

Abstract
The chemical diversity of natural products is fueled by the emergence and ongoing evolution of biosynthetic pathways in secondary metabolism1-5. However, co-evolution of enzymes as functional assemblies for metabolic diversification is not well understood, especially at the biochemical level. Here, two parallel enzyme assemblies with an extraordinarily high sequence identity form a β-branched cyclopropane in the curacin A (Cur), and a vinyl chloride group in the jamaicamide (Jam) pathways, respectively. The assemblies include a halogenase (Hal), a 3-hydroxy-3-methylglutaryl (HMG) enzyme cassette for β-branching and an enoyl reductase domain (ER). Bioinformatic analysis indicated that the corresponding genes were inserted into modular polyketide synthases (PKSs) via acyltransferase (AT) domain replacement. The Hal from CurA, and the dehydratases (ECH1s) and decarboxylases (ECH2s) within the HMG enzyme cassettes and ERs from both Cur and Jam were assessed biochemically to determine the mechanism of cyclopropane and vinyl chloride formation. Unexpectedly, the polyketide β-branching pathway was modified by introduction of a γ-chlorination step on (S)-HMG mediated by Cur Hal, a non-heme FeII, α-ketoglutarate (α-KG)-dependent halogenase6. In a divergent scheme, Cur ECH2 was found to catalyze formation of the α,β C=C enoyl thioester, whereas Jam ECH2 formed a vinyl chloride moiety by selectively generating the corresponding β,γ C=C (enoyl thioester) of the 3-methyl-4-chloroglutaconyl decarboxylation product. A non-conserved Tyr82 residue in Cur ECH2 was crucial to this regiochemical control. CurF ER specifically catalyzed an unprecedented cyclopropanation reaction on the chlorinated product of Cur ECH2. Thus, the combination of chlorination and polyketide β-branching, coupled with mechanistic diversification of ECH2 and ER leads to formation of cyclopropane and vinyl chloride substituents. These results reveal a remarkable parallel interplay of evolutionary events in multienzyme systems leading to functional group diversity in secondary metabolites.
The tremendous biosynthetic capability of nature is well exemplified by structurally diverse secondary metabolites that help their hosts, typically microorganisms and plants, to survive and thrive in environmental niches by mediating a broad range of ecological and physiological interactions1-5. The biosynthesis of secondary metabolites is “diversity-oriented”1,5,7, targeting the variable environment by producing a vast array of complex chemical structures8. This enormous productivity is largely fueled by the rapid evolution of biosynthetic genes and functional alteration of the corresponding enzymes3,4. As such, the evolutionary history of metabolic gene assemblies informs the origin of their biosynthetic diversity. However, tracing the ancestral forms of multiple genes as a functional collective is elusive, especially when they are dispersed in the genome. Biosynthetic genes from microbial hosts are usually clustered, and are ideal for evolutionary and functional studies4. Currently, our understanding of the evolution and function of multienzyme systems in secondary metabolism is largely based on genetic studies, with recent efforts increasingly focused on comparative biochemical analysis9.
Modular PKSs originated from fatty acid synthases (FASs) and serve as a paradigm for secondary metabolic systems that evolve to expand chemical diversity10. These giant biochemical machines are modular assembly-lines that catalyze highly programmed biosynthetic pathways, where final product structures result from variation in chain initiation, extension, termination and tailoring steps11-13. Moreover, PKSs have a propensity to form hybrids such as with nonribosomal-peptide synthetase (NRPS) modules and recently reported HMG enzyme cassettes14,15. The metamorphic properties shown in these composite systems drive metabolic diversification, but relatively few details are currently available to describe the mechanistic details.
The curacin and jamaicamide marine cyanobacterial metabolites from Lyngbya majuscula are mixed-polyketide nonribosomal-peptide natural products with potent anticancer and sodium channel blocking activities, respectively16,17. The parallel components of the Cur and Jam biosynthetic pathways (Fig. 1a) provide an unusual opportunity to investigate the biosynthetic origin of chemical diversity, in the form of cyclopropane ring formation for curacin and vinyl chloride formation for jamaicamide16,18. Studies on the variant function and selectivity of these highly parallel biosynthetic systems form the subject of this report.
Figure 1. Comparison of enzyme assemblies in the Cur and Jam pathways.
a, Formation of cyclopropane and vinyl chloride functional groups. b, Comparative sequence identities of the enzymes encoded by the two highly similar regions in the Cur and Jam pathways. The aligned DNA sequences are located at the boundaries of these two regions. c, Formation of 3-ACP3 in the Cur pathway, and hypothesized reactions for 4-ACP3, 5-ACP3 and 6-ACP3. The hypothetic chlorinated intermediates are shown along with the non-chlorinated ones. The β-branching carbon atoms are highlighted in red.
