

Abstract
A-kinase-anchoring protein 250 (AKAP250; gravin) acts as a scaffold that binds protein kinase A (PKA), protein kinase C and protein phosphatases, associating reversibly with the β2-adrenergic receptor. The receptor-binding domain of the scaffold and the regulation of the receptor–scaffold association was revealed through mutagenesis and biochemical analyses. The AKAP domain found in other members of this superfamily is essential for the scaffold–receptor interactions. Gravin constructs lacking the AKAP domain displayed no binding to the receptor. Metabolic labeling studies in vivo demonstrate agonist-stimulated phosphorylation of gravin and enhanced gravin–receptor association. Analysis of the AKAP domain revealed two canonical PKA sites phosphorylated in response to elevated cAMP, blocked by PKA inhibitor, and essential for scaffold–receptor association and for resensitization of the receptor. The AKAP appears to provide the catalytic PKA activity responsible for phosphorylation of the scaffold in response to agonist activation of the receptor as well as for the association of the scaffold with the receptor, a step critical to receptor resensitization.
Keywords: β-adrenergic receptor/AKAP/gravin/protein kinase A/protein kinase C/scaffold
Introduction
Recent advances in our understanding of the workings of cell signaling have highlighted the roles of molecules, often referred to as ‘scaffolds’, in organizing the interactions of molecules in signaling pathways. In Saccharomyces cerevisiae, for example, STE5 is an essential component of the pheromone-mediated mitogen-activated protein kinase (MAPK) pathway. The STE5 scaffold recruits MAPK cascade members STE11, STE7 and FUS3 to generate specificity for the pheromone pathway (Kim et al., 1998). In vertebrates, A-kinase-anchoring proteins (AKAPs) constitute a diverse family of such scaffold molecules, organizing not only protein kinases, but also protein phosphatases and other molecules, into multivalent signaling complexes (Pawson and Scott, 1997; Colledge and Scott, 1999; Bauman and Scott, 2002). AKAPs bind the RII subunits of the cAMP-dependent kinase (PKA), providing discrete localization of PKA within the cell. The muscle-specific AKAP, termed ‘mAKAP’, coordinates multivalent signaling complexes in both the sarcoplasmic reticulum and perinuclear membrane of muscle cells (Michel and Scott, 2002). Another family of AKAPs are derived from a single gene, producing the splice products AKAP350, AKAP450, CG-NAP and Yotiao that are involved in coordinating multivaltent signaling complexes with N-methyl-d-aspartate (NMDA) receptors, protein phosphatases PP1 and calcineurin, as well as PKA (Michel and Scott, 2002; Bauman and Scott, 2002). Gravin (also known as AKAP250 and AKAP12) is another prominent member of the AKAP family, a family that includes >50 members. As scaffold molecules, the AKAPs display binding sites for several well known protein kinases and phosphatases. Gravin, for example, binds the RII subunit of PKA, protein kinase C (PKC), protein phosphatase 2B (PP2B) (Nauert et al., 1997; Shih et al., 1999; Fan et al., 2001b) and the β2-adrenergic receptor (β2AR) (Fan et al., 2001b), a prototypic member of the superfamily of G-protein-linked receptors (GPCRs) (Morris and Malbon, 1999). Suppression of gravin leads to disruption of internalization and resensitization (Lin et al., 2000), critical stages in GPCR regulation (Morris and Malbon, 1999; Shih et al., 1999; Fan et al., 2001b).
The organization of AKAPs has been revealed by painstaking analysis of the structure/function of these scaffold proteins. Docking sites have been described for several major AKAP signaling partners, including PKC, phosphodiesterase 4D3, protein phosphatases and the RII subunit of PKA (Alto et al., 2002). The site through which the β2AR binds dynamically to AKAP250 has been deduced for this GPCR and is localized predominantly to the C-terminal, cytoplasmic domain of the receptor (Fan et al., 2001b). The site by which AKAPs, such as AKAP79 (Cong et al., 2001) and gravin (Lin et al., 2000), dock GPCRs such as the β2AR remains to be established. Likewise, the functional role of the AKAP domain common to several AKAPs, including gravin, SSeCKS and AKAP79, has not been elucidated. In the current study, we report major progress in both pursuits, identifying the AKAP domain of gravin both as the docking site for the β2AR and as an important target for dynamic regulation. We demonstrate that the AKAP domain is a substrate for phosphorylation by PKA, an event catalyzed locally by the catalytic subunits of PKA bound by gravin and necessary to drive binding of the scaffold to the cell membrane via the embedded β2AR.
Results
Gravin–β2AR interaction enhanced by receptor activation
Gravin–β2AR binding was established by pull-down assays of hemagglutinin (HA)-tagged gravin and subsequent immunoblotting for β2AR in the complex (Figure 1) from extracts of human epidermoid A431 cells expressing endogenous β2AR. β2AR binding of gravin occurs in the absence of added agonist (isoproterenol; Iso), but is increased >2.5-fold by activation of the β2AR or by the plant diterpene FK (Figure 1A), agents that increase intracellular cAMP concentrations and PKA activity. Prior exposure (30 min) to the PKA inhibitor KT5720 blocks scaffold–receptor binding in response to receptor activation (or treatment with FK) (Figure 1A). A 12 h exposure of cells to KT5720 markedly attenuates the basal level of gravin binding by the receptor while completely blocking the ability of agonist to stimulate the association (Figure 1B). Although gravin is a well-known substrate for PKC (Nauert et al., 1997a; Gelman, 2002), inhibition of PKC activity by calphostin C does not alter β2AR–gravin binding stimulated by either receptor activation or FK (Figure 1C). These data implicate increased intracellular concentrations of cAMP and PKA activation as determinants in the association of scaffold with the receptor. We tested the role of cAMP directly in wild-type A431 cells, using either FK or the chlorophenylthio (CPT)-analog of cAMP to activate PKA. CPT-cAMP was as effective as FK in stimulating the association of the scaffold with the receptor (Figure 1D). The pull-downs performed with anti-gravin antibodies isolate gravin in a complex with endogenous β2AR, demonstrating an interaction between endogenous scaffold and receptor that is regulated by cAMP (Figure 1D). Investigation of the distribution of gravin in a low-speed particulate membrane fraction (P) versus the supernatant (S) reveals that Iso stimulated redistribution of gravin from the supernatant fraction to the membrane fraction (Figure 1E). Thus the association of the scaffold gravin with the membrane-localized β2AR can be enhanced dramatically by agonist stimulation (FK) of the direct addition of CPT-cAMP to the medium of the cells.
