

Abstract
Cerebral accumulation of amyloid β-peptide (Aβ) is characteristic of Alzheimer disease and of amyloid precursor protein (APP) transgenic mice. Here, we assessed the efficacy of CI-1011, an inhibitor of acyl-coenzyme A:cholesterol acyltransferase, which is suitable for clinical use, in reducing amyloid pathology in both young (6.5 months old) and aged (16 months old) hAPP transgenic mice. Treatment of young animals with CI-1011 decreased amyloid plaque load in the cortex and hippocampus and reduced the levels of insoluble Aβ40 and Aβ42 and C-terminal fragments of APP in brain extracts. In aged mice, CI-1011 specifically reduced diffuse amyloid plaques with a minor effect on thioflavin S+ dense-core plaques. Reduced diffusible amyloid was accompanied by suppression of astrogliosis and enhanced microglial activation. Collectively, these data suggest that CI-1011 treatment reduces amyloid burden in hAPP mice by limiting generation and increasing clearance of diffusible Aβ.
Keywords: Alzheimer disease, Cholesterol transport, Glia, Lipids, Neurodegeneration, Transgenic
INTRODUCTION
Alzheimer disease (AD), the most common cause of dementia in the elderly, is characterized by the progressive cerebral accumulation of amyloid-β (Aβ) deposits in either dense-core senile plaques or diffuse amorphous plaques (1). In vivo imaging studies strongly support the amyloid hypothesis, which postulates that formation of senile plaques initiates a pathological cascade resulting in recruitment of microglia and induction of local neuritic changes near the plaques (2, 3). Aβ is composed primarily of 40- and 42-amino acid peptides generated from the amyloid precursor protein (APP) by sequential proteolytic cleavages mediated by β- and γ-secretases (4). Many anti-amyloid therapies are currently in development but only a few have successfully reversed existing amyloid pathology (2, 5). In regulatable APP transgenic mice, a conceptual model for therapies targeting Aβ generation, plaque pathology could not be reversed by simply shutting down APP over-expression and Aβ production (6). Thus, suppression of Aβ generation may only be able to cease the progression of the disease without reversing existing amyloid pathology.
Genetic, epidemiological and biochemical studies have suggested that cholesterol is an important risk factor for AD (7, 8). We have previously shown that pharmacological or genetic inhibition of acyl-coenzyme A:cholesterol acyltransferase (ACAT), an enzyme that controls cellular equilibrium between free cholesterol and cholesteryl esters, modulates proteolytic processing of APP in vitro (9, 10). In a transgenic mouse model of AD, a 2-month treatment with the ACAT inhibitor CP-113,818 markedly reduced Aβ generation and amyloid pathology, resulting in reversal of cognitive deficits (11). Recently, ACAT1 gene ablation in triple transgenic 3xTg-AD mice was shown to reduce brain levels of APP and its proteolytic fragments while improving cognitive function (12).
CI-1011, a [(2,4,6-tris(1-methylethyl)phenyl) acetyl]sulfamic acid, 2,6-bis(1-methylethyl)phenyl ester, also known as avasimibe, is an ACAT inhibitor that is suitable for clinical use because of an improved pharmacological and safety profile (13). CI-1011 failed to improve coronary atherosclerosis in phase III clinical trials (14), but it may hold therapeutic potential for AD. Here, we tested the anti-amyloidogenic effects of CI-1011 in 2 age groups of hAPP transgenic mice. We show that CI-1011 partially protects from development of amyloid pathology in young mice and reduces amyloid burden in old animals with preexisting amyloid deposits. Intriguingly, our results suggest that by limiting further Aβ generation, ACAT inhibition may be able to reverse neuronal damage caused by earlier accumulation of oligomeric deposits of Aβ.
MATERIALS AND METHODS
Mice
hAPP transgenic mice overexpress human APP751 with the London (V717I) and Swedish (K670M/N671L) mutations under the regulatory control of the neuron-specific murine Thy-1 promoter (mThy-1-hAPP751; heterozygous with respect to the transgene, on a C57BL/6 F3 background) (15). Mice were handled and treated as previously described (11). CI-1011 was kindly provided by Dr. Lit-Fui Lau (Pfizer, Groton, Connecticut). The drug was compounded in biopolymer release pellets to provide continuous dosing for 60 days by Innovative Research of America (Sarasota, FL). For implantation of pellets, female mice were anesthetized with isofluorane. Sterile pellets containing either CI-1011 or placebo (containing only the biopolymer matrix) were then implanted subcutaneously along the anterolateral aspect of the shoulder with a special precision trocar in accordance with the supplier’s instructions. A single pellet was inserted for placebo and 4.8 mg/kg/day dose of CI-1011. Two 7.2 mg/kg/day pellets were used to achieve the 14.4 mg/kg/day dose.
Tissue and Cerebrospinal Fluid Sampling
Cerebrospinal fluid (CSF) was obtained from anesthetized mice after exsanguination by blunt dissection and exposure of the foramen magnum. Upon exposure, a Pasteur pipette was inserted to the approximate depth of 0.3 to 1 mm into the cisterna magna. CSF was suctioned by capillary action until flow fully ceased. Animals were killed on day 56 of treatment. Brain, liver, kidney, adrenal gland and blood samples were collected. Brains were divided along the sagittal plane and then either frozen in liquid N2 or immersion fixed in 4% paraformaldehyde for histologic evaluation.
Cholesterol Determination
Tissues were homogenized in the presence of trypsin (10 mg/ml) in a Dounce homogenizer on ice. Protein concentration of the homogenate was determined using the BCA protein assay kit (Pierce). The tissue homogenate was extracted in chloroform:methanol (2:1, v/v) overnight. Before drying the chloroform phase, polyoxyethylene-9-lauryl ether (Sigma, St. Louis, MO; 5 μl/ml of extract) was added. Dried lipid pellets were dissolved in water and free cholesterol was measured enzymatically using the Amplex Red Cholesterol Assay kit (Molecular Probes/Invitrogen, Eugene, OR). To measure cholesteryl esters directly in samples, free cholesterol was first converted to cholest-4-ene-3-one by cholesterol oxidase and the resulting hydrogen peroxide decomposed by catalase after which the enzymatic cholesterol assay was performed in the presence of cholesterol esterase (16). Finally, the values were normalized to protein concentration of the tissue homogenate and expressed as mg of cholesterol per g of protein.
Immunohistochemistry and Automated Image Analysis
Amyloid deposition was determined by immunohistochemical analysis of sagittally cut brain slices. Ten-μm-thick paraffin sections from 5 different layers across the brain were stained with 6E10 monoclonal mouse anti-human amyloid-β-antibody (Clone 6E10; 1:1000, Signet, Covance, Princeton, NJ) and visualized with a secondary anti-mouse Cy3 antibody (1:500, Jackson Laboratories, Bar Harbor, ME). β-sheet formations located in the core of dense plaques were determined with a thioflavin S fluorescence staining. Quantification of 6E10 and thioflavin S staining was performed in 5 different sagittal, 5-μm-thick slices. The first slice was chosen randomly according to the full appearance of the dentate gyrus and the following 4 slices derived from 4 uniformly and systematically sampled lateral layers (25 retained, 10 discarded). Thioflavin S and 6E10 were quantified in slice #8 from 5 layers, astrocytosis in slice #7 (glial fibrillary acidic protein [GFAP]), and reactive microglia (Iba-1) in slice #10 from layers 3 and 4. To evaluate plaque load, brain regions were outlined in 100-fold magnified high-resolution images (~80 × 106 pixels) of the whole sagittal slice using Image Pro Plus software (v6.2). The first step was to measure the region area, followed by a macro supported automated (rater independent) setting of constant enhancement (brightness and contrast level), threshold (RGB) and object size criteria (minimum size 7.25 μm2), which were applied constantly to all images. Absolute and relative (related to region area) object area and number and mean object sizes were automatically transported into an Excel spread sheet.
