

Abstract
Analogs of nantenine were docked into a modeled structure of the human 5-HT2A receptor using ICM Pro, GLIDE and GOLD docking methods. The resultant docking scores were used to correlate with observed in vitro apparent affinity (Ke) data. The GOLD docking algorithm when used with a homology model of 5-HT2A, based on a bovine rhodopsin template and built by the program MODELLER, gives results which are most in agreement with the in vitro results. Further analysis of the docking poses among members of a C1 alkyl series of nantenine analogs, indicate that they bind to the receptor in a similar orientation, but differently than nantenine. Besides an important interaction between the protonated nitrogen of the C1 alkyl analogs and residue Asp155, we identified Ser242, Phe234 and Gly238 as key residues responsible for the affinity of these compounds for the 5-HT2A receptor. Specifically, the ability of some of these analogs to establish a H-bond with Ser242 and hydrophobic interactions with Phe234 and Gly238 appears to explain their enhanced affinity as compared to nantenine.
Keywords: Nantenine, aporphine, serotonin, MDMA, 5-HT2A
Introduction
At present there are thirteen distinct serotonin G-protein-coupled receptors that are divided into six families, based on pharmacology, amino acid sequences, gene organization and second messenger coupling pathways.1, 2 The serotonin 5-HT2A receptor is of significant clinical interest because of its involvement in cardiovascular function and in certain mental disorders.3 Agonists at this receptor may be used for treatment of sleep disorders and arousal.4 The utility of antagonists in the treatment of depression and certain psychotic conditions has already been well explored.5, 6 The investigation of 5-HT2A antagonists as potential drug-abuse therapeutics is topical in the recent literature.7, 8
Aporphines have been relatively unexplored as 5-HT2A receptor ligands, but it is known that they bind to several other CNS receptors. The aporphine natural product nantenine is a moderate 5-HT2A receptor antagonist and functions in vivo as an antagonist to a range of behavioral and physiological effects of MDMA (“Ecstasy”).9 Very little is known about the structural requirements for affinity and activity of nantenine at the 5-HT2A receptor. As part of our ongoing efforts to develop novel aporphine-based 5-HT2A antagonists as potential MDMA antagonists, we prepared a series of analogs based on nantenine and evaluated their affinity and activity at the 5-HT2A receptor.10, 11 The analogs were specifically designed to investigate the role of molecular rigidity, N-substitution and substitution at the C1 position on 5-HT2A affinity and activity. This study resulted in the identification of C1 analogs with up to 12-fold increase in 5-HT2A apparent affinity (Ke) as compared to nantenine.11
In order to understand the possible binding modes of nantenine and nantenine analogs at the human 5-HT2A receptor (to complement future drug design efforts), we have conducted a docking study with our library of compounds. Since the crystal structure of the 5-HT2A receptor has not been solved, our approach necessitated the use of a homology modeling paradigm. Although other homology models for the 5-HT2A receptor have been previously described3, 12, 13 we decided to build and evaluate a number of homology models using various in silico tools in order to determine which model was best in line with our in vitro data. Four homology models were built based on bovine rhodopsin (PDB code 1U19) and human β2-adrenoceptor (PDB code: 2RH1) templates using two programs for molecular modeling - MODELLER and ICM Pro. To determine the extent to which our in vitro results correlated with each homology model, we then performed docking/scoring experiments, using three docking programs: ICM Pro, GLIDE and GOLD.
Based on these experiments we have determined that the homology model built by MODELLER and based on a bovine rhodopsin template together with a GOLD docking algorithm is in the best agreement with our in vitro results. These studies have provided useful insights into the possible binding modes of nantenine analogs at the 5-HT2A receptor - a significant prelude in our quest towards potent aporphine-derived 5-HT2A antagonists. Results from our studies are presented herein.
Results and Discussion
Chemistry
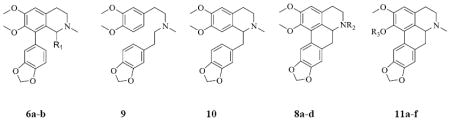
The structures of the nantenine analogs used for our docking study, 6a-b, 9, 10, 8a-d and 11a-f are shown in Table 1. Compounds 9, 10, and 11a-f were prepared as reported by us elsewhere.11
Table 1.
Structures of nantenine analogs evaluated
 | |||
|---|---|---|---|
| R1 | R2 | R3 | Compound |
| -H | 6a | ||
| -CH3 | 6b | ||
| - | - | - | 9 |
| - | - | - | 10 |
| -H | 8a | ||
| -CH2CH3 | 8b | ||
| -COCH3 | 8c | ||
| -SO2CH3 | 8d | ||
| -CH3 | nantenine | ||
| -CH2CH3 | 11a | ||
| -CH2CH2CH3 | 11b | ||
| -CH2CH2CH2CH3 | 11c | ||
| -CH2CH2CH2CH2CH3 | 11d | ||
| -CH2-cyclopropyl | 11e | ||
| -benzyl | 11f | ||
Compounds 6a-b were prepared as outlined in Scheme 1. Commercially available bromo-aldehyde 1 was coupled to 3,4-(methylenedioxyphenyl) boronic acid (2) under Suzuki conditions affording the biaryl aldehyde, 3. A nitro-aldol reaction on aldehyde 3 followed by reduction of the resulting nitrostyrene (4), gave an intermediate amine which was subsequently converted to amides 5a and 5b. These amides were then subjected to Bischler-Napieralski cyclization to afford intermediate dihydroisoquinoline products that were sequentially reduced and N-methlyated to provide the target seco-ring C derivatives 6a and 6b.
Scheme 1. Reagents and conditions.

(a) Pd(PPh3)4, DME, K2CO3, reflux, 24h; (b) CH3NO2, NH4OAc, reflux, 4h; (c) LiBH4, TMSCl, THF, reflux; (d) HCOOEt, Et3N, DCM for 5a; (e) acetyl chloride, Et3N, for 5b (f) POCl3, MeCN, 50 °C, 12h; (g) NaBH4, MeOH, 0 °C, 4h; (h) HCHO, NaBH(OAc)3, DCM, rt, 24h
Compounds 8a-d were prepared as shown in Scheme 2. We initially attempted removal of the Boc group of 7 with TFA.14 However with these conditions only a 50% yield of the amine 8a was obtained. After some experimentation with other acidic cleavage conditions, we found that anhydrous ZnBr2 in DCM gave the expected product in excellent yield. Treatment of 8a with acetaldehyde under reductive amination conditions gave 8b. Compound 8c was prepared by reacting 8a with acetyl chloride, while 8d was obtained after reaction of 8a with methanesulfonyl chloride.
Scheme 2. Reagents and conditions.

(i) ZnBr2, DCM, rt, 24 h (ii) acetaldehyde, NaBH(OAc)3, DCM, rt for 8b (iii) acetyl chloride, Et3N, DCM, 0 °C for 8c (iv) methanesulfonylchloride, Et3N, DCM for 8d.
5-HT2A Receptor Affinities from Functional Inhibition Assays
Apparent 5HT2A receptor affinity (Ke) data were obtained for each of the target nantenine analogs using a FLIPR (Fluorometric Imaging Plate Reader) assay and are presented in Table 2. Ke values give an indirect measure of compound affinity. Ke and Ki values usually display the same trend in rank-order within a series of compounds15 and thus it is acceptable to use Ke (instead of Ki) values in evaluations of compound affinity. Others have shown in binding studies with 5-HT2A antagonists that there is good correlation between antagonist affinity determined in a FLIPR assay and those reported in radioligand binding assays and that the rank order of potency is maintained.16 A functional assay was used instead of receptor binding because we could obtain functional and affinity data from the same assay.
Table 2.
Results of docking experiments with the ICM Pro, GLIDE and GOLD
| HAdrb2_ICMa | BRho_ICMb | HAdrb2_MODELLERc | BRho_MODELLERd | HAdrb2_MODELLER | BRho_MODELLER | ||
|---|---|---|---|---|---|---|---|
| Compound | Ke, nM | ICM score | ICM score | GLIDE score | GLIDE score | GOLD score | GOLD score |
| ketanserin | 32 | -75.82 | -72.28 | -11.43 | -4.30 | 27.34 | 30.63 |
| ± nantenine | 850 ± 6 | -54.93 | N | -11.23 | -8.03 | 27.19 | 23.42 |
| 6a | >10000 | -48.30 | N | -12.03 | -8.50 | 29.72 | N |
| 6b | >10000 | -50.94 | N | N | -8.47 | N | N |
| 9 | 5180 | -54.32 | -47.69 | -13.53 | -8.76 | 31.44 | N |
| 10 | >10000 | Ne | N | -10 | -9.05 | N | N |
| 8a | >10000 | -51.64 | N | -13.81 | -8.31 | 30.84 | N |
| 8b | >10000 | N | N | -13.48 | -8.22 | 33.83 | N |
| 8c | >10000 | N | N | N | N | N | N |
| 8d | >10000 | N | N | N | N | N | N |
| 11a | 890 ± 430 | -55.14 | N | -11.45 | -8.30 | 30.19 | 24.22 |
| 11b | 297 ± 130 | -65.37 | -51.87 | -8.94 | -9.09 | 31.78 | 22.98 |
| 11c | 274 ± 80 | -68.37 | -54.23 | -11.20 | -5.01 | 32.53 | 29.10 |
| 11d | 171 ± 50 | -70.39 | N | -12.06 | -5.05 | 33.17 | 30.52 |
| 11e | 68 ± 8 | -64.75 | N | -12.21 | -8.03 | 33.30 | 29.06 |
| 11f | 4600 | -69.79 | N | -13.44 | -11.67 | 35.64 | 29.48 |
5-HT2A receptor homology model built by ICM Pro based on a human β2- adrenoceptor template
5-HT2A receptor homology model buil by ICM Pro based on a bovine rhodopsin template
5-HT2A receptor homology model built by MODELLER based on a human β2- adrenoceptor template
5-HT2A receptor homology model buil by MODELLER based on a bovine rhodopsin template
N – not in the binding pocket or no key Asp155-NH3+ interaction (determined by visual inspection)
SAR studies
The apparent affinities of seco-C-ring analogs 6a-b were >10,000 nM, suggesting together with the results for compounds 9 (Ke = 5,180 nM) and 10 (Ke > 10,000 nM) that the intact aporphine core is required for 5-HT2A receptor affinity.