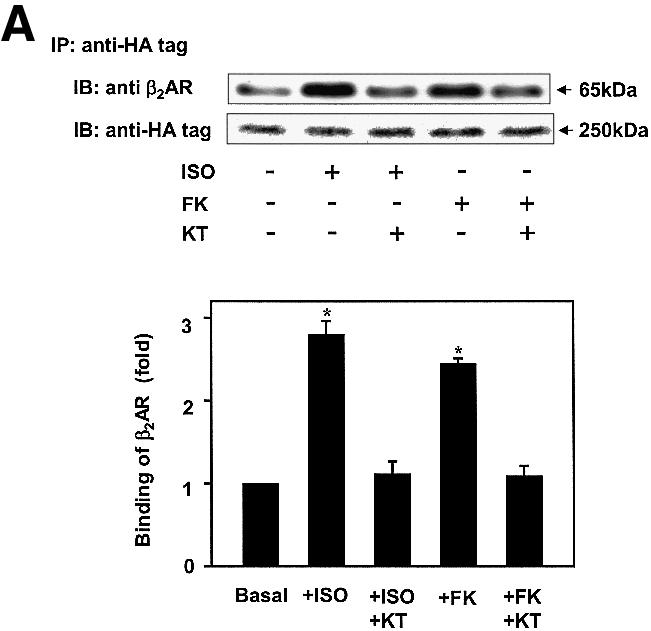

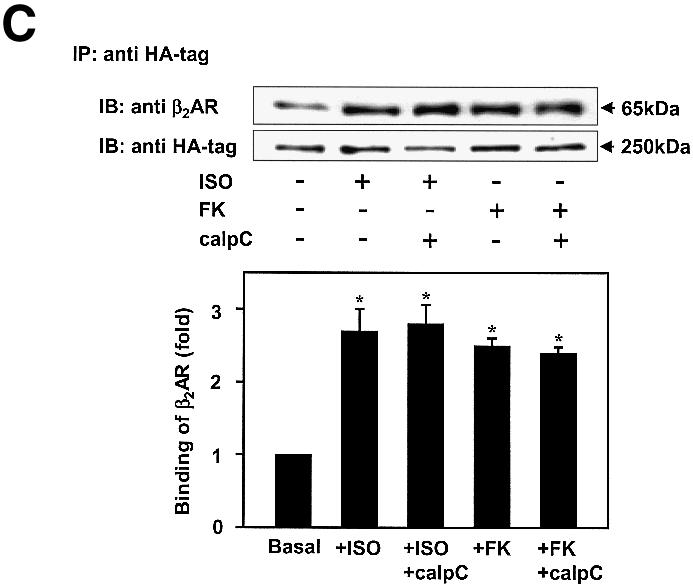

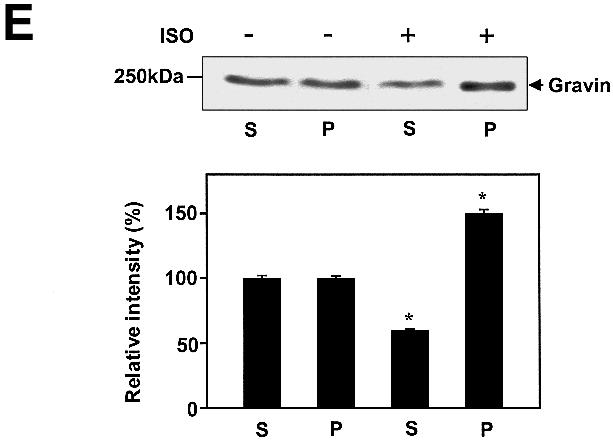
Fig. 1. Gravin associates with the β2AR and cell membrane upon receptor activation. A431 cells were left untreated or pre-treated with either 1 µM KT5720 (KT) (A), 0.1 µM KT5720 (KT) for 12 h (B) or 1 µM calphostin C (calp C) (C) for 30 min. Cells were then challenged with either 10 µM Iso (ISO) or 20 µM FK for 20 min. Whole-cell lysates were incubated with antibodies against the HA tag covalently conjugated to protein A/G– agarose beads. The immune complexes were subjected to SDS–PAGE, transferred to nitrocellulose blots, and the blots probed with an antibody specific for the β2AR or with the anti-HA antibody. Wild-type A431 cells were treated with either 20 µM FK or 20 µM of the chlorophenylthio analog of cAMP (CPT-cAMP) for 20 min, lysed, and the lysates subjected to pull-downs of endogenous gravin, SDS–PAGE, and immunoblotting of gravin and β2AR (D). The distribution of gravin between a low-speed crude membrane pellet (P) and supernatant (S) subcellular fractions was determined by subjecting equivalent amounts of each fraction prepared from cells either left untreated (–) or treated (+) with 10 µM Iso to SDS–PAGE and immunoblotting with anti-gravin antibodies (E). Protein loading for SDS–PAGE of P and S fractions was 50 and 30 µg/lane, respectively. The immune complexes were made visible by the chemiluminescence method. The relative densities of the bands were determined with a Bio-Rad imaging densitometer (GS-700) and MultiAnalyst densitometer software. The images presented are representative of at least three separate determinations performed with different immunoprecipitations. IP, immunoprecipitation; IB, immunoblotting. The results displayed in the bar graphs are from three separate determinations and are presented as the mean values ± SEM. The asterisk denotes statistical significance with P < 0.05.
Gravin binds the β2AR via the AKAP domain
The sites for PKC and PKA binding have been identified (Colledge and Scott, 1999). The domain(s) of AKAPs responsible for GPCR binding to AKAP scaffolds as a group are unknown. A core sequence of ∼31 residues, termed an AKAP ‘motif’, is highly conserved in several AKAPs. Three such homologous motifs appear in the ‘AKAP domain’ of gravin, within regions of residues 603–633, 753–783 and 797–827 (Figure 2A). Mutant, HA-tagged versions of gravin deficient in one or more of the PKC-, AKAP- and PKA-binding domains were expressed individually in A431 cells (Figure 2B) and employed in pull-down assays to evaluate the β2AR binding capacity of each gravin mutant (Figure 2C). Stable transfectant clones expressing constructs at comparable levels were selected for study. On SDS–PAGE analysis, all of the gravin constructs behave as dimeric, unless subjected to electrophoresis in the presence of 8 M urea (Figure 2B). Using 8% rather than 10% acrylamide gels provides sufficient resolution to identify the presence of some gravin dimer with Mr > 400 kDa, also sensitive to dissolution by 8 M urea (data not shown). Even in the presence of 8 M urea, most gravin constructs display some dimerization, providing insight into their nature. The entire AKAP domain of gravin itself was found to be an essential domain, enabling the binding of the scaffold to the β2AR. Deletion of residues 653–938, disrupting the AKAP domain while retaining the single, most N-terminal AKAP motif (residues 603–633), abolished the ability of gravin to bind β2AR (Figure 2C). Thus, the presence of a single AKAP motif was insufficient for receptor binding. Complementary pull-down assay with antibodies to the β2AR and assay of gravin binding confirmed the inability of the gravin constructs lacking the full AKAP domain to bind to the receptor (Figure 2D). Previously we showed that the region Arg329–Leu413 of the C-terminal, cytoplasmic tail of the β2AR is the receptor domain necessary for binding gravin. Herein we show that the AKAP domain of gravin is essential for gravin binding to the β2AR, and perhaps should be re-named the ‘β2AR-binding domain’.



Fig. 2. The AKAP domain of gravin mediates binding between the scaffold and the GPCR. Full-length and truncated mutants of HA-tagged gravin were generated as a schematic representation of the topological organization of the human gravin fragment (A). The proteins were expressed in A431 cells following transient transfection, subjected to SDS–PAGE on 10% acrylamide gels (B) performed in the absence (left-hand panel) or presence of 8 M urea (right-hand panel), subjected to immunoblotting, and then detected using anti-HA antibodies for staining. To detect gravin–GPCR association, we incubated whole-cell lysates with antibodies against either the HA tag or the β2AR (CM-4) covalently linked to protein A/G–agarose beads. The immune complexes were subjected to SDS–PAGE, transferred to nitrocellulose and probed with an antibody specific for either the β2AR or the HA tag. The ability of the full-length and the truncated mutants of gravin to form signaling complexes with endogenous β2AR was probed in whole-cell lysates using pull-down assays of the HA-tagged gravin molecules by anti-HA antibodies and detection of the signaling complex members using SDS–PAGE and immunoblotting (C). The blots were stained with antibodies against the β2AR. Complementary pull-down assays were performed using antibodies to the β2AR to isolate the signaling complexes from whole-cell lysates. The immune complexes were subjected to SDS–PAGE and immunoblotting (D). In this case, the blots were stained with antibodies to the HA-tag to reveal the presence of HA-tagged gravin molecules, if present, in the β2AR-based signaling complexes. The data presented are representative of at least three separate determinations performed on separate occasions. The positions of the molecular weight standards (kDa) are displayed at the left and the molecular weights of the HA-tagged gravin or gravin truncated versions are identified at the right. IP, immune precipitation; IB, immunoblotting.
