

Abstract
CHARGE syndrome (Coloboma of the eye, Heart defects, Atresia of the choanae, Retardation of growth and/or development, Genital and/or urinary abnormalities, and Ear abnormalities (including deafness) is a genetic disorder characterized by a specific and a recognizable pattern of anomalies. De novo mutations in the gene encoding chromodomain helicase DNA binding protein 7 (CHD7) are the major cause of CHARGE syndrome. Here, we review the clinical features of 379 CHARGE patients who tested positive or negative for mutations in CHD7. We found that CHARGE individuals with CHD7 mutations more commonly have ocular colobomas, temporal bone anomalies (semicircular canal hypoplasia/dysplasia), and facial nerve paralysis compared with mutation negative individuals. We also highlight recent genetic and genomic studies that have provided functional insights into CHD7 and the pathogenesis of CHARGE syndrome.
Keywords: CHARGE syndrome, CHD7, chromodomain helicase DNA binding protein 7, review
Introduction
CHARGE syndrome (OMIM #214800) is a rare (incidence ∼1:10,000 [Jongmans et al., 2006; Lalani et al., 2006], nonrandom combination of multiple anomalies. The pattern of anomalies now associated with CHARGE syndrome were described independently in 1979 by Hittner and coworkers [1979] and Hall [1979], though it was Pagon et al., [1981] who formally defined the pattern and coined the acronym CHARGE to summarize its major features. In its early delineation Pagon et al. [1981] referred to the pattern as an “Association”; now with its recognizable pattern and definition CHARGE is referred to as a syndrome. The major clinical features of CHARGE syndrome are ocular Coloboma, Heart malformations, Atresia of the choanae, Retardation of growth, Genital hypoplasia, and Ear abnormalities. Numerous other less consistent features, including hyposmia, cleft lip/palate, and tracheoesophageal fistula, are also reported. Many of these features, including genital hypoplasia, cleft palate, and heart defects, are shared by other multiple anomaly syndromes such as 22q11.2 deletion [Sullivan 2008], Kallmann [Ogata et al., 2006; Kim et al., 2008; Jongmans et al., 2009], and Treacher Collins [Dixon et al., 2007]; however, CHARGE syndrome is considered unique in its combination of these features with distinctive inner and outer ear defects and optic colobomas.
The life expectancy of patients with CHARGE syndrome varies widely, with individuals living anywhere from five days [Issekutz et al., 2005] to at least 46 years [Jongmans et al., 2006]. Actuarial analysis of survival in children with CHARGE showed a 70% survival rate to five years of age, with the highest rate of mortality in the first year of life. The rate of mortality is highest in infants with a combination of choanal atresia and heart defects or tracheoesophagal fistula. Prevalent feeding and swallowing difficulties as well as gastroesophagal reflux also contribute to CHARGE infant mortality and may also contribute to overall mortality in all CHARGE patients [Blake et al., 1998].
Since the discovery in 2004 of CHD7 as a causative gene in CHARGE, several studies have attempted to define genotype-phenotype correlations and to determine the overall contribution of CHD7 mutations to various CHARGE features. In this study, we reviewed all manuscripts published in English and indexed in PubMed from 2004-2009 in which patients with CHARGE features were tested for CHD7 mutation status. Detailed clinical information for mutation-positive patients is listed in Table I and mutation-negative patients in Table II. Only those manuscripts which contained clinical information and mutation status were included, and care was taken to note the presence or absence of a particular clinical feature only when evaluation for that condition was explicitly stated, to minimize effects of under-reporting. We used a chi-square test to identify statistically significant differences in the frequencies of each clinical feature between mutation-positive and mutation-negative individuals (Table III).
Table I.
Clinical features in CHD7 mutation-positive individuals. Frequencies are represented as the number of individuals with a particular feature / the total number of individuals tested. ND = not done.
| 2004 | 2005 | 2006 | 2007 | 2008 | 2009 | |||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Syndrome feature | Vissers | Lalani | Aramaki | Felix | Jongmans | Johnson | Ogata | Sanlaville | Delahaye | Sanka | Udaka | Van de Laar | Writzl | Alazami | Bergman | Chopra | Fujita | Gennery | Jongmans | Jongmans | Kim | Wincent | Hoover-Fong | Jongmans | Jyonouchi | Lee | Pauli | Wright |
| Coloboma | 8/12 | 55 / 62 | 15 / 17 | 2 / 2 | 33 / 47 | 2 / 2 | 1 / 1 | 7 / 10 | 4 / 6 | 1 / 1 | 1 / 1 | 2 / 3 | 0 / 2 | 1 / 1 | 1 / 1 | 3 / 3 | 5 / 7 | 3 / 4 | 6 / 11 | 1 / 3 | ND | 19 / 21 | 0 / 1 | 1 / 3 | 14 / 25 | 2 / 4 | 2 / 2 | 1 / 1 |
| Heart malformations | 8 / 12 | 54 / 59 | 13 / 17 | 3 / 3 | 31 / 47 | 2 / 2 | 1 / 1 | 9 / 10 | 3 / 6 | 1 / 1 | 0 / 1 | 3 / 3 | 2 / 2 | 1 / 1 | ND | 3 / 3 | 4 / 7 | 4 / 4 | 5 / 11 | 0 / 3 | ND | 15 / 21 | 1 / 1 | 0 / 3 | 23 / 25 | 4 / 4 | 2 / 2 | 1 / 1 |
| Choanal atresia | 3 / 12 | 34 / 57 | 5 / 17 | 0 / 2 | 17 / 47 | 2 / 2 | 0 / 1 | 6 / 10 | 0 / 6 | ND | 0 / 1 | 2 / 3 | 1 / 2 | 1 / 1 | 0 / 1 | 1 / 3 | 0 / 7 | 1 / 4 | 2 / 11 | 0 / 3 | ND | 9 / 21 | 1 / 1 | 0 / 3 | 9 / 25 | 0 / 4 | 0 / 2 | 1 / 1 |
| Retarded growth | 12 / 12 | ND | 17 / 17 | 1 / 1 | 21 / 32 | 2 / 2 | ND | ND | 5 / 6 | ND | 1 / 1 | ND | ND | 1 / 1 | ND | 1 / 3 | 7 / 7 | ND | 1 / 8 | 2 / 3 | ND | 16 / 20 | ND | ND | 11 / 25 | ND | 2 / 2 | 1 / 1 |
| Developmental delay | 12 / 12 | ND | 17 / 17 | ND | 24 / 32 | 2 / 2 | ND | ND | 3 / 4 | ND | 1 / 1 | 2 / 3 | ND | 1 / 1 | ND | 1 / 3 | 7 / 7 | ND | 6 / 10 | 1 / 3 | ND | 10 / 14 | ND | ND | 13 / 25 | 4 / 4 | 2 / 2 | 1 / 1 |
| Genital hypoplasia | 5 / 12 | 29 / 53 | 13 / 17 | 2 / 2 | 17 / 22 | ND | ND | 6 / 10 | ND | ND | 1 / 1 | 3 / 3 | 2 / 2 | 0 / 1 | 1 / 1 | ND | 4 / 7 | 2 / 4 | ND | 3 / 3 | 2 / 7 | 11 / 21 | 0 / 1 | ND | 11 / 25 | 3 / 4 | 1 / 2 | ND |
| Ear abnormalities including deafness | 12 / 12 | 54 / 59 | 17 / 17 | 2 / 2 | 37 / 41 | 2 / 2 | 1 / 1 | ND | 5 / 6 | 1 / 1 | 1 / 1 | 2 / 3 | ND | 1 / 1 | 1 / 1 | 3 / 3 | 7 / 7 | ND | 8 / 10 | 3 / 3 | 1 / 7 | 13 / 14 | ND | 3 / 3 | 20 / 25 | 4 / 4 | ND | ND |
| Temporal bone anomalies | 8 / 8 | 21 / 22 | 6 / 6 | ND | 21 / 21 | ND | ND | 10 / 10 | 4 / 4 | ND | 1 / 1 | 1 / 1 | 1 / 1 | ND | 1 / 1 | ND | ND | ND | 5 / 5 | 1 / 3 | ND | 8 / 9 | 1 / 1 | 1 / 1 | ND | 4 / 4 | 1 / 2 | ND |
| External ear malformations | ND | 59 / 62 | 17 / 17 | ND | 47 / 47 | 2 / 2 | ND | 10 / 10 | 4 / 6 | ND | 1 / 1 | 3 / 3 | 2 / 2 | 1 / 1 | 1 / 1 | 1 / 1 | 7 / 7 | 3 / 4 | 10 / 11 | 1 / 3 | ND | 19 / 21 | 1 / 1 | 1 / 3 | 21 / 25 | 2 / 4 | ND | 1 / 1 |
| Facial nerve palsy | 3 / 12 | 36 / 56 | ND | ND | 10 / 47 | ND | 0 / 1 | ND | ND | ND | 1 / 1 | ND | ND | ND | 1 / 1 | 1 / 1 | ND | ND | 2 / 11 | 1 / 3 | ND | 9 / 20 | ND | ND | 5 / 25 | 1 / 4 | 1 / 2 | 1 / 1 |
| Cleft lip and/or Cleft palate | 4 / 12 | 18 / 60 | 8 / 17 | 3 / 4 | 17 / 47 | ND | 0 / 1 | 5 / 10 | 1 / 6 | ND | 0 / 1 | 1 / 3 | ND | ND | ND | ND | 4 / 7 | ND | 2 / 11 | 2 / 3 | 2 / 7 | 4 / 21 | ND | 1 / 3 | 6 / 25 | 0 / 4 | 1 / 2 | ND |
| Tracheoesophageal fistula | ND | 10 / 55 | 3 / 17 | ND | 8 / 47 | ND | ND | 2 / 10 | 1 / 6 | 1 / 1 | ND | ND | 1 / 2 | ND | ND | 1 / 1 | ND | 1 / 4 | 1 / 11 | ND | ND | ND | ND | ND | 5 / 25 | 0 / 4 | 1 / 2 | ND |
| Urogenital abnormalities | ND | 29 / 53 | ND | ND | 17 / 22 | ND | 0 / 1 | 6 / 10 | 2 / 6 | 1 / 1 | 1 / 1 | 3 / 3 | 2 / 2 | ND | ND | ND | ND | ND | 4 / 11 | ND | 7 / 7 | 11 / 21 | ND | ND | ND | 3 / 4 | ND | ND |
Table II.