Removal of the methyl group from position N6 (ie compound 8a) also led to a noticeable decrease in affinity as compared to nantenine. Placing a slightly bulkier ethyl group instead of a methyl group at the same position, (compound 8b), gave similar results as compound 8a. Compounds 8c-d showed decreased affinity, likely because of the absence of a basic nitrogen atom in these compounds; it has been previously proposed that the nitrogen of nantenine is involved in an ionic bond with Asp155 of helix 3 in the 5-HT2A receptor and that this interaction is important for binding of nantenine to the receptor.17 In summary, for the less rigid and N-analogs only compound 9 showed any appreciable (though quite weak) affinity for the receptor.
The effects of substituent changes at the C1 position (ie compounds 11a-f) on the affinity of nantenine have already been reported by us.11 Briefly, these SAR studies indicate that the C1 position may be tolerant of bulky lipohilic groups and that this site may be a key position to modify in order to improve affinity for the 5-HT2A receptor. The enhanced affinity of members of the C1 series of analogs compared with the parent molecule suggested to us that they may interact differently with the 5-HT2A receptor.
Molecular Modeling
Scheme 2 summarizes the general approach we have taken in this study.
Selection of templates
The main challenge in homology modeling is to find a suitable template. Since the crystal structure of the 5-HT2A receptor is still not solved, and the direct determination, by X-ray crystallography of the atomic structure of any member of the serotonin receptor family has not yet been accomplished, the only way to achieve our objective is to use a similar GPCR structure as a template to build a homology model. We searched for templates using BLAST18 (Basic Local Alignment Search Tool), - see Experimental section. Currently there are four available crystal structures of mammalian GPCRs: turkey β1-adrenergic receptor (PDB code: 2VT419), human adenosine A2A receptor (PDB code: 3EML20), human β2-adrenergic receptor (PDB code: 2RH121) and bovine rhodopsin (PDB code: 1U1922). In a recent report, Mobarec et al suggested that a good template to use to build a homology model for the serotonin 5-HT2A receptor is either the crystal structure of a turkey β1-adrenergic receptor or human β2-adrenergic receptor.23
Although other GPCR structures are modeled in the literature using bovine rhodopsin as a template, there are some disadvantages of using this template; for example, it shares a low sequence similarity to 5-HT2A receptor (less than 20%) and it is a retinal-binding protein, functionally completely different from the 5-HT2A receptor, which is a typical representative of the neurotransmitter GPCRs. On the other hand, the high-resolution of its crystal structure and the specific arrangement of the seven transmembrane helices stabilized by a series of intramolecular interactions make bovine rhodopsin a very good structural template for generating molecular models of the 5-HT2A receptor. Until very recently, there were no experimental structures available for any neurotransmitter GPCRs. A high resolution (2.40 Å) of the human β2 adrenergic receptor has been reported.24 It has better sequence similarity with the 5-HT2A receptor (almost 30%) and it is functionally closer to the family of serotonin receptors, since it also belongs to the group of neurotransmitter GPCRs. Disadvantages are that this crystal structure has been solved at lower resolution than the one at bovine rhodopsin, and there are only a few 5-HT2A receptor homology models built using this structure as a template.12
As a template for building the 5-HT2A receptor homology model we selected two different available GPCR crystal structures. The first one is bovine rhodopsin (PDB code: 1U19) since it has been shown in many previous reports 3, 13, 25, 26 that this structure is a reliable template for 5-HT2A receptor homology models and also because its crystal structure is solved at high resolution of 2.20 (x001FA) as compared to other known structures. The structures resolved below 2.5 (x001FA) are normally regarded as very good molecules for docking, yielding good results for most typical applications.27 We also selected the human β2-adrenergic receptor (PDB code: 2RH1) as a second template, because to date 2RH1 is the best resolved structure (2.40 Å) of any GPCR neurotransmitter receptor.23 The other BLASTP hit structures were omitted because of their low resolution.
Only one group (Indra et al17) has tried to explain a binding mode of (±)-nantenine at the 5-HT2A receptor.17 However, this study described a homology model of a rat 5-HT2A receptor. (Since our in vitro data was based on human rather than rat 5-HT2A receptor data, we have constructed homology models based on the human 5-HT2A receptor.) The Indra model proposes that Asp155, a residue in transmembrane helix 3 (TM3) that is strongly conserved across the family of serotonin receptors, forms a strong H-bond via N6 of nantenine. The oxygen atom of the C1 methoxy group interacts via H-bonding with Asn343, a residue from TM6, also strongly conserved in many GPCRs. It is hypothesized that this interaction may be directly involved in 5-HT2A receptor antagonism.17
Alignment
Alignments of the target sequence to the template structures were made using the ClustalX 28 alignment tool and the ICM Pro sequence alignment program 29, both using their default parameters. The sequence alignment of the human 5-HT2A receptor using ClustalX shows good sequence identity with the bovine rhodopsin template (19.5%) with more than 45% sequence similarity. The same human 5-HT2A receptor sequence has, compared with the bovine rhodopsin sequence, a higher sequence identity with the human β2-adrenergic receptor template (almost 28%) with more than 48% sequence similarity. Although the ICM Pro alignment program is based on a different algorithm and method (it uses ZEGA algorithm with zero gap penalties30) than ClustalX, the sequence of the human 5-HT2A receptor nevertheless showed 19% sequence identity with the bovine rhodopsin template which is almost the same as we determined using ClustalX. Also comparable results, (30% identity), were obtained when the ICM Pro alignment program was used for the β2-adrenergic receptor template and 5-HT2A receptor sequence, (see Experimental). Since the secondary structure elements are well-conserved among these sequences, the alignments were then manually refined to ensure a perfect alignment of the highly conserved residues of the GPCR superfamily, according to Baldwin et al.31
Model construction
3D-Model building was performed with the modeling suite, Discovery Studio 2.0, which uses MODELLER 32 for building protein structures. Twenty models were built for each of the templates using the homology module of Discovery Studio and one model for each template was built by the ICM Pro 3.6 model building software.29 The conserved disulfide bond between residues C148 at the beginning of TM3 and C228 in the middle of extracellular loop 2 (ECL2), a feature common in many GPCR receptors, was also created and was kept during the building of homology models in both programs.
Model evaluation
Evaluation methods check whether a model satisfies standard steric and geometric criteria. Each of the tools used in the construction of a model (e.g. template selection, alignment, model building, and refinement) has its own internal measures of quality. In order to evaluate and select the best homology model for the purpose of our study we used several different programs for evaluation that are available via the server of the UCLA-DOE Institute for Genomics and Proteomics. This Institute offers a service called Structural Analysis and Verification Server (SAVES) 33 that encompass five verification tools for model evaluation: PROCHECK 34, WHAT_CHECK33, ERRAT 35, VERIFY_3D 36 and PROVE37. We evaluated each model using the above methods and selected the final model for each template (see Experimental for details), which fits best within the criteria for selection of each method. From twenty 5-HT2A receptor homology models, which were built by MODELLER32 based on a bovine rhodopsin template (1U19), we selected one homology model (hereinafter called BRho_MODELLER). A selected model, out of twenty homology models built by MODELLER, based on human β2-adrenoceptor template (2RH1), was named HAdrb2_MODELLER. Using ICM Pro software, two homology models, one for each template, were built, and both showed good evaluation scores; hence we decided to also use these for the purpose of docking experiments. We named them according to the origin of their templates, bovine rhodopsin and human β2-adrenoceptor - BRho_ICM and HAdrb2_ICM, respectively.
Energy minimization
The four selected homology models were relaxed with 500 steps of steepest descent energy minimization using Charmm and using the Discovery Studio 2.0, we calculated the RMSD values for the each homology model and their appropriate template structure. The structural superposition of Cα trace of the 5-HT2A receptor homology models with respect to the templates, gave us the RMSD values: 1.53 for BRho_MODELLER, 2.69 for HAdrb2_MODELLER, 3.73 for the BRho_ICM and 2.40 for the HAdrb2_ICM (see Experimental). All selected homology models share a close homology with the template as indicated by their RMSD value of their backbone atoms with respect to the template. The resulting, energy minimized homology models were subsequently used for the docking experiments.
Docking
There are many commercially available docking software, each using a different combination of searching methods and algorithms, and scoring functions. We used three programs for docking experiments and compared the docking scores with our in vitro results. We selected the ICM Pro docking29 algorithm, GLIDE38, 39 and GOLD40 methods for the purpose of docking of our analogs. Each compound that was used for the docking experiment was prepared, converted to a 3D structure and energy minimized using Chem3D Ultra41 or LigPrep (developed and distributed by Schrodinger Inc - www.schrodinger.com; described in detail in Experimental section). The results from our docking study are summarized in Table 2. After we performed docking using the different programs and obtained the docking scores, each structure was visually inspected to be assured that the molecules were actually docked into the expected binding site and that interactions with key residues of the active site were maintained. All final receptor-analog complexes, obtained from the three different programs, were saved as PDB files and visualized in Discovery Studio 2.0 and ICM Pro. H-bonds, hydrophobic interactions and distances for each 3D structure were calculated and plotted by LigPlot42 v 4.4.2., a program for schematic 2D diagrams of ligand and receptor plots.