β2AR stimulates phosphorylation of gravin by PKA, not PKC
The C-terminal region of gravin (1540–1553) displays a docking site for the RII subunit of PKA. Since activation of β2AR and of PKA enhances scaffold–β2AR binding (Figure 1), we explored the role of protein phosphorylation of gravin in response to β2AR activation (Figure 3). Gravin and its mouse homolog SSeCKS are well-known substrates for phosphorylation by PKC and PKA. In cells metabolically labeled with [32P]Pi, stimulation of the β2AR with Iso provokes phosphorylation of gravin (Figure 3A). Elevation of intracellular cAMP by treatment with FK also provokes gravin phosphorylation. Treatment of cells with phorbol 12-myristoyl 13-acetate (PMA) activates PKC and provoked an 8- to 10-fold increase in gravin phosphorylation (Figure 3A). Inhibition of PKA with KT5720 (KT) blocks the ability of β2AR activation to stimulate phosphorylation of the AKAP scaffold (Figure 3B). Gravin not only docks PKC, but is a well-known substrate for PKC (Gelman, 2002). Treatment of cells with the PKC inhibitor calphostin C, in contrast to treatment with the PKA inhibitor KT5720, fails to influence the phosphorylation of gravin stimulated by activation of the β2AR (Figure 3C), suggesting a primary role for PKA in receptor-induced phosphorylation of this AKAP. To test the specificity of the read-out, we made use of Ht31 a 24 amino acid, conserved amphipathic helix peptide which blocks the interaction of AKAP family members with PKA (Hausken and Scott, 1996). Previously we have shown that Ht31 blocked the ability of the β2AR to bind PKA and gravin (Shih et al., 1999). Treating the cells with Ht31 effectively blocks the ability of either Iso or FK to stimulate phosphorylation of gravin (Figure 3D). These studies extend earlier studies showing Ht31 blunting the function of gravin (Shih et al., 1999). Ht31 is shown herein to effectively block the phosphorylation of gravin stimulated in response to agonist activation.
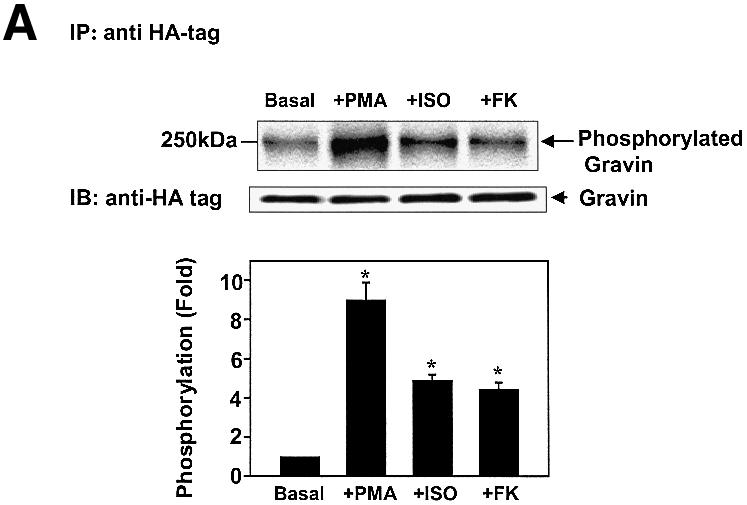



Fig. 3. Phosphorylation of gravin in response to receptor activation is mediated by PKA. Clones stably expressing HA-tagged gravin were labeled metabolically with [32P]orthophosphate for 12 h. HA-tagged gravin was subjected to immunoprecipitation from cell lysates, SDS–PAGE, and analysis for phosphorylation. Following the period of metabolic labeling, cells were stimulated either with 10 µM Iso or with 20 µM FK to activate PKA (A). To stimulate PKC, cells were incubated with 1 µM PMA for 30 min (A). Some of the clones were pre-treated with 1 µM KT7520 (KT), a selective PKA inhibitor (B). Others were pre-treated with 1 µM calphostin C (calp C), a selective inhibitor of PKC (C). To test for specificity, cells were treated with the Ht31 peptide (+Ht31), a 24 amino acid, conserved amphipathic helix peptide which blocks the interaction of AKAP family members with PKA (D). As a control, the Ht31-Pro mutant that has a prolyl insertion to disrupt the AKAP PKA RII-binding motif and is inactive was similarly employed in the cell treatment. Gravin and the molecular weight standards (kDa) are identified on the autoradiograph. The relative densities of the bands were determined with a Bio-Rad imaging densitometer (GS-700) and MultiAnalyst densitometer software. The data shown are representative of three independent experiments. IP, immunoprecipitation; IB, immunoblotting. Representative immunoblots are displayed in each panel. The results displayed in the bar graphs are from three separate determinations and are presented as the mean values ± SEM. The asterisk denotes statistical significance with P < 0.05.
β2AR stimulates phosphorylation of PKA sites in the AKAP domain of gravin
Since inhibition of PKA by KT5720 blocks β2AR–gravin binding as well as β2AR-stimulated phosphorylation of gravin, the AKAP domain of gravin was scanned for canonical sites of PKA phosphorylation. Three canonical sites targeting Ser627, Ser696–698 and Ser772 of gravin were identified (Figure 4A). Alanine substitution of these sites in combination (Ser627Ala, Ser696–698Ala, Ser772Ala) essentially abolishes phosphorylation of gravin in response to either receptor activation or FK (Figure 4B). Alanine substitution of all canonical PKA sites, of Ser696–698Ala or of Ser772Ala sites individually abolishes the ability of gravin to bind β2AR in response to receptor activation (Figure 4C). In addition, alanine substitution of Ser696–698, of Ser772 or of all of the PKA sites in combination reduced the amount of β2AR–gravin complex formation in the basal, unstimulated state (Figure 4C). The Ser627Ala mutant, in contrast, displayed normal levels of β2AR–gravin complex in the agonist-stimulated and basal states. The availability of Ser696–698 and Ser772 in the AKAP domain for phosphorylation by PKA is critical for receptor–scaffold interactions.



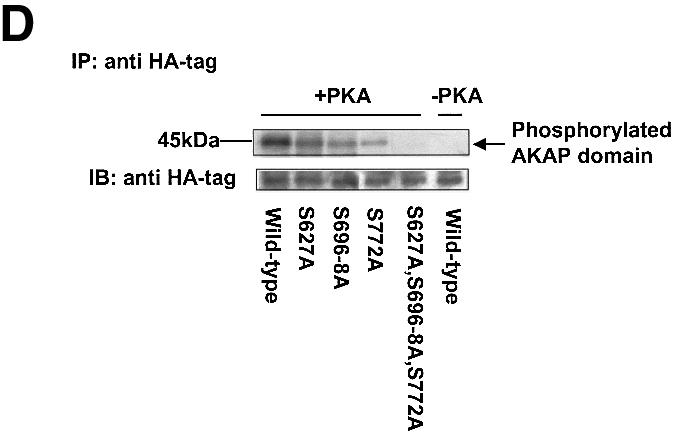
Fig. 4. Receptor activation stimulates phosphorylation of the AKAP domain of the gravin scaffold: mapping of sites of phosphorylation. Canonical sites for phosphorylation of the AKAP domain of gravin by PKA (Ser627, 696–698 and 772) are shown in (A). Cells transfected with wild-type HA-tagged gravin or gravin mutants with serine to alanine triple mutation of the PKA phosphorylation sites within the AKAP domain were subjected to metabolic labeling with [32P]orthophosphate for 12 h. The cells were then stimulated with either 10 µM Iso (ISO) or 20 µM FK for 20 min (B). Whole-cell lysates were prepared and subjected to immunoprecipitation with anti-HA antibody and detected with an antibody specific for the β2AR, as described in Materials and methods (C). The data shown are representative of three independent experiments. IP, immunoprecipitation; IB, immunoblotting. Representative immunoblots are displayed in each panel. The results displayed in the bar graphs are from three separate determinations and are presented as the mean values ± SEM. The asterisk denotes statistical significance with P < 0.05. For in vitro phosphorylation studies, wild-type AKAP domain or AKAP domains with specific serine to alanine substitution were incubated in phosphorylation assay buffer in the absence and presence of purified, catalytic subunits of PKA. Wild-type HA-AKAP domain or the HA-AKAP domain with serine to alanine substitutions at each individual site (S627A, S696,697,698A, S772A) as well as the HA-AKAP domain with triple mutation of the PKA phosphorylation sites were transfected into cells, cell lysates were immunoprecipitated with anti-HA antibody, and in vitro phosphorylation of the AKAP domain was performed as described in Materials and methods (D). The relative densities of the protein bands were determined by use of an imaging densitometer (GS-700, Bio-Rad) and MultiAnalyst densitometer software (Bio-Rad). The data shown are representative of three independent experiments. IP, immunoprecipitation; IB, immunoblotting.
