

Abstract
The anaphase-promoting complex (APC) or cyclosome is a ubiquitin ligase that initiates anaphase and mitotic exit. APC activation is thought to depend on APC phosphorylation and Cdc20 binding. We have identified 43 phospho-sites on APC of which at least 34 are mitosis specific. Of these, 32 sites are clustered in parts of Apc1 and the tetratricopeptide repeat (TPR) subunits Cdc27, Cdc16, Cdc23 and Apc7. In vitro, at least 15 of the mitotic phospho-sites can be generated by cyclin-dependent kinase 1 (Cdk1), and 3 by Polo-like kinase 1 (Plk1). APC phosphorylation by Cdk1, but not by Plk1, is sufficient for increased Cdc20 binding and APC activation. Immunofluorescence microscopy using phospho-antibodies indicates that APC phosphorylation is initiated in prophase during nuclear uptake of cyclin B1. In prometaphase phospho-APC accumulates on centrosomes where cyclin B ubiquitination is initiated, appears throughout the cytosol and disappears during mitotic exit. Plk1 depletion neither prevents APC phosphorylation nor cyclin A destruction in vivo. These observations imply that APC activation is initiated by Cdk1 already in the nuclei of late prophase cells.
Keywords: anaphase-promoting complex/Cdk1/mitosis/phosphorylation/Plk1
Introduction
In mitosis, the anaphase-promoting complex (APC) is activated by the WD40 protein Cdc20, which is thought to recruit substrates to the APC (reviewed in Harper et al., 2002; Peters, 2002). APCCdc20 ubiquitinates numerous regulatory proteins, targets them for destruction by the 26S proteasome and thereby controls key events in mitosis. For example, APC-mediated degradation of securin is essential for sister chromatid separation, and destruction of mitotic A and B type cyclins is required to inactivate Cdk1 and to allow exit from mitosis.
Several mechanisms control APCCdc20 activation. First, the activity of vertebrate APCCdc20 is antagonized by Emi1, a protein that is itself inactivated by degradation in early mitosis (Guardavaccaro et al., 2003; Margottin-Goguet et al., 2003). Emi1 destruction appears to be required for activation of APCCdc20 in prometaphase, when some APCCdc20 substrates such as cyclin A are degraded. Second, although cyclin A degradation is initiated in prometaphase, the proteolysis of other APCCdc20 substrates such as cyclin B and securin is inhibited by the spindle checkpoint until all chromosomes have been attached to both poles of the mitotic spindle (reviewed in Musacchio and Hardwick, 2002). Third, APCCdc20 may be regulated by phosphorylation, because at least 4 of 11 known APC subunits in vertebrates (Apc1, Cdc27, Cdc16 and Cdc23) are hyper-phosphorylated in mitosis (Peters et al., 1996). The precise role of these modifications is controversial and poorly understood, but several observations suggest that they contribute to APC activation. Phosphatase treatment of mitotic Xenopus and clam APC resulted in loss of ubiquitination activity (Lahav-Baratz et al., 1995; Peters et al., 1996), whereas phosphorylation of fractions containing clam APC stimulated activity (Lahav-Baratz et al., 1995). We and others found that Cdc20 binds and activates preferentially mitotically phosphorylated APC (Kramer et al., 1998; Shteinberg et al., 1999; Kramer et al., 2000; Tang et al., 2001), but it has also been reported that Cdc20 can activate interphase APC in vitro (Fang et al., 1998; Kotani et al., 1999). APC phosphorylation is also observed in yeasts (Yamada et al., 1997; Rudner and Murray, 2000; May et al., 2002), although in budding yeast these modifications do not change dramatically between interphase and mitosis (Rudner and Murray, 2000). Simultaneous mutation of 12 putative Cdk1 sites in budding yeast Cdc27, Cdc16 and Cdc23 largely abolished APC phosphorylation, caused a reduction in Cdc20 binding and delayed progression through mitosis (Rudner and Murray, 2000).
In vertebrates, both Cdk1 and the Polo-like kinase Plk1/Plx1 have been implicated in APCCdc20 regulation. In Xenopus extracts, Cdk1 complexes containing the subunit p9/Cks are required for cyclin B degradation and for Apc1 and Cdc27 phosphorylation (Patra and Dunphy, 1998), whereas Plx1 is needed for exit from meiosis II (Descombes and Nigg, 1998) and for maintaining APC activity in mitotic extracts (Brassac et al., 2000). Kotani et al. (1998, 1999) reported that phosphorylation of human interphase APC by Plk1 in vitro is sufficient to stimulate ubiquitination activity even in the absence of Cdc20, provided that Plk1 had been activated by Cdk1. In contrast, Hershko and colleagues found that the activity of clam and human interphase APC can be increased in vitro by both Cdk1 and Plk1 in a Cdc20-dependent manner (Shteinberg et al., 1999; Golan et al., 2002).
The physiological roles of Cdk1 and Plk1 in APC activation are therefore rather unclear. This is in part due to the fact that it is not known which sites on APC are phosphorylated in vivo and which sites are generated by which kinase. It is likewise unknown where and when in mitotic cells these modifications occur. To begin to address these questions, we mapped in vivo phosphorylation sites on APC, compared them to sites that can be generated by Cdk1 or Plk1 in vitro, re-investigated the effects of Cdk1 and Plk1 on APC activation, extended them to analyse their effects on Cdc20 binding, and generated phospho-antibodies to analyse APC phosphorylation in vivo. We have also used RNA interference (RNAi) to address whether Plk1 is required for APC activation. Our data imply that Cdk1 begins to phosphorylate APC already in the nuclei of prophase cells and thereby initiates APCCdc20 assembly and activation. Our analysis revealed an unexpected and unprecedented complexity of mitotic phosphorylation sites and suggests that other kinases than Cdk1 and Plk1 also contribute to APC phosphorylation.
Results
Identification of mitotic phosphorylation sites on human APC
To identify cell cycle regulated phosphorylation sites, APC was immunopurified with Cdc27 antibodies from lysates of HeLa cells that had either been arrested in mitosis with nocodazole or in S-phase with hydroxyurea (Figure 1A). APC was digested with various proteases in solution and the resulting peptides were analysed by HPLC–ESI-MS/MS. Peptides derived from 9 of the 11 known APC subunits were detected in both S- and M-phase samples (Apc1, Apc2, Cdc27, Apc4, Apc5, Cdc16, Apc7 and Cdc23). No peptides from the small subunits Apc10, Apc11 and Cdc26 were recovered. Cdc20 peptides were only found in mitotic APC samples, confirming that Cdc20 associates with APC in a mitosis-specific manner. The detected peptides covered between 56 and 99% of the sequences of APC subunits (Figure 1C; Supplementary table 1 available at The EMBO Journal Online).

Fig. 1. Phosphorylation sites on human APC. (A) APC was immunoprecipitated from extracts of HeLa cells arrested with nocodazole (M) or hydroxyurea (S), subjected to SDS–PAGE and silver staining. Positions of subunits phosphorylated in mitosis are indicated (pApc1, pCdc27, pCdc16 and pCdc23). (B) Schematic phosphorylation site map of APC subunits derived by nano-HPLC–ESI-MS/MS. Black numbers indicate amino acid residues specifically phosphorylated in mitosis, numbers in parentheses indicate sites that could not be assigned with certainty, blue numbers depict sites found both on mitotic and S-phase APC, red refers to sites only phosphorylated in S-phase. Sites found on mitotic APC that were not covered in the S-phase analysis are shown in dark green. (Light green segments, TPRs; blue segments, WD40 repeats; orange segment, cullin domain.) (C) Summary of phosphorylation sites found on human APC.
In total, 51 phosphorylation sites were identified on APC subunits and two on Cdc20 (Table I). Of the 53 sites, 45 could unambiguously be assigned to a specific amino acid residue, whereas in eight peptides the modified residue could not be identified with certainty. Among the clearly identified sites, 22 were phosphorylated on serine, 18 on threonine and three on tyrosine residues. Many of these sites were also identified in an independent analysis of the human APC using a different mass spectrometric technique (J.Jebanathirajah and M.W.Kirschner, personal communication). In our analysis, 34 sites were exclusively found on mitotic APC, four only on S-phase APC and two were found on both. This calculation likely underestimates the number of mitosis-specific sites because mitotic sites where the corresponding peptide could not be recovered from S-phase APC were not counted (Table I). Of the 34 mitosis-specific sites, 30 were found in the subunits Apc1, Cdc27, Cdc16 and Cdc23, i.e. in subunits whose mitotic hyperphosphorylation causes electrophoretic mobility shifts (Figure 1A). Remarkably, most of these sites are clustered in confined regions of the proteins (Figure 1B). Cdc27, Cdc16, Cdc23 and Apc7 are characterized by the presence of multiple tetratricopeptide repeats (TPRs), but only two phosphorylation sites were found within TPRs, whereas 21 mitotic sites were found in clusters outside the repeats (Figure 1B). Most sites are conserved among vertebrate orthologues of APC subunits, but there is little similarity with the sequences from non-vertebrate species, although the known budding yeast phospho-sites in Cdc27 are located in similar regions as the human Cdc27 sites (data not shown; Rudner and Murray, 2000).