A model of the human 5-HT2A receptor has been constructed using an in silico activated bovine rhodopsin template.13 The major difference between our bovine rhodopsin homology model of the 5-HT2A receptor and that previously reported is the presence of a network of polar interactions “ionic lock” that is present between residues Arg135 (in TM3) and Glu247 (in TM6), which bridges these two transmembrane helices, stabilizing the inactive-state conformation. The crystal structures of other GPCRs have revealed that these polar interactions are broken in the ligand-activated GPCR crystal structures eg. human β2-adrenoceptor, turkey β1-andrenoceptor and human adenosine A2A. Recent crystallographic evidence suggests that the conformation of the activated form of bovine rhodopsin does not significantly change in the ligand binding region; thus, rhodopsin represents a good template for homology models of other GPCRs used in docking calculations of both agonists and antagonists because both ground-state and photoactivated rhodopsin are structurally similar.43 Therefore, we did not include this network of polar interactions in our model. Since 3D-coordinates and the information on model quality through any of the verification tools (Procheck, Verify_3D, Errat, etc) were not available for the previously constructed model, we are unable to perform any detailed structural comparisons of the models. Others have also built homology models of BRho with the MODELLER program.44
Recently, a 5-HT2A receptor homology model based on a human β2 adrenoceptor was reported by Bruno et al.45 Using this GPCR as a template is considered more suitable than the bovine rhodopsin template since sequence identity of the human 5-HT2A receptor with human β2 adrenoceptor is much better (around 30%), than with the bovine rhodopsin template (below 20%), and also because both β2 and 5-HT2A receptors are members of the amine group of class A GPCRs. Mobarec et al. proposed that generating a 5-HT2A receptor homology model based on this template will produce a better homology model.23 We decided to use this template as our second template. Comparing the alignment constructed by us with the alignment constructed by Bruno et al. was consistent, including the disulphide bridge between C148 on TM3 and C227 on ECL2. Since Procheck results for their model were included, we compared them with our Procheck results, and our results show that fewer residues of our model fall outside the allowed region of the Ramachandran plot. One major difference in the construction of our model is that we included an artificial intracellular loop 3 (ICL3), whereas this was omitted in the Bruno model. Others have also constructed homology models of the human β2 adrenoceptor with MODELLER.46, 47
As shown in Table 2, the best overall agreement between the in vitro experimental findings and the docking scores are obtained by using a homology model that was built by MODELLER based on a bovine rhodopsin template (BRho_MODELLER) with the GOLD docking program. None of the models gave very good correlation values for the C1 analogs when nantenine and compounds 11a-f were included in the data set (Figures 1 - 2 and Table 3). However, analogs which had low apparent affinity (Ke > 10,000) were predicted not to bind with the BRho_MODELLER/GOLD combination whereas analogs with good to poor apparent affinity (50 < Ke< 5,000, ie nantenine and the C1 analogs 11a-11f) gave reasonable and measurable GOLD scores with BRho_MODELLER. In contrast for example, the HAdrb2_MODELLER/GOLD combination gave binding poses and scores for flexible analogs 6a and 9 as well as the N-analogs 8a and 8b whereas the BRho_MODELLER/GOLD indicated that these analogs would have no affinity, which is in line with the in vitro data. Interestingly, if we considered only analogs 11a-11e (ie C1 alkyl analogs excluding the benzyl analog), we obtained a very good correlation (R2=0.928 in a plot of Ke vs docking score) for the HAdrb2_MODELLER/GOLD combination. Therefore, in the evaluation of a more diverse set of nantenine analogs it appears that the BRho_MODELLER/GOLD combination performs better than the HAdrb2_MODELLER/GOLD combination, although the latter may be more suited for a restricted set of C1 alkyl congeners. Although the correlation is low, the BRho_MODELLER/GOLD combination appears to provide the best gross approximation of affinity or lack thereof in the entire series of compounds and we therefore proceeded to look in detail at potential binding modes with the BRho_MODELLER model.
Figure 1.
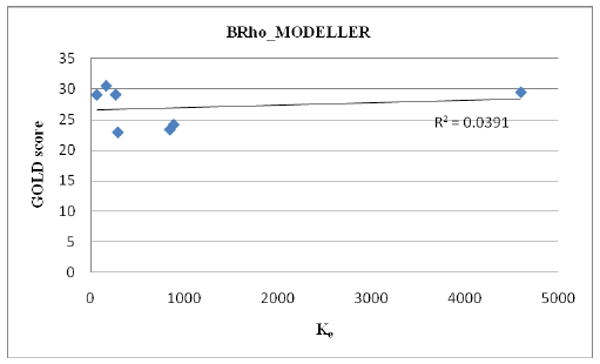
Correlation of GOLD score and affinity data for BRho_MODELLER
Figure 2.
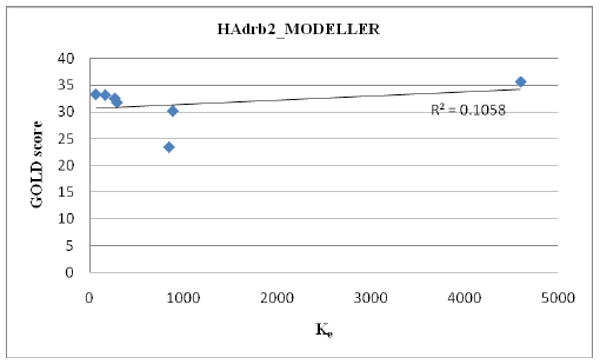
Correlation of GOLD score and affinity data for HAdrb2_MODELLER
Table 3.
R2 values for compoundsa docked with various models
| Model/scoring | r2 | r2 |
|---|---|---|
| HAdrb2_ICM/ICM | 0.043b | 0.747c |
| HAdrb2_MODELLER/GLIDE | 0.357b | 0.012c |
| BRho_MODELLER/GLIDE | 0.576b | 0.106c |
| HAdrb2_MODELLER/GOLD | 0.201b | 0.928c |
| BRho_MODELLER/GOLD | 0.039b | 0.381c |
C1 analogs;
nantenine and 11a-11f;
11a-11e only
Binding modes of nantenine as well as C1 analogs were further examined in order to begin to understand the trend in apparent affinity values observed in this series of compounds. Figures 3-9 show 3D and 2D representations (generated with ICM Pro and LigPlot respectively) of important interactions of nantenine and the analogs in the 5-HT2A receptor (for the BRho_MODELLER homology model). Table 4 summarizes the key interactions for the C1 analogs. We observed that C1 analogs 11a-f are binding in the 5-HT2A receptor pocket differently than nantenine, while still maintaining similarity in orientation among them. Figure 3 shows that nantenine has a H-bond between the protonated N6 atom and Asp155, as well as a second key interaction between the oxygen atom of the C1 methoxy group and Asn343 located in TM6. This is in agreement with a previous study done by Indra et al although we used a different protein source (human vs. rat 5-HT2A receptor) and methods for homology modeling and docking experiments. The congruence of these results is perhaps not surprising given the high degree of sequence similarity between rat and human 5-HT2A receptors.25
Figure 3.
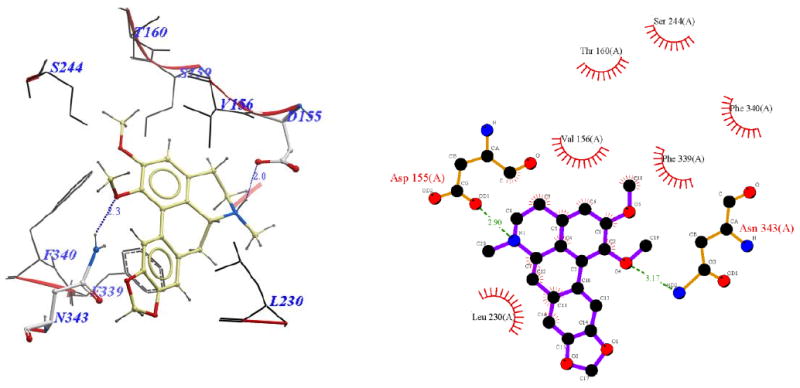
Binding pose of nantenine represented with: a) ICM Pro, and b) Ligplot
Figure 9.
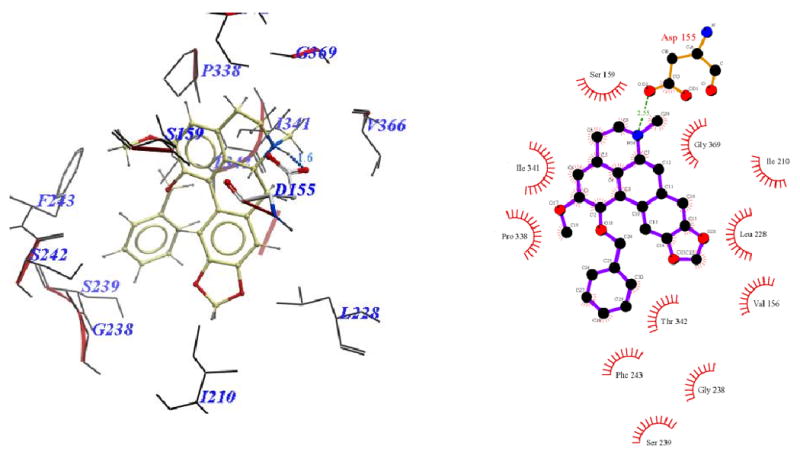
Binding pose of 11f represented with: a) ICM Pro and b) Ligplot
Table 4.
Residues interacting with nantenine and C1 analogs
| TM | Residues | Nantenine | 11a | 11b | 11c | 11d | 11e | 11f |
|---|---|---|---|---|---|---|---|---|
| 3 | ILE 152 | ILE152 | ILE152 | ILE152 | ||||
| 3 | ASP155a,b,c | ASP155 | ASP155 | ASP155 | ASP155 | ASP155 | ASP155 | ASP155 |
| 3 | VAL156 | VAL156 | VAL156 | VAL156 | VAL156 | VAL156 | ||
| 3 | SER159 | SER159 | SER159 | SER159 | SER159 | |||
| 3 | THR160 | THR160 | ||||||
| 3 | ILE163a | ILE163 | ILE163 | ILE163 | ||||
| 4 | ILE206 | ILE206 | ILE206 | ILE206 | ||||
| 4 | ILE210 | ILE210 | ILE210 | ILE210 | ILE210 | |||
| 5 | LEU228 | LEU228 | LEU228 | LEU228 | LEU228 | LEU228 | ||
| 5 | PHE234 | PHE234 | PHE234 | PHE234 | PHE234 | |||
| 5 | GLY238 | GLY238 | GLY238 | GLY238 | GLY238 | |||
| 5 | SER239 | SER239 | ||||||
| 5 | SER242 | SER242 | SER242 | SER242 | SER242 | SER242 | ||
| 5 | PHE243a,b | PHE243 | ||||||
| 5 | SER 244 | SER244 | ||||||
| 6 | PRO338a,b,c | PRO338 | ||||||
| 6 | PHE339a | PHE339 | ||||||
| 6 | PHE340a | PHE340 | ||||||
| 6 | ILE341 | ILE341 | ILE341 | ILE341 | ILE341 | |||
| 6 | THR342 | THR342 | THR342 | |||||
| 6 | ASN343 | ASN343 | ||||||
| 6 | MET345 | MET345 | ||||||
| 7 | VAL366 | VAL366 | ||||||
| 7 | GLY369a | GLY369 |
Visual inspection of the binding mode of the C1 analogs with highest affinity - 11d (Ke = 171 nM) and 11e (Ke = 68 nM), revealed a different orientation of these molecules in the receptor as compared to nantenine (Figures 7 and 8). For both of these compounds there is a protonated N6/Asp155 H-bond and a H-bond between Ser242 and one of the oxygen atoms located in the methylenedioxy ring. Ser242 has also been reported to be important as a H-bonding residue for other high affinity 5-HT2A ligands both as H-bond acceptor and as a H-bond donor. For example, the partial agonist LSD is reported to interact with the hydroxyl group of Ser242 through H-bonding via it's N1 hydrogen atom. Ser242 also plays a H-bonding role in the binding of a series of benzofuranone 5-HT2A antagonists to the receptor via it's H-bond donor capacity to an oxygen atom in the ligand. The H-bonding interaction between the C1 oxygen atom and Asn343 which is present in nantenine is absent in the binding poses of 11d and 11e as well as the other C1 analogs. In addition to the strong stabilizing H-bond interactions, the higher affinity of 11d and 11e might also be accounted for by strong hydrophobic interactions that are formed between the spatially close residues Phe234 and Gly238 (located in TM5) and the C1 alkyl groups in 11d and 11e respectively. Like 11d and 11e, compounds 11b and 11c (Figure 5 and Figure 6 respectively), also form H-bonds with Asp155 and Ser242, and their C1 side chains make important hydrophobic interactions with Phe234 but not with Gly238. Absence of the Gly238 hydrophobic interaction probably has some bearing on the slightly lower in vitro affinity of these two nantenine analogs (Ke = 297 nM for 11b and Ke = 274 nM for 11c), as compared to the most potent analog from this series, compound 11e. Some of the analogs also had contacts with hydrophobic residues in TM4 (Ile206 and Ile210). However, these TM4 interactions by themselves do not account entirely for the higher affinity seen with 11e; they were absent in 11d but did not severely affect its affinity. Compound 11f, which had the lowest affinity among C1 congeners (Ke = 4600 nM), showed only one H-bonding interaction (via protonated N6 and Asp155), and also hydrophobic interactions between its aromatic ring in the C1 benzyl side chain and Phe234 and Gly238 residues (Figure 9). The lack of a strong stabilizing Ser242 H-bonding interaction as seen in other analogs may be partially responsible for its lower affinity. It is interesting that compound 11a has a similar affinity to nantenine but binds in a different manner. It still utilizes Asp155 for H-bonding to the protonated N6 but lacks the Asn343 H-bond seen with nantenine. A number of hydrophobic interactions stabilize the molecule in the binding site including interactions with residues in TM6 and TM7 (Met345 and Val366 respectively) not seen in any of the other analogs. It is also noteworthy that whereas other analogs (11b-e) utilize Ser242 for H-bonding, 11a utilizes Ser242 for hydrophobic interactions.