Under in vitro phosphorylation conditions, the purified catalytic subunit of PKA phosphorylates HA-tagged gravin (Figure 4D). The three individual alanine substitution mutants, Ser627Ala, Ser696–698Ala and Ser772Ala, were phosphorylated by PKA to a lesser extent than wild-type gravin (Figure 4D). The gravin mutant with an AKAP domain devoid of PKA sites displayed no phosphorylation under these conditions. These observations suggest that in vivo each canonical site in the AKAP domain may contribute to the overall phosphorylation of gravin by PKA. Mutation of two PKA substrate sites, Ser696–698Ala and Ser772Ala, alone or in tandem, but not Ser627Ala alone, virtually eliminates receptor–scaffold association in the basal, unstimulated state (Figure 4C).
Phosphorylation of gravin AKAP domain catalyzed by associated PKA
The central role of PKA-catalyzed phosphorylation for gravin binding to the β2AR provoked the question of whether the catalytic subunit of PKA responsible for the phosphorylation is donated by the RII domain of the AKAP itself, or by a PKA localized elsewhere in the cell. A gravin mutant lacking the RII-binding site was constructed and employed for study of this fundamental question. Remarkably, deletion of the PKA RII-binding site of gravin (Δ1540–1553) provoked a loss of the ability of β2AR activation (or FK) to stimulate either gravin phosphorylation (Figure 5A) or gravin binding to the receptor (Figure 5B).


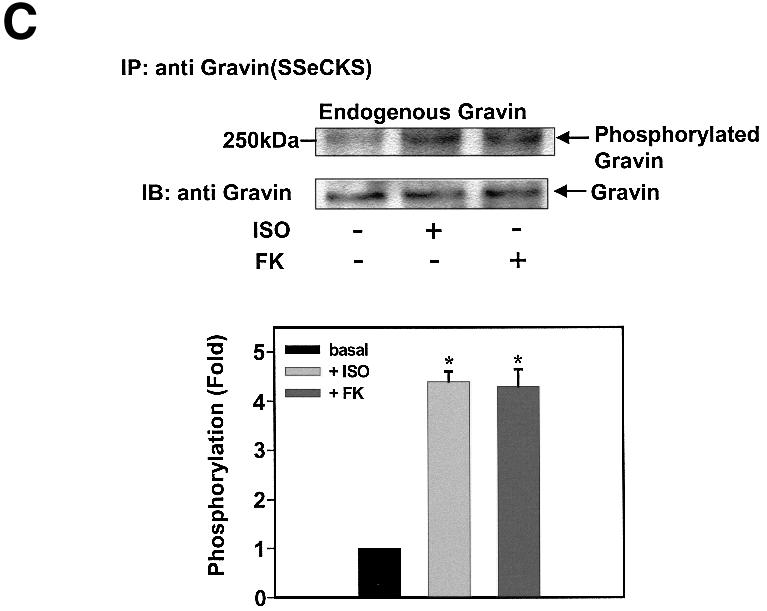

Fig. 5. Deletion of the RII-binding site at HA-gravin abolishes phosphorylation of gravin and gravin binding of receptor. Cells transfected with wild-type HA-tagged gravin (A) or a gravin mutant lacking the RII-binding domain (Δ1540–1553) (A and C) were subjected to metabolic labeling with [32P]orthophosphate for 12 h and then stimulated with either 10 µM Iso (ISO) or 20 µM FK for 20 min (A). The HA-tagged gravin and RII-binding site-deficient gravin were collected from whole-cell extracts by immunoprecipitation with anti-HA antigen antibodies. The extracts depleted of HA-tagged proteins were then subjected to immunoprecipitation with anti-gravin (anti-SSeCKS) antibodies (C), to collect the wild-type gravin. The phosphorylation state of gravin and its mutants was established as described in Materials and methods. Cells transfected with either the HA-tagged gravin or the HA-tagged mutant gravin deficient in the RII-binding domain (Δ1540–1553) were stimulated with either 10 µM Iso (ISO) or 20 µM FK for 20 min. The association of receptor with gravin was analyzed by immunoprecipitation of receptor and immunostaining of blots from SDS–PAGE of the immune complexes (B). Phosphorylation of the β2AR in response to cAMP or Iso stimulation was increased by addition of wild-type gravin (D). Crude cell membranes were incubated in phosphorylation assay buffer supplemented with wild-type or mutant gravin forms prepared as described in Materials and methods, in the absence or presence of 5 µM cAMP or 10 µM Iso (ISO) for 30 min. The phosphorylation of β2AR was detected as described in Materials and methods. The relative densities of the labeled bands were determined by use of an imaging densitometer (GS-700, Bio-Rad) and MultiAnalyst densitometer software (Bio-Rad). The data shown are representative of three independent experiments. IP, immunoprecipitation; IB, immunoblotting. Representative immunoblots are displayed in each panel. The results displayed in the bar graphs are from three separate determinations and presented as the mean values ± SEM. The asterisk denotes statistical significance with P < 0.05.
We tested these same clones to ascertain if endogenous wild-type gravin expressed in these same cells was phosphorylated in response to receptor activation. Although the mutant gravin lacking the RII-binding domain expressed in these cells failed to be phosphorylated in response to receptor activation, the endogenous gravin in these same cells displayed increased phosphorylation (Figure 5C). Under in vitro phosphorylation conditions, wild-type and the RII-deficient forms of gravin are phosphorylated equivalently by exogenously added, purified PKA catalytic subunit (data not shown). Indeed, phosphorylation of the β2AR itself by PKA in crude cell membranes supplemented with exogenous cAMP occurs only in the presence of wild-type gravin (Figure 5D). The addition of the gravin mutant lacking the canonical PKA sites or of the gravin mutant lacking the RII-binding site failed to enable phosphorylation of the β2AR by PKA under these conditions. Thus, phosphorylation of gravin in response to β2AR activation is catalyzed by the PKA catalytic subunit donated by the gravin molecule to which the RII subunit is docked.
