

Abstract
The human AP-endonuclease (APE1/Ref-1), a multifunctional protein central to repairing abasic sites and single-strand breaks in DNA, also plays a role in transcriptional regulation. Besides activating some transcription factors, APE1 is directly involved in Ca2+-dependent downregulation of parathyroid hormone (PTH) expression by binding to negative calcium response elements (nCaREs) present in the PTH promoter. Here we show that APE1 is acetylated both in vivo and in vitro by the transcriptional co-activator p300 which is activated by Ca2+. Acetylation at Lys6 or Lys7 enhances binding of APE1 to nCaRE. APE1 stably interacts with class I histone deacetylases (HDACs) in vivo. An increase in extracellular calcium enhances the level of acetylated APE1 which acts as a repressor for the PTH promoter. Moreover, chromatin immunoprecipitation (ChIP) assay revealed that acetylation of APE1 enhanced binding of the APE1–HDACs complex to the PTH promoter. These results indicate that acetylation of APE1 plays an important role in this key repair protein’s action in transcriptional regulation.
Keywords: acetylation/AP-endonuclease/chromatin immunoprecipitation/histone deacetylase/nCaRE/p300
Introduction
Repair of abasic (AP) sites in DNA, generated either spontaneously by oxygen free radicals or after excision of abnormal bases by monofunctional DNA glycosylases during the DNA base excision repair (BER) process, is initiated by AP-endonucleases (APE) (Mitra et al., 1997; Evans et al., 2000). The major human APE, APE1, plays a central role in BER (Demple and Harrison, 1994), and also functions as a redox activator of AP-1 DNA binding activity, and was named Ref-1 (Xanthoudakis et al., 1992). In vitro studies showed that APE1/Ref-1 reductively activates c-Jun (Walker et al., 1993), and was subsequently shown to activate p53 and several other trans-acting factors similarly (Jayaraman et al., 1997).
A third and distinct function of APE1 was discovered independently as a trans-acting factor involved in Ca2+-dependent repression of the parathyroid hormone (PTH) gene (Okazaki et al., 1994). PTH regulates the extracellular Ca2+ [Ca2+e] level via a negative feedback loop (Okazaki et al., 1992). An increase in [Ca2+e] due to the elevated PTH level triggers downregulation of the PTH gene, which is expressed exclusively in parathyroid cells (Yamamoto et al., 1989). This regulation is mediated by interaction between a negative calcium response element (nCaRE), present in the PTH promoter, and its cognate trans-acting factors (Okazaki et al., 1994). APE1/Ref-1 was found to be a component of protein complexes that bind to nCaRE-A and nCaRE-B (Okazaki et al., 1994). Interestingly, we identified one nCaRE-A and two nCaRE-Bs in the promoter of the APE1 gene itself, among which the proximal nCaRE-B was found to downregulate APE1 expression, and APE1 was identified in the complex bound to it (Izumi et al., 1996). Similar involvement of APE1 was observed recently in Ca2+-dependent downregulation of the human renin gene (Fuchs et al., 2003). The inability of recombinant APE1 alone to bind to nCaRE-A or nCaRE-B in the PTH gene promoter indicated a requirement for additional factors in the complexes (Chung et al., 1996; Kuninger et al., 2002). The Ku70 (Ku86) proteins were identified in the complex bound to nCaRE-A (Chung et al., 1996). We recently have identified APE1 and heterogeneous nuclear ribonucleoprotein L (hnRNP-L) as the major components of the complex bound to nCaRE-B (Kuninger et al., 2002).
A number of recent studies have underscored the key role of CREB-binding protein (CBP) and its homolog p300 in transcriptional regulation by integrating diverse signaling pathways. These proteins function as coactivators for many sequence-specific transcription factors (TFs), e.g. AP-1, p53 and nuclear receptors (Chrivia et al., 1993; Eckner et al., 1994; Goodman and Smolik, 2000). CBP/p300 by themselves do not bind to DNA. However, when brought to DNA by binding to TFs, CBP/p300 are believed to activate transcription via chromatin remodeling by acetylating histones of the nucleosomes with their intrinsic histone acetyltransferase (HAT) activity, and interaction with components of the basal transcription machinery (Bannister and Kouzarides, 1996; Ogryzko et al., 1996). CBP/p300 and their associated factor (P/CAF) acetylate not only histones but also many TFs, and are thus also named FAT (factor acetyltransferase; Sterner and Berger, 2000). Acetylation of p53, the first transcription factor shown to be acetylated by p300, enhances its cis element binding (Gu and Roeder, 1997). Similar acetylation and the resulting increase in affinity for cognate cis elements were observed subsequently for GATA-1 and E2F (Boyes et al., 1998; Martinez-Balbas et al., 2000).
Histone deacetylases (HDACs) reverse HAT action by deacetylating histones, leading to recondensation of the unfolded chromatin, which causes inhibition of transcription (Laherty et al., 1997; Luo et al., 1998). Many transcriptional repressors, e.g. pRb, NcoR and mSin3A, interact with and recruit HDACs to promoter sequences (Laherty et al., 1997; Luo et al., 1998). Complementary to the FAT activity of p300/CBP, HDACs deacetylate acetylated TFs. The activity of many transcription factors is thus regulated via acetylation by p300 and deacetylation by HDAC. Acetylation of TFs could modulate transcription both positively and negatively (Vo and Goodman, 2001). For example, acetylation of the central region of YY1 is required for its full repressor activity, and targets it for active deacetylation by HDACs whose affinity for YY1 is enhanced by its acetylation (Yao et al., 2001).
Many DNA repair and replication proteins, including FEN1 and G·T-specific thymine-DNA glycosylase (TDG), are acetylated by p300/CBP (Hasan et al., 2001; Tini et al., 2002), but the role of acetylation in DNA repair and other DNA transactions remains to be elucidated.
In this report, we show that the human APE1 is acetylated by p300 both in vivo and in vitro. Acetylation markedly enhances APE1’s affinity for nCaRE-B, leading to downregulation of the PTH gene. Ca2+-dependent activation of p300 increases the level of acetylated APE1 (AcAPE1), and enhances binding to nCaRE-B in the PTH promoter in vivo, while repression of the PTH gene is mediated by HDAC which binds APE1. These results suggest acetylation of APE1 as a novel regulatory modification for its transcriptional functions.
Results
In vivo and in vitro acetylation of APE1 by p300
APE1 was identified as a trans-acting factor because of its presence in the nCaRE-bound protein complexes which mediate Ca2+-dependent repression of the PTH gene (Okazaki et al., 1994). Because recombinant APE1 does not bind to nCaREs by itself, we suspected that its binding is dependent on other factors. Furthermore, our serendipitous discovery of two distinct species of APE1 after two-dimensional gel analysis of similar size but with different pI, one of which was identical to the recombinant enzyme (Figure 1A, panel II), suggested the presence of modified APE1 species in the nuclear extract of HeLa cells. This raised the possibility that the modified species is involved in the nCaRE complexes. Because of the small difference in the pIs of the two forms (Figure 1A), we suspected that the modified species was acetylated. To test for such acetylation, HCT116 cells were transfected with either empty vector or expression vector for FLAG-tagged APE1, and pulse-labeled with [3H]Na-acetate. Immunoprecipitation of the total extract with FLAG antibody, followed by SDS–PAGE and fluorography, showed the presence of radioactivity in the FLAG-APE1 band (Figure 1B, lane 2). This provided the first evidence for in vivo acetylation of APE1. To confirm further the presence of AcAPE1, the cell extract was immunoprecipitated with acetyl-lysine (AcLys) antibody. The presence of APE1 in this immunoprecipitate (Figure 1C, lane 2), but not in one with pre-immune sera (lane 1), indicated that a fraction of cellular APE1 is present in acetylated form.