Table I. APC phosphorylation sites determined by mass spectrometry.

Phospho-residues are indicated by the amino acid position in upper case. Sequences used for the generation of phospho-antibodies are shaded in dark grey. In sequences shaded in light grey the identity of the phospho-residues could not be determined with certainty. In these sequences at least one but possibly multiple sites are phosphorylated. In these cases all residues are labelled, where the mass spectrometry data indicated possible phosphorylation. n.d., the corresponding peptide was not detected.
Characterization of selected phosphorylation sites in unperturbed and in spindle assembly checkpoint arrested mitotic cells
We raised antibodies against two mitosis-specific sites on Cdc27 (phospho-S427 and phospho-T447) and one each on Apc1 (phospho-S355), Cdc16 (phospho-S558) and Cdc23 (phospho-T556). In immunoblot experiments, all five antibodies recognized the mitotic form of the corresponding APC subunit but not the subunits of S-phase APC or of mitotic APC that had been treated with λ protein phosphatase (Figure 2A). All antibodies also recognized the corresponding APC subunits in immunoblot analyses of whole cell lysates from nocodazole-arrested HeLa cells but not in lysates from S-phase cells (Figure 2B). Also in immunoprecipitation experiments that were analysed by SDS–PAGE, silver staining and mass spectrometry, all five phospho-antibodies were able to bind APC specifically in whole cell lysates from nocodazole-arrested HeLa cells but not in lysates from S-phase cells (data not shown). These data confirm that the phosphorylation sites identified by mass spectrometry exist in nocodazole-arrested cells in vivo.

Fig. 2. Characterization of phospho-specific APC antibodies. (A) Cdc27 immunoprecipitates from extracts prepared from nocodazole (M) or hydroxyurea (S) arrested HeLa cells were analysed by immunoblotting using phospho-specific APC antibodies. As a control, non-phospho-specific Cdc27 antibodies were used for immunoblotting. Where indicated, APC was treated with λ-protein phosphatase (PPase). (B) Whole cell extracts prepared from mitotic or S-phase HeLa cells were analysed as in (A). Arrows indicate the corresponding APC subunit. (C) HeLa cells were synchronized by a double-thymidine arrest-and-release protocol and cell extracts were prepared at the indicated timepoints after release. Immunoblot analysis was performed using the indicated antibodies. As controls, samples from non-synchronized cells (log) and from nocodazole-arrested cells (noc) were analysed. pCdc23 signals could only be detected in Cdc27 immunoprecipitates (IP *). (D) HeLa cells were treated as in (C) but released into medium containing 100 ng/ml nocodazole. After 12.5 h, mitotic cells were collected by shake-off and transfered to fresh medium (noc release) and samples were taken as indicated.
To analyse whether the phospho-epitopes recognized by the antibodies are also generated during mitosis that has not been perturbed by nocodazole treatment, which activates the spindle checkpoint, HeLa cells were analysed that had been synchronized by a double thymidine arrest-release protocol (Figure 2C). Eight hours after release, entry into mitosis could be detected by an electrophoretic mobility shift of Cdc27. Cyclin A levels began to fall at 8.5 h, indicating that most cells had entered prometaphase and that APCCdc20 had become active. Cyclin B levels began to decrease at 10 h, implying that most cells had reached metaphase. As judged by FACS analysis, most cells reached G1 after 12–13 h (data not shown). All phospho-epitopes recognized by our antibodies appeared specifically during mitosis (Figure 2C). Apc1-phospho-S355 and Cdc27-phospho-T447 appeared early in mitosis (between 8 and 8.5 h), reached maximal levels in metaphase (between 10 and 10.5 h) and disappeared at the end of mitosis (between 12 and 13 h). A similar pattern was observed for the Cdc27 mobility shift. Cdc27-phospho-S427 and Cdc23-phospho-T556 appeared and disappeared with similar kinetics, although they could only be detected with lower intensity, in the case of Cdc23-phospho-T556 only when APC was enriched by immunoprecipitation. In contrast, Cdc16-phospho-S558 could only be detected once cells had reached metaphase (between 10 and 10.5 h), but it then persisted with equal intensity until 12 h, whereas the other phospho-epitopes began to decrease in abundance before that. These data indicate that the five sites analysed are generated during normal progression through mitosis, although possibly to different extents and with different kinetics.
To analyse if a mitotic arrest caused by activation of the spindle checkpoint influences the timing or extent of APC phosphorylation we synchronized HeLa cells by a double thymidine block and released them into media containing nocodazole. We also released cells 12.5 h after the thymidine release from the nocodazole arrest (Figure 2C). Under these conditions all five phospho-epitopes could readily be detected in mitosis, and the bulk of Cdc27 molecules was subject to a mobility shift. The increased abundance of the phospho-epitopes may be caused by the increased synchrony of cells in the nocodazole arrest, but it is also possible that the checkpoint arrest increases the extent of APC phosphorylation. In contrast, the checkpoint arrest had no major effect on the kinetics of APC phosphorylation: all phospho-epitopes began to appear at 10.5 h, 1 h after histone H3-Ser10 phosphorylation became detectable, but before cyclin A proteolysis began at 12.5 h, i.e. presumably in prophase. Four of the five phospho-epitopes disappeared 45–60 min after release from the nocodazole arrest, but as in the previous experiment Cdc16-phospho-S558 persisted longer, until 75 min after the nocodazole release.
APC phosphorylation by Cdk1 and Plk1 in vitro
Because Cdk1 and Plk1 have been implicated in mitotic APC phosphorylation, we analysed whether the mitotic APC phospho-sites could be generated in vitro using recombinant purified Cdk1-cyclin B1-p9 complexes (hereafter called Cdk1) or Plk1. Immunopurified S-phase APC was phosphorylated in vitro using Cdk1, Plk1, or both in the presence of either non-radioactive ATP or [γ-32P]ATP. The latter reactions were analysed by SDS–PAGE followed by silver staining and phosphorimaging (Figure 3A). Both kinases were able to phosphorylate Apc1, Cdc27, Apc5, Cdc16 and Cdc23. Apc1 was phosphorylated to a slightly higher extent by Cdk1 than by Plk1, whereas Cdc16 was phosphorylated more strongly by Plk1 than by Cdk1. Simultaneous APC phosphorylation by both kinases enhanced the incorporation of 32P into Apc1, Apc5 and Cdc23.
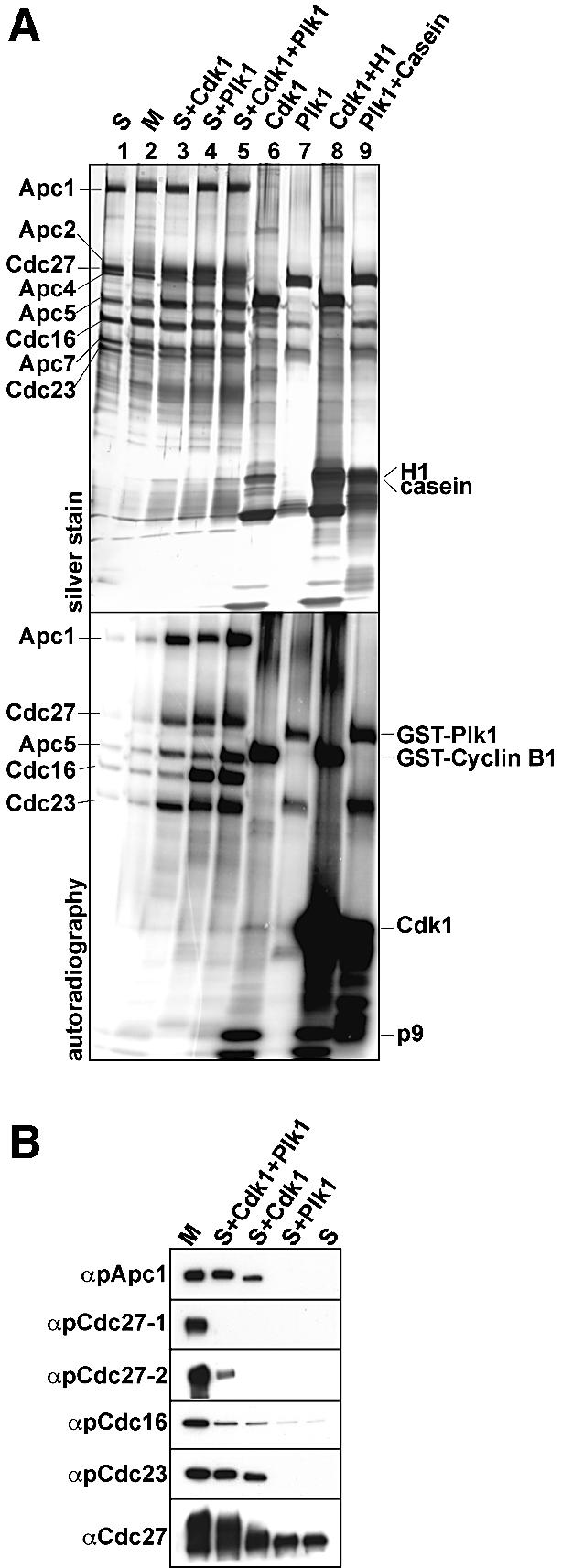
Fig. 3. In vitro phosphorylation of APC by Cdk1 and Plk1. (A) Immunopurified S-phase APC was in vitro phosphorylated using recombinant Cdk1 and Plk1 in the presence of [γ-32P]ATP, as indicated. After stringent washing, the samples were separated by SDS–PAGE, silver stained (upper panel) and analysed by phosphorimaging (lower panel). Lanes 1 and 2, APC incubated in kinase buffer without kinases; lanes 6 and 7, incubation of kinases alone in kinase buffer; lanes 8 and 9, kinases incubated with histone H1 and casein as substrates. (B) APC phosphorylated in vitro as in (A) was analysed by immunoblotting with pApc1-1, pCdc27-1, pCdc27-2, pCdc16 and pCdc23 antibodies. Cdc27 antibodies were used as a control.