For detection of astrocytosis and reactive microglia, accumulated images of DAPI and Cy3 secondary fluorescence were used to count the white addition color in astro- or microglial cells having a nucleus in the counting layer. The threshold was accordingly set to the white color and all other steps were equal to plaque load evaluations.
A stack of 100 images at 400-fold magnification was used for 3D imaging of single plaques. Thioflavin S and 6E10 stacks were recorded separately, deconvolved (inverse filter), arithmetically added and reconstructed using Image Pro 3D Suite (v6) in full color palette.
Aβ1-40 and Aβ1-42 Determinations
For Aβ determinations, frozen hemispheres were extracted in a 4-step protocol using Tris-buffered saline (TBS), 1% Triton X-100, 2% SDS and 70% formic acid, as previously described (17). The plasma and CSF Aβ was analyzed using commercially available ELISA kits (The Genetics Company, Schlieren, Switzerland). Brain Aβ1-40 and Aβ1-42 were assayed by standard sandwich ELISA (Aβ ELISA Core Facility, Center for Neurological Diseases, Harvard Institutes of Medicine, Harvard Medical School, Boston, MA). Measurements were performed at least in duplicate. The following Aβ antibodies were used in the ELISA assays: 2G3/3D6B for Aβ1-40 and 21F12/3D6B for Aβ1-42. These antibodies were kindly provided by Peter Seubert, PhD, and Dale Schenk, PhD (Elan Corp, South San Francisco, CA).
Western Blots
1% Triton X-100 extracts of the brains from Aβ analyses were analyzed on Western blots, as previously described (11). Antibodies used were APP C-terminal (A8717; Sigma), β-tubulin (Sigma), ACAT-1 (Santa Cruz Biotechnology, Santa Cruz, CA), ApoE (BD Pharmingen, San Diego, CA), presenilin-1 and nicastrin (Chemicon, Temecula, CA), ATP-binding cassette transporter A1 (ABCA1) and ATP-binding cassette transporter G1 (ABCG1) (Novus Biologicals, Littleton, CO). Anti-β secretase (BACE1) monoclonal antibody was a kind gift from Dr. Robert Vassar (Northwestern University, IL).
Statistical Analysis
Statistical analyses were performed using Student t-test except for Figure 3B and Figure 7 where ANOVA was used. Significance was placed at p < 0.05.
Figure 3.
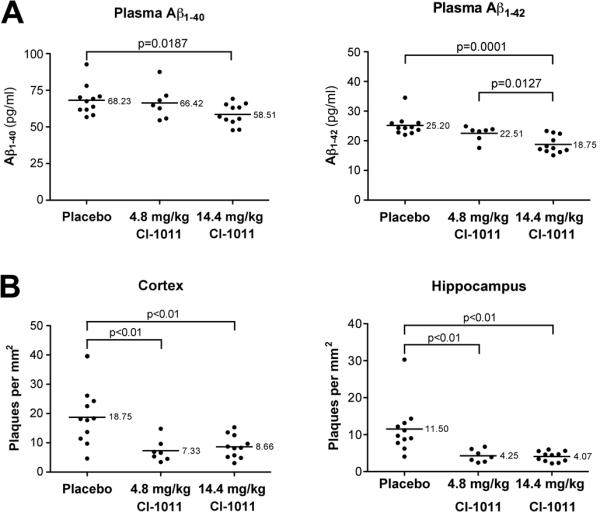
CI-1011 treatment reduces amyloid plaque load in the brains of young hAPP mice. (A) Plasma Aβ1-40 and Aβ1-42 levels were determined by sandwich ELISA. (B) Quantitative analysis of brain plaque load, as assessed by number of plaques per mm2, stained with the 6E10 antibody in the cortex and hippocampus.
Figure 7.

Enhanced clearance of diffuse amyloid deposits in old mice treated with CI-1011. (A) Representative immunostaining of brain slices showing staining of glial fibrillary acidic protein (GFAP)+ astrocytes (red) and a DNA-binding marker, DAPI (blue). Insets show higher magnifications of plaque-like deposits with surrounding GFAP+ cells; these are reduced by CI-1011 treatment. (B) Quantitation of astrogliosis by automated image analysis. CI-1011 treatment reduced the number of GFAP+ astrocytes in the cortex (p = 0.0088) but not in the hippocampus. (C) Representative immunostaining of brain slices showing staining of Iba-1+ microglia (red) and DAPI (blue). Insets show higher magnifications of plaque-like deposits with surrounding Iba-1+ cells; these are increased by CI-1011 treatment. (D) Quantitation of microglial activation by automated image analysis. CI-1011 treatment increased the number of Iba-1+ microglia in the cortex (p>0.05) and in the hippocampus (p = 0.0256). Each bar represents a mean ± SEM of 2 brain slices from 5 control- and 8 CI-1011-treated mice.
RESULTS
ACAT Inhibition by CI-1011 Reduces APP Processing and Aβ Generation in Cells
Micromolar concentrations of CI-1011 reduce cellular cholesteryl ester content in macrophages and secretion of ApoB-containing lipoproteins by hepatocytes in vitro (13). We treated CHO cells expressing human APP751 with CI-1011 for 96 hours and analyzed APP metabolism. CI-1011 decreased cholesteryl ester content of CHO/APP751 cells in a dose-dependent manner (Fig. 1A), while reducing both α- and β APP C-terminal fragments (CTF) (Fig. 1B). The conditioned media from these cells showed that CI-1011 treatment reduced the levels of secreted Aβ in a dose-dependent manner. At 10 μM CI-1011, Aβ1-40 and Aβ1-42 were reduced by 38% and 44%, respectively (Fig. 1C). Thus, CI-1011 has very similar in vitro anti-amyloidogenic properties to those of the structurally different ACAT inhibitors CP-113,818 and Dup128, which are comparable to direct siRNA-mediated knockdown of ACAT1 (9, 10).
Figure 1.

CI-1011 reduces amyloid β peptide generation in CHO/APP751 cells. (A) CHO/APP751 cells were treated with increasing concentrations of CI-1011 for 96 hours. Lipids were extracted and cellular sterol content was analyzed with enzymatic cholesterol assay (n = 3). (B) Western blot of cell extracts after 96-hour CI-1011 treatment shows a decrease in both α- and β amyloid precursor protein (APP) C-terminal fragment (C-term) levels. Changes are expressed numerically after normalization to glyceraldehyde 3-phosphate dehydrogenase (GAPDH) and immature APP holoprotein levels. (C) Levels of Aβ1-40 and Aβ1-42 are decreased in a concentration-dependent manner in conditioned media (last 24 hours media) of CHO/APP751 cells treated with CI-1011 for 96 hours. Each bar represents mean ± SD of 3 experiments. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
CI-1011 Reduces Liver and Brain Cholesterol in hAPP Mice
To determine the in vivo efficacy of CI-1011, we treated hAPP mice with CI-1011 for 2 months. Although CI-1011 has improved oral bioavailability compared to that of CP-113,818, we administered the drug via implanted biopolymer pellets as in our previous study (11). This approach guaranteed constant levels of CI-1011 in the circulation and allowed for direct comparison between the 2 studies. Based on an initial 21-day dose-finding study with CI-1011 in non-transgenic animals (data not shown), we selected 2 doses: 4.8 and 14.4 mg/kg/day. The dose of CI-1011 required to reduce brain cholesteryl esters by >70% in the pilot study was higher than that of CP-113,818 and reflects the lower inhibitory potency of CI-1011 on ACAT (18).
Female 4.5-month-old hAPP transgenic mice were treated with placebo pellets or with pellets releasing 4.8 or 14.4 mg/kg/day of CI-1011. hAPP mice develop detectable plaques in the neocortex and hippocampus beginning at the ages of 4 and 6 months, respectively (Fig. 2A) (15). Because the effect of ACAT inhibition on preformed amyloid plaques was not assessed in our previous study with CP-113,818, we also treated 14-month-old hAPP mice with 14.4 mg/kg/day CI-1011 or placebo.