Metabolic labeling reveals sites of in vivo phosphorylation of gravin as PKA substrates
We sought to establish the precise site(s) of AKAP phosphorylation in response to receptor activation in vivo. Clones expressing HA-tagged gravin were treated with Iso. The HA-tagged gravin was purified from the cell extracts by affinity chromatography (Figure 6A), with the 250 kDa species dominant in a sample, with much lesser amounts of the 140 and 180 kDa cleaved species. The gravin purified from the cells stimulated with Iso was subjected to protease digestion, and then analyzed by matrix-assisted laser desorpton ionization (MALDI) mass spectrometry (Figure 6B–D). The mass spectrum from 700–900 m/z reveals the presence of the tryptic phosphopeptide containing the canonical 625RPSESDK631 (Mr 818 + 79 = 897) from Iso-treated cells that reverts to the non-phosphorylated peak (Mr 818) upon treatment with purified calf intestinal alkaline phosphatase (Figure 6B). Analysis at the mass range of 950–1250 m/z reveals the phosphopeptide 695GSSSDEEGGPK705 harboring the phosphorylated PKA site (Mr 1049 + 158 = 1207) from Iso-treated cells that reverts to the non-phosphorylated peak (Mr 1049) upon treatment with calf alkaline phosphatase (Figure 6C). A mass equivalent to two, but not three, phosphate groups was sensitive to protein phosphatase treatment, suggesting that two PKA sites, probably S696 and S697, are substrates for PKA in the AKAP domain. The mass spectrum from 1800–2500 m/z reveals the endoproteinase Glu-C digest phosphopeptide 761SFKRLVTPRKKSKSKLE777 (Mr 2033 + 79 = 2112) from Iso-treated cells that reverts to the non-phosphorylated product (Mr 2032) upon treatment with calf alkaline phosphatase (Figure 6D). These determinations demonstrate the precise sites of the AKAP domain phosphorylated in response to receptor activation in vivo. The two PKA substrate sites, Ser696–698 and Ser772, alone in the AKAP domain, are essential to provoke gravin binding to this GPCR.
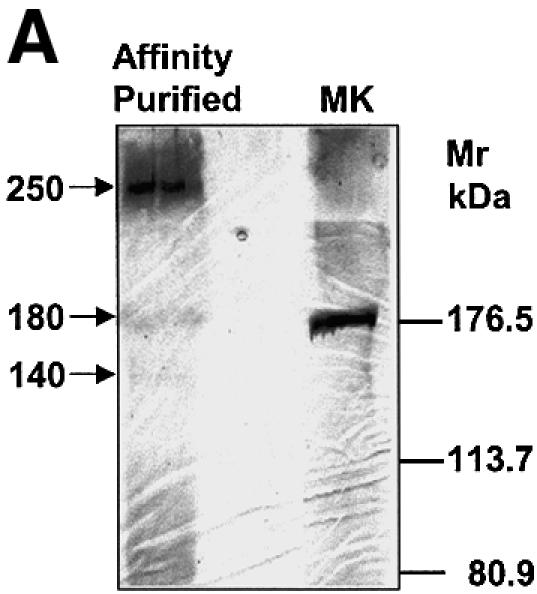



Fig. 6. Phosphorylation of the AKAP domain of gravin in response to receptor activation: mapping of phosphorylation sites by LS MS. Clones stably transfected with HA-tagged gravin were treated with 10 µM Iso to stimulate phosphorylation of gravin. The HA-tagged gravin was isolated from the whole-cell lysates using anti-HA antibody immobilized on an affinity matrix. A portion of the purified HA-gravin isolated from Iso-treated cells was subjected to treatment with alkaline phosphatase to release phosphate groups from sites of phosphorylation. (A) Purified HA-tagged gravin identified by SDS–PAGE and silver staining. Marker proteins (MK) were employed to estimate the Mr of the bands. The gravin-containing bands were excised and subjected to in-gel digestion using either trypsin (B and C) or endoproteinase Glu-C (D), as described in Materials and methods. The samples were processed and then subjected to LS MS. The phosphorylated and subsequently dephosphorylated peptides are identified in bold. The data shown are representative of three independent experiments.
Specific mutations of gravin attenuate its role in resensitization of β2AR
We tested the effects of the two key mutations of gravin that alter the ability of agonist to stimulate gravin phosphorylation and association with the β2AR (Figure 7). The read-outs for these experiments are desensitization and resensitization of the β2AR. Desensitization was measured at 30 min post-stimulation with agonist (Figure 7A), whereas resensitization is the loss of agonist-stimulated desensitization measured at 30, 45 and 60 min following a wash-out (W) of agonist and addition of propranolol (10 µM, Figure 7B). For the wild-type cells, the amount of desensitization declines/resensitization increases from 30 min following washout (W30) to 60 min following washout (W60), when desensitization is no longer observed. Suppression of gravin expression using antisense morpholinos (Morpho) suppresses the resensitization following washout of agonist, as reported earlier (Shih et al., 1999a; Fan et al., 2001b). Transient expression of wild-type gravin rescues the resensitization in antisense morpholino-treated cells, demonstrating levels of resensitization observed in the wild-type cells. Expression of either the RII-deleted gravin (Δ1540–1553) or the triple mutant (S627A, S696–698A, S772A) does not rescue the loss of resensitization in the antisense morpholino-treated cells, even though expression levels of all three forms of gravin were equivalent (not shown). These data provide a compelling, functional understanding of the essential roles of phosphorylation of the β2AR-binding domain and the RII-binding domain of gravin on the ability of the scaffold to participate in receptor resensitization.

Fig. 7. Expression of a mutant gravin with deficiency of the RII-binding site (Δ1540–1553) or of a gravin mutant lacking the AKAP sites for phosphorylation by PKA (S627A, S696–698A, S772A) is functionally impaired in catalyzing receptor resensitization. Wild-type A431 cells were treated or not with antisense morpholinos to suppress expression of endogenous gravin. The cells then were transiently transfected with an expression vector harboring either HA-tagged wild-type or mutant gravin to ascertain if the mutations altered the ability of gravin to regulate the desensitization or resensitization of the β2AR. Receptor desensitization after 30 min of agonist (10 µM Iso) was unaffected by any of these treatments (A). Resensitization of the receptor, initiated by the wash-out of Iso and addition of 10 µM propranolol, was then assayed at 30, 45 and 60 min post-wash-out (B). The time-dependent loss of agonist-induced desensitization reflects the resensitization process. The data displayed are mean values ± SEM from three independent assays.
Discussion
AKAPs, by definition, anchor PKA via its RII subunit to specific cellular locales in which PKA plays a regulatory role, influencing the activities of ion channels and other GPCR-regulated effectors (Pawson and Scott, 1997). The family of AKAPs includes gravin (AKAP250), shown to organize PKA in a multivalent GPCR signaling complex along with PKC, PP2B, Src and other components (Fan et al., 2001b). How the dynamic interaction between AKAPs (e.g. AKAP79 and AKAP250) and GPCRs is regulated remains poorly understood. We made use of the receptor-induced interaction between gravin and β2AR signaling complexes to probe this question. Gravin not only tethers PKA to the β2AR signaling complex, but is shown herein itself to be a substrate for PKA-catalyzed phosphorylation. A primary target of the study was to map the region(s) of gravin important in binding to a GPCR, the β2AR. The AKAP domain of gravin is shown to constitute the site for gravin docking to the β2AR. Following receptor activation, gravin binding to the receptor increases, a process dependent upon PKA-catalyzed phosphorylation of two canonical PKA sites (Ser696–698 and Ser772) located within the AKAP domain of gravin. Mutation (alanine substitution) of these serinyl residues blocks PKA-catalyzed phosphorylation of gravin, binding of gravin to the β2AR and the functional role of gravin in receptor resensitization. These observations provide compelling evidence in support of an essential role for PKA in targeting AKAP250 to the cell membrane-localized β2AR. Interesting, the docking site of the β2AR for gravin, the C-terminal, cytoplasmic tail of the receptor, is also a substrate for phosphorylation by PKA (Morris and Malbon, 1999). Thus, the phosphorylation by PKA appears to play a central role in docking interactions between the AKAP250 scaffold and this GPCR.