After phosphorylation with Cdk1, 22 phosphorylated serine and threonine residues were detected by mass spectrometry that were not present on S-phase APC (Table I). Of these, 15 had also been found on mitotic APC in vivo. Of the 22 serine or threonine residues, 16 were followed by a proline residues, but only two sites conformed to the complete Cdk1 consensus site S/T-P-x-K/R. After phosphorylation with Plk1, eight newly phosphorylated serine and threonine residues were identified, of which only three had also been found on mitotic APC in vivo. Two of the in vivo sites could be generated by either Cdk1 or Plk1, whereas two different in vivo sites were only detected when both kinases were used simultaneously. These data show that many but by no means all in vivo APC phosphorylation sites can be phosphorylated by Cdk1, whereas Plk1 can only phosphorylate a few of these sites. Our data further imply that Cdk1 and in particular Plk1 can phosphorylate sites in vitro that are not phosphorylated in vivo.
To confirm the mass spectrometry results (Table I) we also analysed S-phase APC phosphorylated in vitro with Plk1 or Cdk1 by immunoblotting with the phospho-antibodies. Figure 3B shows that the antibodies pApc1, pCdc16 and pCdc23 recognized their corresponding subunits after treatment with Cdk1, and the pCdc27-2 antibodies recognized Cdc27 when APC had been phosphorylated by both kinases. However, the pCdc27-1 antibodies failed to recognize APC that had been phosphorylated in vitro, although this site was detected after phosphorylation with Cdk1 by mass spectrometry. It is possible that Cdc27-S427 is phosphorylated in vitro only with low efficiency and that this phospho-residue can therefore only be detected by mass spectrometry.
APC phosphorylation by Cdk1 increases Cdc20 binding and is sufficient for APC activation in vitro
To address the role of Cdk1 and Plk1 in APC activation we analysed the ability of interphase APC to bind Cdc20 and to ubiquitinate substrates before and after kinase treatment. We used APC isolated from Xenopus interphase extracts for these experiments because mitotic Xenopus APC can be activated by Cdc20 in vitro (Kramer et al., 2000), whereas in our hands mitotic human APC could not be efficiently activated (data not shown). After in vitro phosphorylation of interphase APC with Plk1, Cdk1 or both, the kinases were washed away, and APC was incubated with purified recombinant Cdc20. After additional washing, APCCdc20 was eluted from the antibody beads with antigenic peptide to eliminate Cdc20 molecules from the analysis that had bound to the beads non-specifically. Cdc20 bound to APC was analysed by immunoblotting (Figure 4A), and APC activity was measured in ubiquitination assays using a purified radiolabelled N-terminal fragment of cyclin B (Figure 4B and C). APC phosphorylation by Cdk1 increased the ability of APC to bind Cdc20 and correlated with a 4-fold increase in APC activity. APC phosphorylation by Plk1 did not increase Cdc20 binding and had no effect on APC activity, but combining Cdk1 and Plk1 resulted in binding of more Cdc20 than was seen after treatment with Cdk1 alone, and increased APC activity 6-fold. The same result was obtained independent of the order in which the two kinases were added, indicating that both kinases act on the APC and not on each other (data not shown). Without Cdc20, none of the kinase treatments stimulated APC activity. Under the in vitro conditions used, APC phosphorylation by Cdk1 alone is therefore sufficient for Cdc20-dependent APC activation.
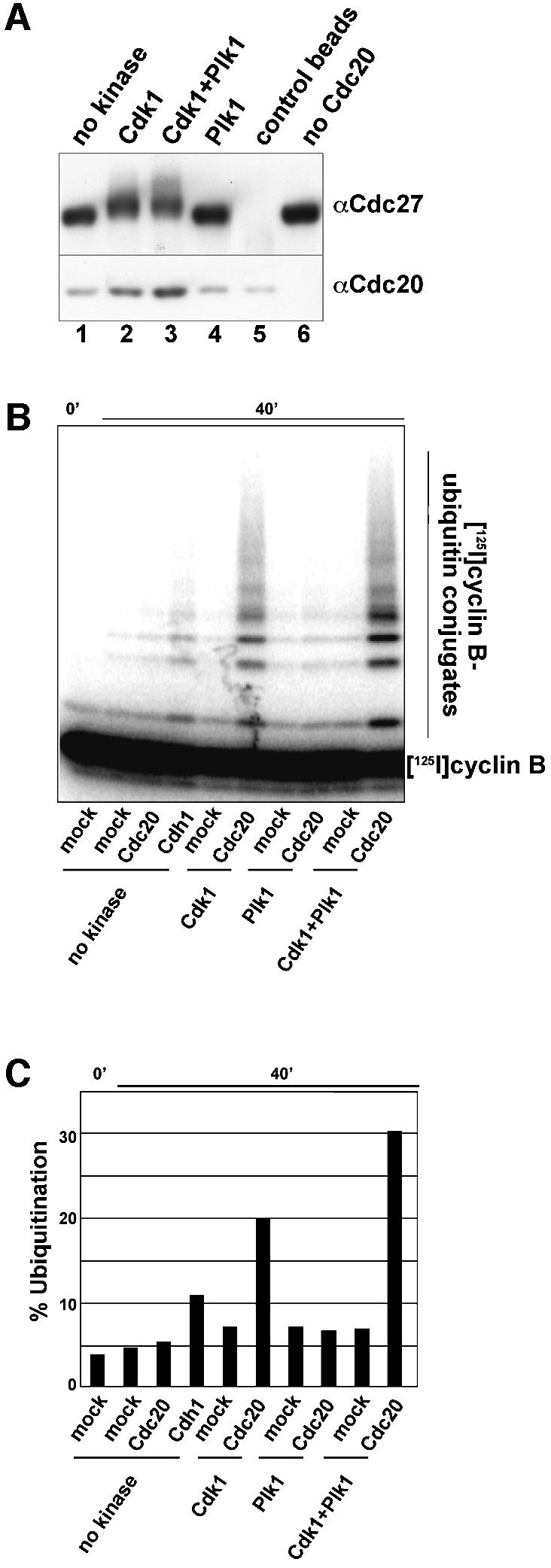
Fig. 4. Cdk1 phosphorylation increases APC activity and Cdc20 binding. (A) Xenopus interphase APC was phosphorylated in vitro by Cdk1 and/or Plk1 and incubated with recombinant Cdc20. Binding of Cdc20 to the APC was monitored by immunoblotting. As a control, beads coupled to non-specific antibodies were treated in the same way (lane 5) and one sample was incubated with buffer instead of Cdc20 (lane 6). (B) Interphase APC purified and treated as in (A) was analysed for the ability to ubiquitinate 125I-cyclin B. (C) The amount of 125I-cyclin B conjugated to ubiquitin is shown as the percentage of the total amount of 125I-cyclin B per reaction.
The onset of APC phosphorylation correlates with nuclear uptake of cyclin B1 in prophase
To analyse when and where in mitotic cells APC is phosphorylated, we stained HeLa cells with the antibodies pApc1 and pCdc27-1, both of which are specific in immunoblot experiments (Figure 2B). Both antibodies stained exclusively mitotic cells (Figure 5A and data not shown), whereas antibodies that also recognize non-phosphorylated APC stained both interphase and mitotic cells (Figure 5B). In prophase cells, the phospho-APC antibodies stained nuclei with the exception of nucleoli where signal intensities were reduced (Figure 5A). After nuclear envelope breakdown, fluorescent signals were seen throughout the cytoplasm, but chromosomes were largely spared. Signal intensities increased up to metaphase, began to decline in anaphase and disappeared when cells completed cytokinesis and re-entered G1. Co-staining with antibodies to the lamina associated protein LAP2β confirmed that pApc1 and pCdc27-1 epitopes appeared before nuclear envelope breakdown (NEBD) at the prophase–prometaphase transition (Figure 5C).