Figure 2.

CI-1011 reduces cholesteryl ester levels in mouse liver and brain. (A) Experimental design of the study. Fifty-six-day treatment was started at either 4.5 or 14 months of age (n = 11 for control and 14.4 mg/kg/day, n = 7 for 4.8 mg/kg/day in the young animal group; n = 5 for control and n = 8 for 14.4 mg/kg/day in the old animal group). Average plaque load is shown as a function of time. (B) Decreased levels of total serum cholesterol in hAPP transgenic mice after 56-d treatment with placebo or CI-1011. (C, D) Decreased levels of cholesteryl esters in the liver of the hAPP transgenic mice (C) and in the brain of non-transgenic littermates (D) after CI-1011 treatment. Each bar represents mean ± SD of triplicate measurements.
After 56 days of treatment, tissues were harvested and analyzed. CI-1011 reduced total serum cholesterol by 18% with both doses (p < 0.0001 for both) in the young (6.5-month-old) animals, and by 25% (p = 0.0014) in 16-month-old animals (Fig. 2B). Liver cholesteryl esters were reduced in young mice (−56%, p = 0.0021 with 4.8 mg/kg/day and −66%, p < 0.0001 with 14.4 mg/kg/day of CI-1011; Fig. 2C). In old mice, liver cholesteryl esters were decreased by 64% (p = 0.0004) with 14.4 mg/kg/day of CI-1011 (Fig. 2C).
To preserve brain tissue from hAPP mice for neuropathological and biochemical analyses, brain cholesteryl esters were extracted and analyzed from brains of 6.5-month-old non-transgenic littermates treated similarly to the transgenics (only 6.5-month-old littermates were available for these studies). CI-1011 reduced brain cholesteryl ester levels by 33% (p = 0.0358) and 67% (p = 0.0022) with 4.8 and 14.4 mg/kg/day doses, respectively (Fig. 2D). These results indicate that CI-1011 had reduced ACAT-mediated generation of cholesteryl esters in the liver as well as in the brain, which is particularly important because little is known about blood brain barrier penetrability of CI-1011.
CI-1011 Reduces Aβ Levels and Amyloid Pathology in 6.5-Month-Old Mice
To evaluate the effects of CI-1011 treatment on amyloid pathology we first analyzed young animals. Plasma Aβ1-40 and Aβ1-42 levels were reduced maximally by 14% and (p = 0.0187) and 26% (p = 0.0001), respectively (Fig. 3A). Brain plaque load was evaluated by immunohistochemical staining with the 6E10 anti-Aβ monoclonal antibody in paraffin sections of the right hemispheres of each animal. Individual heterogeneity of plaque load was high in the placebo-treated control mice, as is typical in early stages of amyloid pathology in APP transgenic mice. Nevertheless, both doses of CI-1011 reduced plaque numbers in the cortex and hippocampus (Fig. 3B). In the cortex, mean plaque number per mm2 was reduced by 61% and 54% in the 4.8 mg/kg/day and 14.4 mg/kg/day groups, respectively (p < 0.01 for both). In the hippocampus, plaque load was reduced by 63% and 65% (p < 0.01 for both) with the respective treatments.
To extract pools of Aβ from the brain, the left hemispheres were homogenized and extracted sequentially in TBS, 1% Triton X-100, 2% SDS and 70% formic acid. The extracts were then analyzed in a sandwich ELISA to measure Aβ1-40 and Aβ1-42 levels. SDS-extractable Aβ1-40 was reduced by 32% (p = 0.0309) and Aβ1-42 by 31% (p = 0.0031) with the highest dose of CI-1011 (Table). Formic acid-extractable Aβ1-40 was reduced by 59% (p = 0.0403) and Aβ1-42 by 66% (p = 0.0130) with 14.4 mg/kg/day of CI-1011. Together with the immunohistochemical data these results show that CI-1011 has potent anti-amyloidogenic effects.
Table. Four-Step Extraction of Aβ from Placebo- and CI-1011-treated 6.5- and 16-month-old hAPP Trangenic Mouse Brains.
| Age (months) | Treatment | N | Aβ | TBS | 1% Triton X-100 | 2% SDS | 70% FA |
|---|---|---|---|---|---|---|---|
| 6.5 | Placebo | 11 | Aβ40 | 1.9 ± 0.7 | 25.2 ± 1.2 | 32.7 ± 5.1 | 7.9 ± 2.5 |
| Aβ42 | 1.9 ± 0.5 | 15.0 ± 1.0 | 17.8 ± 1.6 | 2.8 ± 0.8 | |||
| 6.5 | 4.8 mg/kg/day | 7 | Aβ40 | 0.9 ± 0.1 | 28.2 ± 1.3 (+12%) | 31.7 ± 3.7 (−3%) | 4.5 ± 0.8 (−43%) |
| C1-1011 | Aβ42 | 1.1 ± 0.1 | 16.3 ± 0.7 (+9%) | 17.7 ± 1.7 (−0.1%) | 1.5 ± 0.2 (−48%) | ||
| 6.5 | 14.4 mg/g/day | 11 | Aβ40 | 1.1 ± 0.1 | 27.3 ± 1.5 (+9%) | 22.3 ± 2.4 (−32%) | 3.3 ± 0.5 (−59%) |
| C1-1011 | Aβ42 | 1.1 ± 0.1 | 15.7 ± 1.1 (+5%) | 12.3 ± 0.7 (−31%) | 1.0 ± 0.1 (−66%) | ||
| 16 | Placebo | 5 | Aβ40 | 4.4 ± 2.9 | 31.6 ± 5.9 | 7417.7 ± 2260.5 | 564.4 ± 68.7 |
| Aβ42 | 5.2 ± 1.3 | 14.0 ± 2.1 | 3106.7 ± 836.2 | 73.6 ± 12.8 | |||
| 16 | 14.4 mg/kg/day | 8 | Aβ40 | 6.7 ± 1.9 | 30.0 ± 3.0 (−5%) | 4981.8 ± 770.6 (−33%) | 825.5 ± 119.9 (+46%) |
| C1-1-11 | Aβ4 | 2 3.2 ± 1.8 | 16.6 ± 1.8 (+19%) | 2305.5 ± 351.9 (−26%) | 82.2 ± 12.0 (+12%) |
Average ± standard error of mean (pmol/g). Numbers in parentheses show the change (%) as compared to the placebo-treated controls. Bold values indicate statistically significant changes (p < 0.05).
CP-113,818 specifically reduces proteolytic processing of APP holoprotein resulting in decreased APP-CTFs without affecting β- or γ-secretase levels in vivo or activities in vitro (11). To analyze changes in APP metabolism, we next analyzed brain extracts from CI-1011-treated 6.5-month-old hAPP mice by Western blotting. CI-1011 reduced the levels of both α- and β-CTFs of APP (Fig. 4A). In the brain extracts, APP-C83 levels were reduced by 33% (p = 0.0180) and APP-C99 by 35% (p = 0.0048) in the 14.4 mg/kg/day CI-1011 group (Fig. 4B). Thus, CI-1011 has effects on proteolytic processing of APP that are similar to those of other ACAT inhibitors. CI-1011 also reduced the level of ApoE, whereas other tested proteins were not significantly altered (Fig. 4; Supplemental Fig. 1).
Figure 4.