Clinical features in CHD7 mutation-negative individuals. Frequencies are represented as the number of individuals with a particular feature / the total number of individuals tested. ND = not done.
| 2004 | 2005 | 2006 | 2008 | ||
|---|---|---|---|---|---|
| Syndrome feature | Vissers | Lalani | Felix | Bergman | Wincent |
| Coloboma | 6 / 7 | 30 / 43 | 2 / 2 | 30 / 53 | 6 / 9 |
| Heart malformations | 5 / 7 | 30 / 42 | 9 / 9 | 36 / 53 | 6 / 9 |
| Choanal atresia | 3 / 7 | 23 / 39 | 1 / 2 | 23 / 53 | 2 / 9 |
| Retarded growth | 7 / 7 | ND | 3 / 3 | 14 / 14 | 7 / 9 |
| Developmental delay | 7 / 7 | ND | ND | 31 / 31 | 6 / 9 |
| Genital hypoplasia | 5 / 7 | 26 / 39 | 2 / 3 | 9 / 9 | 4 / 8 |
| Ear abnormalities including deafness | 7 / 7 | 36 / 38 | 3 / 3 | 39 / 47 | 4 / 8 |
| Temporal bone anomalies | 5 / 5 | 9 / 10 | ND | 5 / 11 | 2 / 2 |
| External ear malformations | ND | 39 / 42 | ND | ND | 7 / 9 |
| Facial nerve palsy | 2 / 7 | 13 / 39 | ND | 4 / 47 | 0 / 9 |
| Cleft lip and/or Cleft palate | 4 / 7 | 9 / 41 | 8 / 9 | 12 / 53 | 1 / 9 |
| Tracheoesophageal fistula | ND | 3 / 40 | ND | 5 / 5 | ND |
| Urogenital abnormalities | ND | 26 / 39 | ND | 8 / 8 | 4 / 8 |
Table III.
Comparison of clinical features in CHD7 mutation-positive vs. mutation-negative individuals. Significant p values as calculated by Chi-square test are in bold.
| Syndrome feature | CHD7 Mutation Positive | CHD7 Mutation Negative | p value |
|---|---|---|---|
| Coloboma | 190 / 253 (75%) | 74 / 114 (65%) | 0.044 |
| Heart malformations | 193 / 250 (77%) | 86 / 120 (72%) | 0.247 |
| Choanal atresia | 95 / 247 (38%) | 52 / 110 (47%) | 0.118 |
| Retarded growth | 101 / 141 (72%) | 31 / 33 (94%) | 0.007 |
| Developmental delay | 107 / 141 (76%) | 44 / 47 (94%) | 0.008 |
| Genital hypoplasia | 116 / 187 (62%) | 46 / 66 (70%) | 0.265 |
| Ear abnormalities including deafness | 198 / 223 (89%) | 83 / 103 (86%) | 0.538 |
| Temporal bone anomalies | 94 / 96 (98%) | 21 / 28 (75%) | 0.00004 |
| External ear malformations | 214 / 235 (91%) | 46 / 51 (90%) | 0.845 |
| Facial nerve palsy | 72 / 187 (39%) | 19 / 102 (19%) | 0.0005 |
| Cleft lip and/or Cleft palate | 79 / 242 (33%) | 34 / 119 (29%) | 0.433 |
| Tracheoesophageal fistula | 35 / 185 (19%) | 8 / 45 (18%) | 0.860 |
| Urogenital abnormalities | 86 / 142 (61%) | 38 / 55 (69%) | 0.266 |
We found 25 studies in which 379 individuals with CHARGE features were tested for CHD7 mutations. Of the 379 individuals tested, 254 (67%) were CHD7 mutation-positive, whereas 125 (33%) were mutation-negative. It is important to note that not all patients met full CHARGE diagnosis by Blake or Verloes clinical criteria [Blake et al., 1998; Verloes, 2005], and not all studies provided full clinical information on every individual. Methods of mutation testing varied across sites, but all included full sequencing of coding exons. Not all individuals were tested for CHD7 gene exonic deletions/duplications, so these types of mutations are likely to be under-represented here. Nevertheless, these reports were useful for analyzing potential differences in clinical phenotype between mutation-positive and mutation-negative patients, and for estimating the overall prevalence of each feature within CHARGE.
Most (67%) (Table I) clinically diagnosed CHARGE patients had pathogenic mutations in the gene encoding chromodomain helicase DNA binding protein 7 (CHD7), located on chromosome 8q12.1. Of the CHD7 mutations reported thus far, approximately 72% are nonsense or frameshift, 13% are splice site, and 10% are missense (Table IV). CHD7 mutations are reported throughout the entire coding sequence of the gene and do not appear to cluster in any meaningful way. Recurrent mutations are rare, and clear genotype-phenotype correlations have not been recognized, even among patients with identical CHD7 mutations [Jongmans et al., 2006; Lalani et al., 2006; Jongmans et al., 2008]. The majority of CHD7 mutations are predicted to be loss of function, likely leading to an aberrant mRNA targeted for degradation via nonsense-mediated decay. Therefore, haploinsufficiency for CHD7 is the most likely pathogenic mechanism underlying CHARGE syndrome. This mechanism is supported by mouse studies in which homozygosity for Chd7 loss-of-function mutations result in embryonic lethality and heterozygous Chd7 mice display phenotypic features similar to those associated with CHARGE syndrome [Bosman et al., 2005; Hurd et al., 2007].
Table IV.
Frequencies of the various types of reported CHD7 mutations from studies in which molecular details of the mutations were provided.
| Mutation type | Number of mutations | Frequency (%) |
|---|---|---|
| Nonsense | 112 | 46 |
| Frameshift | 58 | 24 |
| Missense | 37 | 15 |
| Splice site | 23 | 10 |
| Whole-gene or chromosomal deletion | 3 | 1 |
| Exonic deletion | 7 | 3 |
| Chromosomal rearrangement | 2 | 1 |
While the majority of CHD7 disruptions are nonsense or frameshift mutations, chromosomal abnormalities have also been reported, including an interstitial deletion of 8q12.2-q13 [Arrington et al., 2005] and a balanced translocation disrupting 8q12 [Johnson et al., 2006]. Several chromosomal abnormalities not involving CHD7 have also been reported to confer a phenotype similar to those seen in CHARGE patients [Clementi et al., 1991; North et al., 1995; Wieczorek et al., 1997; De Krijger et al., 1999; Lev et al., 2000].
Recent work indicates that CHARGE patients without detectable single-base mutations may have heterozygous deletions of CHD7 [Wincent et al., 2009]. In fact, CHD7 was originally identified as the causative gene in CHARGE syndrome due to detection of a deletion by array-CGH [Vissers et al., 2004]. In one exceptional case, deletion of a portion of CHD7 was caused by an Alu retrotransposon [Udaka et al., 2007]. Alterations in exon copy number have been reported, but they represent only a small fraction of the reported disruptions of CHD7 [Bergman et al., 2008].
The minority (37%) of CHARGE patients have no identifiable mutation in CHD7, thus the underlying etiology remains undetermined. These cases could be explained by mutations in coding (deletions or duplications) or noncoding regions of CHD7 such as 5′ or 3′ untranslated regions, introns, or critical regulatory elements. There may also be locus heterogeneity in CHARGE, but no other convincing chromosomal regions or candidate genes have emerged.
Almost all cases of CHARGE syndrome are sporadic; however, a small number of cases of familial CHARGE syndrome and parent-to-child transmission of CHD7 mutations have been reported [Lalani et al., 2006; Delahaye et al., 2007; Jongmans et al., 2008; Vuorela et al., 2008; Wincent et al., 2008; Pauli et al., 2009]. In these cases, parents generally have a mild CHARGE phenotype or are asymptomatic, whereas their children display more severe defects. Somatic mosaicism for a CHD7 mutation was described in an asymptomatic mother of two affected siblings [Jongmans et al., 2008]. Germ cell mosaicism was also described in an asymptomatic, father of two affected children [Pauli et al., 2009]. It therefore seems that in cases of parental transmission of CHD7 mutations, parents with the mutations are mildly affected or asymptomatic and it is only their children who display the full spectrum of CHARGE features.
CHD7 mutations have also been reported in patients diagnosed with conditions that have significant clinical overlap with CHARGE, including Kallmann [Ogata et al., 2006; Kim et al., 2008; Jongmans et al., 2009], Omenn-like [Gennery et al., 2008], and 22q11.2 deletion syndromes [Sanka et al., 2007]. These findings imply that patients presenting with features of these syndromes, but lacking typical molecular findings, should be examined for clinical signs of CHARGE syndrome and tested for CHD7 mutations. These discoveries also raise the possibility that CHD7 mutations may underlie the pathogenesis of more human phenotypes than previously appreciated, including isolated congenital defects. Large-scale mutational analysis of CHD7 in developmental disorders involving organ systems affected in CHARGE are needed to explore this possibility further.
Clinical Features of Charge Syndrome
From the 254 CHD7 mutation-positive individuals reported since 2004, the most common clinical findings were temporal bone anomalies (98%), external ear malformations (91%), and hearing loss (89%) (Table III). About three fourths of patients had one or more of the following: coloboma, heart malformations, and delayed growth and development (Table III). Between 61-62% of CHARGE individuals had genital hypoplasia and/or urogenital abnormalities, and 38% had choanal atresia/stenosis and/or facial nerve palsy (Table III). Less commonly reported were cleft lip and/or palate (33%) and tracheoesophageal fistula (19%). In the following sections, we compare the clinical findings associated with CHARGE syndrome in CHD7 mutation-positive and mutation-negative individuals.