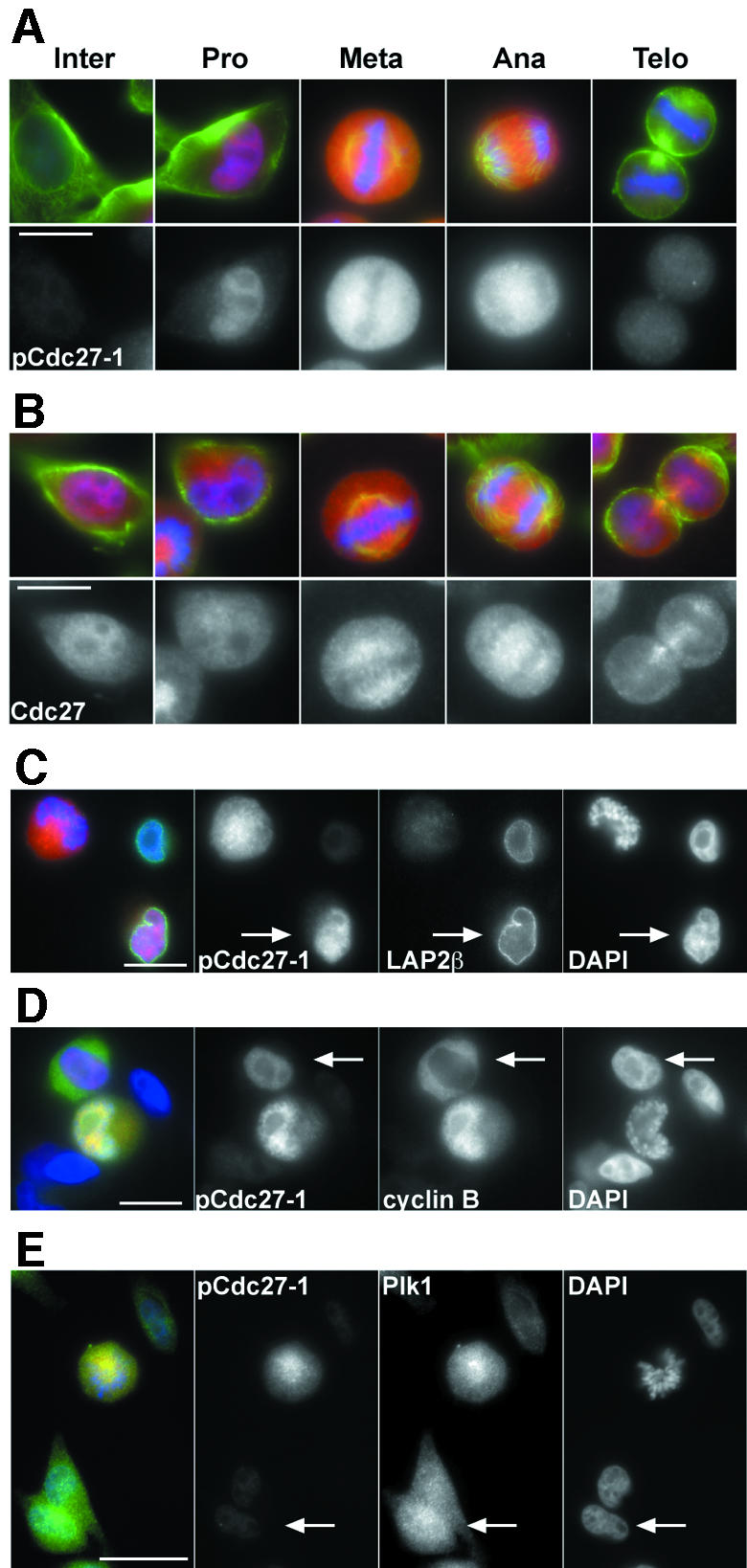
Fig. 5. APC phospho-epitopes appear in prophase when cyclin B1 translocates into the nucleus. (A) HeLa cells were stained with pCdc27-1 antibodies (red), tubulin (green) and DAPI (blue). (B) Staining was performed as in (A) but using non-phospho-specific Cdc27 antibodies (red). (C) HeLa cells treated as in (A) were stained with pCdc27, LAP2β (green) and DAPI (blue). The arrow indicates a nucleus that stains with pCdc27-1 antibodies and is still surrounded by an intact lamina. (D and E) Co-staining of pCdc27-1 with cyclin B1 (D) and Plk1 (E). The arrow in (D) indicates a cell where nuclear uptake of cyclin B1 has just begun and where pCdc27-1 staining can be seen in the nucleus. The arrow in (E) indicates a cell in which Plk1 but no pCdc27-1 staining can be detected in the nucleus. Bar, 10 µm (A and B), 20 µm (C–E).
Cyclin B1 and Plk1 are both cytoplasmic in interphase and enter the nucleus late in prophase (Pines and Hunter, 1991; Jackman et al., 2003). Plk1 enters the nucleus first, about 10 min before NEBD, followed a few minutes later by cyclin B1. Colocalization experiments revealed that cells in which cyclin B1 was exclusively cytoplasmic did not yield detectable staining with phospho-APC antibodies (data not shown). In contrast, all cyclin B1 positive nuclei could also be stained with either pApc1 or pCdc27-1 antibodies (Figure 5D). In some cases, weak nuclear phospho-APC staining was seen in cells in which the bulk of cyclin B1 was still cytoplasmic. However, in these cells weak cyclin B1 signals could already be detected in the nuclei by confocal imaging, implying that nuclear uptake of cyclin B1 had just begun (Figure 5D and data not shown). All phospho-APC positive prophase nuclei could also be stained with Plk1 antibodies, but not all nuclei that contained Plk1 were stained with phospho-APC antibodies (Figure 5E). These observations imply that APC phosphorylation, at least on Apc1-S355 and on Cdc27-S427, is initiated after nuclear uptake of Plk1 in prophase and coincides with entry of cyclin B1 into the nucleus. Similar results were obtained with the other three APC phospho-antibodies (data not shown), consistent with the possibility that other sites on APC are phosphorylated at the same time.
In mitotic cells, APC is enriched on centrosomes and spindle microtubules (Tugendreich et al., 1995). To analyse whether the phosphorylated form of the APC localizes in the same way, we analysed cells from which the bulk of soluble proteins was removed by extraction before cells were fixed. All phospho-APC antibodies revealed centrosomal staining from prophase to telophase, but no spindle staining could be observed, whereas antibodies that recognize both phosphorylated and unphosphorylated Cdc27 showed staining of both centrosomes and the spindle (Figure 6 and data not shown). Phosphorylated APC therefore appears to be enriched at centrosomes but not on spindles.
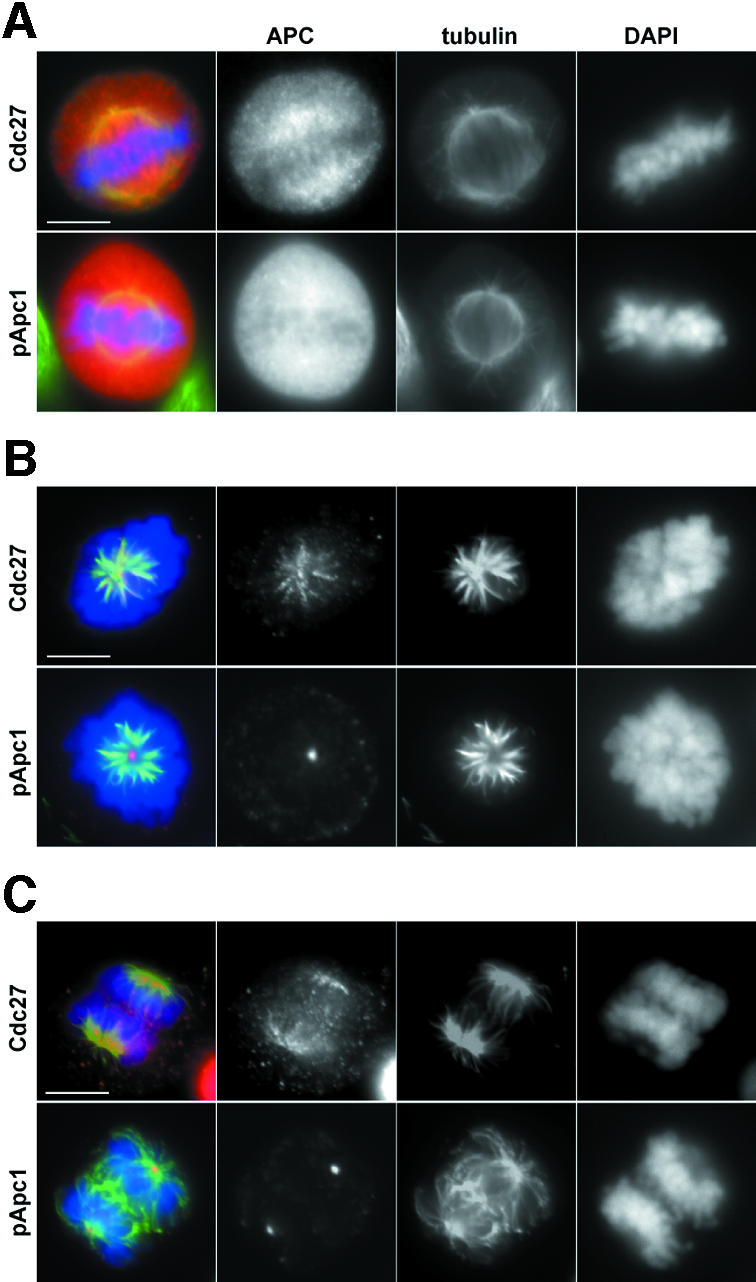
Fig. 6. Phospho-APC is enriched at centrosomes but not at the mitotic spindle. (A) Logarithmically growing HeLa cells were stained with non-phospho Cdc27 or pApc1 antibodies (red), tubulin (green) and DAPI (blue). (B and C) HeLa cells were extracted with 0.1% Triton X-100 prior to fixation to remove soluble proteins and stained as in (A). Bar, 10 µm.