Figure 7.
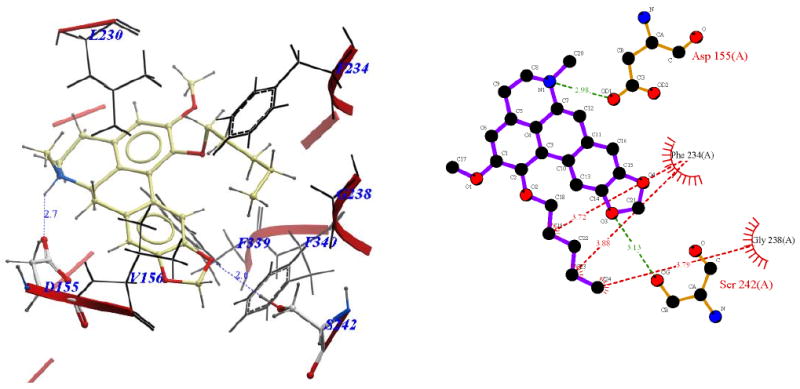
Binding pose of 11d represented with: a) ICM Pro and b) Ligplot
Figure 8.
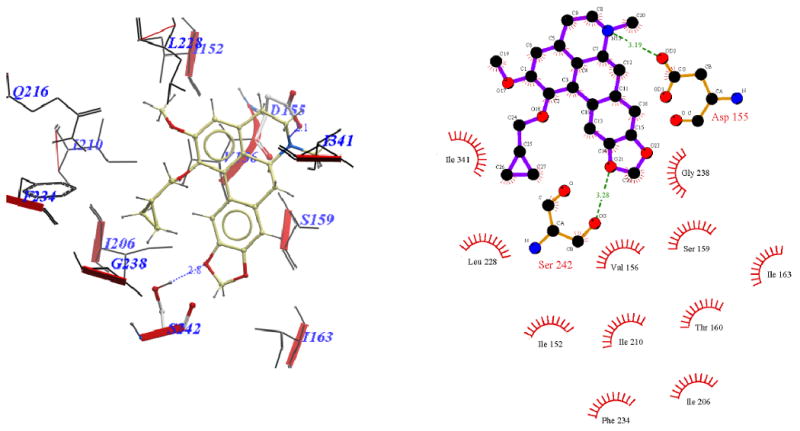
Binding pose of 11e represented with: a) ICM Pro and b) Ligplot
Figure 5.
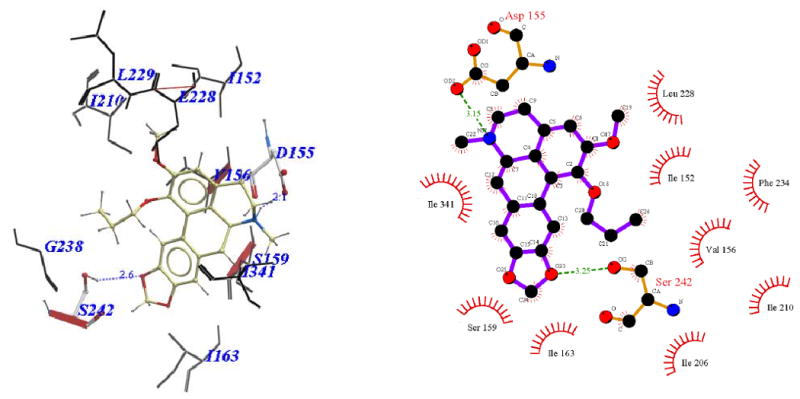
Binding pose of 11b represented with: a) ICM Pro and b) Ligplot
Figure 6.
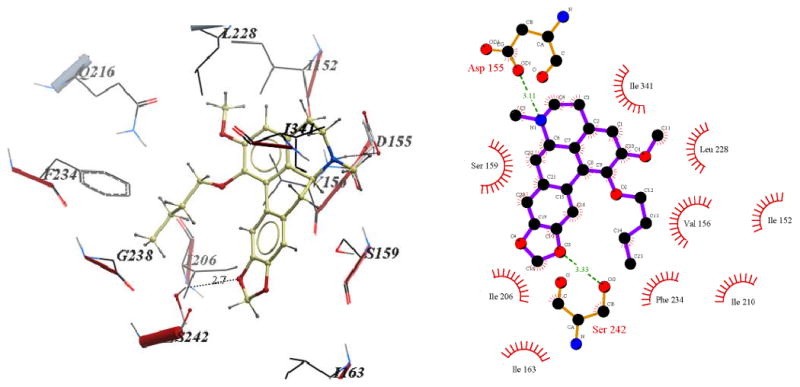
Binding pose of 11c represented with: a) ICM Pro and b) Ligplot
Overall, it is apparent that for this series of compounds, the Asn343 H-bond interaction seen with nantenine is not required for affinity to the receptor. However, it is clear that H-bonding with the highly conserved Asp155 is required for any appreciable affinity to the receptor; in that regard it seems to be necessary but not sufficient for high affinity.
Sowdhamini et al recently reported the construction of a homology model of the human 5-HT2A receptor based on a human β2 adrenoceptor template.48 In this study they docked a number of known 5-HT2A antagonists and inverse agonists (ketanserin, haloperidol, clozapine and risperidone) using the program Autodock 4.0. In the case of the antagonists ketanserin and haloperidol, the helix regions TM3, TM5, TM6, TM7 as well as ECL2 were involved in binding, whereas for the inverse agonists clozapine and risperidone TM3, TM5, TM6, ECL2 and ECL3 were involved. It has been suggested that 5-HT2A ligands may bind in two different spatially-allowed sites of the receptor.3 One site is bordered by transmembrane helices TM3, TM4, TM5 and TM6. A second site is flanked by TM1, TM2, TM3 and TM7. Asp155 (TM3) is a key residue in both sites and forms a H-bond contact to a protonated amine functionality of typical ligands. The results obtained in our work suggest that nantenine and the C1 alkyl analogs bind in the site bordered by TM3-TM6 and utilize the key Asp155 interaction but are oriented differently as shown diagramatically in Figure 10. Based on the residues that are used to establish key interaction to the ligands, the analogs also appear to bind in a manner different from that reported for other known antagonists such as ketanserin and haloperidol which supports the assertion that the structure of the ligand impacts its orientation and preference for residues in certain helices that are utilized for binding.48 Indeed it is clear that the binding pocket is tolerant of a variety of structural classes of ligands which may adopt different orientations between classes as well as within a given class as seen here with nantenine and its analogs.
Figure 10.
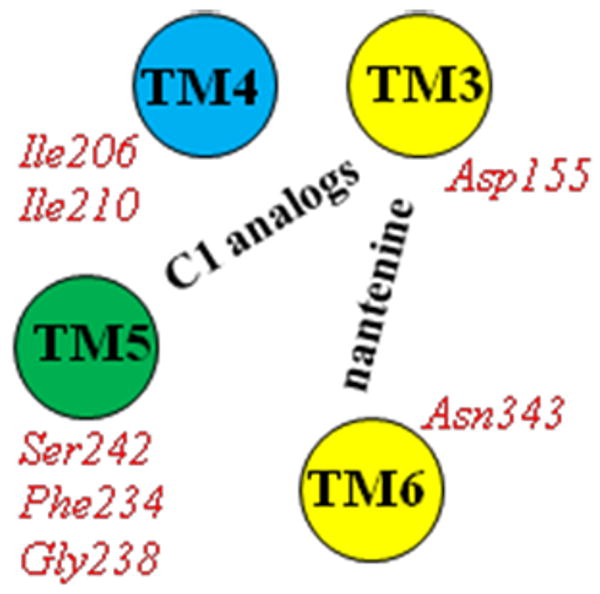
Orientation of nantenine and C1 alkyl analogs in the 5-HT2A receptor with key TM residue interactions (H-bond = yellow; hydrophobic = blue; H-bond plus hydrophobic = green)
Taken together, the results above suggest that the binding orientation of C1 analogs when compared to nantenine may be different, and that both H-bond and hydrophobic interactions are important for achieving high affinity to the receptor in this series. It is apparent that the pendant groups of nantenine C1 analogs project into a hydrophobic region on TM5 which include the residues Phe234 and Gly238. Given these insights, as well as information obtained from our SAR studies, it will be interesting to undertake in silico screening of a series of C1 aporphines with various hydrophobic substituents (branched alkyl, cycloalkyl, substituted aryl etc) and to subsequently synthesize and evaluate the compounds in vitro.