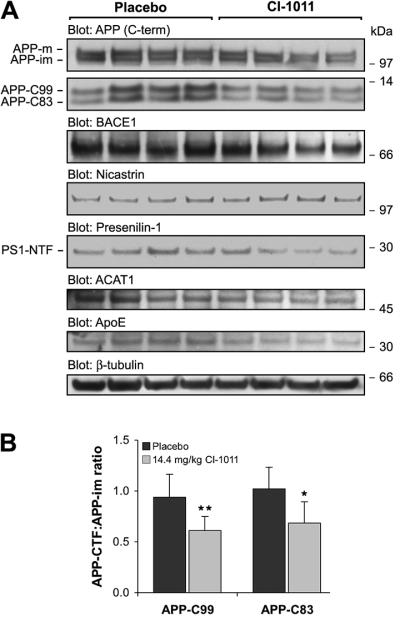
CI-1011 treatment modulates proteolytic processing of brain amyloid precursor protein (APP). (A) Representative Western blots of brain extracts from control- and CI-1011-treated (14.4 mg/kg/day) young hAPP mice analyzed for APP, β-secretase (BACE1), Nicastrin, Presenilin-1, acetyl-Coenzyme A acetyltransferase 1 (ACAT1), ApoE and β-tubulin levels. (B) Decreased levels of both α- and β amyloid precursor protein C terminal fragments (APP-CTFs). APP-CTFs and immature APP (APP-im) holoprotein levels were quantitated from Western blots (A). APP-CTFs were first normalized to β-tubulin and then to immature APP holoprotein levels (p = 0.0180 for APP-C83, p = 0.0048 for APP-C99). Each bar represents a mean ± SD of brain samples from 9 placebo- and 9 CI-1011-treated mice. PS1-NTF = presenilin1 N-terminal fragment.
CI-1011 Reduces Net Amyloid Burden in 16-Month-Old Mice
Due to the slow progression of AD and the current lack of methods for early diagnosis there is a need for therapeutic interventions that can reverse existing amyloid pathology. As compared to 6.5-month-old placebo-treated mice, 16-month-old placebo-treated mice displayed a >20-fold increase in the brain amyloid plaque load. hAPP mice at this age exhibit severe cognitive impairment due to the prominent plaque pathology. Although plasma cholesterol was efficiently reduced by CI-1011 treatment in the old animal cohort (Fig. 2B), there was no effect on plasma Aβ levels (data not shown). Instead, we found 38% (p = 0.0071) and 34% (p = 0.0492) decreases in CSF levels of Aβ1-40 and Aβ1-42, respectively (Fig. 5A). This is in accordance with a previous report that showed that once plaque deposition begins in APP transgenic mice, the correlation between CSF and plasma Aβ levels is lost (19).
Figure 5.

CI-1011 treatment reduces net amyloid burden in the brains of aged hAPP mice. (A) Decreased Aβ1-40 and Aβ1-42 levels in the cerebrospinal fluid (CSF) of CI-1011-treated old hAPP mice. (B, C) Representative staining of old hAPP brains treated with placebo (B) or CI-1011 (C). Red color represents diffuse amyloid β (Aβ) staining (Aβ-specific antibody 6E10) and yellow/green color dense-core plaques (thioflavin S and 6E10). Insets show single plaques from motor cortex. (D) Quantitative analysis of brain plaque load, as assessed by number of plaques per mm2 stained with 6E10 antibody in the cortex and hippocampus. (E) Quantitative analysis of brain plaque load, as assessed by number of plaques per mm2 stained with thioflavin S. (F) 3-dimensional reconstructions of stacks of 100 confocal micrographs from 16-month-old mice stained with monoclonal antibody 6E10 (red) and thioflavin S (green). A 2-month treatment with CI-1011 promoted clearance of small, diffuse amyloid deposits and the halo-like areas surrounding the large, dense-core plaques.
Thioflavin S+ dense-core plaques were barely detectable in the young animal cohort, but brains from placebo-treated 6.5-month-old animals contained large thioflavin S+ dense-core plaques as well as diffuse plaques that were positive only for Aβ-immunostaining (Fig. 5B). Interestingly, in brain sections from CI-1011-treated old mice, thioflavin S+ plaques remained more or less unchanged, whereas 6E10+ (but thioflavin S-negative) plaques were significantly decreased (Fig. 5C-F). Also, diffuse 6E10+ Aβ signal appeared less intense in the CI-1011-treated brains than in the control brains. Closer analysis of plaque morphologies in 6E10/thioflavin S-double stained sections showed that the diffuse 6E10+ red signal (“halo”) surrounding the yellow/green dense core of the plaque was reduced in the CI-1011-treated mice (Figs. 5B and C insets, and 5F). Quantitation of the plaque load in old mice revealed that numbers of diffuse 6E10+ plaques were reduced by 68% (p = 0.0111) in the cortex and by 53% in the hippocampus (p = 0.047) (Fig. 5D). Numbers of thioflavin S+ dense-core plaques were only mildly affected (<15% decrease) and was not statistically significant (Fig. 5E).
The numbers of diffuse amyloid deposits correlates with the amount of SDS-soluble brain Aβ, whereas the numbers of dense-core plaques correlated with formic-acid extractable Aβ, as in previous reports (17, 20). The SDS-extractable Aβ1-40 was reduced 33% (p = 0.0336) and Aβ1-42 by 26% (p = 0.0365) with 14.4 mg/kg/day CI-1011 (Table). There was a slight increase in the formic acid-extracted pool of Aβ, suggesting that there may be a subtle effect on the conversion of remaining diffuse Aβ to dense-core plaques in the old animals; however, this trend was not statistically significant (p = 0.0853 for Aβ1-40 and p = 0.9501 for Aβ1-42).
Next, we analyzed proteolytic processing of APP and changes in the levels of other relevant proteins in the brains of the old mice. Similarly to 6.5-month-old mice treated with CI-1011, the levels of both APP-C99 and APP-C83 were reduced by CI-1011 treatment (Fig. 6A). The decrease was 41.3% for APP-C99 (p = 0.0041) and 35.9% for APP-C83 (p = 0.0066) (Fig. 6B). As in 6.5-month-old mice (Fig. 4A) and in mice treated with CP-113,818 (11), the levels of BACE1, nicastrin and presenilin-1 (i.e. proteins involved in Aβ-generating machinery) were not significantly altered (Fig. 6A). Moreover, the levels of insulin-degrading enzyme, an important Aβ-degrading enzyme in the brain (21), were not significantly different between the control and CI-1011 groups (Fig. 6A; Supplemental Fig. 2).
Figure 6.

CI-1011 treatment decreases proteolytic processing of amyloid precursor protein (APP) while reducing ApoE level in the brains of old mice. (A) Representative Western blots of brain extracts from control- and CI-1011-treated old hAPP mice analyzed for amyloid precursor protein (APP), β-secretase (BACE1), Nicastrin, Presenilin-1, acetyl-Coenzyme A acetyltransferase 1 (ACAT1), ATP-binding cassette transporter A1 (ABCA1), ATP-binding cassette transporter G1 (ABCG1), β-tubulin, ApoE and insulin-degrading enzyme 1 (IDE1) levels. (B) Decreased levels of both α- and β-C terminal fragments (CTF). APP-CTFs and immature APP (APP-im) holoprotein levels were quantitated from Western blots (A). APP-CTFs were first normalized to β-tubulin and then to immature APP holoprotein levels (p = 0.0041 for APP-C99, p = 0.0066 for APP-C83). (C) ApoE level was quantitated from Western blots (TBS homogenates in Fig. 6B) and normalized to β-tubulin levels (p = 0.0129). Each bar represents mean ± SD of brain samples from 5 placebo- and 5 CI-1011-treated mice. PS1-NTF = presenilin1 N-terminal fragment; TBS = Tris-buffered saline
To evaluate whether changes in proteins that regulate brain cholesterol metabolism are altered by ACAT inhibitor treatment, we assessed the levels of ACAT1, ABCA1, ABCG1 and ApoE in brain lysates by Western blotting. ACAT1 levels remained unchanged in brain lysates of 16-month-old animals similarly to young mice. Members of the ATP-binding cassette family transporters mediate the rate-limiting step of reverse cholesterol transport in cells (22). ABCA1 and ABCG1 have been implicated has important regulators of Aβ metabolism through lipidation of ApoE (23-25). On the other hand, brain ApoE levels inversely correlate with the rate of amyloid deposition in AD transgenic mouse models (26). Transgenic over-expression of ABCA1 in PDAPP mice resulted in increased ApoE lipidation, decreased ApoE levels (20%-40%) and significantly reduced amyloid deposition (23). We did not detect changes in the brain levels of ABCA1 or ABCG1 proteins in 16-month-old ACAT inhibitor-treated mice (Fig. 6A; Supplemental Fig. 2). There was, however, a modest reduction in brain ApoE levels (−25.8%, p = 0.0129; Fig. 6A, C), which may indicate an increased rate of reverse cholesterol transport in the absence of ACAT activity in the brain. Altogether, data from the aged animals suggest that CI-1011 treatment allows for removal of existing diffusible Aβ from the brain, possibly by limiting generation of new Aβ.