Fig. 1. In vivo acetylation of APE1. (A) Two-dimensional gel electrophoresis of (I) recombinant APE1 (100 ng) and (II) HeLa nuclear extract (50 µg). After isoelectric focusing on an Immobiline dry strip (pH gradient 3–10) in an IPGphor electrophoresis system (Amersham), the proteins were separated by SDS–PAGE, and APE1 was visualized by western blotting. The asterisk indicates modified APE1. (B) Extracts of HCT116 cells transfected with FLAG-tagged APE1 expression plasmid (lane 2) or empty vector (lane 1), after labeling with [3H]Na- acetate, were immunoprecipated with FLAG antibody and analyzed by SDS–PAGE and fluorography. Lane 3: in vitro acetylated [3H]APE1 marker. (C) Western analysis with APE1 antibody of extracts of HCT116 immunoprecepitated with either AcLys antibody (α-AcLys; lane 2) or pre-immune control IgG (lane 1); total cell extract (lane 3) was used as a reference. (D) Panel I: western analysis with AcLys antibody of extracts of HCT116 cells transfected with FLAG-APE1 (lane 5), FLAG-APE1 plus p300 expression plasmids (lane 4), or empty vector (lane 3) followed by immunoprecipitation with FLAG antibody. The specificity of AcLys antibody for AcAPE1 was shown by western analysis of 0.1 µg of APE (lane 1) or AcAPE1 (lane 2). Western analysis of the same blot with FLAG antibody (panel III) and Coomassie staining (panel II) of duplicate immunoprecipitate in the same experiment to show comparable amounts of APE1 in samples with different levels of AcAPE1.
Because transcriptional co-activators p300/CBP are the major contributors of protein acetylation, we tested whether p300 is involved in in vivo acetylation of APE1. HCT116 cells were co-transfected with expression plasmids for FLAG-tagged APE1 and p300. Western analysis with AcLys antibody showed a significant increase in the level of AcAPE1 (Figure 1D, panel I, lane 4). Comparison of the western blot with FLAG-specific antibody and Coomassie blue staining of duplicate immunoprecipitates showed similar amounts of APE1 in different samples (Figure 1D, panels II and III). We therefore concluded that p300 contributes significantly to in vivo acetylation of APE1.
We then examined in vitro acetylation of APE1 by incubating the p300 immunoprecipitate from extracts of HCT116 cells transfected with the p300 expression plasmid, together with recombinant APE1 in the presence of [3H]acetyl-CoA. Radiolabeling of APE1 indicated that it was acetylated by the p300 immunoprecipitate but not by a control immunoprecipitate (Figure 2A, lanes 1 and 2). A parallel experiment with the same amount of the APE1 mutant lacking 40 N-terminal amino acid residues (NΔ40) showed no acetylation of APE1 (Figure 2A, lane 3), suggesting that the acetyl-acceptor lysine residues are localized within the 40 N-terminal residues. These initial results were confirmed by acetylation studies of APE1 with the recombinant HAT domain of p300 (Chakravarti et al., 1999). Again, full-length APE1, but not the NΔ40 mutant, was acetylated by purified HAT (Figure 2B).
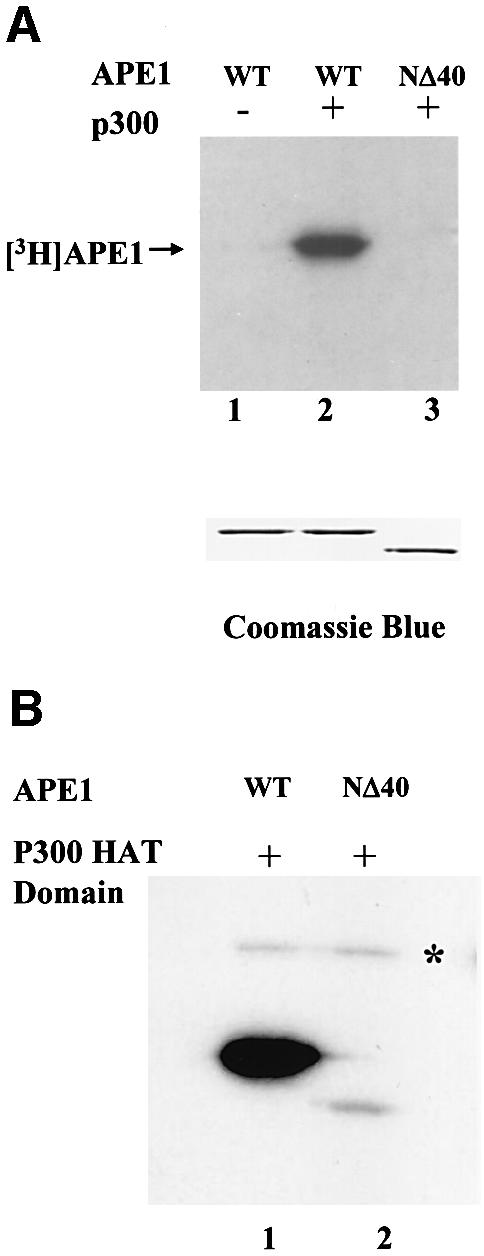
Fig. 2. In vitro acetylation of APE1. (A) Immunoprecipitated p300 from HCT116 extract was used for in vitro acetylation of wild-type (WT) (lane 2) or NΔ40 APE1 (lane 3) with [3H]acetyl-CoA, followed by SDS–PAGE and fluorography. Lane 1: non-specific IgG control with full-length APE1. Lower panel: Coomassie staining after SDS–PAGE. (B) In vitro acetylation with purified p300 HAT domain of wild-type APE1 (lane 1) or NΔ40 APE1 (lane 2) and [3H]acetyl-CoA followed by SDS–PAGE and fluorography. The asterisk indicates autoacetylated p300 HAT domain.
Identification of acetyl-acceptor lysine residues in APE1
To identify the acetyl-acceptor lysine residues localized in the N-terminal domain of APE1, two peptides were chemically synthesized, corresponding to residues 1–20 (N1) and 11–40 (N11) of APE1, and used as substrates for the HAT immunoprecipitate. Figure 3A shows that while both peptides contained multiple lysine residues, only the N1 peptide was acetylated when similar amounts of the peptides were used (data not shown). Mass spectrometric analysis confirmed the presence of one monoacetylated species with an increase of 42 mass units (Figure 3B). N-terminal sequencing of the peptide indicated that Lys6 and Lys7 were preferentially acetylated (data not shown), although a very low level of acetylation was also observed at Lys3. We then confirmed Lys6 and Lys7 residues as acetylation sites in the full-length APE1 (Figure 3C). These results were confirmed further by the lack of significant acetylation with K6R/K7R or K6L/K7L double mutants of APE1 using equal amounts of these proteins (Figure 3D). Thus APE1 is monoacetylated primarily at Lys6 or Lys7, but not both.

Fig. 3. Identification of AcLys residues in APE1. (A) Acetylation of N1 and N11 peptides (sequences in the lower panel) with immunoprecipitated p300 and [3H]acetyl-CoA as described in Materials and methods. (B) Mass spectroscopic analysis of in vitro acetylated N1 peptide. Peak corresponds to the unmodified peptide (m/z 2186.9) and peak Y to the monoacetylated form (m/z 2228.8). (C) Wild-type APE1 (10 µg) was incubated with 0.4 µg of p300 (HAT domain) and 1 mM acetyl-CoA for 1 h at 30°C. N-terminal sequencing in cycle 6 identified AcLys (AcK) residues as PTH-AcLys, which elutes just before PTH-Ala. (D) Acetylation of wild-type APE1 and K7R (KR), K6R/K7R (RR) or K6L/K7L (LL) mutants as described in Materials and methods. Lower panel: Coomassie staining after SDS–PAGE.