We did not obtain correlation between the in vitro affinity data and docking scores, so at this time it is clear that the model cannot be utilized in a predictive fashion for similar aporphine compounds. (This is not completely surprising since no scoring program can correctly rank every protein-ligand complex). The tolerability of the receptor for more than one binding modes of the same compound is one possible reason for this lack of correlation. For example, several possible binding modes have been reported for ketanserin.44, 48 Another possible reason for the low correlation observed is that we used a relatively small library of analogs with measurable affinity. For these reasons, it will be useful to synthesize and evaluate a larger set of nantenine analogs towards refinement of the model. Nevertheless, it will also be valuable to conduct in silico screening of known non-aporphinoid 5-HT2A ligands to test the applicability of our model in identifying agonists, partial agonists, antagonists and inverse agonists at this receptor. In the case of future generations of C1 alkyl analogs, given the good correlation seen with the HAdrb2_MODELLER/GOLD docking/scoring method, it will also be informative to evaluate these analogs using this method as a means of comparison of potential binding modes. For the antagonist ketanserin, we obtained a docking score predictable of high affinity for BRho_Modeller/GOLD combination (Table 2) which is promising. Clearly however, further evaluations on a structurally diverse set of ligands needs to be undertaken to determine the predictive value and robustness of our model for a broader set of compounds. Additionally, site-directed mutagenesis studies are required to shed light on the importance of the key residues identified in the affinity of the analogs. These are worthwhile avenues to pursue in future and the results presented here represent a good foundation for these directions.
Conclusions
We built four homology models and compared results of our in vitro data (5-HT2A apparent affinity of nantenine analogs) with the docking scores obtained for the analogs with each homology model. A bovine rhodopsin template built with MODELLER and evaluated by the GOLD docking algorithm gave docking scores in best gross agreement with our in vitro data although the correlation was poor. Our molecular docking studies showed that C1 nantenine analogs 11a-11f bind in a different manner than nantenine, but in a similar orientation among them. Visual inspection of the final poses of the high affinity compounds revealed two key H-bond interactions - one between Asp155 and protonated nitrogen, and the other between Ser242 and an oxygen atom in the methylenedioxy ring of the nantenine analogs. Hydrophobic interactions between the alkyl group at C1 and the residues Phe234 and Gly238 seem to be crucial for the enhanced affinity of this series at the 5-HT2A receptor. Our findings presented here will be useful in the future design of high affinity 5-HT2A ligands based on the nantenine aporphine core structure.
Experimental
General Methods and Instrumentation
Chemistry
HRESIMS spectra were obtained using an Agilent 6520 Q-TOF instrument. NMR data were collected on a Bruker 500 MHz machine with TMS as internal standard and CDCl3 as solvent unless stated otherwise. Chemical shift (δ) values are reported in ppm and coupling constants in Hertz (Hz). Melting points were obtained on a Mel-Temp capillary electrothermal melting point apparatus. Reactions were monitored by TLC with Analtech Uniplate silica gel G/UV 254 precoated plates (0.2 mm). TLC plates were visualized by UV (254 nm) and by staining with phosphomolybdic acid reagent followed by heating. Flash column chromatography was performed with Silicagel 60 (EMD Chemicals, 230-400 mesh, 0.04-0.063 μm particle size). All compounds evaluated were ≥95% pure as determined by analytical HPLC performed with an Agilent 1200 system equipped with PDA detector, Eclipse XDB-C18 column (4.6 × 150 mm) and eluted with methanol: water (80:20) at a flow rate of 1 mL/min.
Chemistry
3-(benzo[d][1,3]dioxol-5-yl)-4,5-dimethoxybenzaldehyde (3)
A mixture of Pd(PPh3)4 (2.35 g, 2.03 mmol) and commercially available 5-bromoveratraldehyde 1 (5.00 g, 20.60 mmol) in DME (250 mL) was stirred for 15 min at 20 °C under argon. 2M aqueous K2CO3 (71.5 mL, 142.80 mmol) was added to the mixture, followed by boronic acid 8 (6.77 g, 40.80 mmol) in DME. The mixture was refluxed for 18 h and then cooled to rt. The reaction mixture was treated with water and ethyl acetate and the layers separated. The organic extract was washed sequentially with 1M NaOH and water and dried over Na2SO4. The solvent was evaporated to give crude 3, which was purified by column chromatography (hexanes:EtOAc, 4:1). Compound 3 (5.66 g, 19.80 mmol, 96%) was obtained as a pale yellow oil: 1H NMR (500 MHz, CDCl3): δ 9.92 (s, 1H), 7.44 (d, 1H, J=2.0 Hz), 7.42 (d, 1H, J=2.0 Hz), 7.06 (d, 1H, J = 1.7 Hz), 7.00 (dd, 1H, J=8.0, 1.7 Hz), 6.89 (d, 1H, J=8.0 Hz), 6.02 (s, 2H), 3.97 (s, 3H); 3.71 (s, 3H); 13C NMR (125 MHz, CDCl3): δ 191.2, 153.7, 151.9, 147.5, 147.2, 135.7, 132.3, 130.8, 127.2, 122.7, 109.7, 109.3, 108.3, 101.2, 60.7, 56.1; HRESIMS calcd. for C16H14O5 [M]+ 286.0841; found 286.0841.
(E)-5-(2,3-dimethoxy-5-(2-nitrovinyl)phenyl)benzo[d][1,3]dioxole (4)
A mixture of aldehyde 3 (5.31 g, 18.56 mmol), ammonium acetate (1.43 g, 18.56 mmol), nitromethane (4.98 mL, 92.82 mmol), and glacial AcOH (100 mL) was refluxed for 4 h. After cooling to rt, the product was filtered and recrystallized from EtOH to afford 4 as a yellow solid (5.15 g, 15.65 mmol, 84%): mp 124-127 °C. 1H NMR (500 MHz, CDCl3): δ 7.97 (d, 1H, J=13.6 Hz), 7.55 (d, 1H, J=13.6 Hz), 7.14 (d, 1H, J=2.1 Hz), 7.02 (d, 1H, J=1.7 Hz), 7.01 (d, 1H, J=2.1 Hz), 6.96 (dd, 1H, J=8.0, 1.7 Hz), 6.88 (d, 1H, J=8.0 Hz), 6.02 (s, 1H), 3.95 (s, 3H); 3.68 (s, 3H); 13C NMR (125 MHz, CDCl3): δ 153.0, 150.3, 147.8, 147.5, 139.2, 136.6, 136.6. 130.9, 125.9, 125.1, 122.9, 111.0, 109.9, 108.5, 101.4, 61.0, 56.3; HRESIMS calcd. for C17H15NO6 [M+H]+ 330.0899; found 330.0971.
N-(3-(benzo[d][1,3]dioxol-5-yl)-4,5-dimethoxyphenethyl)formamide (5a)
TMSCl (5.00 mL, 24.2 mmol) was added to a vigorously stirred suspension of LiBH4 (0.35 g, 7.62 mmol) in anhyd. THF (15 mL) over a period of 2 min. After the gas evolution had ceased, the trimethylsilane was removed by purging the solution with argon. Then, over a period of 5 min, a solution of 4 (1.00 g, 3.04 mmol) in anhyd. THF (15 mL) was added and the mixture was heated for 18 h at reflux. After cooling to rt, the mixture was quenched carefully with methanol (25 mL) at 0 °C. The solvent was removed with a rotatory evaporator, the resulting residue dissolved in 20% aq KOH (50 mL), and extracted with DCM (3×50 mL). The combined extracts were dried (Na2SO4) and then concentrated under reduced pressure. This gave a crude oily primary amine product. The crude amine, ethyl formate (0.62 mL, 7.8 mmol) and triethylamine (0.86 mL, 6.20 mmol) were heated to reflux for 48 h. Removal of excess ethyl formate and triethylamine under reduced pressure gave a dark-brown oil, that was purified by column chromatography (MeOH:DCM, 1:99). Compound 5a (0.92 g, 2.80 mmol, 92% from 4) was obtained as an orange-red oil: 1H NMR (500 MHz, CDCl3): δ 8.12 (s, 1H), 7.05 (s, 1H), 6.98 (d, 1H, J=8.0 Hz), 6.85 (d, 1H, J=8.0 Hz), 6.73 (br. s, 2H), 5.98 (s, 2H), 3.88 (s, 3H), 3.56 (m, 5H), 2.82 (t, 2H, J=6.9 Hz); 13C NMR (125 MHz, CDCl3): δ 161.4, 153.1, 147.4, 146.8, 135.4, 134.4, 133.6, 131.9, 122.7, 122.6, 111.7, 109.9, 108.2, 101.2, 60.6, 56.1, 39.3, 35.5; HRESIMS calcd. for C18H20NO5 [M+H]+ 330.1263; found 330.1333.
N-(3-(benzo[d][1,3]dioxol-5-yl)-4,5-dimethoxyphenethyl)acetamide (5b)
The crude primary amine (0.95 g), prepared as described above from compound 4 (1.00 g, 3.03 mmol), acetyl chloride (0.35 mL, 5.5 mmol) and triethylamine (1.35 mL, 10.00 mmol) were mixed with dichloromethane (10 mL) and stirred for 6 hours at 0 °C. The reaction was treated with saturated sodium bicarbonate, extracted with DCM (3×35 mL), dried over Na2SO4 and concentrated under reduced pressure to give a yellow oil, that was subsequently purified by column chromatography (MeOH:DCM, 1:99). Compound 5b (0.92 g, 88% from compound 4) was obtained as an orange-red oil: 1H NMR (500 MHz, CDCl3): δ 8.12 (s, 1H), 7.06 (br. s, 1H), 6.98 (d, 1H, J=8.0 Hz), 6.86 (d, 1H, J=8.0 Hz), 6.73 (br. s, 1H), 5.98 (s, 2H), 5.71 (br. s, 1H), 3.88 (s, 3H), 3.58 (s, 3H), 3.51 (dd, 2H, J=13.2, 6.5 Hz), 2.78 (t, 2H, J=7.0 Hz), 1.95 (s, 3H); 13C NMR (125 MHz, CDCl3): δ 170.1, 153.1, 147.4, 146.8, 145.0, 135.4, 134.7, 131.9, 122.6, 122.5, 111.7, 109.8, 108.1, 101.0, 60.5, 56.0, 40.7, 35.6, 23.4; HRESIMS calcd. for C19H22NO5 [M+H]+ 344.1415; found 344.1488.