Ocular defects
Colobomas typically involving the choroid, retina, and optic nerve are the major ophthalmic manifestations associated with CHARGE syndrome. Colobomas were more common in mutation-positive (75%) than in mutation-negative (65%) individuals. Colobomas are commonly bilateral, and can involve chorioretina, and optic nerve [Tellier et al., 1998; Aramaki et al., 2006; Jongmans et al., 2006; Alazami et al., 2008; McMain et al., 2008]. Iris, eyelid, and optic disc colobomas are described with less frequency [Aramaki et al., 2006; Jongmans et al., 2006; McMain et al., 2008]. Microphthalmos, optic nerve hypoplasia, myopia, and strabismus are also reported [Aramaki et al., 2006; Jongmans et al., 2006; Lalani et al., 2006; Delahaye et al., 2007; Alazami et al., 2008; Wincent et al., 2008; Jyonouchi et al., 2009], but frequencies of these malformations were not compared in our study. Typical CHARGE colobomas produce field defects in the upper quadrant and are often associated with significant refractive errors, severe myoptic astigmatism, anisometropia, microphthalmos, microcornea, and reduced visual acuity [Tellier et al., 1998; Aramaki et al., 2006; Jongmans et al., 2006; Delahaye et al., 2007; Alazami et al., 2008; McMain et al., 2008].
Heart defects
A wide variety of heart defects is reported as part of CHARGE syndrome, highlighting the variable nature of its clinical presentation. We found that CHD7 mutation-positive and mutation-negative patients were equally likely to have congenital heart malformations (77% vs. 72%, respectively). Tetralogy of Fallot, characterized by ventricular septal defects (VSD), pulmonary stenosis, right ventricular hypertrophy, and an overriding aorta, is frequently reported in CHARGE [Lin et al., 1987; Wyse et al., 1993; Tellier et al., 1998; Jongmans et al., 2006; Lalani et al., 2006]. Each of the individual constituents of tetralogy of Fallot as well as patent ductus arteriosis (PDA), atrial septal defects (ASD), and solitary septal defects are reported, in various combinations [Lin et al., 1987; Wyse et al., 1993; Tellier et al., 1998; Aramaki et al., 2006; Jongmans et al., 2006; Lalani et al., 2006; Delahaye et al., 2007; Alazami et al 2008; Wincent et al., 2008; Wright et al., 2009]. Double outlet right ventricle and hypoplastic left/right heart are also sometimes noted [Jongmans et al., 2006; Van de Laar et al., 2007]. Aortal defects, including overriding aorta, right descending aorta, and right aortic arch are reported individually and in combination with other heart defects [Aramaki et al., 2006; Jongmans et al., 2006; Van de Laar et al., 2007; Alazami et al., 2008].
The literature regarding the various heart malformations in patients clinically suspected of having CHARGE syndrome is broad. Since the development of CHD7 mutation screening, most information on heart defects draws from CHD7 mutation-positive patients. However, detailed reports of heart defects were published before the availability of CHD7 mutation testing [Lin et al., 1987; Tellier et al., 1998] and the heart defects of mutation-negative individuals are rarely considered in detail in the literature. Therefore, it is difficult at this time to accurately assess the frequencies of specific heart defects between mutation-positive and mutation-negative patients. However, Lalani and coworkers [2006] compared the frequency of several specific heart defects between the two groups and found that mutation-positive individuals were significantly more likely to have patent ductus arteriosis, but not tetralogy of Fallot, septal defects, or outflow tract defects. More studies, with detailed analysis of heart malformations in both mutation-positive and mutation-negative patients, are needed to further address any phenotypic differences in heart defects between these two groups.
Choanal atresia
CHD7 mutation-positive and mutation-negative patients did not differ in the likelihood of having choanal atresia or stenosis (38% vs. 47%, respectively). Interestingly, the incidence of choanal atresia varies depending upon the incidence of cleft lip/palate, as the choanae are usually normal when clefting is present [Aramaki et al., 2006; Jongmans et al., 2006]. Breathing difficulties are reported in approximately half of neonatal cases [Jongmans et al., 2006]. About 19% of CHD7 mutation-positive CHARGE syndrome patients also present with tracheoesophageal (TE) fistula (Table III), an abnormal connection between the trachea and esophagus leading to feeding difficulties and aspiration in babies which is typically corrected surgically [White et al., 2005; Aramaki et al., 2006; Jongmans et al., 2006; Lalani et al., 2006; Jyonouchi et al., 2009].
Growth and developmental delays
Growth defects, including short stature and low birth weight, are reported in CHARGE patients and are typically of postnatal onset [Aramaki et al., 2006; Jongmans et al., 2006; Lalani et al., 2006; Jyonouchi et al., 2009]. Growth defects associated with CHARGE rarely appear to correlate with deficiencies in pituitary hormones including growth hormone (9% of patients) and thyroid stimulating hormone (one incidence) and not with adrenocorticotropic hormone [Pinto et al., 2005; Aramaki et al., 2006; Asakura et al., 2008]. Factors contributing to postnatal growth delays include feeding and swallowing difficulties, gastroesophagal reflux, olfactory dysfunction, cardiac dysfunction, and poor nutrition. Additionally, due to the many medical complications of CHARGE syndrome, patients may be extensively hospitalized for surgery and this may contribute to delayed growth. Surprisingly, we found that delays in growth were more common in CHD7 mutation-negative individuals (94%) than in mutation-positive individuals (72%) (Table III). This difference was unexpected, and may reflect patterns of data collection or reporting. Alternatively, CHD7 mutation negative patients may have different genetic etiologies which predispose them to more significant growth delays.
A wide range of developmental symptoms, from mild speech delay to severe intellectual disability without speech, has also been reported in CHARGE [Jongmans et al., 2006; Delahaye et al., 2007; Udaka et al., 2007; Alazami et al., 2008; Wright et al., 2009]. As with growth delays, we found that significantly fewer mutation-positive individuals (76%) were reported to have developmental delays compared to mutation-negative (94%) individuals. As yet, there is no comprehensive information about long-term cognitive and functional outcomes between mutation-positive vs. mutation-negative individuals. Nonetheless, it will be interesting to determine whether the CHD7 mutation-negative cohort of individuals has distinct genetic or environmental etiologies to predispose them to more severe developmental and growth delays.
Endocrine and urogenitary anomalies
Hypogonadotropic hypogonadism and delayed puberty are commonly reported features in CHARGE patients. Urogenital abnormalities, including genital hypoplasia, were observed in many (61%) CHARGE patients. These defects are less commonly reported in females; thus, the use of urogenital anomalies as a diagnostic feature of CHARGE syndrome is biased towards males. Micropenis and/or cryptorchidism are commonly diagnosed in male patients. Female urogenital defects diagnosed in CHARGE syndrome include hypoplasia of the uteris and labia. Some females also experience pubertal failure [Jongmans et al., 2006]. We found no statistically significant differences in the frequency of genital hypoplasia or urogenital abnormalities in mutation-positive (61%) vs. mutation-negative (69%) individuals. Abnormalities in the gonadotropic axis, including deficiencies in luteinizing hormone (LH), follicle-stimulating hormone (FSH), testosterone, and estradiol, are frequently reported in CHARGE and are likely to contribute to delayed puberty and impaired urogenital development [Jongmans et al., 2006; Udaka et al., 2007; Asakura et al., 2008]. In one study, deficiency for gonadotropins (LH and FSH) was found in 81% of males and in 93% of females analyzed [Jongmans et al., 2006]. Stimulation by gonadotropin releasing hormone (GnRH) had variable effects in males, while females had little to no response to stimulation [Pinto et al., 2005; Ogata et al., 2006].
Developmental abnormalities of the ear
For clarity, we divided the ear abnormalities in individuals with CHARGE into three separate types, based on reporting patterns across all 25 studies: (a) deafness, (b) inner ear malformations including semicircular canal dysplasia/hypoplasia, and (c) external ear abnormalities. Hearing loss was equally common among CHD7 mutation-positive (89%) and mutation-negative (86%) individuals. Hearing loss can be conductive in nature owing to structural anomalies of the inner ear, sensorineural due to deficiencies in cranial nerve VIII function, or mixed conductive/sensorineural [Edwards et al., 2002]. External ear malformations were equally common in mutation-positive (91%) and mutation-negative (90%) individuals. These malformations include lowset ears, asymmetric ear shape or size, and small/absent lobes [Jongmans et al., 2006; Lalani et al., 2006]. The outer ear often takes on a characteristic “cup” shape in affected patients.
Inner ear anomalies detected by temporal bone CT or skull x-ray were much more common in mutation-positive (98%) vs. mutation-negative (75%) individuals (Table III). These anomalies include semicircular canal hypoplasia/aplasia, cochlear hypoplasia, and Mondini malformation [Aramaki et al., 2006; Jongmans et al., 2006; Lalani et al., 2006; Delahaye et al., 2007; Udaka et al., 2007; Writzl et al., 2007]. Inner ear malformations are the most highly penetrant clinical feature reported in CHARGE, with only two individuals having CHD7 mutations and normal temporal bone CT scans [Lalani et al., 2006; Wincent et al., 2008]. Balance disturbances and vestibular areflexia, associated with deficiencies in vestibular function due to malformation or agenesis of the semicircular canals, have been previously noted as one of the most consistent features of CHARGE syndrome [Jongmans et al., 2006; Delahaye et al., 2007].
Central nervous system and cranial nerve anomalies
CNS anomalies are also reported in CHARGE patients and are considered by Blake et al., to be a major diagnostic criterion [Blake et al., 1998]. Unilateral and bilateral hypoplasia of the olfactory bulb and/or arhinencephaly are most commonly reported in CHARGE, and may contribute to hyposmia or anosmia [Sanlaville et al., 2006; Asakura et al., 2008]. Other defects, such as agenesis of the corpus callosum, cerebellar hypoplasia, hydrocephaly, and atrophy of the cerebral cortex have also been reported. Seizures are also observed in CHARGE patients [Jongmans et al., 2006]. There was inadequate information about CNS findings for many individuals in the reports we reviewed, precluding accurate comparison between mutation-positive and mutation-negative cases.
Defects in several cranial nerves have been reported in CHD7 mutation-positive individuals. Although olfactory dysfunction is most likely a result of hypoplasia/absence of the olfactory bulbs, defects in cranial nerve I may also contribute to olfactory dysfunction. Cranial nerve VI defects are reported, and lead to internal strabismus [Bergman et al., 2008]. Facial palsy and asymmetry, indicative of anomalies in cranial nerve VII, are often seen in CHARGE patients [Aramaki et al., 2006; Jongmans et al., 2006; Udaka et al., 2007; Jongmans et al., 2008; Wincent et al., 2008; Pauli et al., 2009]. We found that mutation-positive individuals were far more likely to have facial palsy (39%) than mutation-negative (19%) CHARGE individuals. Further studies will help determine whether this reflects an underlying bias in clinical data reporting or a real biological effect of CHD7 function.
