells (Muchardt et al., 1996), the anti-BrmAcK antibodies did not label mitotic OV1063 cells (Figure 3E and I, bottom cell). We concluded from these experiments that endogenous Brm is also acetylated at its carboxy-terminal sites.

Fig. 3. The carboxy-terminal consensus acetylation sites are acetylated in vivo. (A) The indicated amounts of either non-modified or acetylated peptide were spotted onto nitrocellulose membrane. The peptides corresponded to amino acids 1531–1546 of the Brm sequence. The acetylated peptide was acetylated on all its lysine residues. The peptides were detected using the polyclonal rabbit anti-BrmAcK antibodies raised against the acetylated peptide. (B) 293T cells were transfected with expression vector for either WT Brm (lanes 3 and 4) or Brm mutK mutant carrying non-acetylatable carboxy-terminal acetylation sites (lanes 1 and 2). Extracts were resolved by SDS–PAGE and proteins were detected first using the anti-BrmAcK antibodies (bottom panel) and then using anti-Brm antibodies (top panel). (C) Extracts from either SW13 or OV1063 cells were resolved by SDS–PAGE and western blots were performed with the indicated antibodies. (D–K) OV1063 or SW13 cells were cultured for 12 h in the presence of 10 mM NaBut, and then fixed and stained with anti-BrmAcK antibodies. DNA was stained with DAPI. Scale bar, 10 µm.
Mutation of the carboxy-terminal acetylation sites modifies the growth controlling properties of Brm
To address the function of Brm acetylation, we first immunoprecipitated the SWI–SNF complex from OV1063 cell extracts using anti-Brm antibodies in either the absence or the presence of NaBut. Treatment with the HDAC inhibitor did not dissociate Brm from the BAF155 and SNF5/INI1 subunits, suggesting that acetylation of Brm does not lead to dissociation of the SWI–SNF complex. We then concentrated our efforts on the carboxy-terminal acetylation sites. These sites are located in a protein region that has not been associated with any specific function. However, they are neighboring the acetyl-histone-binding bromo-domain, the DNA binding AT-hook and the LXCXE motif binding the pocket domain of the Retinoblastoma family of protein. Therefore we questioned whether the acetylated region was involved in proper folding of the carboxy-terminal domain of Brm. To address this issue, we expressed in E.coli either GST–Cter, a construct lacking amino acids from 1528 to the end of the protein, and a version of GST–Cter carrying six histidine residues at the carboxy-terminus. These proteins were tested under native conditions for DNA binding using gel mobility shift assays. All constructs bound the labeled DNA probe under these conditions (Figure 4B, lanes 2, 6 and 10). However, when challenged with polydAdT competitor DNA, the additional histidine tail was strongly destabilizing (compare lanes 4–5 and 12–13), whereas deletion of the acetylated domain was moderately stabilizing (compare lanes 5 and 9). These experiments suggested that the carboxy-terminal tail of Brm, substrate for acetylation, plays a role in the folding of a larger area containing several domains of interaction with structural or regulatory chromatin components. To illustrate this issue further, we tested p105Rb pocket binding-activity of Brm expressed in vitro as a 35S-labeled protein. Association of Brm with the p105Rb pocket was not visibly affected by deletion of a large carboxy-terminal region spanning amino acid 1341 to the end, indicating that this region is not strictly required for binding to p105Rb (Figure 4C). However, when Brm binding was challenged with Brm(1531–1546) peptide, we observed partial elution of the p105Rb-associated Brm. Interestingly, elution was not obtained when using either the acetylated version of the peptide or an unrelated peptide (Figure 4D). When coupled to beads, the non-modified Brm(1531–1546) peptide was also found to bind p105Rb weakly independently of the rest of the Brm protein. This interaction was strongly decreased when using acetylated Brm(1531–1546) peptide (Supplementary figure 1 available at The EMBO Journal Online). These observations suggest that the carboxy-terminal acetylation sites are involved in interaction of Brm with Rb, possibly with a stabilizing role. They also suggest that acetylation of Brm may weaken the p105Rb-Brm interaction.