Does PKA anchored to gravin through the RII-binding domain catalyze the phosphorylation of gravin itself? Earlier studies demonstrated that either suppression of gravin or introduction of Ht31 peptide, a peptide that specifically disrupts RII subunit binding of AKAPs, nearly abolishes the resensitization of β2AR following activation and desensitization (Shih et al., 1999; Lin et al., 2000). A complementary strategy used herein employs a gravin mutant deficient in the RII-binding site. The RII-deficient mutant is no longer phosphorylated in response to receptor activation by agonist (or to elevations in cAMP sufficient to activate PKA), although capable of some binding to the β2AR. The RII-docking site-deficient gravin no longer demonstrates, however, agonist-induced association with β2ARs. Remarkably, this loss of AKAP domain phosphorylation and binding to receptor by the RII-binding site-deficient mutant form of gravin occurs in cells that express a complement of wild-type gravin. In these cells, the wild-type gravin is capable of phosphorylation by its PKA and association with β2AR. The PKA docked on this wild-type gravin does not appear to be able to act in trans to phosphorylate the mutant gravin that lacks the RII-docking site, but possesses a normal AKAP domain. This observation provides compelling evidence that the PKA responsible for AKAP domain phosphorylation in gravin is the kinase anchored to the RII-binding site of the same polypeptide. These observations suggest that gravin targets the association with β2AR through activation of the PKA that it binds and directs phosphorylation of two key PKA sites within the AKAP domain. The association of gravin with the β2AR involves an 83 residue region in the C-terminal cytoplasmic tail of this GPCR and the 250 residue AKAP domain with AKAP motifs conserved among several members of the AKAP family of scaffold proteins, both being substrates for phosphorylation by PKA.
These data provide a compelling argument for the tight spatial activation of PKA that occurs in this well-known paradigm of G-protein-mediated signaling. Gravin not only coordinates the multivalent signaling complexes to the cell membrane by dynamic association with GPCRs, but also insures that the activation of the PKA proceeds within a highly restricted spatial domain. Furthermore, we provide the first demonstration of the function of the AKAP domain. The AKAP domain provides a reversible, dynamic docking site for the β2AR and, presumably, other GPCRs. This would explain the basis for the association of both gravin and AKAP79 with β2AR (Lin et al., 2000; Cong et al., 2001). We propose that for gravin, this AKAP domain might be better denoted the ‘β2AR-docking domain’, reflecting its role in docking the β2AR and presumably other GPCRs. Future analyses of AKAP-coordinated, multivalent signaling complexes might include screens for GPCRs, to test further the range of receptors that might provide localization of these multivalent signaling complexes to the cell membrane. The observation that the gravin scaffold appears to behave as a dimer may provide an additional dimension in understanding the organization of AKAP-based, β2AR signaling complexes. Finally, the loss of gravin disrupts the ability of the GPCR-docked signaling complexes to resensitize the β2AR, a process catalyzed in part by the protein phosphatase PP2B present in the complex (Dell’Acqua et al., 2002; Oliveria et al., 2003). The PKA-mediated phosphorylation and enhanced association of gravin with the β2AR may be required for the proper environment in which PP2B dephosphorylates both the receptor and the scaffold. Understanding the operation of these solid-state, multivalent signaling complexes would benefit from higher resolution of their structures, particularly in the regions of the AKAP responsible for the reversible docking of protein kinases, phosphatases and receptors.
Materials and methods
HA-tagged fusion proteins
The HA-tagged gravin fusion protein was generated using PCR-derived DNA fragments from human gravin–green fluorescent protein (GFP) (Nauert et al., 1997). To amplify the N-terminal 2210 bp fragment, the sense primer contained a 5′ NheI site and sequence encoding the HA tag (MYPYDVPDPYA) at the N-terminus of gravin (ACCGCTAGCATGT ACCCATACGATGTTCCAGATTACGCTATGGGCGCCGGGAGCT CCACC), the antisense primer contained a BamHI site corresponding to the sequence of gravin (AGCAAGGATCCCGTCTGTCCCCGTCTC). The 2210 bp fragment was cloned into pCDNA3 (Invitrogen) between the NheI and BamHI sites. The C-terminal 3139 bp fragment of gravin was generated by a pair of primers, the sense primers contain the BamHI site corresponding to the sequence of gravin (GAGACGGGGACAGAC GGGATCCTTGC), and the antisense primers contain sequence of the C-terminus of gravin plus a NotI site following the stop code (ACCGCGGCCGCTTAAGATTCTGTAAGTTCTGACTTTGC). The 3139 bp fragment was subcloned into the vector containing the N-terminal, 2210 bp fragment to generate a 1792 amino acid HA-tagged gravin.
To generate HA-tagged gravin with N-terminal-specific truncations by PCR, the N-terminal sense primer containing a 5′ NheI site and sequence encoding the HA-tagged region was used. The antisense primers, corresponding to amino acids 355–362 (HA 1–362) and 645–652 (HA 1–652) followed by a BamH1 site, and primers corresponding to amino acids 976–983 (HA-1–983) followed by a NotI site, were designed and synthesized. The C-terminal truncated HA-tagged gravin (HA 840–1782) was generated by PCR using a pair of primers, the sense primer containing an 5′ NheI site and the sequence encoding the HA tag region followed by the sequence corresponding to amino acids 840–847, and the antisense primer containing nucleotides of the C-terminus of gravin, plus a NotI site following the stop codon. To generate the AKAP domain, a pair of primers was designed; the sense primer contains a 5′ NheI site and nucleotide sequence encoding the HA tag followed by nucleotides corresponding to amino acids 554–561. The antisense primer contained a BamHI site corresponding to the sequence of gravin. The amplified PCR product was double digested with NheI and BamHI, replaced and subcloned into the NheI–BamHI sites in the construction of the HA-tagged 1–983 N-truncated version of HA-tagged gravin. Serine to alanine substitutions in gravin were performed at serine residues 627, 696, 697, 698 and 772. The RII site-deficient gravin was created by deleting the PKA RII-binding domain (amino acids 1539–1552, ILELETKSSKL VQN) using PCR-mediated mutagenesis. All PCRs were performed using Pfu polymerase (Stratagene). The integrity of the amplified sequences was confirmed by direct DNA sequencing.
Cell culture
Human epidermoid carcinoma cells (A431) were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (HyClone, Logan, UT), penicillin (60 µg/ml) and streptomycin (100 µg/ml) and grown in a humidified atmosphere of 5% CO2 and 95% air at 37°C.
Immunoprecipitation and immunoblotting
For most studies, A431 cells were transiently transfected with expression vector harboring the HA-tagged protein, and pre-incubated with or without KT7520 or calphostin C (Calbiochem) for 30 min, then either left untreated or stimulated with 10 µM Iso or FK for 20 min. Cells were harvested and lysed in a lysis buffer [1% Triton X-100, 0.5% NP-40, 10 mM dithiothreitol (DTT), 5 µg/ml aprotinin, 5 µg/ml leupeptin, 100 µg/ml bacitracin, 100 µg/ml benzamidine, 2 mM sodium orthovanadate, 150 mM NaCl, 5 mM EDTA, 50 mM NaF, 40 mM sodium pyrophosphate, 50 mM KH2PO4, 10 mM sodium molybdate and 20 mM Tris–HCl pH 7.4] at 4°C for 20 min. After centrifugation of the cell debris at 10 000 g for 30 min, the lysates were pre-cleared with protein A/G–agarose for 90 min and then subjected to immunoprecipitation for 2 h with antibodies specific to the β2-adrenergic receptor (CM4) or to the HA tag. The primary antibodies were linked covalently to protein A/G–agarose matrix. Immune complexes were washed three times with immune precipitation buffer (20 mM Tris–HCl, 150 mM NaCl, 1 mM EDTA, 10 mM DTT, 1% Triton X-100 pH 8.0), denatured for 5 min at 95°C and then subjected to SDS–PAGE on 10% acrylamide gels, with the electrophoresis performed in the absence or presence of 8 M urea. Using 8% acrylamide separating gels enabled the detection of some gravin dimer (Mr > 400 kDa), that was reduced to the Mr 250 kDa monomer in the presence of 8 M urea. Immunoblotting and detection of gravin or the β2AR by immunostaining was performed as described (Lin et al., 2000).