Reduced Astrogliosis and Enhanced Microglial Activation in 16-Month-Old mice Treated with CI-1011
Reactive gliosis and chronic inflammation are prominent features of AD (27); activated astrocytes accumulate in the vicinity of both diffuse and dense-core plaques and neurofibrillary tangles (28-31). Astrogliosis may contribute to disease progression in AD through dysregulation of astrocyte-neuron networks. CI-1011 treatment significantly reduced the numbers of GFAP+ cells in the cortex (−55.6%, p = 0.0088) but not the hippocampus (Fig. 7A, B), where CI-1011 treatment was slightly less effective in removing the amyloid burden (Fig. 5D). Reduced GFAP staining in the cortex suggests that reactive gliosis can be reversed in the aged brain by CI-1011 treatment. Moreover, there was a correlation between the 6E10-reactive area and the numbers of GFAP+ cells in individual animals (not shown). We also noted that DAPI, a DNA-binding dye used for counter staining of nuclei, appeared to stain compact plaques, a finding also reported by others (32).
Because CI-1011 might have an effect on clearance of Aβ in aged animals with preformed plaques, we constructed 3-dimensional images of stacks of 100 confocal micrographs taken at 400x magnification. In 16-month-old animals treated with CI-1011 there was a highly significant decrease of small thioflavin S-negative but 6E10+ amyloid deposits (Fig. 5F). The 6E10+ halo-like surroundings of thioflavin S+ dense-core plaques were also markedly reduced. Using Iba-1, a marker for activated microglia (33), there was a significant increase in the number of microglial cells per mm2 in the hippocampus (+38.4%, p = 0.0256; Fig. 7B, C). Microglial cell numbers in the cortex were also increased but this did not reach statistical significance (+31.1%, p > 0.05; Fig. 7B, C). Importantly, Iba-1+ microglial cells appeared to be recruited to the surroundings of large, DAPI+, plaque-like structures in CI-1011-treated brains (Fig. 7C). To determine if there was a relationship between plaque density and microglial activation, we performed a correlation analysis on the immunohistochemical data. In CI-1011-treated animals there was a positive correlation between thioflavin S+ dense-core plaques and the number of microglial cells (Iba-1) that appeared to be tighter and more significant than in the placebo-treated group (Supplemental Fig. 3A). A similar positive correlation was found between the number of microglial cells and 6E10+ diffuse plaque density in placebo-treated mice, but in CI-1011-treated mice the correlation was negative within both cortex and hippocampus (Supplemental Fig. 3B). These data suggest that CI-1011 can improve glial responses in aged plaque-bearing hAPP mice, and that microglia may have contributed to the clearance of diffuse amyloid deposits seen in CI-1011-treated animals.
DISCUSSION
Here we show that a clinically relevant ACAT inhibitor, CI-1011 decreases proteolytic processing of APP and Aβ generation in young mice and in old mice with pre-existing plaque pathology, it appears to reduce the diffuse amyloid burden, likely by limiting generation of new Aβ. This results in partial reversal of amyloid pathology, suppression of astrogliosis and increased microglial activation.
Treatment of young mice with CI-1011 corroborated our previous findings with an older generation ACAT inhibitor, CP-113,818 (11). CI-1011 appears to be slightly less effective than CP-113,818 with respect to effects on brain cholesteryl ester, amyloid plaque load and Aβ levels, (Supplementary Table 1), which is consistent with its lower antagonistic potency on ACAT. Importantly, in both studies all key parameters seem to correlate closely with brain cholesteryl ester levels. Statins, classic inhibitors of cholesterol biosynthesis, lower total cholesterol in cells and result in reduced Aβ production in many cell- and animal models of AD (7). In most animal studies using statins and other inhibitors of the cholesterol biosynthetic pathway, drug administration was started before plaque deposition begins (34, 35), making comparison of CI-1011 treatment to statins complicated. Interestingly, one report showed that lovastatin treatment of 12-month-old Tg2576 mice for 3 weeks did not affect amyloid load or brain Aβ levels in males whereas it increased Aβ pathology in female animals (36). Moreover, the beneficial effects of statins for AD may be at least partially related to their cholesterol-independent, indirect anti-inflammatory and antioxidant effects (37-39). It should be noted, however, that the clinical utility of statins in AD is controversial; the first longitudinal clinical study assessing the efficacy of statins in mild-to-moderate AD failed to show significant differences in cognition or global function (40).
The finding that brain levels of both α- and β-CTFs of APP are reduced by CI-1011 treatment is in accordance with our previous studies (9-11). Importantly, in 3x-TgAD transgenic mice lacking both copies of the ACAT1 gene, significant reductions in brain levels of APP holoprotein, APP proteolytic fragments as well as Aβ40 and Aβ42 were associated with amelioration of the hippocampal- and amygdala-dependent cognitive deficits (12). Thus, reduced ACAT activity in the brain of AD mouse models has direct or indirect beneficial effects, since the results from the ACAT1 gene ablation study agree with the overall outcome of our current and previous ACAT inhibitor studies.
Based on our previous mechanistic analysis in young hAPP mice treated with ACAT inhibitors, we suggest that CI-1011 treatment allows for removal of existing diffusible Aβ from the brain, possibly by limiting generation of new Aβ. Because ACAT resides in the endoplasmic reticulum and both CTFs are similarly affected, it seems plausible that ACAT inhibition affects APP trafficking in the early compartments of the secretory pathway, altering the maturation of APP and thus limiting its availability for Aβ generation (41). Thus, ACAT inhibitors seem to reduce Aβ generation through a different mechanism from γ- and β-secretase inhibitors or statins. It is very likely that inhibition of ACAT activity in cells promotes reverse cholesterol transport (13). Although there likely are some mechanistic differences, both genetic and pharmacological inhibition of ACAT seem to affect APP holoprotein. ACAT1 gene ablation was suggested to reduce APP holoprotein (both immature and mature) levels through increased levels of 24(S)-hydroxycholesterol, the most abundant cholesterol metabolite in the brain (12). We did not assess the brain levels of oxysterols in this study, but non-neuronal cell lines do not produce 24(S)-hydroxycholesterol and yet show reduced Aβ generation when treated with ACAT inhibitors (9). Our results also suggest that pharmacological ACAT inhibition affects mostly a subpopulation of APP molecules, the mature APP. The reasons for these differences are currently unknown. For mechanistic comparison, β- and γ-secretase inhibitors directly target the proteolytic events generating Aβ and statins may act through enhancement of α-secretase cleavage of APP due to inhibition of the isoprenoid pathway (42), whereas ACAT inhibitors seem to form a mechanistically separate class of compounds that affect APP holoprotein and its proteolytic processing.