Fig. 4. The carboxy-terminal region of Brm in its non-acetylated form interferes with the Brm–p105Rb interaction. (A) OV1063 cells were cultured in the absence or presence of 10 mM NaBut and then used for the preparation of total extracts. Immunoprecipitation was performed using polyclonal goat anti-Brm antibodies. Extracts (lanes 1 and 2) and immunoprecipitates (lanes 3 and 4) were resolved by SDS–PAGE and western blots were performed using the indicated antibodies. (B) GST–Cter (lanes 2–5), or a shorter version lacking amino acids from 1528 to the end (lanes 6–9) or a version carrying an additional 6xHIS carboxy-terminal tag (lanes 10–13) were used in gel mobility shift assays. Competition was performed using synthetic polydAdT at the indicated amounts. Migration of the probe in the absence of protein is shown in lane 1. (C) Either GST (lane 2) or GST–Rb(379–792) (lane 3) were bound to glutathione–agarose beads and assayed for binding of either full-length (top panel) or ΔCter (bottom panel) Brm proteins synthesized in vitro and labeled with [35S]methionine. Lane 1 shows a tenth of the Brm/ΔCter input. (D) As in (C), in vitro labeled WT Brm was incubated with beads associated with either GST (lanes 1–3) or GST–Rb(379–792) (lanes 4–6). Brm–Rb binding was then challenged with 2 µg/µl of HA peptide (lanes 1 and 4), Brm 1531–1546 peptide (lanes 2 and 5) or the acetylated version of this peptide (lanes 3 and 6). Eluate was collected (top panel) and the beads were then heated in loading buffer to obtain quantitative elution (middle panel). Bound proteins were detected by autoradiography or Coomassie brilliant blue staining. Lane 8 shows the Brm input.
To further address the role of the carboxy-terminal acetylation sites in vivo, an HA–Brm construct containing the lysine to arginine mutations (HA–Brm mutK) was transfected into 293T cells. These cells were then used to prepare extracts. Immunoprecipitation assays showed that the mutant protein was not impaired in its ability to associate with BAF155 and SNF5/INI1 (data not shown). We next transfected the HA–Brm mutK construct into NIH 3T3 cells, and colony formation was scored as described above (Table I). With this construct harboring non-acetylatable sites, 30-fold less neomycin-resistant colonies were obtained compared with the vector without insert in the absence of NaBut. This corresponded to a 5-fold decrease in the number of colonies compared with wild-type HA–Brm. Western blot analysis showed that none of these colonies expressed the mutant HA–Brm protein. These observations show that mutation of the carboxy-terminal acetylation sites into non-acetylatable sequences potentiates the negative effect of Brm on cell growth. Further, we found that in the presence of NaBut, expression of Brm in neomycin-resistant colonies was not improved when using the HA–Brm mutK construct. This contrasted with the increased number of neomycin-resistant colonies expressing Brm in the presence of NaBut observed with the wild-type HA–Brm construct. These results suggested that, unlike the wild-type Brm construct, NaBut does not reduce the growth-inhibitory effect of the HA–Brm mutK mutant.
Transient accumulation of Brm in NIH 3T3 cells causes downregulation of cyclin D1
As described above, selection of stably transfected clones in the presence of NaBut allowed the isolation of NIH 3T3-derived clones expressing HA–Brm. Such cells have not been previously available because of the negative effect of Brm on cell growth. Removal of the NaBut from the growth medium caused silencing of the transgene within ∼5 days (Figure 5B). This contrasted with the DT cells where exogenous Brm could be detected for several weeks under normal NaBut-free growth conditions. During the transition phase, expression of HA–Brm in NIH 3T3 showed a mosaic pattern, and uniform expression in all cells could be recovered within 4 days after re-addition of NaBut (Figure 5A). Removal of the NaBut from the growth medium also correlated with increased levels of dephosphorylated p105Rb and p130, frequently associated with a prolonged G1 phase of the cell cycle (Figure 5D). However, levels of cyclin A, cyclin E and HDAC1 did not appear to be affected (Figure 5C). A recent study demonstrated that re-expression of the SWI–SNF subunit SNF5/INI1 in tumor cells lacking this subunit induced growth arrest and repression of cyclin D1 transcription (Zhang et al., 2002). Therefore we examined the levels of cyclin D1 during the transition phase after removal of the NaBut from the medium. As shown in Figure 5C, a clear decrease in the level of cyclin D1 was observed in the cell line expressing HA–Brm, but not in the parental NIH 3T3 cells (for cyclin D1 in NIH 3T3, compare lanes 1–2 and 3–4). Interestingly, cyclin D1 was unaffected in the ras-transformed DT cells expressing HA–Brm (Figure 5C, bottom panel). Possibly, stimulation of cyclin D1 transcription by activated ras may overcome the repressive effect of Brm.