Normal Plk1 levels are not essential for cyclin A degradation in vivo
Earlier studies implicated Plk1 in APC activation, but in our experiments only few of APC’s mitosis-specific in vivo sites could be generated by Plk1 in vitro (Table I), and Plk1 was not sufficient to activate Xenopus interphase APC in vitro (Figure 4). We therefore investigated whether Plk1 is essential for APC activity in vivo by knocking down Plk1 expression by RNAi in HeLa cells that had been synchronized by double thymidine treatment. HeLa cells were transfected with Plk1 siRNA, and Plk1 levels were analysed by immunoblotting and immunofluorescence microscopy 29 h after transfection (Figure 7). We recently found that HeLa cells depleted of Plk1 by this protocol enter mitosis but are then delayed in prometaphase by the spindle checkpoint. These cells often contain highly condensed chromosomes in a rosette-like configuration (I.Sumara and J.-M.Peters, unpublished data; see Plk1 negative cells in Figure 7D). Most Plk1 RNAi cells with this morphology showed intense cyclin B1 staining (Figure 7E), whereas 93% of these cells were negative for cyclin A, implying that cyclin A had been degraded (Figure 7C and F). We were unable to compare Plk1 and cyclin levels in the same cells due to technical limitations, but parallel immunoblotting (Figure 7A) and immunofluorescence microscopy (Figure 7C) performed on cells from the same experiment indicated that most of the cyclin A negative prometaphase cells had lost Plk1. When APC was isolated from Plk1 RNAi cells or from control cells 10 h after the second thymidine release, i.e. when control cells were progressing through mitosis and when most Plk1 RNAi cells were in prometaphase, no difference in the ubiquitination activities of the APC samples could be detected, further supporting the notion that APC activation was not compromised in the Plk1 RNAi cells (Figure 7B). To analyse cyclin A degradation directly we injected cyclin A–GFP cDNA into G2 phase cells that had been transfected with Plk1 siRNA and analysed the cells by time-lapse microscopy. We measured cyclin A degradation in 18 cells in which prometaphase was strongly delayed, indicating that Plk1 levels were significantly reduced. Under the conditions used, control HeLa cells enter anaphase 26 ± 9 min after NEBD (den Elzen and Pines, 2001). In contrast, 11 of the Plk1 RNAi cells entered anaphase between 60 and 346 min after NEBD (x = 151 min) and 7 cells did not enter anaphase before end of the experiment (between 72 and 1440 min after NEBD; x = 384 min). Importantly, cyclin A degradation was initiated at or just after NEBD in all 18 Plk1 RNAi cells, i.e. at the same time as in control cells (Figure 7G). These observations indicate that normal Plk1 levels are not essential for APC-mediated degradation of cyclin A. Plk1-depleted cells in mitosis also did not show any detectable reduction in fluorescence intensity after staining with any of the phospho-APC antibodies (Figure 7A and data not shown), indicating that the generation of the phospho-epitopes recognized by these antibodies does not depend on Plk1.
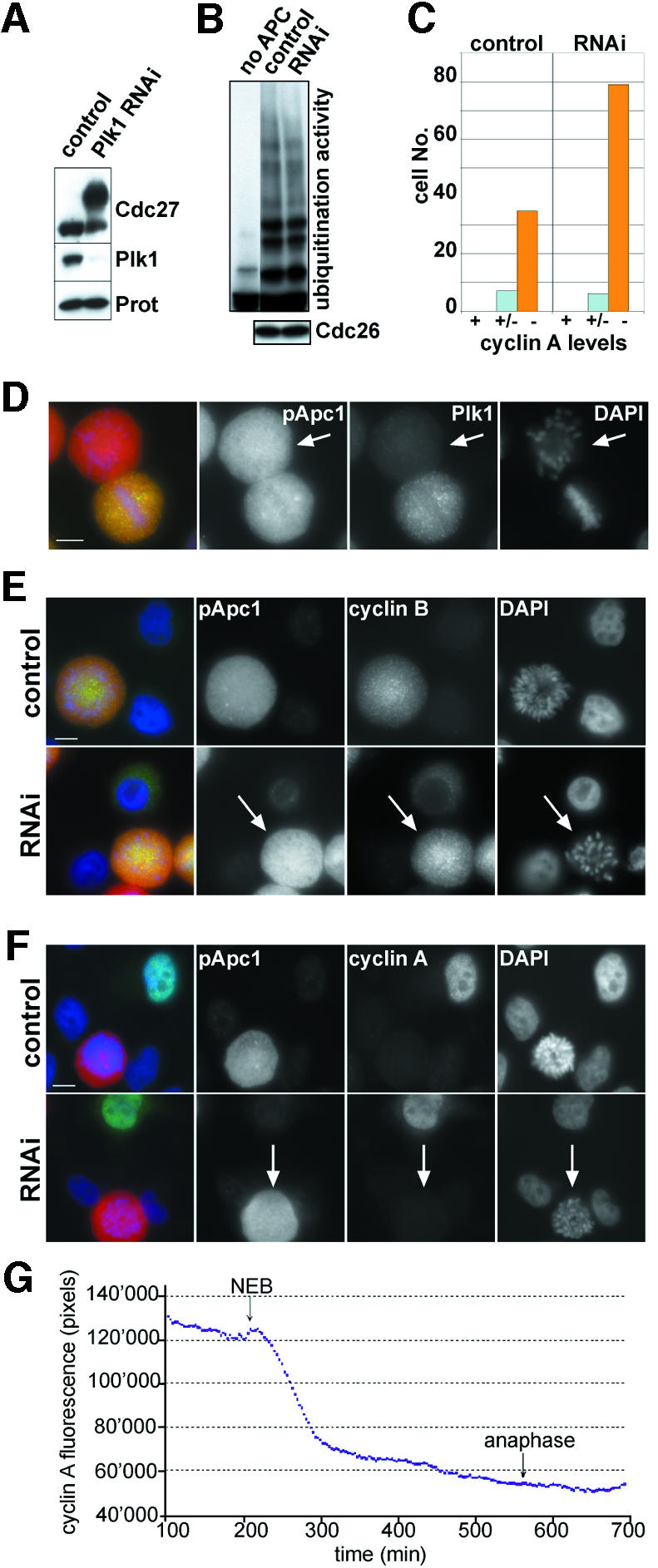
Fig. 7. Knockdown of Plk1 expression does not prevent APC activation. (A) HeLa cells were synchronized by a double thymidine arrest-release protocol and subjected to Plk1-RNAi treatment. After releasing cells from the second thymidine block for 13 h, cells were collected and analysed by immunoblotting using Plk1, Cdc27 and proteasome antibodies. Note that no Cdc27 mobility shift can be detected in the control cells because they had exited mitosis by this time (see Figure 2C). (B) Cells were treated as in (A), except that extracts were prepared 10 h after the second thymidine release when control cells progress through mitosis (see Figure 2C). The ubiquitination activity of APC immunopurified from these extracts was analysed as described for Figure 4, using myc-tagged cyclin B as a substrate (top panel). The amounts of APC used in these assays were compared by immunoblotting with Cdc26 antibodies (bottom panel). (C–F) Plk1 RNAi cells as in (A) were stained with antibodies to Plk1 (D; green), cyclin B1 (E; green) or cyclin A (F; green) and DNA was visualized with DAPI (blue). Note that cells which show the rosette-like arrangement of chromosomes typical for loss of Plk1 expression are positive for cyclin B but negative for cyclin A (indicated by arrows in E and F). The number of cells with prometaphase chromosome morphology that had lost cyclin A staining was counted in both control and Plk1 RNAi cells (C). Forty-two control cells and 85 RNAi cells were analysed and classified into cyclin A negative (–), weakly cyclin A postive (+/–) or cyclin A positive cells (+). Of all Plk1 RNAi cells with prometaphase morphology, 93% were cyclin A negative, whereas 83% of all control prometaphase cells were cyclin A negative. The increased frequency of cyclin A negative Plk1 RNAi cells may be due to the prolonged period of time these cells spend in prometaphase (see text). Bar, 10 µm. (G) HeLa cells treated as in (A) were microinjected in the nucleus with cyclin A–GFP cDNA (1 mg/ml), and followed by time-lapse fluorescence and DIC microscopy. Images (200-ms exposure) were taken at 3-min intervals. The total cell fluorescence minus background was quantified for each cell in successive images of a time series and plotted over time. A graph of a single cell, representative of 18 cells analysed, is shown. The start of NEBD and anaphase are marked.