AD is typically a slowly progressing condition that is difficult to diagnose, especially in the early stages. At the beginning of the CI-1011 treatment, the aged mice had abundant amyloid pathology but CI-1011 treatment reduced the total amyloid burden in their brains. Dense-core plaques were only mildly affected, whereas diffuse plaques were more significantly reduced in CI-1011-treated mice. This result is similar to those in tet-off APP mice suggesting that dense-core plaques, containing β-pleated sheet amyloid structures, are particularly stable structures (6). Therefore, effective therapies for AD may require a combination of reduced Aβ generation and enhanced clearance of existing plaques. In CI-1011-treated aged mice, the diffuse, 6E10+ peripheral areas of the dense-core plaques were almost completely dissolved leaving only the dense cores intact whereas nearly complete suppression of new Aβ generation in tet-off APP mice after development of plaque pathology was not sufficient to promote clearance of diffuse or dense-core plaques, even after 6 months (6). Thus, it is possible that in addition to inhibiting Aβ production, CI-1011 may enhance endogenous Aβ clearance. Moreover, the level and lipidation status of brain ApoE strongly affects Aβ deposition (23, 26). Our finding of reduced brain ApoE in CI-1011-treated hAPP mice suggests that in addition to reduced Aβ generation, deposition of existing Aβ into plaques may be reduced upon ACAT inhibition.
The involvement of microglia in the clearance of brain amyloid plaques remains controversial and seems to depend on their activation phenotype (2, 3, 6, 43). We show immunohistochemical evidence of microglial activation that coincided with reduced amyloid burden in CI-1011-treated old hAPP mice. The specificity of CI-1011-induced clearance effect towards diffuse amyloid is somewhat reminiscent of clearance of diffuse amyloid deposits by topical application of anti-Aβ antibodies in Tg2576 mice (43). Interestingly, in studies where intra-hippocampal lipopolysaccharide injections were used to enhance microglial activation in plaque-bearing 11- and 16-month-old APP+PS1 mice, efficient region-specific clearance of diffuse amyloid deposits was observed while dense-core plaques remained intact (44, 45). These results are very similar to our current results and support the conclusion that clearance of diffuse amyloid deposits is likely mediated by activated microglia. Although our data suggest recruitment of activated microglia in plaque surroundings, we assessed activated microglia solely on the basis of Iba-1 immunoreactivity, which has no bearing on the functional phenotype of microglia (i.e. phagocytic M2 vs. proinflammatory M1). The specific effect of ACAT inhibitors on the state of activation of microglial cells is a subject for future studies. Moreover, it will be interesting to see if longer treatments of aged hAPP mice with CI-1011 reveal a stronger effect on clearance of dense-core plaques.
The efficacy of various therapeutic approaches for AD may depend critically on the timing of the treatment relative to the stage of plaque evolution. For example, a study using Vitamin E in both young and aged Tg2576 mice suggests that antioxidant therapy may be beneficial only if given at a very early stage of the disease process (46). Compounds specifically targeting Aβ generation, such as γ-secretase inhibitors, have been shown to reduce amyloid pathology in both young and aged Tg2576 mice (47, 48) but may require additional amyloid clearance enhancing therapies for clinical efficacy (49). ACAT inhibitor CI-1011 fits into the same category with γ-secretase inhibitors with its anti-amyloidogenic effect and efficacy in both young and old animals. Our data suggest that ACAT inhibitors may enhance clearance of Aβ from the brain making this approach even more clinically applicable. Other compounds with actions similar to CI-1011 have been used in aged mouse models of AD. A 6-month treatment of Tg2576 mice with curcumin was found to decrease amyloid plaque burden and soluble Aβ levels, while specifically promoting recruitment of microglia adjacent to plaques (50). In a related study, a diet enriched with the omega-3 fatty acid docosahexaenoic acid (DHA) markedly reduced amyloid burden in aged Tg2576 mice while decreasing insoluble Aβ as well as both α- and β-APP-CTF levels in the brain (51). A recent study also suggested that DHA might directly bind and (weakly) inhibit ACAT1 (52). Whether the in vivo neuroprotective effects of DHA involve inhibition of ACAT remains to be determined.
Chronically elevated expression of APP and/or β-CTF may be associated with the development neurodegenerative pathology in some AD patients and also in Down syndrome (DS). Although elevated APP mRNA or protein levels may be found only in a subset of AD patients, for example due to promoter mutations or gene duplication (53, 54), increased gene dosage of APP due to triplication of the APP gene in DS is strongly associated with development of neuropathology and cognitive deficits (55, 56). Interestingly, it seems that APP and β-CTF, but not Aβ or α-CTF, may cause the typical endocytic pathway dysfunction characteristic of DS (57), and which has also been implicated as one of the earliest neuropathological changes in late-onset AD (58, 59). In this context, our results suggest that reduction of APP holoprotein and/or β-CTF levels in the brain via modulation of ACAT activity or other similarly acting APP-reducing compounds could also be used therapeutically in DS.
Future studies will be necessary to characterize the mechanisms of CI-1011 action and efficacy on cognitive decline in aged mouse models of AD, but our study shows that a clinically safe and efficacious ACAT inhibitor has the potential to reverse preformed diffuse amyloid pathology in aged hAPP mice. Inasmuch as cognitive disturbances in mild to moderate AD appear to be mediated mostly by diffusible forms of Aβ (60, 61), our results strongly encourage further studies on the potential use of CI-1011 and other ACAT inhibitors for AD treatment.
Supplementary Material
ACKNOWLEDGMENTS
BACE1 monoclonal antibody was a kind gift from Dr. Robert Vassar (Northwestern University, Evanston, IL).
This study was supported by grants from the Cure Alzheimer’s Fund and NIH (R01 NS45860). CI-1011 and CP-113,818 were kind gifts from Lit-Fui Lau and James Harwood (Pfizer, Groton, CT), respectively.
Footnotes
The authors declare no conflict of interest.