Fig. 5. Overexpression of Brm in non-transformed mouse fibroblasts. (A) NIH 3T3-derived clones expressing WT HA–Brm were cultured in either the absence or the presence of NaBut. Cells were fixed with paraformaldehyde and indirectly stained with anti-HA antibodies. DNA was stained with DAPI. Identical results were obtained with two different clones. The panel shows clone NIH5. (B) NIH5 cells were transferred from medium containing 1 mM NaBut to medium without NaBut. Extracts were prepared at the indicated time-points (lanes 1–4). NIH5 cells were also cultured for 6 days in the absence of NaBut (lane 5) and then the medium was supplemented with 1 mM NaBut for 4 days. Extracts were analyzed by western blot using either anti-HA tag or anti-BAF155 antibodies as indicated. (C) Parental NIH 3T3 or DT cells (lanes 3–4) or derived clones expressing WT HA–Brm (lanes 1–2) were cultured in the presence of 1 mM NaBut (lanes 1 and 3). Then the NaBut was removed from the medium and cells were harvested after 4 days (lanes 2 and 4). Extracts were analyzed by western blot using the indicated antibodies. (D) As in (C), cells expressing WT HA–Brm were cultured in the presence of NaBut (lane 1) and then NaBut was removed from the medium for 4 days (lane 2). Extracts were analyzed by western blot using the indicated antibodies. An extract from parental NIH 3T3 cells cultures in the absence of NaBut is shown in lane 3.
HDAC inhibitors negatively regulate transcriptional activation by Brm
Growth control by Brm has been associated with negative regulation of E2F-mediated transcription in cooperation with Rb family members. However, Brm also functions as a transcriptional activator and can cooperate with the glucocorticoid receptor (GR) to activate the MMTV LTR (Muchardt and Yaniv, 1993). To determine whether the activator function of Brm is also affected by HDAC inhibitors, we cotransfected human cervical carcinoma C33A cells with GR and Brm expression vectors in either the absence or the presence of NaBut. The C33A cells were chosen because they are efficiently transfected and fail to express endogenous Brm and Brg1 (Muchardt and Yaniv, 1993; Dunaief et al., 1994). As shown in Figure 6, GR activation itself was moderately increased by NaBut treatment. In the presence of Brm, GR activation was further stimulated 10–fold. This stimulation was entirely inhibited in the presence of NaBut and activation became similar to that observed with GR alone. A Brm construct mutated in the ATP-binding site stimulated GR-mediated activation less efficiently. This activity was also decreased in the presence of NaBut. These observations indicate that NaBut does not affect the Brm-independent activation by GR, but inhibits the ability of Brm to cooperate with the nuclear receptor. A similar result was obtained when NaBut was replaced by TSA (data not shown). Finally, we tested a Brm construct mutated in the two carboxy-terminal acetylation sites. This construct was more active than wild-type Brm and produced an 18–fold stimulation of GR-mediated activation. However, activation by this construct remained inhibited by NaBut, even though the resulting activity was higher than that of wild-type Brm under the same conditions. These observations suggest that the carboxy-terminal acetylation sites are involved in the control of the transcriptional activity of Brm. However, they also clearly indicate that other regions of Brm, or molecular partners of this protein, are affected by the HDAC inhibitor treatment.

Fig. 6. Increased transcriptional activity of a non-acetylatable Brm mutant. (A) An MMTV–luciferase reporter construct (1 µg) was cotransfected in C33A cells in either the absence or the presence of expression vectors for the glucocorticoid receptor (GR, 50 ng), WT Brm (3 µg), Brm mutated in the ATP binding site within the catalytic domain (ΔATP, 3 µg) or Brm mutated within two carboxy-terminal consensus acetylation sequences (mutK, 3 µg) as indicated. Transfections were performed in either the absence (black bars) or the presence (shaded bars) of 10 mM NaBut. Results were averaged from three independent experiments. Insert: expression levels WT Brm (lanes 1 and 2) and mutK (lanes 3 and 4) in transfected C33 cells in the absence (lanes 1 and 3) or the presence (lanes 2 and 4) of 10 mM NaBut were analyzed by western blot using anti-Brm antibodies. (B) Transfections were performed as in (A) using 1 µg of expression vectors for GCN5 or PCAF, or 3 µg of expression vector for p300 as indicated.
To address the effect of different FATs on Brm activity, we tested expression vectors for GCN5, PCAF and p300 for their ability to repress the transcriptional activity of Brm. All constructs had little or no repressing effect on activation by GR in the absence of Brm. p300 reduced Brm-mediated activation ∼4-fold. Best repression was obtained with the two related acetyl-transferases GCN5 and PCAF that lead to 7- to 8-fold repression. To determine whether the repressing effect of GCN5 and PCAF was associated with their FAT activity, we tested GCN5 and PCAF constructs mutated in their catalytic domain. Surprisingly, the GCN5 mutant construct retained most of its repressing activity, suggesting that it represses by sequestering Brm cofactors rather than by acetylating the protein. Mutation in the PCAF construct was more deleterious and this construct repressed transcription only 2-fold. These experiments suggested that only PCAF repressed Brm activity through a mechanism involving Brm acetylation.