Discussion
APC phosphorylation: required, sufficient or irrelevant for APCCdc20 activation?
Since its discovery, the vertebrate APC has been known to be mitotically phosphorylated on multiple subunits (King et al., 1995; Peters et al., 1996), but the functional relevance of these modifications and the identity of the kinases involved have remained controversial. This situation is largely due to the fact that no in vivo APC phosphorylation sites have been identified so far and that, with the exception of budding yeast APC (Rudner and Murray, 2000), no mutational analyses of APC phosphorylation sites have been performed. The latter approach will remain a challenge for the future because all attempts to reconstitute vertebrate APC from recombinant subunits in vitro or from ectopically expressed proteins in vivo have failed so far. Our ability to analyse APC phosphorylation in vertebrates is therefore presently limited to correlative approaches in which we can compare the in vitro effects of APC phosphorylation with APC’s modification state in vivo. The results of this approach have nevertheless yielded clear implications on how phosphorylation may regulate APCCdc20.
Several of our observations suggest an important role for Cdk1 in phosphorylating and activating APCCdc20: in vitro, Cdk1 could generate many of the APC phosphorylation sites that are found in vivo (Table I), the sites generated in vitro were sufficient to increase Cdc20 binding and stimulate APC’s ubiquitination activity (Figure 4), and at least some of the in vivo sites could first be detected in prophase cells when cyclin B1 enters the nucleus (Figure 5D). In contrast, in vitro phosphorylation of APC by Plk1 did not increase Cdc20 binding or APC activity (Figure 4), and knockdown of Plk1 by RNAi in living cells did not prevent destruction of cyclin A in prometaphase (Figure 7), a process that is known to be mediated by APCCdc20 (Dawson et al., 1995; Sigrist et al., 1995; Lorca et al., 1998; Geley et al., 2001). Cyclin B was not degraded in these cells, but this phenotype appears to be caused by activation of the spindle checkpoint following Plk1 depletion. Consistent with this hypothesis we found that the Aurora B inhibitor Hesperadin (Hauf et al., 2003) is able to override the mitotic arrest of Plk1 RNAi cells and to allow cyclin B and securin degradation (I.Sumara, C.Kraft and J.-M.Peters, unpublished observations). In vitro, we observed that Plk1 could further increase Cdc20 binding and APC activity when added simultaneously or sequentially with Cdk1 to interphase APC (Figure 4). These observations imply that Cdk1 is the major kinase that phosphorylates APC, thereby increasing its affinity for Cdc20 and thus activating APC’s ubiquitin ligase activity. In contrast, Plk1 may have a stimulatory but non-essential role, perhaps by generating phosphorylation sites which facilitate the recognition of APC by Cdk1 (Elia et al., 2003).
Where and when is APCCdc20 phosphorylated and activated?
If the model outlined above is correct, the appearance of mitotic phospho-epitopes on APC could be considered as a surrogate marker for Cdc20 binding and APC activation. APC phosphorylation can first be detected in the nuclei of prophase cells (Figure 5A); however, the degradation of cyclin A, APC’s earliest known substrate, is only initiated after NEBD in prometaphase (den Elzen and Pines, 2001; Geley et al., 2001). APC phosphorylation may therefore be required but may not be sufficient for APCCdc20 activation. Consistent with this possibility, degradation of the APC inhibitor Emi1 in early mitosis has recently been reported to be required for timely cyclin A and B degradation (Guardavaccaro et al., 2003; Margottin-Goguet et al., 2003). It is therefore conceivable that the presence of Emi1 in prophase causes a lag between APC phosphorylation and the initiation of cyclin A degradation. Likewise, APC phosphorylation is likely required but certainly not sufficient for cyclin B and securin destruction in vivo because the spindle checkpoint selectively inhibits the ubiquitination of these substrates until metaphase (reviewed in Musacchio and Hardwick, 2002).
Live cell imaging analysis of cyclin B degradation revealed that this APC substrate first disappears from spindle poles in human cells (Clute and Pines, 1999), and similar observations have been made in Drosophila embryos (Su et al., 1998; Huang and Raff, 1999). Interestingly, we observed that phospho-APC is enriched at spindle poles (Figure 6B and C), consistent with the idea that active APCCdc20 initiates cyclin B ubiquitination at this location. The enrichment of phospho-APC at spindle poles depended on Plk1, although the appearance of phospho-APC in the cytosol and cyclin A proteolysis did not (Figure 7; Supplementary figure 2). Plk1 may therefore directly or indirectly be required to enrich phospho-APC at spindle poles, but this localization does not seem to be essential for APC phosphorylation and activation. APC is also known to associate with microtubules of the mitotic spindle (Tugendreich et al., 1995), but surprisingly no phospho-APC could be detected on spindles (Figure 6C and D). The functional relevance of this phenomenon is unclear, but it is worth noting that cell fusion experiments suggest that the spindle checkpoint inhibits APCCdc20 in a spindle autonomous manner, implying that APCCdc20 could be regulated locally on spindle microtubules (Rieder et al., 1997).
The intensity of phospho-APC signals in immunofluorescence microscopy increase until metaphase and then decline throughout anaphase and telophase (Figure 5A) when degradation of B-type cyclins is completed (Clute and Pines, 1999). These observations are consistent with the possibility that APC-mediated Cdk1 inactivation allows protein phosphatases to dephosphorylate APC. Our biochemical data indicate that these reactions decrease Cdc20’s ability to bind to the APC (Kramer et al., 2000) (Figure 4) and APC dephosphorylation could therefore be sufficient for its inactivation, as is the case in vitro (Lahav-Baratz et al., 1995; Peters et al., 1996). In living somatic cells, however, Cdc20 itself is also degraded in anaphase and replaced by a related activator protein, called Cdh1 (Kramer et al., 2000; Hagting et al., 2002). APCCdc20 may therefore be inactivated by both dephosphorylation and Cdc20 degradation.
Why is APC phosphorylated on so many sites in mitosis?
Protein phosphorylation is one of the most widely studied regulatory mechanisms in biology, which has essential functions in signalling, metabolism, and cell cycle control. The analyses of phosphoproteins have so far typically revealed the presence of a few phosphorylation sites, but not several dozen, as are found on the APC. Why is the phosphorylation pattern of the APC so complex? The answer to this question is unknown, but one clue could come from the observation that most of the sites are found in clusters on TPR subunits. It is conceivable that some of the phosphorylation sites function as priming sites, which facilitate the recruitment of additional kinase molecules, which then would phosphorylate more sites in nearby locations. Since clusters of multiple phosphate groups are predicted to change the surface charge distribution significantly, these modifications could either induce structural changes within APC by altering subunit-interactions or they could change the affinity for molecules that are only temporarily associated with the APC. A candidate for a protein that is subject to this type of regulation is Cdc20 because it binds preferentially to mitotically phosphorylated APC, both in vitro and in vivo (Kallio et al., 1998; Kramer et al., 1998, 2000). We have recently found that Cdc20 and Cdh1 interact with the APC by associating with its TPR subunits (Vodermaier et al., 2003). The phosphorylation of the TPR subunits in mitosis may therefore directly regulate the association between Cdc20 and APC’s TPR subunits.
We identified 34 in vivo phosphorylation sites on APC that are clearly mitosis specific, but only 18 of these could be generated by Cdk1 and Plk1 in vitro (Table I). This observation implies that other kinases must also contribute to APC phosphorylation. One kinase that has been implicated in APC regulation is protein kinase A (PKA). In fission and budding yeast, PKA negatively regulates APC-mediated proteolysis, but it is unknown if the APC is directly phosphorylated by PKA (Yamashita et al., 1996; Irniger et al., 2000). In vitro, PKA can phosphorylate and inhibit human APC (Kotani et al., 1998, 1999), but the physiological relevance of this reaction is unclear. Among the 34 mitotic in vivo sites that we identified, only five contain the R-x1-2-S/T consensus motif described for PKA, implying that maximally a few sites on APC are phosphorylated by PKA in vivo, at least in nocodazole-arrested cells. Since we analysed APC isolated from cells in which the spindle checkpoint was activated, it is also possible that checkpoint kinases such as Bub1, BubR1, Mps1 or MAP kinase contributed to APC phosphorylation. Consistent with this possibility, we identified BubR1 by mass spectrometry in APC samples that were immunopurified from extracts of nocodazole-arrested cells (data not shown). Unexpectedly, we also identified three phosphorylated tyrosine residues on mitotic APC (Table I). It will be interesting to determine if these sites are generated by mitosis-specific tyrosine kinases such as Src (Roche et al., 1995). In the future, we are planning to combine mass spectrometry experiments with in vivo inhibition of mitotic kinases by RNAi or chemical inhibitors to identify which kinases are required for APC phosphorylation in vivo.