This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Gandy S. The role of cerebral amyloid beta accumulation in common forms of Alzheimer disease. J Clin Invest. 2005;115:1121–9. doi: 10.1172/JCI25100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bacskai BJ, Kajdasz ST, Christie RH, et al. Imaging of amyloid-beta deposits in brains of living mice permits direct observation of clearance of plaques with immunotherapy. Nat Med. 2001;7:369–72. doi: 10.1038/85525. [DOI] [PubMed] [Google Scholar]
- 3.Meyer-Luehmann M, Spires-Jones TL, Prada C, et al. Rapid appearance and local toxicity of amyloid-beta plaques in a mouse model of Alzheimer’s disease. Nature. 2008;451:720–4. doi: 10.1038/nature06616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Haass C. Take five-BACE and the gamma-secretase quartet conduct Alzheimer’s amyloid beta-peptide generation. EMBO J. 2004;23:483–8. doi: 10.1038/sj.emboj.7600061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wilcock DM, DiCarlo G, Henderson D, et al. Intracranially administered anti-Abeta antibodies reduce beta-amyloid deposition by mechanisms both independent of and associated with microglial activation. J Neurosci. 2003;23:3745–51. doi: 10.1523/JNEUROSCI.23-09-03745.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jankowsky JL, Slunt HH, Gonzales V, et al. Persistent amyloidosis following suppression of Abeta production in a transgenic model of Alzheimer disease. PLoS Med. 2005;2:e355. doi: 10.1371/journal.pmed.0020355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wolozin B. Cholesterol and the biology of Alzheimer’s disease. Neuron. 2004;41:7–10. doi: 10.1016/s0896-6273(03)00840-7. [DOI] [PubMed] [Google Scholar]
- 8.Puglielli L, Tanzi RE, Kovacs DM. Alzheimer’s disease: The cholesterol connection. Nat Neurosci. 2003;6:345–51. doi: 10.1038/nn0403-345. [DOI] [PubMed] [Google Scholar]
- 9.Puglielli L, Konopka G, Pack-Chung E, et al. Acyl-coenzyme A: cholesterol acyltransferase modulates the generation of the amyloid beta-peptide. Nat Cell Biol. 2001;3:905–12. doi: 10.1038/ncb1001-905. [DOI] [PubMed] [Google Scholar]
- 10.Huttunen HJ, Greco C, Kovacs DM. Knockdown of ACAT-1 reduces amyloidogenic processing of APP. FEBS Lett. 2007;581:1688–92. doi: 10.1016/j.febslet.2007.03.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hutter-Paier B, Huttunen HJ, Puglielli L, et al. The ACAT inhibitor CP-113,818 markedly reduces amyloid pathology in a mouse model of Alzheimer’s disease. Neuron. 2004;44:227–38. doi: 10.1016/j.neuron.2004.08.043. [DOI] [PubMed] [Google Scholar]
- 12.Bryleva EY, Rogers MA, Chang CC, et al. ACAT1 gene ablation increases 24(S)-hydroxycholesterol content in the brain and ameliorates amyloid pathology in mice with AD. Proc Natl Acad Sci U S A. 2010;107:3081–6. doi: 10.1073/pnas.0913828107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Llaverias G, Laguna JC, Alegret M. Pharmacology of the ACAT inhibitor avasimibe (CI-1011) Cardiovasc Drug Rev. 2003;21:33–50. [PubMed] [Google Scholar]
- 14.Tardif JC, Gregoire J, L’Allier PL, et al. Effects of the acyl coenzyme A:cholesterol acyltransferase inhibitor avasimibe on human atherosclerotic lesions. Circulation. 2004;110:3372–7. doi: 10.1161/01.CIR.0000147777.12010.EF. [DOI] [PubMed] [Google Scholar]
- 15.Rockenstein E, Mallory M, Mante M, et al. Early formation of mature amyloid-beta protein deposits in a mutant APP transgenic model depends on levels of Abeta(1-42) J Neurosci Res. 2001;66:573–82. doi: 10.1002/jnr.1247. [DOI] [PubMed] [Google Scholar]
- 16.Mizoguchi T, Edano T, Koshi T. A method of direct measurement for the enzymatic determination of cholesteryl esters. J Lipid Res. 2004;45:396–401. doi: 10.1194/jlr.D300024-JLR200. [DOI] [PubMed] [Google Scholar]
- 17.Kawarabayashi T, Younkin LH, Saido TC, et al. Age-dependent changes in brain, CSF, and plasma amyloid (beta) protein in the Tg2576 transgenic mouse model of Alzheimer’s disease. J Neurosci. 2001;21:372–81. doi: 10.1523/JNEUROSCI.21-02-00372.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Alegret M, Llaverias G, Silvestre JS. Acyl coenzyme A:cholesterol acyltransferase inhibitors as hypolipidemic and antiatherosclerotic drugs. Methods Find Exp Clin Pharmacol. 2004;26:563–86. doi: 10.1358/mf.2004.26.7.863738. [DOI] [PubMed] [Google Scholar]
- 19.DeMattos RB, Bales KR, Parsadanian M, et al. Plaque-associated disruption of CSF and plasma amyloid-beta (Abeta) equilibrium in a mouse model of Alzheimer’s disease. J Neurochem. 2002;81:229–36. doi: 10.1046/j.1471-4159.2002.00889.x. [DOI] [PubMed] [Google Scholar]
- 20.Murphy MP, Beckett TL, Ding Q, et al. Abeta solubility and deposition during AD progression and in APPxPS-1 knock-in mice. Neurobiol Dis. 2007;27:301–11. doi: 10.1016/j.nbd.2007.06.002. [DOI] [PubMed] [Google Scholar]
- 21.Qiu WQ, Walsh DM, Ye Z, et al. Insulin-degrading enzyme regulates extracellular levels of amyloid beta- protein by degradation. J Biol Chem. 1998;273:32730–38. doi: 10.1074/jbc.273.49.32730. [DOI] [PubMed] [Google Scholar]
- 22.Kim WS, Weickert CS, Garner B. Role of ATP-binding cassette transporters in brain lipid transport and neurological disease. J Neurochem. 2008;104:1145–66. doi: 10.1111/j.1471-4159.2007.05099.x. [DOI] [PubMed] [Google Scholar]
- 23.Wahrle SE, Jiang H, Parsadanian M, et al. Overexpression of ABCA1 reduces amyloid deposition in the PDAPP mouse model of Alzheimer disease. J Clin Invest. 2008;118:671–82. doi: 10.1172/JCI33622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim WS, Rahmanto AS, Kamili A, et al. Role of ABCG1 and ABCA1 in regulation of neuronal cholesterol efflux to apolipoprotein E discs and suppression of amyloid-beta peptide generation. J Biol Chem. 2007;282:2851–61. doi: 10.1074/jbc.M607831200. [DOI] [PubMed] [Google Scholar]
- 25.Tansley GH, Burgess BL, Bryan MT, et al. The cholesterol transporter ABCG1 modulates the subcellular distribution and proteolytic processing of beta-amyloid precursor protein. J Lipid Res. 2007;48:1022–34. doi: 10.1194/jlr.M600542-JLR200. [DOI] [PubMed] [Google Scholar]
- 26.Bales KR, Verina T, Dodel RC, et al. Lack of apolipoprotein E dramatically reduces amyloid ß-peptide deposition. Nat Genet. 1997;17:263–4. doi: 10.1038/ng1197-263. [DOI] [PubMed] [Google Scholar]
- 27.Wyss-Coray T. Inflammation in Alzheimer disease: Driving force, bystander or beneficial response? Nat Med. 2006;12:1005–15. doi: 10.1038/nm1484. [DOI] [PubMed] [Google Scholar]
- 28.Beach TG, Walker R, McGeer EG. Patterns of gliosis in Alzheimer’s disease and aging cerebrum. Glia. 1989;2:420–36. doi: 10.1002/glia.440020605. [DOI] [PubMed] [Google Scholar]
- 29.Benzing WC, Wujek JR, Ward EK, et al. Evidence for glial-mediated inflammation in aged APP(SW) transgenic mice. Neurobiol Aging. 1999;20:581–9. doi: 10.1016/s0197-4580(99)00065-2. [DOI] [PubMed] [Google Scholar]
- 30.Dudal S, Krzywkowski P, Paquette J, et al. Inflammation occurs early during the Abeta deposition process in TgCRND8 mice. Neurobiol Aging. 2004;25:861–71. doi: 10.1016/j.neurobiolaging.2003.08.008. [DOI] [PubMed] [Google Scholar]
- 31.Oddo S, Caccamo A, Kitazawa M, et al. Amyloid deposition precedes tangle formation in a triple transgenic model of Alzheimer’s disease. Neurobiol Aging. 2003;24:1063–70. doi: 10.1016/j.neurobiolaging.2003.08.012. [DOI] [PubMed] [Google Scholar]
- 32.Sotgiu S, Musumeci S, Marconi S, et al. Different content of chitin-like polysaccharides in multiple sclerosis and Alzheimer’s disease brains. J Neuroimmunol. 2008;197:70–3. doi: 10.1016/j.jneuroim.2008.03.021. [DOI] [PubMed] [Google Scholar]