Discussion
Negative regulation of Brm by acetylation
In the present study, we show that the activity of the Brm protein is regulated by acetylation. Although the protein is acetylated at multiple locations, two sites of acetylation, clustered in the carboxy-terminal region of the protein, appear to play a central role in this mechanism of regulation. Mutation of these sites into non-acetylatable versions creates a Brm protein with increased activity in terms of inhibition of colony formation and transcriptional activation. Therefore acetylation of Brm appears to be a negative regulatory event. Negative regulation by acetylation has previously been reported for other proteins like Sp3, HMGI/Y and Drosophila TCF (for review see Kouzarides, 2000; Sterner and Berger, 2000; Braun et al., 2001).
Mouse fibroblasts transformed by ras can be partially de-transformed in the presence of HDAC inhibitors (Sugita et al., 1992; Lim et al., 2002). This process of de-transformation can be correlated with an increased accumulation of Brm protein. However, as Brm appears to be partially inactive in its hyper-acetylated form, it is likely that increased levels of Brm protein play only a minor role in the process of de-transformation induced by TSA or NaBut. This is consistent with the apparent increased tolerance for exogenous Brm that we observe in both transformed and non-transformed cells in the presence of HDAC inhibitors.
Studies using chromatin immunoprecipitations have shown that the SWI–SNF complex is present on certain promoters together with histone acetyltransferases. Histone acetylation appears to facilitate recruitment of Brm and Brg1 that both contain bromo-domains believed to form acetyl-histone binding modules (Agalioti et al., 2002). We find that Brm can interact with the PCAF acetyltransferase and, in vitro, this enzyme can acetylate Brm. Furthermore, PCAF, but not GCN5, when cotransfected with Brm, reduces its transactivation potential through a mechanism that is dependent on the acetyl-transferase domain. In the same assay, p300 was less efficient than PCAF at repressing Brm transcriptional activity. However, we can not rule out that this FAT is also involved in acetylation of Brm. At the level of the promoters, the association of Brm with FATs and its subsequent inactivation by acetylation may limit the activity of the SWI–SNF complex in time and space, and may restrict chromatin remodeling once the pre-initiation complex has been recruited.
As mentioned earlier, the carboxy-terminal acetylation sites are located in the vicinity of several domains interacting with molecular partners of Brm. Our DNA binding assays further suggest that the carboxy-terminal region of Brm has a three-dimensional organization that can easily be perturbed by modifying the most carboxy-terminal residues. It is possible that one of the functions of Brm acetylation is the modification of this carboxy-terminal structure. Along that line, we observed that a small peptide encompassing the sites of acetylation could function as a competitor for Brm binding to the p105Rb pocket domain. Folding of the carboxy-terminal domain of Brm may bring the acetylatable region in proximity of the associated p105Rb and stabilize its interaction with Brm. Interestingly, the competition was only observed when the lysines of the peptide were not acetylated, suggesting that acetylation could destabilize Brm interaction with Rb family members. It is noteworthy here that p105Rb is involved in both the growth-controlling effect of Brm (Strober et al., 1996) and its activation of the MMTV promoter (Singh et al., 1995). Therefore it is possible that a similar mechanism mediated by Rb family members allows the mutK Brm construct not acetylatable on the carboxy-terminal sites to be more active than WT in both the colony formation assays and in the transcription assays.
We have not yet investigated the possible acetylation of other SWI–SNF subunits. However, we note that the carboxy-terminal acetylation sites identified in Brm are not present in the otherwise highly homologous Brg1 protein. Further studies will be required to determine whether acetylation sites are located elsewhere in the Brg1 sequence, for example within its TSA-sensitive repressive domain that was mapped to the helicase homology region (Lee et al., 2002b). Brg1 may also be indirectly regulated by Brm acetylation as the abundant inactive acetylated Brm present in cells treated with HDAC inhibitors may compete with Brg1 for binding to the rest of the complex and thereby function as a dominant negative.
Interestingly, several SWI–SNF subunits, including Brg1 and SNF5/INI1, are also found associated with HDACs within a corepressor complex known as Nco-R (Underhill et al., 2000). In addition, repression of the c-Fos promoter by Brg1 is favored in the presence of HDAC1 (Lee et al., 2002b). Similarly, we have observed that HDAC1 is co-immunoprecipitated with Brm (data not shown). It is likely that the main function of the HDAC activities associated with the SWI–SNF complex is the regulation of histone acetylation. However, the association of the HDACs and SWI–SNF subunits within the same complex suggests that the HDACs could also favor the catalytic activity of the complex by maintaining Brm in a de-acetylated state.