Phosphorylation of gravin in vivo
A431 cells were transiently transfected in DMEM with an expression vector harboring either wild-type gravin or a mutant, HA-tagged version. The cells then were metabolically labeled in phosphate-free DMEM supplemented with [32P]orthophosphate (to a concentration of label of 1 mCi/ml) for 12 h at 37°C. At the end of the incubation, cells were pre-incubated with or without an enzyme inhibitor (e.g. KT 5720 or calphostin C) for 30 min and thereafter stimulated with either Iso or FK for 20 min, at the final concentrations indicated in the figure legends. To terminate the metabolic labeling, the cells were washed free of label and treated with the lysis buffer. After centrifugation of the cell debris at 10 000 g for 30 min, each sample was subjected to immunoprecipitation with anti-HA tag antibody. For studies of the phosphorylation of endogenous gravin, cell lysates were pre-cleared with anti-HA tag antibody twice and then subjected to immunoprecipitation using anti-gravin antibody (anti-SSeCKS antibody). The immunoprecipitated proteins were solubilized (5% SDS, 20 mM DTT, 20% glycerol), denatured for 5 min at 95°C, and then subjected to SDS–PAGE. The 32P-phosphorylated proteins were made visible by autoradiography. After autoradiography, the separated proteins were transferred electrophoretically to nitrocellulose blots and stained with antibodies to HA tag or to SSeCKS.
In vitro phosphorylation of HA-AKAP domain by PKA
A431 cells were transiently transfected in DMEM with an expression vector harboring either wild-type gravin or the mutant, HA-tagged AKAP domain of gravin possessing specific serine to alanine substitutions. Cells were treated as indicated above, lysed, and subjected to immunoprecipitation reactions with anti-HA antigen antibodies. The immunoprecipitated proteins were phosphorylated at 37°C for 30 min in a reaction mixture containing 50 mM Tris–HCl pH 7.4, 0.1% NP-40, 0.15 M NaCl, 5 mM MgCl2, 0.1 mM ATP (0.5–2.0 × 1015 c.p.m./mol) and 0.1 U/µl catalytic subunit of PKA (Sigma). For a negative control, heat-inactivated PKA was substituted for PKA under the same conditions. At the end of the labeling period, the immunoprecipitated proteins were collected, denatured, and subjected to SDS–PAGE and autoradiography. After autoradiography, the separated proteins were transferred electrophoretically to nitrocellulose blots and stained with antibodies to the HA tag or to SSeCKS.
Affinity purification of gravin and protein in-gel digestion
A431 cells were transiently transfected with expression vector harboring the HA-tagged protein, and pre-incubated with or without KT7520 or calphostin C (Calbiochem) for 30 min, then either left untreated or stimulated with 10 µM Iso or FK for 20 min. Cells were harvested and lysed in a lysis buffer (1% Triton X-100, 0.5% NP-40, 10 mM DTT, 5 µg/ml aprotinin, 5 µg/ml leupeptin, 100 µg/ml bacitracin, 100 µg/ml benzamidine, 2 mM sodium orthovanadate, 150 mM NaCl, 5 mM EDTA, 50 mM NaF, 40 mM sodium pyrophosphate, 50 mM KH2PO4, 10 mM sodium molybdate and 20 mM Tris–HCl pH 7.4) at 4°C for 20 min. After centrifugation of the cell debris at 10 000 g for 30 min, the lysates were pre-cleared with protein A/G–agarose for 90 min and then subjected to immunoprecipitation for 2 h using anti-HA antibody affinity matrix, prepared according to the manufacturer’s instructions (Roche Molecular Biochemical). The eluted proteins were dialyzed with 10 mM Tris–HCl pH 7.4 buffer to remove extra salt and concentrated in a SpeedVac centrifuge. The purified proteins were separated by one-dimensional 10% SDS–PAGE. Silver staining of the protein was performed according to the commercial supplier (Bio-Rad). Protein bands were excised from the gel and an in-gel digestion was performed with either trypsin or endoproteinase Glu-C (Karoor and Malbon, 1996). To dephosphorylate the target protein, the purified proteins were incubated at 37°C for 3 h with protease-free, calf intestinal alkaline phosphatase (Sigma) in 50 mM NH4HCO3. The treated samples then were subjected to SDS–PAGE and to in-gel digestion.
LC MS for analysis of HA-gravin phosphorylation
Liquid chromatography/mass spectrometric analysis of digests of HA-tagged gravin was performed using the API QSTAR Pulsar I LC/MS/MS system. Peptides were selected in the mass range of 700–3000 m/z. All mass spectra were externally calibrated with the Sequazyme peptide mass standards kit (PerSeptive Biosystems) and internally with trypsin or endoproteinase Glu-C autolysis peaks.
In vitro phosphorylation of β2AR by endogenous, gravin-associated PKA
A431 cells were transiently transfected with expression vector harboring the human β2AR and then serum-starved for 12 h. Cells were collected in ice-cold phosphate-buffered saline (PBS) medium. Crude cell membranes were prepared in HME buffer (20 mM HEPES pH 7.0, 2 mM MgCl2, 1 mM EDTA) supplemented with protease inhibitors [5 µg/ml aprotinin, 5 µg/ml leupeptin, 0.1 mM phenylmethylsulfonyl fluoride (PMSF)] (Lin et al., 2000). The cell membranes were incubated in a phosphorylation labeling reaction with either HA-tagged gravin or HA-tagged gravin possessing the triple serine to alanine mutations of the PKA phosphorylation sites in the AKAP domain, or with a HA-tagged gravin mutant lacking the RII-binding site for PKA. These gravin proteins had been first collected by immunoprecipitation to an anti-HA affinity matrix from extracts prepared from stably transfected A431 clones expressing each protein. The gravin proteins were released from the affinity matrix using the HA-peptide (Roche Molecular Biochemical). The labeling reaction was conducted at 37°C in 50 mM Tris–HCl pH 7.5, 0.12% Triton X-100, 10 mM MgCl2, 1 mM EGTA and 0.1 mM ATP (0.5–2.0 × 1015 c.p.m./mol), in the presence or absence of either 5 µM cAMP or 10 µM Iso plus 10 µM GTP for 30 min. The phosphorylated proteins were solubilized, and subjected to SDS–PAGE on 10% (w/v) acrylamide gels. The phosphorylated proteins were made visible by autoradiography of the gel on X-Omat AR film (Kodak) at –20°C. After autoradiography, the separated proteins were transferred electrophoretically from the gel to the nitrocellulose membrane and the β2AR was detected by immunostaining, as previously described (Fan et al., 2001a, b).
Suppression of gravin by antisense morpholino oligodeoxynucleotides
Antisense morpholino oligonucleotides (morpholinos) were synthesized and purified to cell culture grade (Gene Tools, Llc). Prior to addition to cells, the morpholinos were mixed at a ratio of 1:1 (w/w) with a proprietary EPEI, special delivery solution (Gene Tools, Llc). Typically, A431 cells/clones were treated with the anti-gravin morpholinos (5 µg/ml) for 3 days. This treatment effectively suppresses gravin expression. The cells/clones were treated again with freshly prepared morpholinos just prior to transient transfection of the cells with wild-type gravin or specific gravin mutants. After 48 h, the cells/clones were analyzed for agonist (Iso, 10 µM)-stimulated desensitization after 30 min of agonist. Resensitization, i.e. the loss of desensitization following removal of the agonist, was then measured at 30, 45 and 60 min after wash-out of agonist and addition of propranolol (10 µM).