- 33.Ito D, Imai Y, Ohsawa K, et al. Microglia-specific localisation of a novel calcium binding protein, Iba1. Brain Res Mol Brain Res. 1998;57:1–9. doi: 10.1016/s0169-328x(98)00040-0. [DOI] [PubMed] [Google Scholar]
- 34.Fassbender K, Simons M, Bergmann C, et al. Simvastatin strongly reduces levels of Alzheimer’s disease beta - amyloid peptides Abeta 42 and Abeta 40 in vitro and in vivo. Proc Natl Acad Sci U S A. 2001;98:5856–61. doi: 10.1073/pnas.081620098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Refolo LM, Pappolla MA, LaFrancois J, et al. A Cholesterol-Lowering Drug Reduces beta-Amyloid Pathology in a Transgenic Mouse Model of Alzheimer’s Disease. Neurobiol Dis. 2001;8:890–9. doi: 10.1006/nbdi.2001.0422. [DOI] [PubMed] [Google Scholar]
- 36.Park IH, Hwang EM, Hong HS, et al. Lovastatin enhances Abeta production and senile plaque deposition in female Tg2576 mice. Neurobiol Aging. 2003;24:637–43. doi: 10.1016/s0197-4580(02)00155-0. [DOI] [PubMed] [Google Scholar]
- 37.Cordle A, Landreth G. 3-Hydroxy-3-methylglutaryl-coenzyme A reductase inhibitors attenuate beta-amyloid-induced microglial inflammatory responses. J Neurosci. 2005;25:299–307. doi: 10.1523/JNEUROSCI.2544-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kwak B, Mulhaupt F, Myit S, et al. Statins as a newly recognized type of immunomodulator. Nat Med. 2000;6:1399–1402. doi: 10.1038/82219. [DOI] [PubMed] [Google Scholar]
- 39.Menge T, Hartung HP, Stuve O. Statins--a cure-all for the brain? Nat Rev Neurosci. 2005;6:325–31. doi: 10.1038/nrn1652. [DOI] [PubMed] [Google Scholar]
- 40.Jones RW, Kivipelto M, Feldman H, et al. The Atorvastatin/Donepezil in Alzheimer’s Disease Study (LEADe): design and baseline characteristics. Alzheimers Dement. 2008;56:933–44. doi: 10.1016/j.jalz.2008.02.001. [DOI] [PubMed] [Google Scholar]
- 41.Huttunen HJ, Peach C, Bhattacharyya R, et al. Inhibition of acyl-coenzyme A: cholesterol acyl transferase modulates amyloid precursor protein trafficking in the early secretory pathway. FASEB J. 2009;23:3819–28. doi: 10.1096/fj.09-134999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pedrini S, Carter TL, Prendergast G, et al. Modulation of statin-activated shedding of Alzheimer APP ectodomain by ROCK. PLoS Med. 2005;2:e18. doi: 10.1371/journal.pmed.0020018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bacskai BJ, Kajdasz ST, McLellan ME, et al. Non-Fc-mediated mechanisms are involved in clearance of amyloid-beta in vivo by immunotherapy. J Neurosci. 2002;22:7873–8. doi: 10.1523/JNEUROSCI.22-18-07873.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.DiCarlo G, Wilcock D, Henderson D, et al. Intrahippocampal LPS injections reduce Abeta load in APP+PS1 transgenic mice. Neurobiol Aging. 2001;22:1007–12. doi: 10.1016/s0197-4580(01)00292-5. [DOI] [PubMed] [Google Scholar]
- 45.Herber DL, Roth LM, Wilson D, et al. Time-dependent reduction in Abeta levels after intracranial LPS administration in APP transgenic mice. Exp Neurol. 2004;190:245–53. doi: 10.1016/j.expneurol.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 46.Sung S, Yao Y, Uryu K, et al. Early vitamin E supplementation in young but not aged mice reduces Abeta levels and amyloid deposition in a transgenic model of Alzheimer’s disease. FASEB J. 2004;18:323–5. doi: 10.1096/fj.03-0961fje. [DOI] [PubMed] [Google Scholar]
- 47.Barten DM, Guss VL, Corsa JA, et al. Dynamics of {beta}-amyloid reductions in brain, cerebrospinal fluid and plasma of {beta}-amyloid precursor protein transgenic mice treated with a {gamma}-secretase inhibitor. J Pharmacol Exp Ther. 2005;312:635–43. doi: 10.1124/jpet.104.075408. [DOI] [PubMed] [Google Scholar]
- 48.Best JD, Smith DW, Reilly MA, et al. The novel gamma secretase inhibitor N-[cis-4-[(4-chlorophenyl)sulfonyl]-4-(2,5-difluorophenyl)cyclohexyl]-1,1, 1-trifluoromethanesulfonamide (MRK-560) reduces amyloid plaque deposition without evidence of notch-related pathology in the Tg2576 mouse. J Pharmacol Exp Ther. 2007;320:552–8. doi: 10.1124/jpet.106.114330. [DOI] [PubMed] [Google Scholar]
- 49.Garcia-Alloza M, Subramanian M, Thyssen D, et al. Existing plaques and neuritic abnormalities in APP:PS1 mice are not affected by administration of the gamma-secretase inhibitor LY-411575. Mol Neurodegener. 2009;4:19. doi: 10.1186/1750-1326-4-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lim GP, Chu T, Yang F, et al. The curry spice curcumin reduces oxidative damage and amyloid pathology in an Alzheimer transgenic mouse. J Neurosci. 2001;21:8370–7. doi: 10.1523/JNEUROSCI.21-21-08370.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lim GP, Calon F, Morihara T, et al. A diet enriched with the omega-3 fatty acid docosahexaenoic acid reduces amyloid burden in an aged Alzheimer mouse model. J Neurosci. 2005;25:3032–40. doi: 10.1523/JNEUROSCI.4225-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Antalis CJ, Arnold T, Lee B, et al. Docosahexaenoic acid is a substrate for ACAT1 and inhibits cholesteryl ester formation from oleic acid in MCF-10A cells. Prostaglandins Leukot Essent Fatty Acids. 2009;80:165–71. doi: 10.1016/j.plefa.2009.01.001. [DOI] [PubMed] [Google Scholar]
- 53.Theuns J, Brouwers N, Engelborghs S, et al. Promoter mutations that increase amyloid precursor-protein expression are associated with Alzheimer disease. Am J Hum Genet. 2006;78:936–46. doi: 10.1086/504044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rovelet-Lecrux A, Hannequin D, Raux G, et al. APP locus duplication causes autosomal dominant early-onset Alzheimer disease with cerebral amyloid angiopathy. Nat Genet. 2006;38:24–6. doi: 10.1038/ng1718. [DOI] [PubMed] [Google Scholar]
- 55.Lott IT, Head E. Down syndrome and Alzheimer’s disease: a link between development and aging. Ment Retard Dev Disabil Res Rev. 2001;7:172–8. doi: 10.1002/mrdd.1025. [DOI] [PubMed] [Google Scholar]
- 56.Salehi A, Delcroix JD, Belichenko PV, et al. Increased App expression in a mouse model of Down’s syndrome disrupts NGF transport and causes cholinergic neuron degeneration. Neuron. 2006;51:29–42. doi: 10.1016/j.neuron.2006.05.022. [DOI] [PubMed] [Google Scholar]
- 57.Jiang Y, Mullaney KA, Peterhoff CM, et al. Alzheimer’s-related endosome dysfunction in Down syndrome is A{beta}-independent but requires APP and is reversed by BACE-1 inhibition. Proc Natl Acad Sci U S A. 2010;107:1630–5. doi: 10.1073/pnas.0908953107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cataldo AM, Barnett JL, Pieroni C, et al. Increased neuronal endocytosis and protease delivery to early endosomes in sporadic Alzheimer’s disease: neuropathologic evidence for a mechanism of increased beta-amyloidogenesis. J Neurosci. 1997;17:6142–51. doi: 10.1523/JNEUROSCI.17-16-06142.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cataldo AM, Peterhoff CM, Troncoso JC, et al. Endocytic pathway abnormalities precede amyloid beta deposition in sporadic Alzheimer’s disease and Down syndrome: differential effects of APOE genotype and presenilin mutations. Am J Pathol. 2000;157:277–86. doi: 10.1016/s0002-9440(10)64538-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mattson MP. Pathways towards and away from Alzheimer’s disease. Nature. 2004;430:631–9. doi: 10.1038/nature02621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Walsh DM, Selkoe DJ. Deciphering the molecular basis of memory failure in Alzheimer’s disease. Neuron. 2004;44:181–93. doi: 10.1016/j.neuron.2004.09.010. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.