Brm regulates expression of cyclin D1
In earlier studies, we described ras-transformed cell lines expressing exogenous Brm. However, we never succeeded in expressing this transgene in the absence of activated ras. Our current studies on Brm acetylation led us to select Brm-expressing cells in the presence of NaBut. These growth conditions that apparently caused reversible inactivation of Brm activity allowed us to isolate numerous NIH 3T3-derived clones expressing the Brm protein. Unexpectedly, removal of the NaBut from the medium did not cause growth arrest of these clones, but rather a rapid downregulation of the Brm expression. Therefore it is likely that the cells have means of neutralizing the Brm pathway downstream of Brm, allowing them to overcome transitory deregulation of Brm expression. During the transition period, when the NaBut is removed from the medium but Brm still persists, we observed that levels of cyclin A and cyclin E were unchanged, consistent with the fact that the cells were still cycling. However, we detected decreased levels of cyclin D1, as well as its likely consequence: decreased phosphorylation of the pocket proteins p105Rb and p130. Interestingly, ras is a well-characterized activator of cyclin D1 activity (Stacey and Kazlauskas, 2002) and, in our cells, it appears to overrule the negative regulation of the cyclin D1 by the SWI–SNF complex. This mechanism may explain why Brm can be overexpressed in ras-transformed but not non-transformed cells.
Several lines of evidence now converge to suggest that the SWI–SNF complex regulates cyclin D1. For instance, in SW13 adrenal cortex carcinoma cells that lack endogenous Brm and Brg1, cyclin D1 can overcome the formation of flat growth-arrested cells induced by re-introduction of Brg1 (Shanahan et al., 1999). Similarly, one study on overexpression of SNF5/INI1, another SWI–SNF subunit, shows that the growth arrest induced by this protein in rhabdoid tumor-derived cell lines is overcome by overexpression of cyclin D1. The study also provides evidence that the SWI–SNF complex is recruited to the cyclin D1 promoter in vivo, leading to its repression (Zhang et al., 2002). These observations all point toward repression of the cyclin D1 promoter by the SWI–SNF complex. However, we cannot rule out that, upon activation of ras, SWI–SNF also serves as an activator of this promoter. In particular, we note that the Jun–Fos AP1 complex that is downstream of the ras signal transduction pathway and regulates the cyclin D1 promoter positively, is known to cooperate with the SWI–SNF complex in transcriptional activation (Ito et al., 2001). More indirectly, PIP2, an end-product of the ras/PI3K pathway, was shown to target the SWI–SNF complex to chromatin, suggesting that ras activates rather than represses SWI–SNF activity (Zhao et al., 1998). The possible dual role of the SWI–SNF complex on the cyclin D1 promoter depending on activation of ras is currently under investigation.
Materials and methods
Cell culture
C33A, SW13, OV1063, NIH 3T3, DT and derived cell lines were grown at 37°C under 7% CO2 in Dulbeco’s modified Eagle’s medium (DMEM) supplemented with 7% fetal calf serum and antibiotics (penicillin and streptomycin). When indicated, cells were cultured in the presence of 1 mM NaBut or 16.5 nM TSA. Drugs and medium were renewed regularly. Neomycin-resistant clones were selected in medium supplemented with G418 (Muchardt et al., 1998) in either the presence or the absence of NaBut. Brm expression in these clones was detected by western blot.
Transient transfection assays
C33 cells were transfected by calcium phosphate precipitation as previously described (Muchardt and Yaniv, 1993). Plasmid constructs were previously described (Bourachot et al., 1999). When the cells were transfected with GR expression vector, 10–6 M dexamethasone was added to the medium. When indicated, 10 mM NaBut was added to the medium 12 h after the transfection. Forty hours post-transfection, luciferase assays were performed using the Promega luciferase kit according to the manufacturer’s instructions.
Cell extracts, immunoprecipitation, western blot analysis and gel mobility shift assays
Whole-cell extracts were prepared in p300 buffer containing 20 mM NaH2PO4, 250 mM NaCl, 30 mM NaPPi, 0.1% NP-40, 5 mM EDTA, 5 mM dithiothreitol and protease inhibitors (Complete from Roche). After lysis, protein concentrations were determined with Bio-Rad Bradford reagent. For western blot analysis, unless otherwise indicated, 20 µg of protein were fractionated by SDS–PAGE and transferred to nitrocellulose membranes. Enhanced chemiluminescence (ECL) reagents were used for detection. For immunoprecipitations, after a pre-clear the extracts were incubated with the indicated antibodies bound to Protein A (with the 12CA5 anti-HA antibodies) or Protein G (with the goat anti-Brm N19 antibodies) in p300 buffer. After washing, immunoprecipitated proteins were eluted in SDS–PAGE sample buffer. The immunoprecipitates were then analyzed by western blot analysis with the indicated antibodies. Various antibodies are described in the Supplementary data. Gel mobility shift assays were performed as previously described (Bourachot et al., 1999).