Impact of acetylation on APE1’s trans-acting activity
Because no difference was observed in the AP-endonuclease activity of acetylated and unmodified APE1 (data not shown), we tested its involvement in the transcriptional regulation of PTH. Following identification of APE1 in the protein complexes bound to nCaREs in the PTH promoter (Okazaki et al., 1994), we showed that APE1 and hnRNP-L are the major proteins in a stable complex bound to nCaRE-B (Kuninger et al., 2002). The lack of binding of recombinant APE1 to nCaRE oligos raised the possibility that acetylation of APE1 enhances its affinity for nCaREs, which we then tested with AcAPE1. APE1 was incubated with the recombinant HAT domain of p300 and acetyl-CoA, followed by purification of AcAPE1 by FPLC. We confirmed the identity of AcAPE1 by Edman degradation and immunoblotting with AcLys antibody (data not shown). EMSA showed that AcAPE1 did not bind to nCaRE-B by itself (data not shown).
The earlier observation that APE1 is present in the nCaRE-B-bound complex raised the possibility that APE1 has affinity not for nCaRE-B itself but for the proteins bound to it, which is enhanced by its acetylation. We tested this possibility by adding AcAPE1 to the HeLa nuclear extract in EMSA with nCaRE-B. It is evident that AcAPE1 stimulated the nCaRE-B binding activity more strongly than did the unacetylated, recombinant enzyme at the same concentration (Figure 4A). Interestingly, the dose–response was non-linear for both AcAPE1 and APE1, suggesting involvement of multiple molecules of APE1 in the binding complex. In any event, our results clearly show that APE1 is limiting in the formation of the nCaRE-B complex, for which AcAPE1 is preferred over APE1.

Fig. 4. AcAPE1 enhances binding of HCT116 nuclear extract to 5-32P-labeled nCaRE-B oligonucleotide. (A) EMSA with extract alone (lane 2), supplemented with 50 or 100 ng of APE1 (lanes 3 and 4) or AcAPE1 (lanes 5 and 6). Lane 1: free probe. The arrow indicates specific complex. (B) EMSA with nuclear extracts of HCT116 cells transfected with plasmids for wild-type APE1 (lane 3), K6R/K7R (RR) APE1 (lane 4), p300 (lane 5), p300 HAT mutant (p300 HM; lane 6), wild-type APE1 + p300 (lane 7) and wild-type APE1 + p300 mutant (lane 8), and no extract in lane 1. (C) EMSA with nuclear extracts of HCT116 cells transfected with K6R/K7R (RR) APE1 (lane 2), RR + p300 (lane 3), RR + p300 HM (lane 4) and HDAC1 (lane 5). (D) Western analysis for the APE1 level in the extracts used in (B).
To determine whether these in vitro observations have in vivo relevance, we examined the effects of p300 overexpression on the nCaRE-B binding activity in HCT116 cells. Strong enhancement of binding with the HCT116 extract was observed after ectopic expression of p300 alone or together with APE1 (Figure 4B, lanes 5 and 7). That this was a direct effect of APE1 acetylation was indicated by the observation that the binding was not significantly enhanced when p300 was overexpressed together with the non-acetylable APE1 K6R/K7R mutant or barely affected in cells expressing a mutant p300 lacking the HAT domain (Figure 4C, lane 3, and B, lane 6). Moreover, the observation that comparable overexpression of wild-type APE1 alone but not of the K6R/K7R mutant enhanced nCaRE-B binding (Figure 4B, lane 4, and D) supports our conclusion that the stimulation of binding was primarily due to acetylation of APE1.
Effect of APE1 acetylation on nCaRE-B-dependent transcription
Because the PTH gene is downregulated in response to an increase in [Ca2+e], an effect mediated by nCaRE-A and nCaRE-B, we examined modulation of PTH promoter activity due to APE1 acetylation. The PTH promoter is exclusively active in parathyroid cells which, unfortunately, are not available as cell lines. We therefore used the Ca2+-responsive baby hamster kidney cell line BHK-21 in which Ca2+-dependent downregulation of PTH promoter activity was observed earlier (Okazaki et al., 1994). BHK-21 cells were transfected with a luciferase reporter fused to a 4.5 kb promoter region of the PTH gene containing both nCaRE-A and nCaRE-B. Addition of 2 mM Ca2+ to the medium reduced the basal luciferase expression by 2- to 3-fold (Figure 5A). Ectopic expression of both wild-type and K6R/K7R mutant APE1 enhanced luciferase expression by 4- to 5-fold in Ca2+-free medium. However, Ca2+ abolished the enhancement due to overexpression of the wild-type APE1 but not of the non-acetylable mutant. Thus repression of transcription in the presence of Ca2+ appears to be specifically due to acetylation of APE1. Moreover, overexpression of p300 reduced luciferase expression by ∼2.5-fold in the presence of Ca2+. This again appears to be the direct effect of acetylation by p300, because co-expression of wild-type APE1, but not the K6R/K7R mutant, and p300 reduced PTH promoter activity by 3-fold in the presence of Ca2+, while co-expression of wild-type APE1 and p300 HAT mutant (p300 HM) did not (Figure 5A). Western blot analysis using APE1 antibody showed that overexpression of p300 or p300 HAT mutant did not change the APE1 level in the transfected cells (Figure 5A, lower panel).

Fig. 5. Repression of the PTH gene by AcAPE1. (A) BHK-21 cells were co-transfected with a PTH-luciferase reporter plasmid (1 µg) and 1 µg of expression plasmid for APE1 (wild-type or K6R/K7R mutant), p300 (wild-type or a HAT mutant) or equivalent amounts of empty vector. At 8 h after transfection, the cells were incubated in fresh medium containing no or 2 mM Ca2+ for 36 h. Luciferase activity was normalized with co-expressed β-galactosidase. The basal promoter activity of PTH-luc reporter in the presence of empty expression vector was normalized to 10. The bar graph shows the average of three independent experiments performed in duplicate. Cells were treated with TSA (100 ng/ml) in two sets. Western analysis for the APE1 level (lower panel) in the transfected cell extracts used in (A). (B) HCT116 cells transfected with FLAG-APE1 were incubated in medium containing 100 ng/ml TSA (lane 2) for 8 h. Immunoprecipitate of extracts with FLAG antibody were analyzed by western blotting using AcLys antibody. (C) BHK-21 cells were transfected with 5 nM (lane 2) and 10 nM (lane 3) p300 duplex siRNA oligo, and cell extracts were analyzed by western blotting using p300 (upper panel) and APE1 antibody (lower panel). (D) BHK-21 cells were co-transfected with a PTH-luciferase reporter plasmid (1 µg) and 10 nM p300 siRNA oligo along with either 1 µg of APE1 expression plasmid or an equivalent amount of empty expression vector. Luciferase activity was measured as described in (A). (E) BHK-21 cells were co-transfected with SV40-luciferase reporter plasmid (1 µg) containing PTH nCaRE-B upstream of the promoter, and 1 µg of the indicated expression plasmid for wild-type APE1 or K6R/K7R mutant, or p300 or equivalent amounts of empty vector. Luciferase activity was assayed after incubating cells in the presence of Ca2+ as described before.
To assess the role of p300 in Ca2+-induced repression of the PTH promoter, the cellular p300 level was reduced in the presence of p300 small interfering RNA (siRNA) oligo. A strong reduction in the p300 level was observed in BHK-21 cells at 48 h after transfection with duplex p300-specific RNA oligo (Figure 5C, upper panel) without any change in the APE1 level (Figure 5C, lower panel). As a control, an RNA oligo of random sequence was used. The residual p300 expression present in these cells could be due to non-quantitative transfection of these cells.