Materials and methods
Antibodies
Phospho-antibodies (indicated in Table I) were raised against 13mer synthetic peptides in rabbits and affinity purified. The following antibodies were used: cyclin B1 (GNS1, sc-245; Santa Cruz Biotechnology), α-tubulin (B512; Sigma), Plk1 (33-1700; Zymed), myc (9E10; Sigma) Histone H3phospho-Ser10 (06-570; Upstate), Cdc27 and Cdc20 (Gieffers et al., 1999; Kramer, 2000). Mouse monoclonal cyclin A antibodies were kindly provided by Tim Hunt (Imperial Cancer Research Fund, Hertfordshire, UK), rat polyclonal centrin 3 antibodies by Tomotoshi Marumoto (Kumamoto University School of Medicine, Japan) and LAP2β antibodies by Roland Foisner (University of Vienna, Austria).
Kinase and phosphatase assays
Kinase reactions were carried out in 20 mM Tris–HCl pH 7.5, 150 mM NaCl, 10 mM MgCl2, 10 mM β-glycerophosphate, 1 mM EGTA, 1 mM DTT, 1 µM ocadaic acid and either 2 mM ATP for mass spectrometric analysis or 0.2 mM ATP and 1 µCi/µl [γ-32P]ATP. Incubation was for 30 min at 30°C. 40 ng/µl Cdk1 and/or Plk1 were used as indicated. Following the incubation, the complex was washed twice with low salt buffer (BL: 20 mM Tris pH 7.5, 150 mM NaCl, 0.02% Tween-20), twice with HBS (20 mM HEPES pH 7.4, 150 mM NaCl) containing 0.5% NP-40, three times with BL containing 450 mM NaCl and twice more with BL. For mass spectrometric analysis, the last washing step was performed without detergent (20 mM Tris, 150 mM NaCl). The beads were eluted as described in the Supplementary data. 200 U of λ-protein phosphatase (New England Biolabs) per 10 µl APC beads were incubated for 30 min at 30°C in the buffer provided by the manufacturer. For APC isolation see Supplementary data.
Mass spectrometric analysis
Immunopurified APC was digested in solution with different proteases (trypsin, chymotrypsinogen, subtilisin or trypsin and Glu-C) according to Yates et al. (2000). Proteolytic peptides were separated by nano-HPLC (LC Packings) using an analytical column (PepMAP C18, 75 µm × 150 mm; DIONEX) at a flowrate of 200 nl/min, and were introduced via a nanospray ion source interface (Protana) into an ion trap mass spectrometer (Finnigan). The mass spectrometer cycled through five scans, one full mass scan followed by four tandem mass scans of the four most intense ions. All tandem mass spectra were searched against the human non-redundant protein database by using the Sequest program (Flicka). Any phosphopeptide matched by computer searching algorithms was verified manually. TPRs were analysed by database searches against hidden Markov models.
Cdc20 binding assay
5 µl Xenopus interphase APC on antibody beads was treated with recombinant purified kinases (see Supplementary data) followed by two washes with high salt buffer (BH: 20 mM Tris pH 7.5, 500 mM NaCl, 0.1% Tween-20) and two with BL. 200 ng His6-Cdc20 in 100 µl BL containing 3% BSA was added and incubated 30 min at 22°C on a thermomixer. Beads were washed as before and the APC was eluted from the antibody-beads using 7.5 µl of BL containing 0.5 mM DTT and 1 µg/µl of the immunogenic peptide for 2 h rotating at 4°C.
Ubiquitination assay
5 µl of peptide-eluted APC was added to BL containing purified E1 (80 µg/ml), UBC4 and UBCx (50 µg/ml each), ubiquitin (1.25 mg/ml), ATP regenerating system [7.5 mM creatine phosphate, 1 mM ATP, 1 mM MgCl2, 0.1 mM EGTA, 30 U/ml rabbit creatine phosphokinase type I (Sigma-Aldrich)] and 3 µg/ml iodinated sea-urchin cyclin B fragment (amino acids 13–110) (Figure 4) or myc-tagged cyclin B (Figure 7) in a total volume of 10 µl. The reaction mix was incubated for 40 min at 22°C on a thermoshaker and stopped by the addition of SDS–PAGE sample buffer. The samples were analysed by phosphorimaging (Figure 4) or immunoblotting (Figure 7). The ratio of polyubiquitinated substrate to total substrate was calculated after quantification using ImageQuant software (Molecular Dynamics).
Plk1-RNAi
RNAi was carried out using siRNA as described (Elbashir et al., 2001). Transfections were performed at the time of the second thymidine addition and the transfection medium was removed 4 h later. Cells were released from the second thymidine arrest and collected after 13 h. The targeted region in the human Plk1 cDNA is 5′-AACGAGCTGCTTAATGACGAGTT-3′. Synthetic sense and antisense oligonucleotides were obtained from VBC Genomics (Vienna). Annealing of siRNA oligos was performed according to Dharmacon’s instructions. For control transfections the same annealing reaction was set up using H2O instead of siRNA oligos.
Supplementary data
Supplementary data are available at The EMBO Journal Online.
Acknowledgments
Acknowledgements
We thank M.Madalinski for peptide synthesis and help with antibody purification, T.Hunt and T.Marumoto for antibodies, W.Dunphy and H.Yu for baculoviruses and members of the Peters laboratory for discussions and critical comments. Work in the laboratory of J.-M.P. is supported by Boehringer Ingelheim and grants from FWF, WWFF and EMBO.
References
- Brassac T., Castro,A., Lorca,T., Le Peuch,C., Doree,M., Labbe,J.C. and Galas,S. (2000) The polo-like kinase Plx1 prevents premature inactivation of the APC(Fizzy)-dependent pathway in the early Xenopus cell cycle. Oncogene, 19, 3782–3790. [DOI] [PubMed] [Google Scholar]
- Clute P. and Pines,J. (1999) Temporal and spatial control of cyclin B1 destruction in metaphase. Nat. Cell Biol., 1, 82–87. [DOI] [PubMed] [Google Scholar]
- Dawson I.A., Roth,S. and Artavanis-Tsakonas,S. (1995) The Drosophila cell cycle gene fizzy is required for normal degradation of cyclins A and B during mitosis and has homology to the CDC20 gene of Saccharomyces cerevisiae. J. Cell Biol., 129, 725–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- den Elzen N. and Pines,J. (2001) Cyclin A is destroyed in prometaphase and can delay chromosome alignment and anaphase. J. Cell Biol., 153, 121–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Descombes P. and Nigg,E.A. (1998) The polo-like kinase Plx1 is required for M phase exit and destruction of mitotic regulators in Xenopus egg extracts. EMBO J., 17, 1328–1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elbashir S.M., Harborth,J., Lendeckel,W., Yalcin,A., Weber,K. and Tuschl,T. (2001) Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature, 411, 494–498. [DOI] [PubMed] [Google Scholar]
- Elia A.E., Cantley,L.C. and Yaffe,M.B. (2003) Proteomic screen finds pSer/pThr-binding domain localizing Plk1 to mitotic substrates. Science, 299, 1228–1231. [DOI] [PubMed] [Google Scholar]
- Fang G., Yu,H. and Kirschner,M.W. (1998) Direct binding of CDC20 protein family members activates the anaphase-promoting complex in mitosis and G1. Mol. Cell, 2, 163–171. [DOI] [PubMed] [Google Scholar]
- Geley S., Kramer,E., Gieffers,C., Gannon,J., Peters,J.M. and Hunt,T. (2001) Anaphase-promoting complex/cyclosome-dependent proteo lysis of human cyclin A starts at the beginning of mitosis and is not subject to the spindle assembly checkpoint. J. Cell Biol., 153, 137–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gieffers C., Peters,B.H., Kramer,E.R., Dotti,C.G. and Peters,J.M. (1999) Expression of the CDH1-associated form of the anaphase-promoting complex in postmitotic neurons. Proc. Natl Acad. Sci. USA, 96, 11317–11322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gieffers C., Dube,P., Harris,J.R., Stark,H. and Peters,J.M. (2001) Three-dimensional structure of the anaphase-promoting complex. Mol. Cell, 7, 907–913. [DOI] [PubMed] [Google Scholar]
- Golan A., Yudkovsky,Y. and Hershko,A. (2002) The cyclin-ubiquitin ligase activity of cyclosome/APC is jointly activated by protein kinases Cdk1/cyclin B and Plk. J. Biol. Chem., 277, 15552–15557. [DOI] [PubMed] [Google Scholar]
- Guardavaccaro D. et al. (2003) Control of meiotic and mitotic progression by the F box protein β-Trcp1 in vivo. Dev. Cell, 4, 799–812. [DOI] [PubMed] [Google Scholar]