Immunofluorescence
The cells, grown on polylysine-treated coverslips, were fixed with 3.7% paraformaldehyde for 10 min and then permeabilized with PBS Triton X-100 0.5% for 15 min at room temperature. After washing with PBS–Tween 20 0, 1%, the cells were incubated with the indicated primary antibodies in PBS–Tween 20 0,1% supplemented with 10% serum. Fluorescein-linked anti-rat or anti-rabbit secondary antibodies were used for detection. The DNA was labeled with DAPI at 150 ng/ml.
RNA preparation and RT–PCR
Total RNA from DT cells was purified on CsCl cushion as previously described (Sambrook et al., 1989). cDNA was synthesized from 2 µg of RNA as described (Muchardt et al., 1998). One tenth was amplified for 30 cycles in a two-step PCR using Taq polymerase and mouse Brm and β-actin-specific primers to a total of 100 µl. Ten microliters of the PCR were resolved on a 1.5% agarose gel and analyzed by Southern blotting using a BamH1–Sac1 restriction fragment from the Brm cDNA and an EcoRI–BamH1 restriction fragment from β-actin cDNA.
In vitro acetylation assays
Acetylation assays were performed as previously described (Gu and Roeder, 1997). A typical reaction was performed in 50 µl containing 1–2 µg of purified recombinant protein, 200 ng of PCAF HAT, 1.25 nmol 80 mCi/mmol [14C]acetyl-CoA (NEN) in a buffer containing 50 mM Tris–HCl pH 8, 1 mM EDTA and protease inhibitors. After incubation for 1 h at 30°C, the reactions were stopped by addition of SDS–PAGE sample buffer and analyzed by SDS–PAGE. The gels were stained with Coomassie brilliant blue and autoradiographed.
Supplementary data
Supplementary data are available at The EMBO Journal Online.
Acknowledgments
Acknowledgements
We thank P.Chambon, A.Caillaud, J.Bartek, T.Kouzarides, M.Noda, J.Lukas, W.Wang and B.Wasylyk for a gift of antibodies, plasmids and cell lines. We also thank J.-C. Dantonel for sharing information prior to publication and F.Mechta-Grigoriou, E. Batsché and B.Mateescu for valuable discussion. Finally, we thank S.Garbay for help and advice in microscope imaging, and J.Weitzman and J.Seeler for critical reading of the manuscript. The work was supported by grants from the Human Frontier Science Program, L’Association pour la Recherche sur le Cancer and La Ligue contre le Cancer, Ile-de-France.
References
- Abdollahi A., Pisarcik,D., Roberts,D., Weinstein,J., Cairns,P. and Hamilton,T.C. (2003) LOT1 (PLAGL1/ZAC1), the candidate tumor suppressor gene at chromosome 6q24–25, is epigenetically regulated in cancer. J. Biol. Chem., 278, 6041–6049. [DOI] [PubMed] [Google Scholar]
- Agalioti T., Chen,G. and Thanos,D. (2002) Deciphering the transcriptional histone acetylation code for a human gene. Cell, 111, 381–392. [DOI] [PubMed] [Google Scholar]
- Bannister A.J., Miska,E.A., Gorlich,D. and Kouzarides,T. (2000) Acetylation of importin-α nuclear import factors by CBP/p300. Curr. Biol., 10, 467–470. [DOI] [PubMed] [Google Scholar]
- Bourachot B., Yaniv,M. and Muchardt,C. (1999) The activity of mammalian brm/SNF2α is dependent on a high-mobility-group protein I/Y-like DNA binding domain. Mol. Cell. Biol., 19, 3931–3939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun H., Koop,R., Ertmer,A., Nacht,S. and Suske,G. (2001) Transcription factor Sp3 is regulated by acetylation. Nucleic Acids Res., 29, 4994–5000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bultman S., Gebuhr,T., Yee,D., La Mantia,C., Nicholson,J., Gilliam,A., Randazzo,F., Metzger,D., Chambon,P. et al. (2000) A Brg1 null mutation in the mouse reveals functional differences among mammalian SWI/SNF complexes. Mol. Cell, 6, 1287–1295. [DOI] [PubMed] [Google Scholar]
- Decristofaro M.F., Betz,B.L., Rorie,C.J., Reisman,D.N., Wang,W. and Weissman,B.E. (2001) Characterization of SWI/SNF protein expression in human breast cancer cell lines and other malignancies. J. Cell. Physiol., 186, 136–145. [DOI] [PubMed] [Google Scholar]
- Dunaief J.L., Strober,B.E., Guha,S., Khavari,P.A., Ålin,K., Luban,J., Begemann,M., Crabtree,G.R. and Goff,S.P. (1994) The retinoblastoma protein and BRG1 form a complex and cooperate to induce cell cycle arrest. Cell, 79, 119–130. [DOI] [PubMed] [Google Scholar]