We then determined the effect of p300 silencing on Ca2+-mediated repression of the PTH promoter by co-transfecting BHK-21 cells with the PTH promoter reporter and p300 siRNA. Addition of 2 mM Ca2+ to the culture medium reduced basal luciferase expression by ∼1.5-fold (Figure 5D) compared with a 2- to 3-fold decrease in the control cells (Figure 5A). In contrast, ectopic expression of wild-type APE1 enhanced luciferase expression by 3- to 4-fold in the Ca2+-free medium. However, addition of Ca2+ to the culture medium reduced luciferase expression by 1.3-fold (Figure 5D) compared with a 3- to 4-fold reduction in the control cells (Figure 5A). These results show that the silencing of p300 partially blocked Ca2+-mediated repression of the PTH promoter.
The unexpected enhancement of PTH promoter activity due to ectopic overexpression of APE1 raised the possibility that unknown cis elements, in addition to Ca2+-responsive nCaREs, are present in the 4.5 kb promoter fragment and activated by APE1. We therefore decided to examine the specific repressing function of nCaRE by placing the PTH nCaRE-B upstream of the SV40 promoter in a luciferase expression plasmid. Inclusion of nCaRE-B caused an ∼2.5-fold decrease in promoter activity in the presence of Ca2+ relative to the control (Figure 5E). Additionally, overexpression of wild-type APE1 further reduced luciferase expression while ectopic expression of the K6R/K7R mutant had no effect. Expression of neither wild-type nor mutant APE1 affected the control SV40 promoter activity, indicating that Ca2+-dependent repression is mediated by AcAPE1 due to its binding to nCaRE-B. We have observed, consistent with an earlier report (Espinos et al., 1999), that overexpression of p300, together with APE1, activated control SV40 promoter by 3- to 3.5-fold. However, such enhancement was absent when the promoter was linked to nCaRE-B (Figure 5E). In any event, these results further strengthen our conclusion that AcAPE1 functions as a repressor by binding to nCaRE-B.
Effect of [Ca2+e] on acetylation of APE1 and HAT activity of p300
The results presented so far suggest that Ca2+-dependent repression of the PTH promoter is caused by enhanced acetylation of APE1. We therefore tested whether p300, the major enzyme responsible for acetylating APE1, is activated by Ca2+. BHK-21 cells were transfected with FLAG-tagged APE1 and, after incubation for 36 h in Ca2+-free medium followed by 4 h incubation in the presence of 2 mM Ca2+, FLAG-tagged APE1 was immunoprecipitated from the cell extract using FLAG antibody. Western analysis with AcLys antibody showed an ∼2- to 3-fold increase in the level of AcAPE1 compared with the control in which the cells were incubated in Ca2+-free medium (Figure 6A). However, the total APE1 level remained unchanged after Ca2+ treatment (Figure 6A, bottom panel).
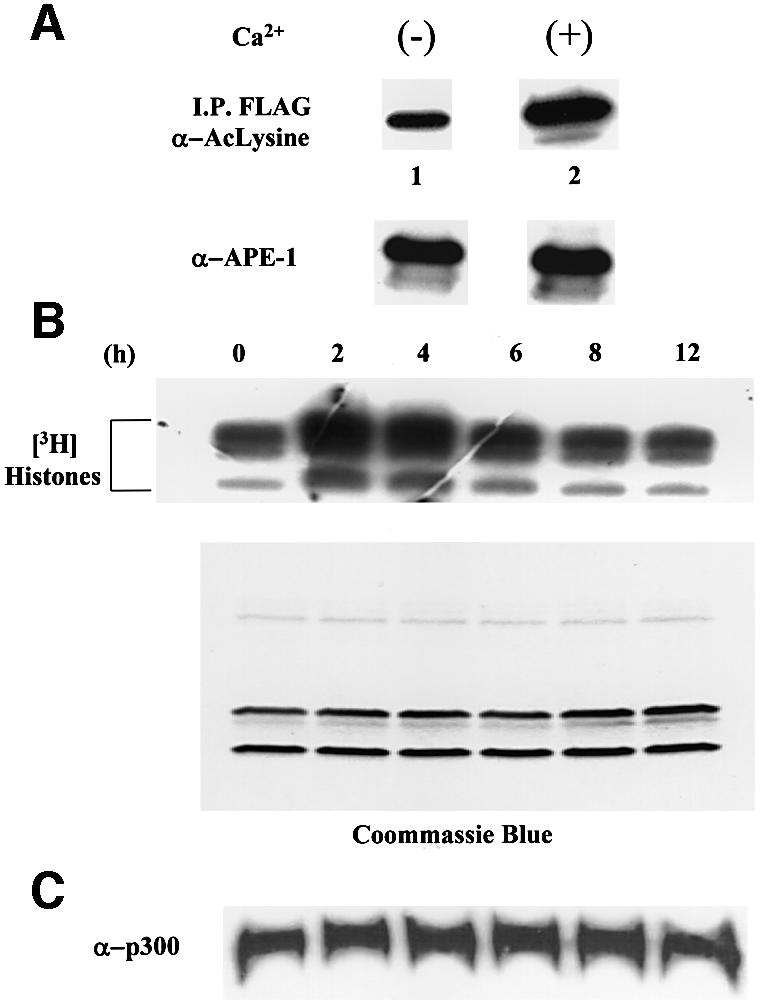
Fig. 6. Enhancement of acetylated APE1 and HAT activity of p300. (A) HCT116 cells transfected with FLAG-APE1 were incubated in medium containing no Ca2+ for 36 h and then treated with no (lane 1) or 2 mM Ca2+ (lane 2) for 4 h. Immunoprecipitates of extracts with FLAG antibody were analyzed by western blotting using AcLys antibody. Lower panel: western analysis for APE1. (B) BHK-21 cells were grown in Ca2+-free medium for 36 h, and then treated with 2 mM Ca2+ for 0, 2, 4, 6, 8 and 12 h. Immunoprecipitated endogenous p300 with [3H]acetyl-CoA was used for in vitro acetylation of core histones, followed by SDS–PAGE and fluorography. Lower panel: Coomassie staining after SDS–PAGE. (C) Western analysis for p300 level.
We then tested whether the increase in the level of AcAPE1 is due to Ca2+-induced activation of p300. In vitro assays with histone substrate and p300 immunoprecipates from the extracts of BHK-21 cells, treated with 2 mM Ca2+, showed that the p300 HAT activity was increased by ∼2.5- to 3-fold after 2 h of Ca2+ treatment, and that the activity gradually decreased after 6 h (Figure 6B). Coomassie staining confirmed equal loading of the histone substrate for various time points (Figure 6B, lower panel). At the same time, western analysis showed no change in the level of the p300 polypeptide (Figure 6C). Thus Ca2+-dependent activation of p300, which appears to be responsible for increasing the level of AcAPE1, is due to post-translational modification.
Repressor activity of APE1 due to HDAC binding
Trichostatin A (TSA), a specific inhibitor of HDAC, enhances the acetylation level of many proteins (Yoshida et al., 1990). Interestingly, we found a moderate effect of TSA on the acetylation level of APE1 as determined by immunoprecipitation of FLAG-tagged APE1 followed by immunoblotting with AcLys antibody (Figure 5B). However, treatment with TSA enhanced PTH promoter-dependent luciferase expression by 2-fold in Ca2+-free medium, and 5- to 6-fold in the presence of Ca2+, suggesting that Ca2+-dependent repression of the PTH gene is mediated by recruitment of HDACs (Figure 5A). Because HDACs have been shown to function directly as components of repressor complexes for various genes, we tested whether one or more HDACs is involved in downregulating the PTH gene by interacting with APE1. After co-transfecting HCT116 cells with expression plasmids of APE1, and FLAG-tagged HDACs 1–6, one at a time, we immunoprecipitated HDACs with FLAG antibody. The levels of ectopic expression of HDACs and APE1 were significant in all independently transfected cells (Figure 7A, panels II and III). Western analysis showed the presence of APE1 in the immunoprecipitates of only class I HDACs (HDACs 1–3) but not of class II HDACs (HDACs 4–6; Figure 7A, panel I) (Gray and Ekstrom, 2001). We therefore conclude that APE1 forms stable complexes only with class I HDACs.