- Hagting A., Den Elzen,N., Vodermaier,H.C., Waizenegger,I.C., Peters,J.M. and Pines,J. (2002) Human securin proteolysis is controlled by the spindle checkpoint and reveals when the APC/C switches from activation by Cdc20 to Cdh1. J. Cell Biol., 157, 1125–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper J.W., Burton,J.L. and Solomon,M.J. (2002) The anaphase-promoting complex: it’s not just for mitosis any more. Genes Dev., 16, 2179–2206. [DOI] [PubMed] [Google Scholar]
- Hauf S. et al. (2003) The small molecule Hesperadin reveals a role for Aurora B in correcting kinetochore-microtubule attachment and in maintaining the spindle assembly checkpoint. J. Cell Biol., 161, 281–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J. and Raff,J.W. (1999) The disappearance of cyclin B at the end of mitosis is regulated spatially in Drosophila cells. EMBO J., 18, 2184–2195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irniger S., Baumer,M. and Braus,G.H. (2000) Glucose and ras activity influence the ubiquitin ligases APC/C and SCF in Saccharomyces cerevisiae. Genetics, 154, 1509–1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackman M., Lindon,C., Nigg,E.A. and Pines,J. (2003) Active cyclin B1-Cdk1 first appears on centrosomes in prophase. Nat. Cell Biol., 5, 143–148. [DOI] [PubMed] [Google Scholar]
- Kallio M., Weinstein,J., Daum,J.R., Burke,D.J. and Gorbsky,G.J. (1998) Mammalian p55CDC mediates association of the spindle checkpoint protein Mad2 with the cyclosome/anaphase-promoting complex and is involved in regulating anaphase onset and late mitotic events. J. Cell Biol., 141, 1393–1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King R.W., Peters,J.M., Tugendreich,S., Rolfe,M., Hieter,P. and Kirschner,M.W. (1995) A 20S complex containing CDC27 and CDC16 catalyzes the mitosis-specific conjugation of ubiquitin to cyclin B. Cell, 81, 279–288. [DOI] [PubMed] [Google Scholar]
- Kotani S., Tugendreich,S., Fujii,M., Jorgensen,P.M., Watanabe,N., Hoog,C., Hieter,P. and Todokoro,K. (1998) PKA and MPF-activated polo-like kinase regulate anaphase-promoting complex activity and mitosis progression. Mol. Cell, 1, 371–380. [DOI] [PubMed] [Google Scholar]
- Kotani S., Tanaka,H., Yasuda,H. and Todokoro,K. (1999) Regulation of APC activity by phosphorylation and regulatory factors. J. Cell Biol., 146, 791–800. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Kramer E.R., Gieffers,C., Holzl,G., Hengstschlager,M. and Peters,J.M. (1998) Activation of the human anaphase-promoting complex by proteins of the CDC20/Fizzy family. Curr. Biol., 8, 1207–1210. [DOI] [PubMed] [Google Scholar]
- Kramer E.R., Scheuringer,N., Podtelejnikov,A.V., Mann,M. and Peters,J.M. (2000) Mitotic regulation of the APC activator proteins CDC20 and CDH1. Mol. Biol. Cell, 11, 1555–1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahav-Baratz S., Sudakin,V., Ruderman,J.V. and Hershko,A. (1995) Reversible phosphorylation controls the activity of cyclosome-associated cyclin-ubiquitin ligase. Proc. Natl Acad. Sci. USA, 92, 9303–9307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorca T., Castro,A., Martinez,A.M., Vigneron,S., Morin,N., Sigrist,S., Lehner,C., Doree,M. and Labbe,J.C. (1998) Fizzy is required for activation of the APC/cyclosome in Xenopus egg extracts. EMBO J., 17, 3565–3575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margottin-Goguet F., Hsu,J.Y., Loktev,A., Hsieh,H., Reimann,J.D. and Jackson,P.K. (2003) Destruction of Emi1 in prophase requires the SCF(β-TrCP/Slimb) ubiquitin ligase and triggers activation of the Anaphase Promoting Complex to allow progression beyond prometaphase. Dev. Cell, in press. [DOI] [PubMed] [Google Scholar]
- May K.M., Reynolds,N., Cullen,C.F., Yanagida,M. and Ohkura,H. (2002) Polo boxes and Cut23 (Apc8) mediate an interaction between polo kinase and the anaphase-promoting complex for fission yeast mitosis. J. Cell Biol., 156, 23–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musacchio A. and Hardwick,K.G. (2002) The spindle checkpoint: structural insights into dynamic signalling. Nat. Rev. Mol. Cell Biol., 3, 731–741. [DOI] [PubMed] [Google Scholar]
- Patra D. and Dunphy,W.G. (1998) Xe-p9, a Xenopus Suc1/Cks protein, is essential for the Cdc2-dependent phosphorylation of the anaphase-promoting complex at mitosis. Genes Dev., 12, 2549–2559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters J.M. (2002) The anaphase-promoting complex: proteolysis in mitosis and beyond. Mol. Cell, 9, 931–943. [DOI] [PubMed] [Google Scholar]
- Peters J.M., King,R.W., Hoog,C. and Kirschner,M.W. (1996) Identification of BIME as a subunit of the anaphase-promoting complex. Science, 274, 1199–1201. [DOI] [PubMed] [Google Scholar]
- Pines J. and Hunter,T. (1991) Human cell division: the involvement of cyclins A and B1 and multiple cdc2s. Cold Spring Harb. Symp. Quant. Biol., 56, 449–463. [DOI] [PubMed] [Google Scholar]
- Rieder C.L., Khodjakov,A., Paliulis,L.V., Fortier,T.M., Cole,R.W. and Sluder,G. (1997) Mitosis in vertebrate somatic cells with two spindles: implications for the metaphase/anaphase transition checkpoint and cleavage. Proc. Natl Acad. Sci. USA, 94, 5107–5112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roche S., Fumagalli,S. and Courtneidge,S.A. (1995) Requirement for Src family protein tyrosine kinases in G2 for fibroblast cell division. Science, 269, 1567–1569. [DOI] [PubMed] [Google Scholar]
- Rudner A.D. and Murray,A.W. (2000) Phosphorylation by Cdc28 activates the Cdc20-dependent activity of the anaphase-promoting complex. J. Cell Biol., 149, 1377–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shteinberg M., Protopopov,Y., Listovsky,T., Brandeis,M. and Hershko,A. (1999) Phosphorylation of the cyclosome is required for its stimulation by Fizzy/cdc20. Biochem. Biophys. Res. Commun., 260, 193–198. [DOI] [PubMed] [Google Scholar]
- Sigrist S., Jacobs,H., Stratmann,R. and Lehner,C.F. (1995) Exit from mitosis is regulated by Drosophila fizzy and the sequential destruction of cyclins A, B and B3. EMBO J., 14, 4827–4838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su T.T., Sprenger,F., DiGregorio,P.J., Campbell,S.D. and O’Farrell,P.H. (1998) Exit from mitosis in Drosophila syncytial embryos requires proteolysis and cyclin degradation and is associated with localized dephosphorylation. Genes Dev., 12, 1495–1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Z., Bharadwaj,R., Li,B. and Yu,H. (2001) Mad2-independent inhibition of APCCdc20 by the mitotic checkpoint protein BubR1. Dev. Cell, 1, 227–237. [DOI] [PubMed] [Google Scholar]
- Tugendreich S., Tomkiel,J., Earnshaw,W. and Hieter,P. (1995) CDC27Hs colocalizes with CDC16Hs to the centrosome and mitotic spindle and is essential for the metaphase to anaphase transition. Cell, 81, 261–268. [DOI] [PubMed] [Google Scholar]
- Vodermaier H.C., Gieffers,C., Maurer-Stroh,S., Eisenhaber,F. and Peters,J.M. (2003) TPR subunits of the anaphase-promoting complex mediate binding to the activator protein CDH1. Curr. Biol., 13, 1459–1468. [DOI] [PubMed] [Google Scholar]
- Vorlaufer E. and Peters,J.M. (1998) Regulation of the cyclin B degradation system by an inhibitor of mitotic proteolysis. Mol. Biol. Cell, 9, 1817–1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waizenegger I.C., Hauf,S., Meinke,A. and Peters,J.M. (2000) Two distinct pathways remove mammalian cohesin from chromosome arms in prophase and from centromeres in anaphase. Cell, 103, 399–410. [DOI] [PubMed] [Google Scholar]
- Yamada H., Kumada,K. and Yanagida,M. (1997) Distinct subunit functions and cell cycle regulated phosphorylation of 20S APC/cyclosome required for anaphase in fission yeast. J. Cell Sci., 110, 1793–1804. [DOI] [PubMed] [Google Scholar]
- Yamashita Y.M., Nakaseko,Y., Samejima,I., Kumada,K., Yamada,H., Michaelson,D. and Yanagida,M. (1996) 20S cyclosome complex formation and proteolytic activity inhibited by the cAMP/PKA pathway. Nature, 384, 276–279. [DOI] [PubMed] [Google Scholar]
- Yates J.R. III, Link,A.J. and Schieltz,D. (2000) Direct analysis of proteins in mixtures. Application to protein complexes. Methods Mol. Biol., 146, 17–26. [DOI] [PubMed] [Google Scholar]