- Farias E.F., Arapshian,A., Bleiweiss,I.J., Waxman,S., Zelent,A. and Mira,Y.L.R. (2002) Retinoic acid receptor α2 is a growth suppressor epigenetically silenced in MCF-7 human breast cancer cells. Cell Growth Differ., 13, 335–341. [PubMed] [Google Scholar]
- Gu W. and Roeder,R.G. (1997) Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell, 90, 595–606. [DOI] [PubMed] [Google Scholar]
- Guo W.H., Weng,L.Q., Ito,K., Chen,L.F., Nakanishi,H., Tatematsu,M. and Ito,Y. (2002) Inhibition of growth of mouse gastric cancer cells by Runx3, a novel tumor suppressor. Oncogene, 21, 8351–8355. [DOI] [PubMed] [Google Scholar]
- Ito T., Yamauchi,M., Nishina,M., Yamamichi,N., Mizutani,T., Ui,M., Murakami,M. and Iba,H. (2001) Identification of SWI.SNF complex subunit BAF60a as a determinant of the transactivation potential of Fos/Jun dimers. J. Biol. Chem., 276, 2852–2857. [DOI] [PubMed] [Google Scholar]
- Klochendler-Yeivin A., Muchardt,C. and Yaniv,M. (2002) SWI/SNF chromatin remodeling and cancer. Curr. Opin. Genet. Dev., 12, 73–79. [DOI] [PubMed] [Google Scholar]
- Kouzarides T. (2000) Acetylation: a regulatory modification to rival phosphorylation? EMBO J., 19, 1176–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee D., Kim,J.W., Seo,T., Hwang,S.G., Choi,E.J. and Choe,J. (2002a) SWI/SNF complex interacts with tumor suppressor p53 and is necessary for the activation of p53-mediated transcription. J. Biol. Chem., 277, 22330–22337. [DOI] [PubMed] [Google Scholar]
- Lee D., Lim,C., Seo,T., Kwon,H., Min,H. and Choe,J. (2002b) The viral oncogene human papillomavirus E7 deregulates transcriptional silencing by Brm-related gene 1 via molecular interactions. J. Biol. Chem., 277, 48842–48848. [DOI] [PubMed] [Google Scholar]
- Lim Y., Han,I., Kwon,H.J. and Oh,E.S. (2002) Trichostatin A-induced detransformation correlates with decreased focal adhesion kinase phosphorylation at tyrosine 861 in ras-transformed fibroblasts. J. Biol. Chem., 277, 12735–12740. [DOI] [PubMed] [Google Scholar]
- Liu Y., Colosimo,A.L., Yang,X.J. and Liao,D. (2000) Adenovirus E1B 55-kilodalton oncoprotein inhibits p53 acetylation by PCAF. Mol. Cell. Biol., 20, 5540–5553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muchardt C. and Yaniv,M. (1993) A human homologue of Saccharomyces cerevisiae SNF2/SWI2 and Drosophila brm genes potentiates transcriptional activation by the glucocorticoid receptor. EMBO J., 12, 4279–4290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muchardt C. and Yaniv,M. (2001) When the SWI/SNF complex remodels..the cell cycle. Oncogene, 20, 3067–3075. [DOI] [PubMed] [Google Scholar]
- Muchardt C., Reyes,J.C., Bourachot,B., Leguoy,E. and Yaniv,M. (1996) The hbrm and BRG-1 proteins, components of the human SNF/SWI complex, are phosphorylated and excluded from the condensed chromosomes during mitosis. EMBO J., 15, 3394–3402. [PMC free article] [PubMed] [Google Scholar]
- Muchardt C., Bourachot,B., Reyes,J.C. and Yaniv,M. (1998) ras transformation is associated with decreased expression of the brm/SNF2α ATPase from the mammalian SWI–SNF complex. EMBO J., 17, 223–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy D.J., Hardy,S. and Engel,D.A. (1999) Human SWI–SNF component BRG1 represses transcription of the c-fos gene. Mol. Cell. Biol., 19, 2724–2733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reisman D.N., Strobeck,M.W., Betz,B.L., Sciariotta,J., Funkhouser,W., Jr., Murchardt,C., Yaniv,M., Sherman,L.S., Knudsen,E.S. et al. (2002) Concomitant downregulation of BRM and BRG1 in human tumor cell lines: differential effects on RB-mediated growth arrest vs CD44 expression. Oncogene, 21, 1196–1207. [DOI] [PubMed] [Google Scholar]
- Reisman D.N., Sciarrotta,J., Wang,W., Funkhouser,W.K. and Weissman,B.E. (2003) Loss of BRG1/BRM in human lung cancer cell lines and primary lung cancers: correlation with poor prognosis. Cancer Res., 63, 560–566. [PubMed] [Google Scholar]