Deficiencies in cranial nerve VIII function result in sensorineural deafness and disturbances in balance and vestibular function [Johnson et al., 2006; Udaka et al., 2007; Bergman et al., 2008; Lee et al., 2009]. Swallowing difficulties due to dysfunction of cranial nerves IX, X, and/or XI are also a common feature of CHARGE syndrome [Udaka et al., 2007; Wincent et al., 2008; Jyonouchi et al., 2009].
Facial anomalies and clefting
Typical facial features of CHARGE syndrome include a square-shaped face with a wide nasal bridge and small mouth [Sanlaville and Verloes 2007]. We found that cleft lip and/or palate were similarly present in mutation-positive (33%) vs. mutation-negative (29%) individuals [Aramaki et al., 2006; Jongmans et al., 2006; Lalani et al., 2006; Wincent et al., 2008; Jyonouchi et al., 2009].
Spinal defects and limb/skeletal abnormalities
Scoliosis is reported in many children with CHARGE, and often includes structural abnormalities of the vertebrae [Jongmans et al., 2006]. Polymorphisms in CHD7 have been associated with susceptibility to idiopathic scoliosis [Gao et al., 2007]. Other spinal defects, including spina bifida occulta and kyphosis, are also present in CHARGE [Jongmans et al., 2006; Delahaye et al., 2007].
Recent reports have shed light into some of the limb abnormalities present in CHARGE patients. Two reports have confirmed tibial agenesis, both with CHD7 mutations [Van de Laar et al., 2007; Alazami et al., 2008]. The authors of one of these studies proposed expanding the “E” of the CHARGE acronym to encompass anomalies of the Extremities in order to draw attention to CHARGE syndrome as a possible diagnosis for patients with limb defects [Alazami et al., 2008]. Other limb defects, including triphalangeal thumb, polydactyly of the foot, monodactyly, radial aplasia, and ectrodactyly have been seen in a minority of patients [Jongmans et al., 2006; Van de Laar et al., 2007; Wright et al., 2009]. Owing to small numbers of reported individuals, skeletal differences between mutation-positive and mutation-negative individuals were not examined in our study.
Immunological abnormalities
Immunological problems are a rare feature of CHARGE syndrome, with a small number of CHARGE patients described as having thymic aplasia or hypoplasia, (sometimes called DiGeorge “syndrome”) IgG2 subclass deficiency, and T-cell lymphopenia [Writzl et al., 2007; Chopra 2009; Jyonouchi et al., 2009]. Severe combined immune deficiency (SCID) has also been reported in rare patients [Gennery et al., 2008]. Based on the small number of cases, these clinical features were not included in our statistical analysis.
Can Clinical Presentation Predict CHD7 Mutation Status?
Here we summarized data from 379 individuals with clinical information and CHD7 mutation status. We found that mutation-positive individuals are more likely to have inner ear malformations including semicircular canal aplasia/dysplasia, facial nerve palsy, and ocular colobomas, and less likely to have delayed growth and development. These data strongly support the use of temporal bone CT as a diagnostic tool for evaluation of CHARGE patients, and confirm previous reports that inner ear malformations should be considered a diagnostic criterion.
In an earlier study of 110 clinically diagnosed CHARGE patients, Lalani et al., [2006] compared phenotypic data from mutative-positive and mutation-negative CHARGE patients and found that mutation-positive patients were significantly more likely to have cardiovascular defects, coloboma, and facial asymmetry resulting from defects in cranial nerve VII. In contrast, no significant differences were observed between the two groups in terms of inner or outer ear anomalies, choanal atresia, cleft lip/palate, deafness, tracheoesophageal fistula, and urogenital anomalies. Interestingly, all 10 patients in the study who presented with the combination of coloboma, choanal atresia, and hypoplastic semicircular canals were found to have mutations in CHD7. Our review of clinical data from 379 patients suggests that there is no difference in frequency of heart defects, choanal atresia/stenosis, genitourinary abnormalities, clefting, or trecheoesophageal fistula between these two groups of individuals. Interestingly, CHD7 mutation-positive patients also appear less likely to have growth and developmental delays, and more likely to have facial palsy compared with mutation-negative individuals.
Previous diagnostic criteria for CHARGE by Blake et al. included ocular coloboma or microphthalmia, choanal atresia or stenosis, characteristic inner and external ear anomalies, and cranial nerve dysfunction as major criteria [Blake et al., 1998]. Revised diagnostic criteria by Verloes include ocular coloboma, choanal atresia/stenosis and hypoplasia of the semicircular canals as major criteria [Verloes, 2005]. While both sets of diagnostic criteria are used clinically, the utility of subtypes such as atypical or partial CHARGE may be relatively low, since these individuals can also present with a CHD7 mutation. In that regard, it may be more helpful to estimate whether an individual has an identifiable CHD7 mutation given the presence or absence of the various CHARGE related clinical features. The full spectrum of CHD7 mutation related phenotypes remain to be determined, but can be explored using current massively parallel rapid sequencing technologies.
CHD7
CHD7 belongs to the chromodomain helicase DNA binding (CHD) family, one of four major families of ATP-dependent chromatin remodeling proteins [Martens and Winston, 2003; Corona and Tamkun, 2004; Denslow and Wade, 2007; Conaway and Conaway, 2009]. The CHD protein family is evolutionarily conserved in eukaryotes from the sole member, CHD1, in the yeast Saccharomyces cerevisiae to the nine CHD proteins found in vertebrates. CHD7 has homologs in several model organisms, including Drosophila, zebrafish, and mouse. Studies of the CHD proteins in a variety of in vivo and in vitro systems have yielded insights into many functions of CHD proteins, including methylated histone binding [Flanagan et al., 2005; Rodriguez-Paredes et al., 2009; Schnetz et al., 2009], DNA binding [Stokes and Perry, 1995; Bouazoune et al., 2002; Shur and Benayahu, 2005], transcriptional regulation [Surapureddi et al., 2002; Ishihara et al., 2006; Surapureddi et al., 2006; Yuan et al., 2007; Rodriguez-Paredes et al., 2009; Schnetz et al., 2009], cell cycle regulation [Biswas et al., 2008; Rodriguez-Paredes et al., 2009], regulation of apoptosis [Bagchi et al., 2007; Nishiyama et al., 2009], chromatin remodeling [Shur and Benayahu 2005; Lutz et al., 2006; Stockdale et al., 2006; Denslow and Wade 2007; Biswas et al., 2008; Thompson et al., 2008], and embryonic stem cell pluripotency [Gaspar-Maia et al., 2009]. Two motifs define members of this family: tandem N-terminal chromodomains, responsible for binding methylated histones [Brehm et al., 2004], and a central SNF2-like ATPase/helicase domain that contains chromatin remodeling activity. While all nine members share these domains, the family can be subdivided into three subgroups based on the presence of additional domains (for review, see [Hall and Georgel, 2007; Marfella and Imbalzano, 2007]).
The CHD7 gene is located on chromosome 8q12.1. Its genomic structure spans 188kb and encompasses 38 exons, the first of which is noncoding. The encoded CHD7 protein (Fig 1) is 2997 amino acids in length and has the tandem N-terminal chromodomains and central helicase domain characteristic of members of the CHD family. It also contains a C-terminal DNA binding domain that shares a low degree of homology with the histone/DNA-binding SANT domain [Cruz et al., 2005] as well as two C-terminal BRK domains, the functions of which are currently unknown. Nuclear localization is predicted due to the presence of these chromatin-related domains as well as five consensus nuclear localization signals (NLS). The nuclear localization of CHD7 has been experimentally confirmed [Schnetz et al., 2009].
Figure 1.
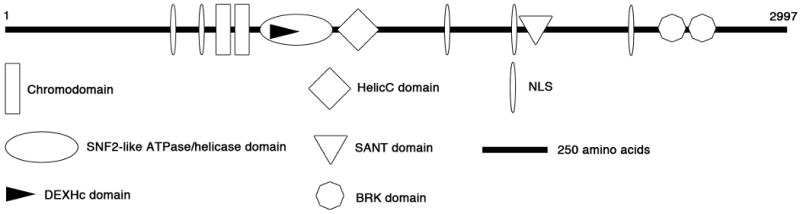
A schematic representation of the CHD7 protein. Domains are depicted approximately to scale.
Expression analysis of CHD7 has been performed in human [Sanlaville et al., 2006] and mouse embryos by in situ hybridization [Bosman et al., 2005; Lalani et al., 2006], β-galactosidase expression [Hurd et al., 2007] and immunofluorescence [Adams et al., 2007; Layman et al., 2009]. Expression of CHD7 is ubiquitous in human fetal tissues by 22 days of development. Later in development, CHD7 localizes to several tissues affected in CHARGE syndrome including the developing ear, eye, and olfactory system. Expression of Chd7 in the mouse embryo is generally consistent with the patterns seen in human fetuses. Expression is widespread and high early in development, with progressive restriction in CHARGE-relevant tissues. In both mice and humans, the expression patterns of CHD7 are dynamic as development progresses, suggesting that CHD7 expression during development is not only tissue-specific but also temporally regulated.
CHD7 Mutant Mice: Models of Charge Syndrome
The first Chd7 mutant mice were reported by Bosman and colleagues, who characterized a set of nine mouse lines with ENU-induced mutations on chromosome 4 that displayed dominant circling behavior owing to inner ear defects [Bosman et al., 2005]. Sequence analysis revealed that all mutations fell within the Chd7 gene. Interestingly, this sequence analysis identified the mutations as truncating mutations or splice site changes that were predicted to be loss-of-function. Detailed analysis of Whirligig (Chd7Whi/+), which carries a heterozygous nonsense mutation in exon 11 (2918G→A leading to W973X), demonstrated the presence of several CHARGE-like features in adult mice and embryos. Chd7Whi/+ mice showed genital defects including vulval hypoplasia and clitoral abnormalities in 94% of adult females, though no genital abnormalities were found in mutant males. 45% of the mutant embryos displayed interventricular septal defects in the heart, a feature of human CHARGE syndrome. 35% of the mutant mice displayed cleft palate or related palatal defects and choanal defects were also seen in some mutant embryos. No optic colobomas were found, though approximately 50% of the mutant mice developed keratoconjunctivitis sicca. Truncation of the lateral semicircular canal and circling behaviors were observed, consistent with the vestibular dysfunction seen in human CHARGE patients. The mean body weight of both male and female Chd7Whi/+ mice was significantly lower than wild-type controls, consistent with the postnatal growth retardation observed in human CHARGE patients.