Fig. 7. In vivo interaction of APE1 and HDACs. (A) HCT116 cells were co-transfected with expression plasmid for wild-type APE1 and individually for FLAG-tagged HDACs 1–6. The immunoprecipitates of cell extracts with FLAG antibody were analyzed for APE1. Panel I: lane 1, cell extract; lane 8, recombinant APE1 as a marker. Panel II: western analysis of cell extracts to show similar levels of APE1 in different transfected cells. Panel III: western analysis for the expression levels of HDACs. (B) Panel I: HCT116 cells were co-transfected with expression plasmid for wild-type APE1, or K6R/K7R or NΔ40 APE1 mutant and FLAG-tagged HDAC1. The FLAG immunoprecipitates of cell extracts were analyzed for APE1. Lane 1: immunoprecipitates from only APE1-transfected cell extracts. Panel II: western analysis of whole-cell extracts showing similar expression of APE1 in different transfected cells. Panel III: western analysis with FLAG antibody indicating significant expression of HDAC1.
Because APE1 interacts with class I HDACs and acetylation of APE1 represses the PTH promoter, it was important to establish whether it is the acetylated APE1 which recruits HDACs to nCaREs. We tested the requirement of APE1 acetylation for its in vivo interaction with HDACs by co-transfecting cells with expression plasmids for wild-type APE1, K6R/K7R or NΔ40 mutants of APE1, and FLAG-tagged HDAC1. Western analysis of the HDAC1 immunoprecipitates showed that wild-type APE1, as well as non-acetylable K6R/K7R and NΔ40 mutants form similar stable complexes with HDAC1 (Figure 7B). Western analysis confirmed comparable levels of ectopic expression of HDAC1 and APE1 in independently transfected cells (Figure 7B, panels II and III). These results thus indicate that acetylation of APE1 is not required for its interaction with HDAC1. We could not directly measure the level of AcAPE1 in the HDAC1 immunoprecipitate, because its amount in the complex was too small to quantitate with low affinity AcLys antibody (data not shown).
In order to demonstrate the presence of APE1 and HDAC1 in the nCaRE-B complex and acetylation-mediated enhancement of promoter occupancy, we utilized the chromatin immunoprecipitation (ChIP) assay. In the absence of an established parathyroid cell line, we used BHK-21 cells for analyzing such complexes. The cells were co-transfected with PTH plasmid and FLAG-tagged APE1 or FLAG-tagged APE1 plus p300 or FLAG-tagged HDAC1 (FLAG expression plasmids were used for better immunoprecipitation). After incubation for 24 h in Ca2+-free medium followed by 4 h incubation in the presence of 2 mM Ca2+, or 8 h in the presence of 100 ng/ml TSA, ChIP analysis was carried out with FLAG antibody as described in Materials and methods. The amount of immunoprecipitated PTH promoter sequence in each sample was quantitated by real-time PCR analysis. Figure 8A shows that the PTH promoter sequence was significantly enriched in the immunocomplex from APE1-FLAG- (12-fold) or HDAC1-FLAG- (9-fold) transfected cells compared with that from the control cells transfected with the empty vector (Figure 8A). Appropriate controls used in these experiments further validated our results. Thus, little or no PTH promoter sequence was detected by the PCR assay in the empty vector-transfected cells (Figure 8A). Furthermore, as expected, no PCR product was observed when the formaldehyde cross-linking step was omitted (data not shown). Importantly, overexpression of p300 enhanced the promoter occupancy by 4-fold. Treatment with Ca2+ caused a significant increase (3.5-fold) in the amount of the PTH promoter sequence (Figure 8A) in the immunocomplex, while TSA treatment moderately (2.5-fold) increased the promoter binding. However, TSA significantly enhanced promoter occupancy (5-fold) of APE1 in the PTH promoter in p300-overexpressing cells. Quantitation of the level of AcAPE1 (Figure 8B) in the cells used in ChIP assays further confirmed that acetylation of APE1 enhanced the promoter occupancy in the PTH promoter. These results suggest that APE1 or HDAC1 are normally associated with nCaRE-B in the PTH promoter, and that acetylation of APE1 enhances the promoter occupancy, at least in the episomal state.

Fig. 8. (A) ChIP assay for in vivo association of APE1 and HDAC1 with PTH promoter in transfected plasmids. The amount of immuno precipitated PTH promoter sequence was reported as pg/unit volume, using an absolute standard curve (0.001–1000 pg) of PTH-luc standards. Fold enrichment was calculated after normalizing with the absolute amount of input chromatin. (B) Western analysis with AcLys antibody of FLAG immunoprecipitates from cell extracts used in the ChIP assay.
Discussion
Multiple functions of APE1 in DNA repair and transcriptional regulation have raised the possibility that a molecular mechanism exists in vivo which directs this protein to different functions as needed. This report provides the first evidence for in vivo modification, namely acetylation of APE1, and suggests that such acetylation provides the novel regulatory switch for APE1’s various functions.
Once we confirmed the cellular presence of AcAPE1, we carried out a series of in vitro studies that identified the acetyl-acceptor site as Lys6 or Lys7, and p300 as the major HAT responsible for acetylation of APE1. The absence of diacetylated APE1 could be explained by the possible steric effects of acetyl groups attached to ε-amino groups of successive lysine residues. It is interesting that X-ray crystallographic studies suggest an unstructured state of the N-terminal segment (encompassing 40 residues) of APE1 (Mol et al., 2000). The N-terminal region, which is not conserved among APEs of various species, is also dispensable for its endonuclease/exonuclease activity in DNA repair (Izumi and Mitra, 1998).
Our initial results, showing strong enhancement of nCaRE-B binding activity due to exogenous APE1 after its acetylation, indicated a role for p300 in modulating the trans-acting function of APE1. Similar effects of acetylation on the activity of other transcription factors have been observed previously. For example, acetylation of p53 by p300 and P/CAF was shown to enhance its binding to the cis element (Gu and Roeder, 1997), and its association with coactivators leading to transcription activation (Barlev et al., 2001). Similarly, acetylation of GATA-1 and E2F increases their affinity for a consensus binding site and enhances their transcriptional activity (Boyes et al., 1998; Martinez-Balbas et al., 2000). Acetylation may also affect protein–protein interaction as indicated by a tighter binding of the MDM2 oncoprotein to the acetylated retinoblastoma protein (pRB) than to the unacetylated form (Chan et al., 2001).
The fact that AcAPE1 alone did not bind to nCaRE-B suggested a requirement for additional factor(s) with which AcAPE1 must interact in order to form a stable complex with this element. Previous affinity chromatography studies identified specific factors present in the nCaREs complexes. We showed that APE1 interacts with hnRNP-L in the absence of DNA, and that hnRNP-L alone could bind to nCaRE-B (Kuninger et al., 2002). The present studies suggest that AcAPE1 is limiting in the cell for binding to nCaRE-B, so that exogenous AcAPE1 could enhance the level of the nCaRE-B complex.