- Reyes J.C., Barra,J., Muchardt,C., Camus,A., Babinet,C. and Yaniv,M. (1998) Altered control of cellular proliferation in the absence of mammalian brahma (SNF2α). EMBO J., 17, 6979–6991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth S.Y., Denu,J.M. and Allis,C.D. (2001) Histone acetyltransferases. Annu. Rev. Biochem., 70, 81–120. [DOI] [PubMed] [Google Scholar]
- Sambrook J., Fritsch,E.F. and Maniatis,T. (1989) Molecular cloning: A laboratory manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- Shanahan F., Seghezzi,W., Parry,D., Mahony,D. and Lees,E. (1999) Cyclin E associates with BAF155 and BRG1, components of the mammalian SWI–SNF complex and alters the ability of BRG1 to induce growth arrest. Mol. Cell. Biol., 19, 1460–1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh P., Coe,J. and Hong,W. (1995) A role for retinoblastoma protein in potentiating transcriptional activation by the glucocorticoid receptor. Nature, 374, 562–565. [DOI] [PubMed] [Google Scholar]
- Stacey D. and Kazlauskas,A. (2002) Regulation of Ras signaling by the cell cycle. Curr. Opin. Genet. Dev., 12, 44–46. [DOI] [PubMed] [Google Scholar]
- Sterner D.E. and Berger,S.L. (2000) Acetylation of histones and transcription-related factors. Microbiol. Mol. Biol. Rev., 64, 435–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strobeck M.W., Knudsen,K.E., Fribourg,A.F., DeCristofaro,M.F., Weissman,B.E., Imbalzano,A.N. and Knudsen,E.S. (2000) BRG-1 is required for RB-mediated cell cycle arrest. Proc. Natl Acad. Sci. USA, 97, 7748–7753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strober B.E., Dunaief,J.L., Guha and Goff,S.P. (1996) Functional interactions between the hBRM/hBRG1 transcriptional activators and the pRB family of proteins. Mol. Cell. Biol., 16, 1576–1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugita K., Koizumi,K. and Yoshida,H. (1992) Morphological reversion of sis-transformed NIH 3T3 cells by trichostatin A. Cancer Res., 52, 168–172. [PubMed] [Google Scholar]
- Trouche D., Le Chalony,C., Muchardt,C., Yaniv,M. and Kouzarides,T. (1997) RB and hbrm cooperate to repress the activation functions of E2F1. Proc. Natl Acad. Sci. USA, 94, 11268–11273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Underhill C., Qutob,M.S., Yee,S.P. and Torchia,J. (2000) A novel nuclear receptor corepressor complex, N-CoR, contains components of the mammalian SWI/SNF complex and the corepressor KAP-1. J. Biol. Chem., 275, 40463–40470. [DOI] [PubMed] [Google Scholar]
- Versteege I., Sevenet,N., Lange,J., Rousseau-Merck,M.F., Ambros,P., Handgretinger,R., Aurias,A. and Delattre,O. (1998) Truncating mutations of hSNF5/INI1 in aggressive paediatric cancer. Nature, 394, 203–206. [DOI] [PubMed] [Google Scholar]
- Wong A.K., Shanahan,F., Chen,Y., Lian,L., Ha,P., Hendricks,K., Ghaffari,S., Iliev,D., Penn,B. et al. (2000) BRG1, a component of the SWI-SNF complex, is mutated in multiple human tumor cell lines. Cancer Res., 60, 6171–6177. [PubMed] [Google Scholar]
- Yamamichi-Nishina M., Ito,T., Mizutani,T., Yamamichi,N., Watanabe,H. and Iba,H. (2003) SWI3 cells can transition between two distinct subtypes by switching expression of Brg1 and Brm at the post-transcriptional level. J. Biol. Chem., 278, 7422–7430. [DOI] [PubMed] [Google Scholar]
- Zhang H.S., Gavin,M., Dahiya,A., Postigo,A.A., Ma,D., Luo,R.X., Harbour,J.W. and Dean,D.C. (2000) Exit from G1 and S phase of the cell cycle is regulated by repressor complexes containing HDAC-Rb-hSWI/SNF and Rb-hSWI/SNF. Cell, 101, 79–89. [DOI] [PubMed] [Google Scholar]
- Zhang Z.K., Davies,K.P., Allen,J., Zhu,L., Pestell,R.G., Zagzag,D. and Kalpana,G.V. (2002) Cell cycle arrest and repression of cyclin D1 transcription by INI1/hSNF5. Mol. Cell. Biol., 22, 5975–5988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao K., Wang,W., Rando,O.J., Xue,Y., Swiderek,K., Kuo,A. and Crabtree,G.R. (1998) Rapid and phosphoinositol-dependent binding of the SWI/SNF-like BAF complex to chromatin after T lymphocyte receptor signaling. Cell, 95, 625–636. [DOI] [PubMed] [Google Scholar]