Use of gene-trap technology to induce Chd7 deficiency in mice has also yielded insights into some of the developmental defects of CHARGE syndrome [Hurd et al., 2007]. The gene-trap strategy used to disrupt Chd7 introduced a β-galactosidase expression vector between exons 1 and 2 of the gene, resulting in a loss-of-function Chd7 allele. Analysis of mice heterozygous for the gene-trapped Chd7 allele (Chd7Gt/+) showed that they recapitulate the circling behavior observed with the ENU-induced mutants, providing further support for vestibular dysfunction as a consequence of Chd7 mutation. Chd7Gt/+ mice also display severe inner ear defects, including truncation or hypoplasia of both the lateral and posterior semicircular canals [Adams et al., 2007]. In addition to inner ear defects in the Chd7Gt/+ mouse line, defects in the olfactory system including olfactory dysfunction and olfactory bulb hypoplasia were reported [Layman et al., 2009]. Chd7 is expressed in the olfactory basal cells which are the neural stem cell population of the olfactory epithelium. BrdU incorporation and immunofluorescence showed that CHD7 is expressed in proliferating neural stem cells, and that Chd7Gt/+ mice have defects in neural stem cell proliferation leading to a reduction in olfactory sensory neuron production and regeneration. These results suggest that CHD7 is required for the development and maintenance of the olfactory epithelium. The olfactory anomalies seen in Chd7Gt/+ mice also suggest that the olfactory dysfunction in humans with CHARGE syndrome may be in part due to defects of the peripheral olfactory system.
Analysis of the reproductive and olfactory abilities of Chd7Whi/+ mice has shown further correlation with human CHARGE phenotypes [Bergman et al., 2009]. Chd7Whi/+ mice have severely reduced olfaction in a smell test and hypoplasia of the olfactory bulb. Chd7Whi/+ mice have a reduction in GnRH neurons in the hypothalamus and median eminence. A reduction in GnRH neurons may contribute to the decrease in gonadotropins commonly found in human CHARGE patients. Interestingly, the mean testis weight of Chd7Whi/+ adult males was significantly lower than wild-type controls and two males also showed severe testicular hypoplasia. These results contrast a previous study of Chd7Whi/+ mice, which showed no genital abnormalities in males [Bosman et al., 2005]. Fertility was also significantly reduced in both male and female Chd7Whi/+ mice.
It is currently unclear why mice with mutations in Chd7 display some, but not all, features of human CHARGE syndrome. The phenotypic discrepancies between humans and mice with CHD7 mutations may simply reflect species-specific differences in the developmental requirement for CHD7. However, it is also likely that the genetic background of the mice plays a role. Genetic modifiers in the genome may alter the penetrance and expressivity of certain phenotypic features in both mice and in humans. To address this question, the mutation could be crossed onto several genetic backgrounds and the mutant progeny examined for penetrance and expressivity of the various CHARGE features. Interestingly, Chd7Gt/+mice maintained on a C57BL/6J background display a more severe ear phenotype while mice on a 129S1/Sv1mJ background display a more severe olfactory phenotype than Chd7Gt/+ mice maintained on a mixed background (D.M.M., unpublished data).
Molecular Analysis of CHD7 Function
In the eukaryotic cell, DNA is wrapped around basic protein octamers composed of histones. In addition to a central globular DNA-binding domain, histones have flexible N-terminal tails that can be modified on many residues by a wide range of enzymes. Modification of histone tails by methylation, acetylation, ubiquitination, and other mechanisms exerts differential effects on transcription [Kouzarides, 2007]. The structure of the CHD7 protein, which includes NLSs, domains for binding methylated histones and DNA, and altering chromatin structure, indicates that it is likely a nuclear protein with transcriptional regulatory activity, acting as an effector for histone modifications. Accordingly, molecular studies of CHD7 have thus far focused on its role as a transcriptional regulator involved in recognizing histone modifications and altering chromatin structure.
Recent genomic studies have provided functional insights into CHD7 [Schnetz et al., 2009]. The chromodomains of CHD7 were found to be functional and able to bind all methylated forms of histone H3 lysine 4 (H3K4me) in vitro. The genomic distribution of CHD7 on chromatin was mapped by chromatin immunoprecipitation on tiled microarrays (ChIP-chip), a technique designed to detect specific protein-DNA interactions. CHD7 binding was correlated closely with regions of H3K4 methylation, characteristic of regions undergoing active transcription. A large number of CHD7 binding sites also displayed characteristics similar to enhancer elements, i.e. they were marked with H3K4me1, predominantly distal to transcriptional start sites, often contained within regions of open chromatin, and located near genes with relatively high levels of expression [Heintzman et al., 2007]. Moreover, several of the CHD7 binding sites were shown to enhance the activity of luciferase reporter genes [Schnetz et al., 2009]. Notably, binding of CHD7 to sites of H3K4me1 was much stronger than binding to sites of H3K4me3, suggesting that CHD7 binds strongly to enhancer elements and transiently loops chromatin to transcription start sites to promote transcriptional regulation (Fig 2). This provides a possible mechanism by which dysregulation of CHD7 target genes during embryonic development could lead to CHARGE syndrome.
Figure 2.

A model for CHD7 function. CHD7 binds to enhancer elements in the presence of protein cofactors and facilitates looping of chromatin to bring the enhancer in close proximity with transcription start sites and allowing CHD7 and associated cofactors to modulate the transcriptional output of the gene.
Studies of the Drosophila melanogaster ortholog of CHD7, Kismet, also support a role for CHD7 as a transcriptional regulator [Srinivasan et al., 2008]. Kismet was found to bind to chromatin at transcriptional start sites and antagonize the establishment of H3K27 trimethylation, a well-characterized repressive histone modification [Schwartz and Pirrotta, 2007], to facilitate early elongation by RNA polymerase II. Interestingly, the chromodomains of Kismet were unable to bind methylated H3K4 in vitro, unlike the chromodomains of human CHD7 [Schnetz et al., 2009]. While Kismet and mammalian CHD7 both appear to function in transcriptional regulation, the mechanisms appear to be different. Kismet binds to transcriptional start sites and facilitates transcriptional elongation, while CHD7 binds to distal enhancer elements and may loop chromatin to transcriptional start sites to promote transcription and facilitate elongation. This may reflect differences in CHD protein function between mammals and insects. However, an alternative explanation for this discrepancy is that Kismet is the Drosophila melanogaster ortholog of not only CHD7, but also CHD8 and CHD9 and that the loss of one CHD protein may have a different functional consequence than loss of all three. Further studies in mammalian cells with knockdown of various combinations of CHD7, CHD8, and CHD9 are needed to elucidate the functional conservation of these proteins with Kismet.
Discussion and Future Directions
Perhaps the most important question regarding the pathogenesis of CHARGE syndrome is how haploinsufficiency of CHD7 gives rise to the specific defects seen in the disorder. Some insight into this issue may come from the function of CHD7. Studies of CHD7 suggest that it has a role in regulating transcription through enhancer elements. Enhancers are known to act with both tissue and temporal specificity; thus, their activity may not be conserved between cell types or developmental stages. It therefore seems likely that CHD7 is required in the developing cell types that give rise to CHARGE-affected tissues to regulate the expression of genes affecting cell proliferation, differentiation, and migration. One possibility is that 50% of the total CHD7 protein in these cell types is insufficient to modulate transcription of these developmentally important genes. However, this possibility may be simplistic, as enhancer-mediated regulation of CHD7 target genes is likely to be cofactor dependent. Recent studies in murine embryonic stem cells have shown that up to five factors may be bound at certain enhancer elements [Chen et al., 2008]. Transcriptional modulation by enhancement may also be dependent on multiple enhancers, as demonstrated for genes within the β-globin cluster and the imprinted IGF2 and H19 loci [Tuan et al., 1985; Dhar et al., 1990; Leighton et al., 1995; Webber et al., 1998]. Current knowledge about CHD7 binding to enhancer elements predicts that loss of 50% of CHD7 protein may impair binding of cofactors to chromatin, looping of enhancers to transcriptional start sites, and/or modulation of chromatin structure at transcriptional start sites. However, further developmental and biochemical studies are needed to delineate the roles of CHD7 in enhancer-mediated transcriptional regulation in various tissues and developmental stages.
Based on analysis of human and mouse embryos, CHD7 expression is variable and dependent on tissue and developmental time. This observation suggests that the requirements for CHD7 are dynamic, depending both on the tissue in question and the developmental stage, and is consistent with the implication of CHD7 in enhancer-mediated transcriptional regulation. To address this challenging problem, mice with conditional knockouts of Chd7 are being created to allow tissue- and temporal-specific reduction of CHD7 levels and assessment of specific developmental consequences of its loss (D.M.M., unpublished data).
Another important component to understanding the pathogenesis of CHARGE syndrome will be discovering regulatory targets of CHD7. Because current evidence suggests that CHD7 regulates transcription via enhancer elements, which may be specific to a certain cell type, it stands to reason that CHD7 may have a distinct set of target genes in individual cell types in specific stages and tissues development. Uncovering these targets in the cell types that give rise to CHARGE-affected tissues will be most informative with regard to the regulatory role of CHD7 in development.
Further molecular analysis of CHD7 is also needed to advance our understanding of CHARGE syndrome. Chromatin remodeling enzymes are generally found in large multiprotein complexes [Vignali et al., 2000], and it seems likely that CHD7 interacts with a specific set of proteins to achieve its regulatory effects. Biochemical purification of this complex will provide insights into the mechanism of transcriptional regulation by CHD7. As mentioned previously, it is likely that CHD7 has different protein partners in different cell types, allowing for diversity in its transcriptional regulatory activity.