Protocols for desensitization and resensitization of β2AR
Two days prior to the analysis of agonist-induced desensitization, the A431 cells were seeded in 96-well microtiter plates at a density of 25 000–50 000 cells/well. Details of the desensitization protocol have been published elsewhere (Shih et al., 1999a). For resensitization, the cells challenged with Iso for 30 min prior were washed free of agonist and maintained in fresh buffer containing propranolol (10 µM) for an additional 30–60 min. Five minutes before the second challenge of the agonist, cells were incubated again in the presence of Ro-20-1724 and adenosine deaminase. The amount of cAMP accumulated by cells in response to agonist was measured in the naive cells (i.e. activation), in the cells treated with agonist for 30 min prior to a second challenge with Iso (i.e. desensitization) and in cells treated with agonist for 30 min prior to a second challenge with Iso, and washed free of agonist for 30–60 min (i.e. resensitization). The cAMP accumulated by the cells was determined as described (Shih and Malbon, 1994). ‘0% desensitization’ denotes a cAMP response to a second challenge with agonist that is equivalent in magnitude to that obtained in naive cells in response to the first challenge. ‘100% desensitization’ represents the maximal level of desensitization obtained by a 30 min pre-incubation of the cells with Iso.
Preparation of subcellular fractions
Cells were washed twice with PBS, scraped off the plates with a rubber policeman using ice-cold PBS and collected for 5 min at 300 g. The pellet was resuspended in HME buffer (20 mM HEPES pH 7.4, 2 mM MgCl2, 1 mM EDTA, supplemented with 5 µg/ml aprotinin, 5 µg/ml leupeptin and 0.2 mM PMSF) and incubated for 15 min at 4°C. Cells were sonicated for 10 s three times, then centrifuged for 10 min at 2000 g to remove nuclei. This supernatant was then centrifuged for 30 min at 13 000 g. The subcellular fraction containing crude membranes was collected and termed the pellet (P) fraction. The low-speed, cytosol-enriched supernatant was collected and termed the supernatant (S) fraction. Equivalent amounts of P and S fractions were subjected to SDS–PAGE and immunoblotting. The resultant blots were stained for gravin content with anti-gravin antibodies.
Acknowledgments
Acknowledgement
Supported by a USPHS grant from the NIH.
References
- Alto N., Carlisle Michel,J.J., Dodge,K.L., Langeberg,L.K. and Scott,J.D. (2002) Intracellular targeting of protein kinases and phosphatases. Diabetes, 51, Suppl. 8. [DOI] [PubMed] [Google Scholar]
- Bauman A.L. and Scott,J.D. (2002) Kinase- and phosphatase-anchoring proteins: harnessing the dynamic duo. Nature Cell Biol., 4, 203–206. [DOI] [PubMed] [Google Scholar]
- Colledge M. and Scott,J.D. (1999) AKAPs: from structure to function. Trends Cell Biol., 9, 216–221. [DOI] [PubMed] [Google Scholar]
- Cong M., Perry,S.J., Lin,F.T., Fraser,I.D., Hu,L.A., Chen,W., Pitcher,J.A., Scott,J.D. and Lefkowitz,R.J. (2001) Regulation of membrane targeting of the G protein-coupled receptor kinase 2 by protein kinase A and its anchoring protein AKAP79. J. Biol. Chem., 276, 15192–15199. [DOI] [PubMed] [Google Scholar]
- Dell’Acqua M.L., Dodge,K.L., Tavalin,S.J. and Scott,J.D. (2002) Mapping the protein phosphatase-2B anchoring site on AKAP79. Binding and inhibition of phosphatase activity are mediated by residues 315–360. J. Biol. Chem., 277, 48796–48802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan G., Shumay,E., Malbon,C.C. and Wang,H. (2001a) c-Src tyrosine kinase binds the β2-adrenergic receptor via phospho-Tyr-350, phosphorylates G-protein-linked receptor kinase 2 and mediates agonist-induced receptor desensitization. J. Biol. Chem., 276, 13240–13247. [DOI] [PubMed] [Google Scholar]
- Fan G., Shumay,E., Wang,H.H. and Malbon,C.C. (2001b) The scaffold protein gravin (AKAP250) binds the β2-adrenergic receptor via the receptor cytoplasmic R329 to L413 domain and provides a mobile scaffold during desensitization. J. Biol. Chem., 276, 24005–24014. [DOI] [PubMed] [Google Scholar]
- Gelman I.H. (2002) The role of SSeCKS/gravin/AKAP12 scaffolding proteins in the spaciotemporal control of signaling pathways in oncogenesis and development. Front. Biosci., 7, 1782–1797. [DOI] [PubMed] [Google Scholar]
- Hausken Z.E. and Scott,J.D. (1996) Properties of A-kinase anchoring proteins. Biochem. Soc. Trans, 24, 986–991. [DOI] [PubMed] [Google Scholar]
- Karoor V. and Malbon,C.C. (1996) Insulin-like growth factor receptor-1 stimulates phosphorylation of the β2-adrenergic receptor in vivo on sites distinct from those phosphorylated in response to insulin. J. Biol. Chem., 271, 29347–29352. [DOI] [PubMed] [Google Scholar]
- Kim S.H., Lee,S.K. and Choi,K.Y. (1998) Saccharomyces cerevisiae STE11 may contribute to the stabilities of a scaffold protein, STE5, in the pheromone signaling pathway. Mol. Cell, 8, 130–137. [PubMed] [Google Scholar]
- Lin F., Wang,H. and Malbon,C.C. (2000) Gravin-mediated formation of signaling complexes in β2-adrenergic receptor desensitization and resensitization. J. Biol. Chem., 275, 19025–19034. [DOI] [PubMed] [Google Scholar]
- Michel J.J. and Scott,J.D. (2002) AKAP mediated signal transduction. Annu. Rev. Pharmacol. Toxicol., 42, 235–257. [DOI] [PubMed] [Google Scholar]
- Morris A.J. and Malbon,C.C. (1999) Physiological regulation of G protein-linked signaling. Physiol. Rev., 79, 1373–1430. [DOI] [PubMed] [Google Scholar]
- Nauert J.B., Klauck,T.M., Langeberg,L.K. and Scott,J.D. (1997) Gravin, an autoantigen recognized by serum from myasthenia gravis patients, is a kinase scaffold protein. Curr. Biol., 7, 52–62. [DOI] [PubMed] [Google Scholar]
- Oliveria S.F., Gomez,L.L. and Dell’Acqua,M.L. (2003) Imaging kinase–AKAP79–phosphatase scaffold complexes at the plasma membrane in living cells using FRET microscopy. J. Cell Biol., 160, 101–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawson T. and Scott,J.D. (1997) Signaling through scaffold, anchoring and adaptor proteins. Science, 278, 2075–2080. [DOI] [PubMed] [Google Scholar]
- Shih M. and Malbon,C.C. (1994) Oligodeoxynucleotides antisense to mRNA encoding protein kinase A, protein kinase C and β-adrenergic receptor kinase reveal distinctive cell-type-specific roles in agonist-induced desensitization. Proc. Natl Acad. Sci. USA, 91, 12193–12197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih M., Lin,F., Scott,J.D., Wang,H.Y. and Malbon,C.C. (1999) Dynamic complexes of β2-adrenergic receptors with protein kinases and phosphatases and the role of gravin. J. Biol. Chem., 274, 1588–1595. [DOI] [PubMed] [Google Scholar]