The functional consequences of enhanced APE1 binding to nCaRE-B after its acetylation were tested in PTH promoter-dependent reporter expression assays. Although APE1 was implicated in repression of the PTH gene in a Ca2+-dependent fashion (Okazaki et al., 1994), overexpression of both wild-type APE1 and the (K6R/K7R) mutant unexpectedly activated the PTH promoter when the 4.5 kb promoter sequence of the PTH gene containing both nCaRE-A and nCaRE-B was used. It appears that APE1 acts as an activator of some unidentified transcription factors which recognize unknown cis elements, distinct from nCaREs, present in the 4.5 kb promoter segment. This possibility was supported by subsequent studies in which we used only the nCaRE-B of the PTH gene linked to a heterologous SV40 promoter. In this case, we did not observe activation of the promoter dependent on overexpression of either wild-type or mutant APE1. However, exogenous Ca2+ significantly reduced PTH promoter activity when the wild-type but not non-acetylable (K6R/K7R) mutant APE1 was overexpressed. This result strongly supports our conclusion that AcAPE1 is involved in repression of the PTH promoter. Moreover, co-expression of APE1 and wild-type but not mutant p300 strongly reduced the PTH promoter-dependent luciferase expression in both the presence and absence of Ca2+, consistent with enhanced binding of nuclear extracts to nCaRE-B. We have also shown that inhibition of p300 partially blocked the Ca2+-mediated repression of the PTH promoter. Moreover, such repression was not pronounced when we overexpressed wild-type APE1 in p300 siRNA-transfected cells. These observations support our conclusion that p300-dependent acetylation of APE1 plays a role in Ca2+-mediated repression of the PTH promoter. That Ca2+-mediated repression of PTH promoter was not completely blocked during p300 inhibition suggests that the residual HAT activity of p300 or other HATs such as CBP could also acetylate APE1 (Goodman and Smolik, 2000).
Treatment with Ca2+ significantly increased APE1 acetylation in BHK-21 cells, which could explain the original observation that an increase in [Ca2+e] enhanced the binding activity of BHK-21 cell extract for nCaRE-B (Okazaki et al., 1994). The increased level of AcAPE1 in vivo could be due to Ca2+-mediated activation of p300, or inhibition of HDAC. We observed that Ca2+ enhanced the HAT activity of p300. This HAT activity is enhanced during the S phase due to phosphorylation (Ait-Si-Ali et al., 1998), possibly by one of several kinases including Ca2+-dependent calmodulin (CAM) kinase IV, mitogen-activated protein kinase (MAPK) and cyclin E–cdk2, which are important in either cell cycle regulation or various signal transduction pathways (Vo and Goodman, 2001). It is thus possible that CAM kinase IV is responsible for Ca2+-dependent activation of p300 HAT. In any case, this is the first report that the p300 HAT activity is enhanced due to Ca2+-dependent modification.
Interaction of APE1 with HDACs 1–3 could at least partially explain how APE1 acts as a transcriptional repressor. We have shown that acetylation is not essential for the stable binding of APE1 to HDACs. We therefore postulate that APE1 constitutively interacts with HDACs, and that the regulation occurs primarily at the level of recruitment of the APE1–HDAC complex to the promoter, which is controlled by APE1 acetylation. Although we have observed a moderate increase in the level of AcAPE1 after TSA treatment, such treatment stimulated PTH promoter activity and abolished Ca2+-dependent repression. This suggests that inhibition of HDACs with TSA prevents recondensation of the local chromatin containing the PTH promoter. This effect appears to be more dominant in the luciferase reporter plasmid than a moderate TSA-dependent increase in AcAPE1 level which would lead to its enhanced binding to the repressor complex. The ChIP assay provided direct evidence that APE1 and HDAC1 are bound to nCaRE-B in the episomal PTH promoter, and that acetylation of APE1 significantly increases this binding. Based on these results, we propose the following model for Ca2+-dependent repression of the PTH gene. APE1, present partly as the APE1–HDAC complex in vivo, is unacetylated. A rise in [Ca2+e] increases the HAT activity of p300, which then acetylates APE1. Acetylation stimulates association of APE1 with its partner hnRNP-L, and binding to nCaRE-B in the PTH promoter. This promoter-bound repressor complex turns off PTH transcription by deacetylating histones in the promoter region. Finally, deacetylation of AcAPE1 by HDAC in the nCaRE complex reduces APE1’s affinity for hnRNP-L (and possibly other proteins) in the nCaRE-B complex; subsequent release of APE1 from the complex leads to restoration of transcription. Thus the negative feedback regulation is mediated by the acetylation/deacetylation cycle of APE1.
Our discovery of APE1 acetylation and the studies described here have raised the possibility that subtle post-translational modifications provide a means for channeling multifunctional proteins such as APE1 to different activities and interactions, and thus acts as a regulatory switch in performing different functions. Whether this is a general mechanism for a variety of proteins remains to be determined.
Materials and methods
Cell culture, DNA transfection and luciferase assays
HCT116 (p53 –/–) cells (a gift from B.Vogelstein) and BHK-21 cells (ATCC No. CCL-10) were transfected using LipofectAMINE 2000 (Life Technologies, Inc.) according to the manufacturer’s instructions. BHK-21 cells were transfected with duplex p300 siRNA (5′-AAC CCC UCC UCU UCA GCA CCA-3′; Dharmacon Research, Inc.) at 5 or 10 nM per 60 mm plate using LipofectAMINE 2000. Luciferase activity, assayed as described earlier (Bhakat and Mitra, 2000), was normalized to β-galactosidase activity expressed from a co-transfected plasmid. In some cases, where the promoter activity of β-galactosidase expression plasmid was also affected, luciferase activity was normalized with total protein in the extract.
In vitro acetylation assays
Recombinant wild-type and mutant APE1 (5 µg) were incubated with 0.2 µg of recombinant p300 (HAT domain) or with an immunoprecipitate of p300 together with 1 µCi of [3H]acetyl-CoA (200 mCi/mM; NEN) in 50 µl of HAT buffer [50 mM Tris–HCl pH 8.0, 0.1 mM EDTA, 10% (v/v) glycerol, 1 mM dithiothreitol (DTT) and 10 mM Na-butyrate] at 30°C for 1 h, followed by SDS–PAGE and fluorography (Amplify, Amersham).
In vivo acetylation of APE1
Forty hours after transfecting HCT116 cells with 1 µg of FLAG-tagged APE1 expression plasmid, the cells were incubated in McCoy 5A medium (Life Technologies, Inc.) containing 1 mCi/ml [3H]Na-acetate (2–10 Ci/mmol; NEN) for 1 h before the cells were collected for lysis. The cell extract was immunoprecipitated for 3 h at 4°C with FLAG M2 antibody cross-linked to agarose beads (Sigma). After washing the beads with cold TBS (50 mM Tris–HCl pH 7.5, 150 mM NaCl), FLAG-APE1 was eluted from the immunocomplex by gently shaking with TBS containing 300 ng/µl FLAG peptide (Sigma) for 30 min, followed by SDS–PAGE (12% polyacrylamide) and fluorography. AcAPE1 from cell extracts was sometimes immunoprecipitated using AcLys antibody (New England Biolabs) and p300 immunoprecipitated with N15 p300 antibody (Santa Cruz Biotechnology).
Identification of acetyllysine residues
Two peptides corresponding to amino acid residues 1–20 and 11–40 of APE1 were chemically synthesized and HPLC-purified, and then acetylated at 30°C for 1 h with immunoprecipitated p300 in the acetylation buffer containing 2 mM acetyl-CoA. After desalting, the peptides were analyzed by MALDI-TOF (Applied Biosystems) or by Edman degradation. AcLys residues were identified as PTH-AcLys, which eluted immediately before PTH-Ala.