Together, mouse models of CHARGE syndrome and functional studies of CHD7 have begun to provide molecular explanations for the diverse clinical manifestations of CHARGE syndrome. Continued study of the molecular functions of CHD7 and its requirement in the development of embryonic tissues will be important in advancing our understanding of this complex condition.
Acknowledgments
We thank Dr. Shawn McCandless for critical reading of the manuscript, Cindy Bartels for assistance with compiling reported CHD7 mutations, and the anonymous reviewers for their comments. G.E.Z. is supported by a training grant from the National Institute of General Medical Sciences (5T32GM008613-14). W.S.L. is supported by a training grant from the National Institute of Deafness and Other Communication Disorders (T32DC00011). D.M.M. is supported by grants from the National Institutes of Health (R01DC009410 and R01NS054784). P.C.S. is supported by grants from the National Institute of Child Health and Development (R01HD056369) and the National Human Genome Research Institute (5R01HG004722).
References
- Adams ME, Hurd EA, Beyer LA, Swiderski DL, Raphael Y, Martin DM. Defects in vestibular sensory epithelia and innervation in mice with loss of Chd7 function: implications for human CHARGE syndrome. J Comp Neurol. 2007;504(5):519–32. doi: 10.1002/cne.21460. [DOI] [PubMed] [Google Scholar]
- Alazami MA, Alzahrani F, Alkuraya FS. Expanding the “E” in CHARGE. Am J Med Genet A. 2008;146A(14):1890–1892. doi: 10.1002/ajmg.a.32376. [DOI] [PubMed] [Google Scholar]
- Aramaki M, Udaka T, Kosaki R, Makita Y, Okamoto N, Yoshihashi H, Oki H, Nanao K, Moriyama N, Oku S, Hasegawa T, Takahashi T, Fukushima Y, Kawame H, Kosaki K. Phenotypic spectrum of CHARGE syndrome with CHD7 mutations. J Pediatr. 2006;148(3):410–414. doi: 10.1016/j.jpeds.2005.10.044. [DOI] [PubMed] [Google Scholar]
- Arrington CB, Cowley BC, Nightingale DR, Zhou H, Brothman HR, Viskochil DH. Interstitial deletion 8q11.2-q13 with congenital anomalies of CHARGE association. Am J Med Genet A. 2005;133A(3):326–330. doi: 10.1002/ajmg.a.30562. [DOI] [PubMed] [Google Scholar]
- Asakura Y, Toyota Y, Muroya K, Kurosawa K, Fujita K, Aida N, Kawame H, Kosaki K, Adachi M. Endocrine and Radiological Studies in Patients with Molecularly Confirmed CHARGE Syndrome. J Clin Endocrinol Metab. 2008;93(3):920–924. doi: 10.1210/jc.2007-1419. [DOI] [PubMed] [Google Scholar]
- Bagchi A, Papazoglu C, Wu Y, Capurso D, Brodt M, Francis D, Bredel M, Vogel H, Mills AA. CHD5 Is a Tumor Suppressor at Human 1p36. Cell. 2007;128(3):459–475. doi: 10.1016/j.cell.2006.11.052. [DOI] [PubMed] [Google Scholar]
- Bergman JE, de Wijs I, Jongmans MC, Admiraal RJ, Hoefsloot LH, van Ravenswaaij-Arts CM. Exon copy number alterations of the CHD7 gene are not a major cause of CHARGE and CHARGE-like syndrome. Eur J Med Genet. 2008;51(5):417–425. doi: 10.1016/j.ejmg.2008.03.003. [DOI] [PubMed] [Google Scholar]
- Bergman JEH, Bosman EA, van Ravenswaaij-Arts CMA, Steel KP. Study of smell and reproductive organs in a mouse model for CHARGE syndrome. Eur J Hum Genet. 2009 doi: 10.1038/ejhg.2009.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biswas D, Takahata S, Xin H, Dutta-Biswas R, Yu Y, Formosa T, Stillman DJ. A Role for Chd1 and Set2 in Negatively Regulating DNA Replication in Saccharomyces cerevisiae. Genetics. 2008;178(2):649–659. doi: 10.1534/genetics.107.084202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blake KD, Hall BD, Hefner MA, Pagon RA, Williams MS, Lin AE, Graham JMJ. CHARGE association: an update and review for the primary pediatrician. Clin Pediatr (Phila) 1998;37(3):159–173. doi: 10.1177/000992289803700302. [DOI] [PubMed] [Google Scholar]
- Bosman EA, Penn AC, Ambrose JC, Kettleborough R, Stemple DL, Steel KP. Multiple mutations in mouse Chd7 provide models for CHARGE syndrome. Hum Mol Genet. 2005;14(22):3463–3476. doi: 10.1093/hmg/ddi375. [DOI] [PubMed] [Google Scholar]
- Bouazoune K, Mitterweger A, Langst G, Imhof A, Akhtar A, Becker PB, Brehm A. The dMi-2 chromodomains are DNA binding modules important for ATP-dependent nucleosome mobilization. EMBO J. 2002;21(10):2430–2440. doi: 10.1093/emboj/21.10.2430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brehm A, Tufteland KR, Aasland R, Becker PB. The many colours of chromodomains. BioEssays. 2004;26(2):133–140. doi: 10.1002/bies.10392. [DOI] [PubMed] [Google Scholar]
- Chen X, Xu H, Yuan P, Fang F, Huss M, Vega VB, Wong E, Orlov YL, Zhang W, Jiang J, Loh YH, Yeo HC, Yeo ZX, Narang V, Govindarajan KR, Leong B, Shahab A, Ruan Y, Bourque G, Sung WK, Clarke ND, Wei CL, Ng HH. Integration of External Signaling Pathways with the Core Transcriptional Network in Embryonic Stem Cells. Cell. 2008;133(6):1106–1117. doi: 10.1016/j.cell.2008.04.043. [DOI] [PubMed] [Google Scholar]
- Chopra C, Baretto R, Duddridge M, Browning MJ. T-Cell Immunodeficiency in CHARGE syndrome. Acta Pediatrica. 2009;98(2):408–410. doi: 10.1111/j.1651-2227.2008.01077.x. [DOI] [PubMed] [Google Scholar]
- Clementi C, Tenconi R, Turolla L, Silvan C, Bortotto L, Artifoni L. Apparent CHARGE association and chromosome anomaly: Chance or contiguous gene syndrome. Am J Med Genet. 1991;41(2):246–250. doi: 10.1002/ajmg.1320410223. [DOI] [PubMed] [Google Scholar]
- Conaway RC, Conaway JW. The INO80 chromatin remodeling complex in transcription, replication and repair. Trends in Biochemical Sciences. 2009;34(2):71–77. doi: 10.1016/j.tibs.2008.10.010. [DOI] [PubMed] [Google Scholar]
- Corona DFV, Tamkun JW. Multiple roles for ISWI in transcription, chromosome organization and DNA replication. Biochem Biophys Res Comm. 2004;1677(1-3):113–119. doi: 10.1016/j.bbaexp.2003.09.018. [DOI] [PubMed] [Google Scholar]
- Cruz Xdl, Lois S, Sánchez-Molina S, Martínez-Balbás MA. Do protein motifs read the histone code? BioEssays. 2005;27(2):164–175. doi: 10.1002/bies.20176. [DOI] [PubMed] [Google Scholar]
- De Krijger RR, Mooy CM, Van Hemel JO, Sulkers EJ, Kros JM, Bartelings MM, Govaerts LCP. CHARGE Association-related Ocular Pathology in a Newborn with Partial Trisomy 19q and Partial Monosomy 21q, from a Maternal Translocation (19;21) (q13.1;q22.3) Pediatr Dev Pathol. 1999;2(6):577–581. doi: 10.1007/s100249900165. [DOI] [PubMed] [Google Scholar]
- Delahaye A, Sznajer Y, Lyonnet S, Elmaleh-Berges M, Delpierre I, Audollent S, Wiener-Vacher S, Mansbach AL, Amiel J, Baumann C, Bremond-Gignac D, Attie-Bitach T, Verloes A, Sanlaville D. Familial CHARGE syndrome because of CHD7 mutation: clinical intra- and interfamilial variability. Clin Genet. 2007;72(2):112–121. doi: 10.1111/j.1399-0004.2007.00821.x. [DOI] [PubMed] [Google Scholar]
- Denslow SA, Wade PA. The human Mi-2/NuRD complex and gene regulation. Oncogene. 2007;26(37):5433–5438. doi: 10.1038/sj.onc.1210611. [DOI] [PubMed] [Google Scholar]
- Dhar V, Nandi A, Schildkraut CL, Skoultchi AI. Erythroid-specific nuclease-hypersensitive sites flanking the human beta-globin domain. Mol Cell Biol. 1990;10(8):4324–4333. doi: 10.1128/mcb.10.8.4324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon J, Trainor P, Dixon MJ. Treacher Collins syndrome. Orthod Craniofac Res. 2007;10(2):88–95. doi: 10.1111/j.1601-6343.2007.00388.x. [DOI] [PubMed] [Google Scholar]
- Edwards BM, Kileny PR, Van Riper LA. CHARGE Syndrome: A Window of Opportunity for Audiologic Intervention. Pediatrics. 2002;110(1):119–126. doi: 10.1542/peds.110.1.119. [DOI] [PubMed] [Google Scholar]
- Flanagan JF, Mi LZ, Chruszcz M, Cymborowski M, Clines KL, Kim Y, Minor W, Rastinejad F, Khorasanizadeh S. Double chromodomains cooperate to recognize the methylated histone H3 tail. Nature. 2005;438(7071):1181–1185. doi: 10.1038/nature04290. [DOI] [PubMed] [Google Scholar]