General procedure for synthesis of compounds 6a and 6b
To a stirred solution of amide 5 (1.40 mmol) in acetonitrile (5 mL) was added POCl3 (0.65 mL, 7.00 mmol) at rt, and the resulting mixture was heated at 50 °C for 12 h. The reaction mixture was concentrated, and the quaternary salt was dissolved in DCM (25 mL). After cooling to 0 °C, the mixture was diluted with water, made basic with 5% aqueous NH4OH, and extracted with DCM. The organic solution was washed with water, dried with anhydrous Na2SO4 and concentrated to give yellow crude dihydroisoquinolines. Sodium borohydride (1.04 g, 27.60 mmol) was added portion-wise to a stirred solution of the crude dihydroisoquinoline in methanol (20 mL) and the mixture was stirred at 0 °C for 3 h. The reaction mixture was concentrated, and excess NaBH4 was destroyed by adding water and glacial acetic acid. The mixture was then extracted with DCM (3×25 mL), dried over Na2SO4 and concentrated to give a crude oily product. This crude secondary amine product and formaldehyde solution, 37% (2.59 mL, 2.55 mmol) were mixed in anhydrous DCM (10 mL) and then treated with NaBH(OAc)3 (1.35 g, 6.38 mmol). The mixture was allowed to stir at rt for 24 h. The reaction was quenched with 5% aq sodium bicarbonate (25 mL), and extracted with ethyl acetate (2×25 mL), dried over Na2SO4 and concentrated to dryness. The residue was purified via silica flash column chromatography (MeOH:DCM, 2:98) to provide 6a and 6b.
8-(benzo[d][1,3]dioxol-5-yl)-6,7-dimethoxy-2-methyl-1,2,3,4-tetrahydroisoquinoline (6a)
Prepared from 5a in 88% overall yield as bright yellow crystals: mp 82-84 °C. 1H NMR (500 MHz, CDCl3): δ 6.87 (d, 1H, J=7.9 Hz), 6.70-6.65 (m, 3H), 6.01 (d, 2H, J=8.4 Hz), 3.88 (s, 3H), 3.54 (s, 3H), 3.15 (br. s, 2H), 2.94 (br. s, 2H), 2.65 (br. s, 2H), 2.35 (s, 3H); 13C NMR (125 MHz, CDCl3): δ 151.0, 147.3, 146.5, 144.8, 134.0, 129.8, 129.5, 125.8, 122.6, 111.6, 110.0, 108.2, 101.0, 60.8, 56.5, 55.8, 52.6, 46.3, 29.8; HRESIMS calcd. for C19H21NO4 [M]+ 327.1478; found 327.1471.
8-(benzo[d][1,3]dioxol-5-yl)-6,7-dimethoxy-1,2-dimethyl-1,2,3,4-tetrahydroisoquinoline (6b)
Mixture of atropisomers. Prepared from 5b in 90% overall yield as bright yellow crystals: mp 101-104 °C. 1H NMR (500 MHz, CDCl3): δ 6.87-6.84 (m, 1H), 6.76-6.67 (m, 1H), 6.66-6.65 (m, 2H), 6.03-6.00 (m, 2H), 3.87 (s, 3H), 3.78-3.71 (m, 1H), 3.53-49 (m, 3H), 3.08-3,07 (m, 2H), 2.77-2.71 (m, 2H), 2.38-2.46 (m, 3H), 0.97-0.94 (m, 3H); 13C NMR (125 MHz, CDCl3) (doubling of signals observed; average values for atropisomeric shifts reported): δ 151.0, 147.3, 146.5, 145.1, 134.6, 130.8, 129.9, 128.6, 123.3, 112.0, 110.6, 108.1, 101.0, 60.8, 55.7, 55.0, 44.1, 42.0, 26.3, 16.2; HRESIMS calcd. for C20H23NO4 [M]+ 341.1627; found 341.1628.
1,2-dimethoxy-5,6,6a,7-tetrahydro-4H-benzo[de][1,3]benzodioxolo[5,6-g]quinoline (8a)
Compound 7 (0.50 g, 1.18 mmol) in dry dichloromethane (10 mL) was stirred with anhydrous ZnBr2 (1.06 g, 4.71 mmol) under argon at room temperature for 4h. The reaction mixture was then quenched by adding a solution of saturated sodium bicarbonate (50 mL) and was extracted with DCM (2 × 60 mL), dried over Na2SO4 and concentrated to give 8a as yellow oil. We found this compound to be unstable; for long-term storage 8a was converted to its hydrochloride salt by treatment with HCl in ether. Data for the hydrochloride salt follows: 1H NMR (500 MHz, DMSO-d6): δ 7.76 (s, 1H), 7.03 (s, 1H), 6.90 (s, 1H), 6.06 (s, 2H), 4.20 (br. d, 1H, J = 11.0 Hz), 3.83 (s, 3H), 3.60 (s, 3H), 3.33 (s, 2H), 3.23 (dt, 1H, J = 12.6, 4.5 Hz), 3.13 (m, 1H), 2.97 (dd, 1H, J = 14.2, 4.5 Hz), 2.90 (dd, 1H, J = 14.2, 1.1 Hz), 2.80 (t, 1H, J = 14.2 Hz); 13C NMR (125 MHz, DMSO-D6): δ 152.8, 146.7, 146.6, 144.5, 128.2, 126.8, 125.8, 124.5, 121.6, 111.6, 108.7, 108.1, 101.3, 59.9, 55.9, 51.8, 40.3, 33.0, 25.1; HRESIMS: calcd. for C19H19NO4 [M]+ 325.1314; found 325.1313.
1,2-dimethoxy-6-ethyl-5,6,6a,7-tetrahydro-4H-benzo[de][1,3]benzodioxolo[5,6-g]quinoline (8b)
Compound 8a (0.30 g, 0.92 mmol) and acetaldehyde solution, (5.4 mL, 1.84 mmol) were mixed in anhydrous DCM (20 mL) and then treated with sodium triacetoxy-borohydride (0.98 g, 4.61 mmol). The mixture was allowed to stir at room temperature overnight. The reaction was quenched with 5% aq sodium bicarbonate (25 mL), and extracted with dichloromethane (2 × 25 mL), dried over sodium sulfate and concentrated to dryness. The residue was purified via flash chromatography (MeOH:DCM, 1:99) to provide 8b (0.23 g, 0.65 mmol, 71%) as a bright red oil: 1H NMR (500 MHz, CDCl3): δ 7.92 (s, 1H), 6.75 (s, 1H), 6.59 (s, 1H), 5.97 (s, 1H), 5.96 (s, 1H), 3.86 (s, 3H), 3.64 (s, 3H), 3.24 (dd, 1H, J = 13.7, 3.1 Hz), 3.16 (dd, 1H, J = 11.3, 5.1 Hz), 3.12-3.04 (m, 2H), 2.97 (dd, 1H, J = 13.7, 3.8 Hz), 2.70-2.67 (dd, 1H, J = 13.4, 3.0 Hz), 2.62-2.43 (m, 3H), 1.14 (t, 3H, J = 7.1 Hz); 13C NMR (125 MHz, CDCl3): δ 151.8, 146.4, 146.3, 144.4, 131.0, 129.0, 127.9, 127.1, 125.6, 110.6, 108.9, 108.2, 100.8, 60.2, 59.2, 55.8, 48.3, 47.8, 35.0, 29.3, 10.8; HRESIMS: calcd. for C21H24NO4 [M+H]+ 354.1627; found 354.1650.
1,2-dimethoxy-6-acetyl-5,6,6a,7-tetrahydro-4H-benzo[de][1,3]benzodioxolo[5,6-g]quinoline (8c)
Compound 8a (0.10 g, 0.31 mmol) and acetyl chloride (0.01 mL, 0.41 mmol) were mixed in anhydrous DCM (20 mL) and then treated with triethylamine (0.12 mL, 0.86 mmol). The mixture was allowed to stir at 0 °C for 3h under argon. The reaction was quenched with saturated sodium bicarbonate (20 mL), extracted with dichloromethane (3 × 20 mL), dried over sodium sulfate and concentrated to dryness. The residue was recrystallized from ethyl acetate to obtain 8c (0.07 g, 0.19 mmol, 61%) as a white solid (mixture of rotamers; NMR data for major rotamer provided); mp 190-192 °C. 1H NMR (500 MHz, CDCl3): δ 8.01 (s, 1H), 6.76 (s, 1H), 6.64 (s, 1H), 6.01 (s, 1H), 5.99 (s, 1H), 5.04 (dd, 1H, J = 13.8, 3.8 Hz), 4.00 (dd, 1H, J = 13.3, 2.3 Hz), 3.89 (s, 3H), 3.69 (s, 3H), 3.01-2.64 (m, 5H), 2.29 (s, 3H); 13C NMR (125 MHz, CDCl3): δ 169.7, 169.1, 152.2, 151.9, 146.8, 146.7, 146.6, 145.1, 144.9, 131.3, 130.5, 128.7, 127.9, 125.8, 124.9, 110.9, 108.9, 108.8, 101.1, 60.0, 55.9, 53.0, 50.4, 41.9, 36.6, 34.0, 30.7, 29.8, 22.6, 21.6; HRMS: m/z (%) [M+H]+ 404 (100).
1,2-dimethoxy-6-methanesulfonyl-5,6,6a,7-tetrahydro-4H-benzo[de][1,3]benzodioxolo[5,6-g]quinoline (8d)
Compound 8a (0.10 g, 0.31 mmol) and methanesulfonyl chloride (0.04 mL, 0.52 mmol) were mixed in anhydrous DCM (20 mL) and then treated with triethylamine (0.12 mL, 0.86 mmol). The mixture was allowed to stir at 0 °C − rt overnight. The reaction was quenched with saturated sodium bicarbonate (25 mL), and extracted with dichloromethane (2×25 mL), dried over sodium sulfate and concentrated to dryness. The residue was recrystallized in diethyl ether to obtain 8d (0.07 g, 0.17 mmol, 55%) as a white solid; mp 197-199 °C. 1H NMR (500 MHz, CDCl3): δ 7.98 (s, 1H), 6.76 (s, 1H), 6.63 (s, 1H), 6.00 (s, 1H), 5.98 (s, 1H), 4.10 (br. d, 1H, J = 11.3 Hz), 3.90 (s, 3H), 3.66 (s, 3H), 3.25-3.15 (m, 4H), 2.96-2.2.94 (m, 2H), 2.88 (s, 3H); 13C NMR (125 MHz, CDCl3): δ 152.4, 146.8, 146.7, 145.2, 130.8, 128.5, 128.0, 124.8, 124.5, 110.8, 108.9, 108.8, 101.0, 60.06, 55.9, 53.0, 46.1, 40.4, 37.4, 29.4; HRESIMS: calcd. for C20H22NO6S [M+H]+ 404.1090; found 404.1160.
5HT2A Ke determination in FLIPR assay
The Ke data were obtained by monitoring the ability of test compounds to inhibit serotonin-mediated stimulation of the human 5-HT2A receptor heterologously expressed in Chinese hamster ovary (CHO) cells. Agonist dose response curves were run in the presence or absence of a single concentration of test compound. The Ke values are a measure of antagonist affinity determined in a functional assay and these values were calculated using the formula: Ke=[L]/(DR − 1), where [L] is the concentration of test compound and DR is the ratio of agonist EC50 in the presence or absence of test compound, respectively. At least two different concentrations of test compound were used to calculate the Ke, and their concentrations were chosen such that the agonist EC50 exhibited at least a 4-fold shift to the right.