Electrophoretic mobility shift assay
The oligonucleotides encompassing the PTH nCaRE-B (the palindromic core sequence is underlined), 5′-TTTTTGAGACAGGGTCTCACTCTG-3′, were used for EMSA with 10 µg of nuclear extract (Izumi et al., 1996).
In vitro acetylation assay using p300 immunoprecipitate
BHK-21 cells were grown in S-MEM (Life Technologies), treated with 2 mM CaCl2 for various times and then lysed in lysis buffer (50 mM Tris–HCl pH 7.5, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, 1 mM NaF, 1 mM Na-orthovanadate, 10 mM Na-butyrate and protease inhibitor mixture). The lysate were immunoprecipitated with p300 antibody (5 µg/ml) for 2–3 h at 4°C. The immunocomplexes were collected by adding 40 µl of protein A–agarose beads at 4°C for 1 h, which were pelleted by centrifugation and washed three times with cold phosphate-buffered saline (PBS), and then once with HAT buffer. A standard HAT assay was performed containing 5 µg of purified histones or APE1 as the substrate at 30°C for 1 h, followed by SDS–PAGE and fluorography.
Chromatin immunoprecipitation (ChIP) assay
Immunoprecipitation of chromatin was performed as described earlier, with some modifications (Bhakat and Mitra, 2000). BHK-21 cells were co-transfected with PTH promoter reporter plasmid and FLAG-tagged APE1, FLAG-tagged APE1 and p300, or FLAG-tagged HDAC1. After incubation for 24 h in Ca2+-free medium followed by 4 h incubation in the medium containing 2 mM in Ca2+, or 8 h incubation in the medium containing 100 ng/ml TSA, cells were incubated in 1% formaldehyde at 37°C for 10 min to allow reversible cross-linking of proteins including histones to DNA. The cells were harvested and lysed in 0.5 ml of lysis buffer (150 mM Tris–HCl pH 7.5, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100) and the lysate was incubated overnight at 4°C with 80 µl of agarose-conjugated FLAG M2 antibody. The beads were collected by centrifugation, washed three times with TBS then once with TBS buffer containing 500 mM NaCl. Finally the beads were washed with TE, and the complexes eluted with two 250 µl aliquots of elution buffer (1% SDS, 0.1 M NaHCO3) at room temperature. After reversal of cross-links and DNA extraction from the immunocomplex, the PTH promoter containing the nCaRE-B element was amplified by real-time PCR in an Applied Biosystems cycler ABI7000 and TaqMan MGB probe (FAM™ dye-labeled) for quantitating the human PTH promoter sequences. The primers and probe sequences used were as follows: 5′ primer (5′-TGTATCTTATGGTACTGTAACTG-3′) corresponding to PGL2 basic vector, and 3′ primer (5′-TTGACCTTCCCCACAAAGCAGTAAT-3′) corresponding to the PTH promoter, and probe ACCCGGAGG TACCTAT (assays-by-designSM; P/N 4331348). Real-time PCR was performed with the same volume of DNA in duplicate using TaqMan universal PCR master mix reagent kit (P/N 4304437). The cycling parameters were: Amplitaq activation at 95°C for 10 min, denaturation at 95°C for 15 s and annealing/extension at 60°C for 1 min (repeated 40 times). The results of the real-time PCR assay for each sample were reported as pg/unit volume, using an absolute standard curve (0.001, 0.01. 0.1, 1, 10, 100 and 1000 pg of PTH-luc plasmid standards; Applied Biosystems). Linearity was observed over the concentration range 0.001 pg to 1 ng of PTH-luc plasmid.
Proteins
The wild-type and mutant APE1s were purified as described (Izumi et al., 1999). FLAG-tagged p300 (HAT domain) expressed from recombinant baculovirus was purified from virus-infected Sf9 cells using FLAG antibody affinity matrix according to the manufacturer’s instructions (Sigma). Recombinant APE1 (0.5 mg) was acetylated with the p300 HAT domain, and the acetylated protein was eluted from a Mono-S column at 280 mM NaCl. The identity of AcAPE1 was confirmed by sequencing and western analysis with anti-AcLys antibody.
Plasmids
Wild-type APE1 was cloned into the mammalian expression plasmid pcDNA 3.1 Zeo (Invitrogen Corp.) by PCR. The K7R, K6R/K7R and K6L/ K7L mutants of APE1 were also generated by PCR and cloned into an Escherichia coli expression plasmid. The expression plasmid pCMV-p300, p300 HAT mutant, Flag-p300 HAT domain in the baculovorus expression system and FLAG-tagged HDACs 1–6 were used in this study. The 4.5 kb sequence upstream of the PTH promoter was generated by PCR and then cloned into pGL2 basic vector (Promega). A 400 bp promoter region of the PTH gene containing the nCaRE-B element was generated by PCR. The identity of all recombinant DNAs generated by PCR was confirmed by sequencing.
Acknowledgments
Acknowledgements
We are grateful to Dr T.Okazaki for providing PTH-CAT expression plasmid, Drs D.Chakravarty, S.Grossman, B.Bernstein and J.Wang for providing various HDAC and p300 plasmids, Drs Sutapa Ray and Dora Bocangel for help, Dr Istvan Boldogh for guidance and materials, Ms W.Smith for secretarial assistance, and Dr D.Konkel for editing the manuscript. We acknowledge the expert services of the UTMB Proteomics and Protein Expression, Cancer Cell Biology and Toxicology Cell Biology. The research was supported by USPHS grants R01 ES08457, R01 CA53791, P01 AG10514, and ES06676.