- Gao X, Gordon D, Zhang D, Browne R, Helms C, Gillum J, Weber S, Devroy S, Swaney S, Dobbs M, Morcuende J, Sheffield V, Lovett M, Bowcock A, Herring J, Wise C. CHD7 gene polymorphisms are associated with susceptibility to idiopathic scoliosis. Am J Hum Genet. 2007;80(5):957–965. doi: 10.1086/513571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaspar-Maia A, Alajem A, Polesso F, Sridharan R, Mason MJ, Heidersbach A, Ramalho-Santos Jo, McManus MT, Plath K, Meshorer E, Ramalho-Santos M. Chd1 regulates open chromatin and pluripotency of embryonic stem cells. Nature. 2009;460(7257):863–868. doi: 10.1038/nature08212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gennery AR, Slatter MA, Rice J, Hoefsloot LH, Barge D, McLean-Tooke A, Montgomery T, Goodship JA, Burt AD, Flood TJ, Abinun M, Cant AJ, Johnson D. Mutations in CHD7 in patients with CHARGE syndrome cause T-B + natural killer cell + severe combined immune deficiency and may cause Omenn-like syndrome. Clin Exp Immunol. 2008;153(1):75–80. doi: 10.1111/j.1365-2249.2008.03681.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall BD. Choanal atresia and associated multiple anomalies. J Pediatr. 1979;95(3):395–398. doi: 10.1016/s0022-3476(79)80513-2. [DOI] [PubMed] [Google Scholar]
- Hall JA, Georgel PT. CHD proteins: a diverse family with strong ties. Biochem Cell Biol. 2007;85:463–476. doi: 10.1139/O07-063. [DOI] [PubMed] [Google Scholar]
- Heintzman ND, Stuart RK, Hon G, Fu Y, Ching CW, Hawkins RD, Barrera LO, Van Calcar S, Qu C, Ching KA, Wang W, Weng Z, Green RD, Crawford GE, Ren B. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat Genet. 2007;39(3):311–318. doi: 10.1038/ng1966. [DOI] [PubMed] [Google Scholar]
- Hittner HM, Hirsch NJ, Kreh GM, Rudolph AJ. Colobomatous micropthalmia, heart disease, hearing loss, and mental retardation-a syndrome. J Pediatr Ophth Strab. 1979;16(2):122–128. doi: 10.3928/0191-3913-19790301-10. [DOI] [PubMed] [Google Scholar]
- Hurd E, Capers P, Blauwkamp M, Adams M, Raphael Y, Poucher H, Martin D. Loss of Chd7 function in gene-trapped reporter mice is embryonic lethal and associated with severe defects in multiple developing tissues. Mamm Genome. 2007;18(2):94–104. doi: 10.1007/s00335-006-0107-6. [DOI] [PubMed] [Google Scholar]
- Ishihara K, Oshimura M, Nakao M. CTCF-Dependent Chromatin Insulator Is Linked to Epigenetic Remodeling. Mol Cell. 2006;23(5):733–742. doi: 10.1016/j.molcel.2006.08.008. [DOI] [PubMed] [Google Scholar]
- Issekutz KA, Graham JM, Jr, Prasad C, Smith IM, Blake KD. An epidemiological analysis of CHARGE syndrome: preliminary results from a Canadian study. Am J Med Genet A. 2005;133A(3):309–317. doi: 10.1002/ajmg.a.30560. [DOI] [PubMed] [Google Scholar]
- Johnson D, Morrison N, Grant L, Turner T, Fantes J, Connor JM, Murday V. Confirmation of CHD7 as a cause of CHARGE association identified by mapping a balanced chromosome translocation in affected monozygotic twins. J Med Genet. 2006;43(3):280–284. doi: 10.1136/jmg.2005.032946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jongmans MC, Admiraal RJ, van der Donk KP, Vissers LE, Baas AF, Kapusta L, van Hagen JM, Donnai D, de Ravel TJ, Veltman JA, Geurts van Kessel A, De Vries BB, Brunner HG, Hoefsloot LH, van Ravenswaaij CM. CHARGE syndrome: the phenotypic spectrum of mutations in the CHD7 gene. J Med Genet. 2006;43(4):306–314. doi: 10.1136/jmg.2005.036061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jongmans MC, Hoefsloot LH, van der Donk KP, Admiraal RJ, Magee A, van de Laar I, Hendriks Y, Verheij JB, Walpole I, Brunner HG, van Ravenswaaij CM. Familial CHARGE syndrome and the CHD7 gene: a recurrent missense mutation, intrafamilial recurrence and variability. Am J Med Genet A. 2008;146A(1):43–50. doi: 10.1002/ajmg.a.31921. [DOI] [PubMed] [Google Scholar]
- Jongmans MC, van Ravenswaaij-Arts CM, Pitteloud N, Ogata T, Sato N, Claahsen-van der Grinten HL, van der Donk K, Seminara S, Bergman JE, Brunner HG, Crowley WF, Jr, Hoefsloot LH. CHD7 mutations in patients initially diagnosed with Kallmann syndrome--the clinical overlap with CHARGE syndrome. Clin Genet. 2009;75(1):65–71. doi: 10.1111/j.1399-0004.2008.01107.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jyonouchi S, McDonald-McGinn DM, Bale S, Zackai EH, Sullivan KE. CHARGE (coloboma, heart defect, atresia choanae, retarded growth and development, genital hypoplasia, ear anomalies/deafness) syndrome and chromosome 22q11.2 deletion syndrome: a comparison of immunologic and nonimmunologic phenotypic features. Pediatrics. 2009;123(5):871–877. doi: 10.1542/peds.2008-3400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HG, Kurth I, Lan F, Meliciani I, Wenzel W, Eom SH, Kang GB, Rosenberger G, Tekin M, Ozata M, Bick DP, Sherins RJ, Walker SL, Shi Y, Gusella JF, Layman LC. Mutations in CHD7, Encoding a Chromatin-Remodeling Protein, Cause Idiopathic Hypogonadotropic Hypogonadism and Kallmann Syndrome. Am J Hum Genet. 2008;83(4):511–519. doi: 10.1016/j.ajhg.2008.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouzarides T. Chromatin Modifications and Their Function. Cell. 2007;128(4):693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- Lalani SR, Safiullah AM, Fernbach SD, Harutyunyan KG, Thaller C, Peterson LE, McPherson JD, Gibbs RA, White LD, Hefner M, Davenport SL, Graham JM, Bacino CA, Glass NL, Towbin JA, Craigen WJ, Neish SR, Lin AE, Belmont JW. Spectrum of CHD7 mutations in 110 individuals with CHARGE syndrome and genotype-phenotype correlation. Am J Hum Genet. 2006;78(2):303–314. doi: 10.1086/500273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Layman WS, McEwen DP, Beyer LA, Lalani SR, Fernbach SD, Oh E, Swaroop A, Hegg CC, Raphael Y, Martens JR, Martin DM. Defects in neural stem cell proliferation and olfaction in Chd7 deficient mice indicate a mechanism for hyposmia in human CHARGE syndrome. Hum Mol Genet. 2009:ddp112. doi: 10.1093/hmg/ddp112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YW, Kim S, Shin Y, Kim JW, Hong H, Lee Y, Ki CS. Clinical and genetic analysis of the CHD7 gene in Korean patients with CHARGE syndrome. Clinical Genetics. 2009;75(3):290–293. doi: 10.1111/j.1399-0004.2008.01127.x. [DOI] [PubMed] [Google Scholar]
- Leighton P, Saam J, Ingram R, Stewart C, Tilghman S. An enhancer deletion affects both H19 and Igf2 expression. Genes Dev. 1995;9(17):2079–2089. doi: 10.1101/gad.9.17.2079. [DOI] [PubMed] [Google Scholar]
- Lev D, Nakar O, Bar-Am I, Zudik A, Watemberg N, Finkelstein S, Katzin N, Lerman-Sagie T. CHARGE association in a child with de novo chromosomal aberration 46,X,der(X)t(X;2)(p22.1;q33) detected by spectral karyotyping. J Med Genet. 2000;37(12):e47. doi: 10.1136/jmg.37.12.e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin AE, Chin AJ, Devine W, Park SC, Zackal E. The pattern of cardiovascular malformation in the CHARGE association. Am J Dis Child. 1987;141(9):1010–1013. doi: 10.1001/archpedi.1987.04460090087034. [DOI] [PubMed] [Google Scholar]
- Lutz T, Stöger R, Nieto A. CHD6 is a DNA-dependent ATPase and localizes at nuclear sites of mRNA synthesis. FEBS Letters. 2006;580(25):5851–5857. doi: 10.1016/j.febslet.2006.09.049. [DOI] [PubMed] [Google Scholar]
- Marfella CGA, Imbalzano AN. The Chd family of chromatin remodelers. Mutat Res. 2007;618(1-2):30–40. doi: 10.1016/j.mrfmmm.2006.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martens JA, Winston F. Recent advances in understanding chromatin remodeling by Swi/Snf complexes. Curr Opin Genet Dev. 2003;13(2):136–142. doi: 10.1016/s0959-437x(03)00022-4. [DOI] [PubMed] [Google Scholar]
- Nishiyama M, Oshikawa K, Tsukada Yi, Nakagawa T, Iemura Si, Natsume T, Fan Y, Kikuchi A, Skoultchi AI, Nakayama KI. CHD8 suppresses p53-mediated apoptosis through histone H1 recruitment during early embryogenesis. Nat Cell Biol. 2009;11(2):172–182. doi: 10.1038/ncb1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- North KN, Wu BL, Cao BN, Whiteman DAH, Korf BR. CHARGE association in a child with de novo inverted duplication (14)(q22 rarr q24.3) Am J Med Genet. 1995;57(4):610–614. doi: 10.1002/ajmg.1320570419. [DOI] [PubMed] [Google Scholar]
- Ogata T, Fujiwara I, Ogawa E, Sato N, Udaka T, Kosaki K. Kallmann syndrome phenotype in a female patient with CHARGE syndrome and CHD7 mutation. Endocr J. 2006;53(6):741–3. doi: 10.1507/endocrj.k06-099. [DOI] [PubMed] [Google Scholar]
- Pagon RA, Graham HM, Zonana J, Yong SL. Coloboma, congenital heart disease, and choanal atresia with multiple anomalies: CHARGE association. J Pediatr. 1981;99(2):223–227. doi: 10.1016/s0022-3476(81)80454-4. [DOI] [PubMed] [Google Scholar]