Molecular modeling and docking
Sequence Alignment, Disulfide Bond Assignment, Model Building, Evaluation/Selection of the best Model and Energy Minimization
Amino acid sequence of the human 5-HT2A receptor was retrieved from NCBI protein database49. BLASTP was used to search for a suitable template from the Protein Structure Database (PDB) for homology modeling. Sequence alignment was carried out with the ClustalX50 software (using the Gonnet series matrix with the “gap open” and “gap elongation” penalties of 10 and 0.2 respectively) and ICM Pro 3.6 (based on ZEGA sequence alignment - Needleman and Wunsch algorithm with zero gap and penalties). The alignment was then manually refined to ensure a perfect alignment of the highly conserved residues of the GPCR superfamily, according to Baldwin et al. The conserved disulfide bond between residue C148 at the beginning of TM3 and the cysteine C228 in the middle of extracellular loop 2 (a feature common to many GPCR receptors) was also created and was kept as a constraint in the geometric optimization. To make the homology models, the programs MODELLER (Discovery Studio 2.0 of Accelrys Inc) and ICM Pro 3.6 were used. From several crystal structures available in PDB, as a template, we selected bovine rhodopsin (PDB code 1U19) since in many previous reports it has been used as a good template for the 5-HT2A receptor and also because its crystal structure is solved at high resolution 2.20Å. We also selected β2-adrenergic receptor (PDB code 2RH1) as a template, because this is presently the best resolved structure (2.40Å) of the available GPCR neurotransmitter receptors. Twenty models were built for each of the templates using MODELLER and one model for each template using ICM Pro 3.6. All homology models were evaluated using several different model evaluation tools such as PROCHECK,37 Verify3D,36 ERRAT,35 WHAT_CHECK51 and PROVE37 from SAVES33 metaserver. After evaluating each model using the above methods, the final models were selected - one from each template, which fits best by criteria of selection in each method. The best docking solution was energy minimized with Charmm as implemented in Discovery Studio 2.0 of Accerlys Inc., by applying 500 cycles of Smart Minimizer Algorithm, followed by gradient minimization until RMS gradient was lower than 0.01 kcal/molÅ. The models were renumbered, according to their original sequence. The four selected homology models were than superimposed with their templates using the protocol in Discovery Studio 2.0: structure superimposing by sequence alignment, and RMSD determined for each model.
Preparation of the Analogs and Receptors, Grid Generation and Docking Experiments
Nantenine and all analogs were drawn as 2D structures with ChemDraw Ultra version 9.0 with a formal positive charge centered on the nitrogen, and then energy minimized through Chem3D Ultra version 9.0/MOPAC, Job Type: Minimum RMS Gradient of 0.010 kcal/mol and RMS distance of 0.1 Å, and saved as MDL MolFiles (*.mol) for purpose of docking with ICM Pro, or as SYBYL2 files (*.mol2) for the purpose of docking with the GLIDE and the GOLD software. Before docking with the GLIDE and the GOLD each analog was prepared using LigPrep, an application that is available through Maestro 7.5 (a Linux graphical interface for Schrödinger06). After employing the energy minimization through MacroModel application, each 3D structure was saved as an SD file (*.sdf), and combined into a single analogs library SD file, to be used for docking experiments.
ICM docking
First the binding site of homology receptors was identified. The binding site was reviewed and adjusted: ICM made a box around the ligand binding site based on the information entered in the receptor setup section. The position of the box encompassed the residues expected to be involved in ligand binding. Then the receptor maps were made: Energy maps of the environment within the docking box were constructed. Flexibility of the receptor residues was set to 4.0. Interactive docking was used to dock nantenine and the other analogs. The thoroughness level was set to the maximum value of 10. ICM scores were obtained after this procedure.
GLIDE docking
Each homology receptor was prepared by the Protein Preparation mode of GLIDE, and then the receptor grid was generated by specifying Asp155 as a central residue and selecting Extra Precision docking within 20 (x001FA) of Asp155. After this step the G-scores were determined and the docking poses of each analog were visually inspected using the GLIDE Pose Viewer.
GOLD docking
As input file for the GOLD docking, both homology models were used as a PDB file. In the GOLD Wizard, setup hydrogens were added and the binding site was defined as the residues that are falling within 15(x001FA) of Asp155. The GOLD Score was chosen as fitness function. After docking was finished the top-scoring receptor-analog complexes were visually inspected. High-scoring complexes that did not meet H-bond Asp155-NH3+ requirement were discarded.
Visualization of Selected Docking Posses
All final receptor-analog complexes, obtained from three different programs were visualized using 3D Window of Discovery Studio 2.0. H-bonds, hydrophobic interactions and distances for each 3D structure were calculated and plotted by LigPlot v4.4.2. H-bonds are indicated by dashed lines between the atoms involved, while hydrophobic contacts are represented by an arc with spokes radiating towards the ligand atoms they contact.
Supplementary Material
Figure 4.
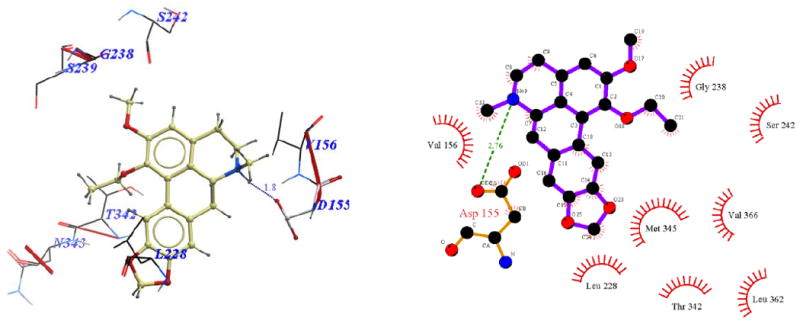
Binding pose of 11a represented with: a) ICM Pro, b) Ligplot
Scheme 3.
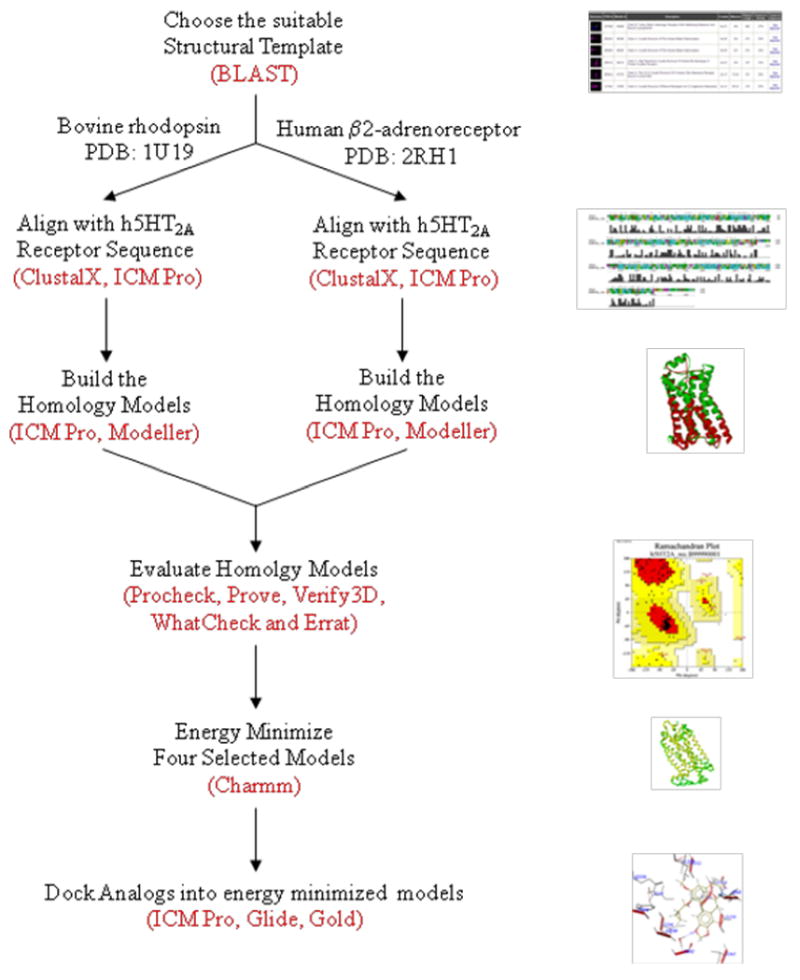
Flowchart summarizing the approach used in this study
Acknowledgments
This publication was made possible by Grant Number RR03037 and R03DA025910 from the National Center for Research Resources (NCRR) and National Institute of Drug Abuse (NIDA) respectively, components of the National Institutes of Health. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIH or its divisions.