References
- Ait-Si-Ali S. et al. (1998) Histone acetyltransferase activity of CBP is controlled by cycle-dependent kinases and oncoprotein E1A. Nature, 396, 184–186. [DOI] [PubMed] [Google Scholar]
- Bannister A.J. and Kouzarides,T. (1996) The CBP co-activator is a histone acetyltransferase. Nature, 384, 641–643. [DOI] [PubMed] [Google Scholar]
- Barlev N.A., Liu,L., Chehab,N.H., Mansfield,K., Harris,K.G., Halazonetis,T.D. and Berger,S.L. (2001) Acetylation of p53 activates transcription through recruitment of coactivators/histone acetyltransferases. Mol. Cell, 8, 1243–1254. [DOI] [PubMed] [Google Scholar]
- Bhakat K.K. and Mitra,S. (2000) Regulation of the human O(6)-methylguanine-DNA methyltransferase gene by transcriptional coactivators cAMP response element-binding protein-binding protein and p300. J. Biol. Chem., 275, 34197–34204. [DOI] [PubMed] [Google Scholar]
- Boyes J., Byfield,P., Nakatani,Y. and Ogryzko,V. (1998) Regulation of activity of the transcription factor GATA-1 by acetylation. Nature, 396, 594–598. [DOI] [PubMed] [Google Scholar]
- Chakravarti D., Ogryzko,V., Kao,H.Y., Nash,A., Chen,H., Nakatani,Y. and Evans,R.M. (1999) A viral mechanism for inhibition of p300 and PCAF acetyltransferase activity. Cell, 96, 393–403. [DOI] [PubMed] [Google Scholar]
- Chan H.M., Krstic-Demonacos,M., Smith,L., Demonacos,C. and La Thangue,N.B. (2001) Acetylation control of the retinoblastoma tumour-suppressor protein. Nature Cell Biol., 3, 667–674. [DOI] [PubMed] [Google Scholar]
- Chrivia J.C., Kwok,R.P., Lamb,N., Hagiwara,M., Montminy,M.R. and Goodman,R.H. (1993) Phosphorylated CREB binds specifically to the nuclear protein CBP. Nature, 365, 855–859. [DOI] [PubMed] [Google Scholar]
- Chung U. et al. (1996) The interaction between Ku antigen and REF1 protein mediates negative gene regulation by extracellular calcium. J. Biol. Chem., 271, 8593–8598. [DOI] [PubMed] [Google Scholar]
- Demple B. and Harrison,L. (1994) Repair of oxidative damage to DNA: enzymology and biology. Annu. Rev. Biochem., 63, 915–948. [DOI] [PubMed] [Google Scholar]
- Eckner R., Ewen,M.E., Newsome,D., Gerdes,M., DeCaprio,J.A., Lawrence,J.B. and Livingston,D.M. (1994) Molecular cloning and functional analysis of the adenovirus E1A-associated 300-kD protein (p300) reveals a protein with properties of a transcriptional adaptor. Genes Dev., 8, 869–884. [DOI] [PubMed] [Google Scholar]
- Espinos E., Le Van Thai,A., Pomies,C. and Weber,M.J. (1999) Cooperation between phosphorylation and acetylation processes in transcriptional control. Mol. Cell. Biol., 19, 3474–3484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans A.R., Limp-Foster,M. and Kelley,M.R. (2000) Going APE over ref-1. Mutat. Res., 461, 83–108. [DOI] [PubMed] [Google Scholar]
- Fuchs S., Philippe,J., Corvol,P. and Pinet,F. (2003) Implication of Ref-1 in the repression of renin gene transcription by intracellular calcium. J. Hypertens., 21, 327–335. [DOI] [PubMed] [Google Scholar]
- Goodman R.H. and Smolik,S. (2000) CBP/p300 in cell growth, transformation and development. Genes Dev., 14, 1553–1577. [PubMed] [Google Scholar]
- Gray S.G. and Ekstrom,T.J. (2001) The human histone deacetylase family. Exp. Cell Res., 262, 75–83. [DOI] [PubMed] [Google Scholar]
- Gu W. and Roeder,R.G. (1997) Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell, 90, 595–606. [DOI] [PubMed] [Google Scholar]
- Hasan S., Stucki,M., Hassa,P.O., Imhof,R., Gehrig,P., Hunziker,P., Hubscher,U. and Hottiger,M.O. (2001) Regulation of human flap endonuclease-1 activity by acetylation through the transcriptional coactivator p300. Mol. Cell, 7, 1221–1231. [DOI] [PubMed] [Google Scholar]
- Izumi T. and Mitra,S. (1998) Deletion analysis of human AP-endonuclease: minimum sequence required for the endonuclease activity. Carcinogenesis, 19, 525–527. [DOI] [PubMed] [Google Scholar]
- Izumi T., Henner,W.D. and Mitra,S. (1996) Negative regulation of the major human AP-endonuclease, a multifunctional protein. Biochemistry, 35, 14679–14683. [DOI] [PubMed] [Google Scholar]
- Izumi T., Malecki,J., Chaudhry,M.A., Weinfeld,M., Hill,J.H., Lee,J.C. and Mitra,S. (1999) Intragenic suppression of an active site mutation in the human apurinic/apyrimidinic endonuclease. J. Mol. Biol., 287, 47–57. [DOI] [PubMed] [Google Scholar]
- Jayaraman L., Murthy,K.G., Zhu,C., Curran,T., Xanthoudakis,S. and Prives,C. (1997) Identification of redox/repair protein Ref-1 as a potent activator of p53. Genes Dev., 11, 558–570. [DOI] [PubMed] [Google Scholar]
- Kuninger D.T., Izumi,T., Papaconstantinou,J. and Mitra,S. (2002) Human AP-endonuclease 1 and hnRNP-L interact with a nCaRE-like repressor element in the AP-endonuclease 1 promoter. Nucleic Acids Res., 30, 823–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laherty C.D., Yang,W.M., Sun,J.M., Davie,J.R., Seto,E. and Eisenman,R.N. (1997) Histone deacetylases associated with the mSin3 corepressor mediate mad transcriptional repression. Cell, 89, 349–356. [DOI] [PubMed] [Google Scholar]
- Luo R.X., Postigo,A.A. and Dean,D.C. (1998) Rb interacts with histone deacetylase to repress transcription. Cell, 92, 463–473. [DOI] [PubMed] [Google Scholar]
- Martinez-Balbas M.A., Bauer,U.M., Nielsen,S.J., Brehm,A. and Kouzarides,T. (2000) Regulation of E2F1 activity by acetylation. EMBO J., 19, 662–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra S., Hazra,T.K., Roy,R., Ikeda,S., Biswas,T., Lock,J., Boldogh,I. and Izumi,T. (1997) Complexities of DNA base excision repair in mammalian cells. Mol. Cell, 7, 305–312. [PubMed] [Google Scholar]
- Mol C.D., Izumi,T., Mitra,S. and Tainer,J.A. (2000) DNA-bound structures and mutants reveal abasic DNA binding by APE1 and DNA repair coordination. Nature, 403, 451–456. [DOI] [PubMed] [Google Scholar]
- Ogryzko V.V., Schiltz,R.L., Russanova,V., Howard,B.H. and Nakatani,Y. (1996) The transcriptional coactivators p300 and CBP are histone acetyltransferases. Cell, 87, 953–959. [DOI] [PubMed] [Google Scholar]
- Okazaki T., Ando,K., Igarashi,T., Ogata,E. and Fujita,T. (1992) Conserved mechanism of negative gene regulation by extracellular calcium. Parathyroid hormone gene versus atrial natriuretic polypeptide gene. J. Clin. Invest., 89, 1268–1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okazaki T., Chung,U., Nishishita,T., Ebisu,S., Usuda,S., Mishiro,S., Xanthoudakis,S., Igarashi,T. and Ogata,E. (1994) A redox factor protein, ref1, is involved in negative gene regulation by extracellular calcium. J. Biol. Chem., 269, 27855–27862. [PubMed] [Google Scholar]
- Sterner D.E. and Berger,S.L. (2000) Acetylation of histones and transcription-related factors. Microbiol. Mol. Biol. Rev., 64, 435–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tini M., Benecke,A., Um,S.J., Torchia,J., Evans,R.M. and Chambon,P. (2002) Association of CBP/p300 acetylase and thymine DNA glycosylase links DNA repair and transcription. Mol. Cell, 9, 265–277. [DOI] [PubMed] [Google Scholar]
- Vo N. and Goodman,R.H. (2001) CREB-binding protein and p300 in transcriptional regulation. J. Biol. Chem., 276, 13505–13508. [DOI] [PubMed] [Google Scholar]
- Walker L.J., Robson,C.N., Black,E., Gillespie,D. and Hickson,I.D. (1993) Identification of residues in the human DNA repair enzyme HAP1 (Ref-1) that are essential for redox regulation of Jun DNA binding. Mol. Cell. Biol., 13, 5370–5376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xanthoudakis S., Miao,G., Wang,F., Pan,Y.C. and Curran,T. (1992) Redox activation of Fos-Jun DNA binding activity is mediated by a DNA repair enzyme. EMBO J., 11, 3323–3335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto M., Igarashi,T., Muramatsu,M., Fukagawa,M., Motokura,T. and Ogata,E. (1989) Hypocalcemia increases and hypercalcemia decreases the steady-state level of parathyroid hormone messenger RNA in the rat. J. Clin. Invest., 83, 1053–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Y.L., Yang,W.M. and Seto,E. (2001) Regulation of transcription factor YY1 by acetylation and deacetylation. Mol. Cell. Biol., 21, 5979–5991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida M., Kijima,M., Akita,M. and Beppu,T. (1990) Potent and specific inhibition of mammalian histone deacetylase both in vivo and in vitro by trichostatin A. J. Biol. Chem., 265, 17174–17179. [PubMed] [Google Scholar]