- Pauli S, Pieper L, Haberle J, Grzmil P, Burfeind P, Steckel M, Lenz U, Michelmann HW. Proven germline mosaicism in a father of two children with CHARGE syndrome. Clin Genet. 2009;75(5):473–9. doi: 10.1111/j.1399-0004.2009.01151.x. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Paredes M, Ceballos-Chavez M, Esteller M, Garcia-Dominguez M, Reyes JC. The chromatin remodeling factor CHD8 interacts with elongating RNA polymerase II and controls expression of the cyclin E2 gene. Nucleic Acids Res. 2009:gkp101. doi: 10.1093/nar/gkp101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanka M, Tangsinmankong N, Loscalzo M, Sleasman JW, Dorsey MJ. Complete DiGeorge syndrome associated with CHD7 mutation. J Allergy Clin Immunol. 2007;120(4):952–954. doi: 10.1016/j.jaci.2007.08.013. [DOI] [PubMed] [Google Scholar]
- Sanlaville D, Etchevers HC, Gonzales M, Martinovic J, Clement-Ziza M, Delezoide AL, Aubry MC, Pelet A, Chemouny S, Cruaud C, Audollent S, Esculpavit C, Goudefroye G, Ozilou C, Fredouille C, Joye N, Morichon-Delvallez N, Dumez Y, Weissenbach J, Munnich A, Amiel J, Encha-Razavi F, Lyonnet S, Vekemans M, Attie-Bitach T. Phenotypic spectrum of CHARGE syndrome in fetuses with CHD7 truncating mutations correlates with expression during human development. J Med Genet. 2006;43(3):211–217. doi: 10.1136/jmg.2005.036160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanlaville D, Verloes A. CHARGE syndrome: an update. Eur J Hum Genet. 2007;15(4):389–99. doi: 10.1038/sj.ejhg.5201778. [DOI] [PubMed] [Google Scholar]
- Schnetz MP, Bartels CF, Shastri K, Balasubramanian D, Zentner GE, Balaji R, Song L, Wang Z, LaFramboise T, Crawford GE, Scacheri PC. Genomic distribution of CHD7 on chromatin tracks H3K4 methylation patterns. Genome Res. 2009;19:590–601. doi: 10.1101/gr.086983.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz YB, Pirrotta V. Polycomb silencing mechanisms and the management of genomic programmes. Nat Rev Genet. 2007;8(1):9–22. doi: 10.1038/nrg1981. [DOI] [PubMed] [Google Scholar]
- Shur I, Benayahu D. Characterization and Functional Analysis of CReMM, a Novel Chromodomain Helicase DNA-binding Protein. J Mol Biol. 2005;352(3):646–655. doi: 10.1016/j.jmb.2005.06.049. [DOI] [PubMed] [Google Scholar]
- Srinivasan S, Dorighi KM, Tamkun JW. Drosophila Kismet Regulates Histone H3 Lysine 27 Methylation and Early Elongation by RNA Polymerase II. PLoS Genetics. 2008;4(10):e1000217. doi: 10.1371/journal.pgen.1000217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stockdale C, Flaus A, Ferreira H, Owen-Hughes T. Analysis of Nucleosome Repositioning by Yeast ISWI and Chd1 Chromatin Remodeling Complexes. J Biol Chem. 2006;281(24):16279–16288. doi: 10.1074/jbc.M600682200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stokes D, Perry R. DNA-binding and chromatin localization properties of CHD1. Mol Cell Biol. 1995;15(5):2745–2753. doi: 10.1128/mcb.15.5.2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan KE. Chromosome 22q11.2 Deletion Syndrome: DiGeorge Syndrome/Velocardiofacial Syndrome. Immunol Allergy Clin North Am. 2008;28(2):353–366. doi: 10.1016/j.iac.2008.01.003. [DOI] [PubMed] [Google Scholar]
- Surapureddi S, Viswakarma N, Yu S, Guo D, Rao MS, Reddy JK. PRIC320, a transcription coactivator, isolated from peroxisome proliferator-binding protein complex. Biochem Biophys Res Comm. 2006;343(2):535–543. doi: 10.1016/j.bbrc.2006.02.160. [DOI] [PubMed] [Google Scholar]
- Surapureddi S, Yu S, Bu H, Hashimoto T, Yeldandi AV, Kashireddy P, Cherkaoui-Malki M, Qi C, Zhu YJ, Rao MS, Reddy JK. Identification of a transcriptionally active peroxisome proliferator-activated receptor α-interacting cofactor complex in rat liver and characterization of PRIC285 as a coactivator. Proc Natl Acad Sci U S A. 2002;99(18):11836–11841. doi: 10.1073/pnas.182426699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tellier AL, Cormier-Daire V, Abadie V, Amiel J, Sigaudy S, Bonne D, de Lonlay-Debeney P, Morrisseau-Durand MP, Hubert P, Michel JL, Jan D, Dollfus H, Baumann C, Labrune P, Lacombe D, Philip N, LeMerrer M, Briard ML, Munnich A, Lyonnet S. CHARGE Syndrome: Report of 47 Cases and Review. Am J Med Genet. 1998;76(5):402–409. doi: 10.1002/(sici)1096-8628(19980413)76:5<402::aid-ajmg7>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- Thompson BA, Tremblay V, Lin G, Bochar DA. CHD8 Is an ATP-Dependent Chromatin Remodeling Factor That Regulates β-Catenin Target Genes. Mol Cell Biol. 2008;28(12):3894–3904. doi: 10.1128/MCB.00322-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuan D, Solomon W, Li Q, London I. The “beta-like-globin” gene domain in human erythroid cells. Proc Natl Acad Sci U S A. 1985;82(19):6384–6388. doi: 10.1073/pnas.82.19.6384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Udaka T, Okamoto N, Aramaki M, Torii C, Kosaki R, Hosokai N, Hayakawa T, Takahata N, Takahashi T, Kosaki K. An Alu retrotransposition-mediated deletion of CHD7 in a patient with CHARGE syndrome. Am J Med Genet A. 2007;143(7):721–6. doi: 10.1002/ajmg.a.31441. [DOI] [PubMed] [Google Scholar]
- Van de Laar I, Dooijes D, Hoefsloot L, Simon M, Hoogeboom J, Devriendt K. Limb anomalies in patients with CHARGE syndrome: an expansion of the phenotype. Am J Med Genet A. 2007;143A(22):2712–5. doi: 10.1002/ajmg.a.32008. [DOI] [PubMed] [Google Scholar]
- Verloes A. Updated diagnostic criteria for CHARGE syndrome: a proposal. Am J Med Genet A. 2005;133A(3):306–8. doi: 10.1002/ajmg.a.30559. [DOI] [PubMed] [Google Scholar]
- Vignali M, Hassan A, Neely K, Workman J. ATP-dependent chromatin-remodeling complexes. Mol Cell Biol. 2000;20:1899–1910. doi: 10.1128/mcb.20.6.1899-1910.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vissers LE, van Ravenswaaij CM, Admiraal R, Hurst JA, de Vries BB, Janssen IM, van der Vliet WA, Huys EH, de Jong PJ, Hamel BC, Schoenmakers EF, Brunner HG, Veltman JA, van Kessel AG. Mutations in a new member of the chromodomain gene family cause CHARGE syndrome. Nat Genet. 2004;36(9):955–7. doi: 10.1038/ng1407. [DOI] [PubMed] [Google Scholar]
- Vuorela PE, Penttinen MT, Hietala MH, Laine JO, Huoponen KA, Kaariainen HA. A familial CHARGE syndrome with a CHD7 nonsense mutation and new clinical features. Clin Dysmorphol. 2008;17(4):249–253. doi: 10.1097/MCD.0b013e328306a704. [DOI] [PubMed] [Google Scholar]
- Webber AL, Ingram RS, Levorse JM, Tilghman SM. Location of enhancers is essential for the imprinting of H19 and Igf2 genes. Nature. 1998;391(6668):711–715. doi: 10.1038/35655. [DOI] [PubMed] [Google Scholar]
- White DR, Giambra BK, Hopkin RJ, Daines CL, Rutter MJ. Aspiration in children with CHARGE syndrome. Int J Pediatr Otorhinolaryngol. 2005;69(9):1205–1209. doi: 10.1016/j.ijporl.2005.03.030. [DOI] [PubMed] [Google Scholar]
- Wieczorek D, Bolt J, Schwechheimer K, Gillessen-Kaesbach G. A patient with interstitial deletion of the short arm of chromosome 3 (pterrarrp21.2∷p12rarrqter) and a CHARGE-like phenotype. Am J Med Genet A. 1997;69(4):413–417. doi: 10.1002/(sici)1096-8628(19970414)69:4<413::aid-ajmg15>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- Wincent J, Holmberg E, Stromland K, Soller M, Mirzaei L, Djureinovic T, Robinson K, Anderlid B, Schoumans J. CHD7 mutation spectrum in 28 Swedish patients diagnosed with CHARGE syndrome. Clin Genet. 2008;74(1):31–38. doi: 10.1111/j.1399-0004.2008.01014.x. [DOI] [PubMed] [Google Scholar]
- Wincent J, Schulze A, Schoumans J. Detection of CHD7 deletions by MLPA in CHARGE syndrome patients with a less typical phenotype. Eur J Med Genet. 2009 doi: 10.1016/j.ejmg.2009.02.005. [DOI] [PubMed] [Google Scholar]
- Wright EM, O'Connor R, Kerr BA. Radial aplasia in CHARGE syndrome: A new association. Eur J Med Genet. 2009 doi: 10.1016/j.ejmg.2009.03.017. [DOI] [PubMed] [Google Scholar]
- Writzl K, Cale CM, Pierce CM, Wilson LC, Hennekam RC. Immunological abnormalities in CHARGE syndrome. Eur J Med Genet. 2007;50(5):338–345. doi: 10.1016/j.ejmg.2007.05.002. [DOI] [PubMed] [Google Scholar]
- Yuan CC, Zhao X, Florens L, Swanson SK, Washburn MP, Hernandez N. CHD8 Associates with Human Staf and Contributes to Efficient U6 RNA Polymerase III Transcription. Mol Cell Biol. 2007;27(24):8729–8738. doi: 10.1128/MCB.00846-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyse RK, al-Mahdawi S, Burn J, Blake K. Congenital heart disease in CHARGE association. Pediatr Cardiol. 14(2):75–81. doi: 10.1007/BF00796983. [DOI] [PubMed] [Google Scholar]