List of Abbreviations
- 5-HT
5-hydroxytryptamine, serotonin
- AcOH
acetic acid
- BOC
t-butoxycarbonyl
- BLAST
Basic Local Alignment Search Tool
- BRho
bovine rhodopsin
- CDI
1,1′-carbonyldiimidazole
- D
aspartic acid
- DCM
dichloromethane, methylene chloride
- DMA
N,N-dimethylacetamide
- DMAP
4-(dimethylamino)pyridine
- DME
1,2-dimethoxyethane
- DMF
dimethylformamide
- DMSO
dimethyl sulfoxide
- ECL
extracellular loop
- ee
enantiomeric excess
- ESI-MS
electrospray ionization mass spectrometer
- GA
Genetic Algorithm
- GPCR
G-protein-coupled receptor
- ICM
Internal Coordinate Mechanics
- HPLC
high-performance liquid chromatography
- HAda2a
human adenosine A2A receptor
- HAdrb2
human β2-adrenergic receptor
- ICL
intracellular loop
- MC
Monte Carlo
- MDMA
3,4-methylenedioxy-N-metamphetamine
- PDB
Protein Data Bank
- Phe F
phenylalanine
- RMSD
root-mean-square deviation
- SAR
structure-activity relationship
- SAVES
Structural Analysis and Verification Server
- Ser, S
serine
- SRho
squid rhodopsin
- TAdrb1
turkey β1-adrenergic receptor
- THF
tetrahydrofuran
- TM
transmembrane
- TMSCl
t-methylsilyl chloride
- Trp W
tryptophan
- Tyr Y
tyrosine
- Val V
valine
- ZEGA
Zero Gap
- Zrms
Z-score RMS
- Zsm
Z-score standard mean
- Zstd
Z-score standard deviation
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kroeze WK, Kristiansen K, Roth BL. Curr Top Med Chem. 2002;2:507. doi: 10.2174/1568026023393796. [DOI] [PubMed] [Google Scholar]
- 2.Bojarski AJ. Curr Top Med Chem. 2006;6:2005. doi: 10.2174/156802606778522186. [DOI] [PubMed] [Google Scholar]
- 3.Runyon SP, Mosier PD, Roth BL, Glennon RA, Westkaemper RB. J Med Chem. 2008;51:6808. doi: 10.1021/jm800771x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sanger DJ, Soubrane C, Scatton B. Ann Pharm Fr. 2007;65:268. doi: 10.1016/s0003-4509(07)90046-2. [DOI] [PubMed] [Google Scholar]
- 5.Shelton RC, Papakostas GI. Acta Psychiatr Scand. 2008;117:253. doi: 10.1111/j.1600-0447.2007.01130.x. [DOI] [PubMed] [Google Scholar]
- 6.Meltzer HY, Li Z, Kaneda Y, Ichikawa J. Prog Neuropsychopharmacol Biol Psychiatry. 2003;27:1159. doi: 10.1016/j.pnpbp.2003.09.010. [DOI] [PubMed] [Google Scholar]
- 7.Levin ED, Slade S, Johnson M, Petro A, Horton K, Williams P, Rezvani AH, Rose JE. Eur J Pharmacol. 2008;600:93. doi: 10.1016/j.ejphar.2008.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nic Dhonnchadha BA, Fox RG, Stutz SJ, Rice KC, Cunningham KA. Behav Neurosci. 2009;123:382. doi: 10.1037/a0014592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fantegrossi WE, Kiessel CL, Leach PT, Van Martin C, Karabenick RL, Chen X, Ohizumi Y, Ullrich T, Rice KC, Woods JH. Psychopharmacology (Berl) 2004;173:270. doi: 10.1007/s00213-003-1741-2. [DOI] [PubMed] [Google Scholar]
- 10.Legendre O, Pecic S, Chaudhary S, Zimmerman SM, Fantegrossi WE, Harding WW. Bioorg Med Chem Lett. 2010;20:628. doi: 10.1016/j.bmcl.2009.11.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chaudhary S, Pecic S, Legendre O, Navarro HA, Harding WW. Bioorg Med Chem Lett. 2009;19:2530. doi: 10.1016/j.bmcl.2009.03.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Aranda R, Villalba K, Ravina E, Masaguer CF, Brea J, Areias F, Dominguez E, Selent J, Lopez L, Sanz F, Pastor M, Loza MI. J Med Chem. 2008;51:6085. doi: 10.1021/jm800602w. [DOI] [PubMed] [Google Scholar]
- 13.Chambers JJ, Nichols DE. J Comput Aided Mol Des. 2002;16:511. doi: 10.1023/a:1021275430021. [DOI] [PubMed] [Google Scholar]
- 14.Lafrance M, Blaquiere N, Fagnou K. Eur J Org Chem. 2007:811. [Google Scholar]
- 15.Schmidhammer H, Jennewein HK, Krassnig R, Traynor JR, Patel D, Bell K, Froschauer G, Mattersberger K, Jachs-Ewinger C, Jura P, Fraser GL, Kalinin VN. J Med Chem. 1995;38:3071. doi: 10.1021/jm00016a010. [DOI] [PubMed] [Google Scholar]
- 16.Jerman JC, Brough SJ, Gager T, Wood M, Coldwell MC, Smart D, Middlemiss DN. Eur J Pharmacol. 2001;414:23. doi: 10.1016/s0014-2999(01)00775-0. [DOI] [PubMed] [Google Scholar]
- 17.Indra B, Matsunaga K, Hoshino O, Suzuki M, Ogasawara H, Ishiguro M, Ohizumi Y. Can J Physiol Pharmacol. 2002;80:198. doi: 10.1139/y02-019. [DOI] [PubMed] [Google Scholar]
- 18.Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Nucleic Acids Res. 1997;25:3389. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Warne T, Serrano-Vega MJ, Baker JG, Moukhametzianov R, Edwards PC, Henderson R, Leslie AG, Tate CG, Schertler GF. Nature. 2008;454:486. doi: 10.1038/nature07101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jaakola VP, Griffith MT, Hanson MA, Cherezov V, Chien EY, Lane JR, Ijzerman AP, Stevens RC. Science. 2008;322:1211. doi: 10.1126/science.1164772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cherezov V, Rosenbaum DM, Hanson MA, Rasmussen SG, Thian FS, Kobilka TS, Choi HJ, Kuhn P, Weis WI, Kobilka BK, Stevens RC. Science. 2007;318:1258. doi: 10.1126/science.1150577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Okada T, Sugihara M, Bondar AN, Elstner M, Entel P, Buss V. J Mol Biol. 2004;342:571. doi: 10.1016/j.jmb.2004.07.044. [DOI] [PubMed] [Google Scholar]
- 23.Mobarec JC, Sanchez R, Filizola M. J Med Chem. 2009;52:5207. doi: 10.1021/jm9005252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rasmussen SG, Choi HJ, Rosenbaum DM, Kobilka TS, Thian FS, Edwards PC, Burghammer M, Ratnala VR, Sanishvili R, Fischetti RF, Schertler GF, Weis WI, Kobilka BK. Nature. 2007;450:383. doi: 10.1038/nature06325. [DOI] [PubMed] [Google Scholar]
- 25.Westkaemper RB, Glennon RA. Curr Top Med Chem. 2002;2:575. doi: 10.2174/1568026023393741. [DOI] [PubMed] [Google Scholar]
- 26.Evers A, Hessler G, Matter H, Klabunde T. J Med Chem. 2005;48:5448. doi: 10.1021/jm050090o. [DOI] [PubMed] [Google Scholar]
- 27.Sousa SF, Fernandes PA, Ramos MJ. Proteins. 2006;65:15. doi: 10.1002/prot.21082. [DOI] [PubMed] [Google Scholar]
- 28.Higgins DG, Sharp PM. Gene. 1988;73:237. doi: 10.1016/0378-1119(88)90330-7. [DOI] [PubMed] [Google Scholar]
- 29.Abagyan R, Totrov M, Kuznetsov D. J Comput Chem. 1994;15:488. [Google Scholar]
- 30.Abagyan RA, Batalov S. J Mol Biol. 1997;273:355. doi: 10.1006/jmbi.1997.1287. [DOI] [PubMed] [Google Scholar]
- 31.Baldwin JM, Schertler GF, Unger VM. J Mol Biol. 1997;272:144. doi: 10.1006/jmbi.1997.1240. [DOI] [PubMed] [Google Scholar]
- 32.Sali A, Blundell TL. J Mol Biol. 1993;234:779. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 33.Li M, Wang B. J Mol Model. 2007;13:1237. doi: 10.1007/s00894-007-0245-0. [DOI] [PubMed] [Google Scholar]
- 34.Morris AL, MacArthur MW, Hutchinson EG, Thornton JM. Proteins. 1992;12:345. doi: 10.1002/prot.340120407. [DOI] [PubMed] [Google Scholar]
- 35.Colovos C, Yeates TO. Protein Sci. 1993;2:1511. doi: 10.1002/pro.5560020916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Eisenberg D, Luthy R, Bowie JU. Methods Enzymol. 1997;277:396. doi: 10.1016/s0076-6879(97)77022-8. [DOI] [PubMed] [Google Scholar]
- 37.Pontius J, Richelle J, Wodak SJ. J Mol Biol. 1996;264:121. doi: 10.1006/jmbi.1996.0628. [DOI] [PubMed] [Google Scholar]
- 38.Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, Repasky MP, Knoll EH, Shelley M, Perry JK, Shaw DE, Francis P, Shenkin PS. J Med Chem. 2004;47:1739. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- 39.Halgren TA, Murphy RB, Friesner RA, Beard HS, Frye LL, Pollard WT, Banks JL. J Med Chem. 2004;47:1750. doi: 10.1021/jm030644s. [DOI] [PubMed] [Google Scholar]
- 40.Jones G, Willett P, Glen RC. J Mol Biol. 1995;245:43. doi: 10.1016/s0022-2836(95)80037-9. [DOI] [PubMed] [Google Scholar]
- 41.Stortz CA. J Comput Chem. 2005;26:471. doi: 10.1002/jcc.20185. [DOI] [PubMed] [Google Scholar]
- 42.Wallace AC, Laskowski RA, Thornton JM. Protein Eng. 1995;8:127. doi: 10.1093/protein/8.2.127. [DOI] [PubMed] [Google Scholar]
- 43.Salom D, Lodowski DT, Stenkamp RE, Le Trong I, Golczak M, Jastrzebska B, Harris T, Ballesteros JA, Palczewski K. Proc Natl Acad Sci U S A. 2006;103:16123. doi: 10.1073/pnas.0608022103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dezi C, Brea J, Alvarado M, Ravina E, Masaguer CF, Loza MI, Sanz F, Pastor M. J Med Chem. 2007;50:3242. doi: 10.1021/jm070277a. [DOI] [PubMed] [Google Scholar]
- 45.Bruno A, Guadix AE, Costantino G. J Chem Inf Model. 2009;49:1602. doi: 10.1021/ci900067g. [DOI] [PubMed] [Google Scholar]
- 46.Shah JR, Mosier PD, Roth BL, Kellogg GE, Westkaemper RB. Bioorg Med Chem. 2009;17:6496. doi: 10.1016/j.bmc.2009.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Selent J, Lopez L, Sanz F, Pastor M. ChemMedChem. 2008;3:1194. doi: 10.1002/cmdc.200800074. [DOI] [PubMed] [Google Scholar]
- 48.Kanagarajadurai K, Malini M, Bhattacharya A, Panicker MM, Sowdhamini R. Mol Biosyst. 2009;5:1877. doi: 10.1039/b906391a. [DOI] [PubMed] [Google Scholar]
- 49.Wheeler DL, Barrett T, Benson DA, Bryant SH, Canese K, Chetvernin V, Church DM, Dicuccio M, Edgar R, Federhen S, Feolo M, Geer LY, Helmberg W, Kapustin Y, Khovayko O, Landsman D, Lipman DJ, Madden TL, Maglott DR, Miller V, Ostell J, Pruitt KD, Schuler GD, Shumway M, Sequeira E, Sherry ST, Sirotkin K, Souvorov A, Starchenko G, Tatusov RL, Tatusova TA, Wagner L, Yaschenko E. Nucleic Acids Res. 2008;36:D13. doi: 10.1093/nar/gkm1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Thompson JD, Plewniak F, Poch O. Nucleic Acids Res. 1999;27:2682. doi: 10.1093/nar/27.13.2682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Feldman HJ, Hogue CW. Proteins. 2000;39:112. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.