Two highly similar enzyme assemblies
The two parallel, highly conserved Cur and Jam enzyme assemblies are incorporated into the early PKS modules, and are predicted to catalyze β-branching reactions in the growing chain elongation intermediate16,18. These unusual embedded domains and discrete enzymes span from CurA to CurF and from JamE to JamJ, and are grouped into three subsets (Fig. 1a): (1) Hals embedded in CurA and JamE; (2) HMG enzyme cassettes containing a tandem acyl carrier protein (ACP) tridomain (ACP3), including ACPI, ACPII and ACPIII embedded in CurA and JamE, discrete CurB and JamF ACPIVs, CurC and JamG KSs, CurD and JamH HMG-CoA synthase-like enzymes (HCSs), CurE and JamI ECH1s, ECH2s embedded in CurF and JamJ; and (3) ERs embedded in CurF and JamJ (Fig. 1a). Comparative analysis of these Cur and Jam enzymes revealed that the sequence identities of the Hals, ACP3s, ACPIVs, KSs, HCSs and ECH1s are extraordinarily high (∼90%), whereas the ECH2s and ERs are substantially lower (∼60% identity) (Fig. 1b).
Cur and Jam Hals were predicted to be α-KG-dependent non-haem halogenases (less than 20% sequence identity to characterized homologs)19-21, that catalyze halogenation of unactivated carbon atoms20-24 through a non-haem FeIV=O intermediate25,26. HMG enzyme cassettes have been demonstrated to catalyze polyketide on-assembly-line β-branching to generate a pendant methyl or ethyl group from a polyketide β-carbonyl14,15,27. Cur and Jam ERs show ∼50% sequence identity to other ERs in Cur and Jam PKS modules, and belong to the acyl-CoA reductase family that catalyzes NADPH-dependent reduction of α,β C=C (enoyl thioester) in acyl-CoAs or acyl-ACPs28. These two ERs are located upstream of CurF and JamJ KS, an unusual location as ERs typically reside between AT and ACP domains in PKS modules.
AT replacement-mediated PKS hybridization
Bioinformatic analyses of Cur and Jam pathway sequences suggested that the parallel AT-Hal-ACPI-ACPII-ACPIII-ACPIV-KS-HCS-ECH1-ECH2-ER-KS-AT gene assembly (Fig. 1b) might have been introduced into the polyketide pathway by AT domain replacement. Based on the DNA and amino acid alignments of CurA—CurF and JamE—JamJ, we found that the highly similar regions, extend from the N-termini of the ATs in CurA and JamE, through the C-terminal “post-AT linkers”29 of the ATs in CurF and JamJ (Fig. 1b, and Supplementary Fig. 1). Recent bioinformatic studies indicate that these highly similar sequences could promote AT domain replacement by homologous recombination30,31. Thus, a “di-AT domain replacement” might have occurred in Cur or Jam pathways through insertion of the above gene assembly into a pre-existing cluster, which could serve as an efficient strategy for PKS pathway expansion or contraction. This hypothesis is supported by phylogenetic analysis for the KS, AT and dehydratase (DH) domains of the sequenced pathways from L. majuscula (Supplementary Fig. 2).
HMG β-branching with ER saturation
HMG β-branching includes a series of modifications on the β-carbonyl group of polyketide intermediates typically tethered to the tandem ACPs. As shown for curacin A (Fig. 1c), the AT domain loads a malonyl group onto CurB ACPIV, and the KS catalyzes subsequent decarboxylation to acetyl-ACPIV. HCS then catalyzes condensation of C-2 from acetyl-ACPIV and acetoacetyl-ACP3, to form (S)-HMG-ACP3 (1-ACP3). As we have shown previously, ECH1 catalyzes dehydration of 1-ACP3 to 3-methylglutaconyl-ACP3 (2-ACP3), followed by ECH2 mediated decarboxylation to generate 3-methylcrotonyl-ACP3 (3-ACP3)14, a presumed precursor for (1R, 2S)-2-methylcyclopropane-1-carboxyl-ACP3 (5-ACP3) (Fig. 1c).
This initial study raised two important questions regarding the role of the Cur and Jam HMG enzyme cassettes and ERs in formation of cyclopropane and vinyl chloride moieties: (1) How is the CurF ER involved in cyclopropyl ring formation based on its predicted function as an enoyl reductase, and is Cur ER involved in reduction of 3-ACP3 to 4-ACP3 (Fig. 1c)? (2) How is the unusual β,γ C=C of the pendant vinyl chloride group formed in the Jam pathway? As previously proposed, is 3-methyl-3-butenoyl-ACP3 (6-ACP3) generated from 3-ACP3 isomerization6, or by differential regiochemical control of double bond formation during ECH2 decarboxylation32 (Fig. 1c)?
First, we sought to test whether CurF ER can saturate 3-ACP3, the previously established product of Cur ECH214. Thus, the embedded domain was excised and cloned as an N-terminal GST-tagged fusion protein. We also overexpressed and purified the CurA ACP3 tridomain and each excised single domain (ACPI, ACPII and ACPIII) as apo proteins (Supplementary Fig. 3). 1-ACPs were generated as previously described12, and substrate loading was examined by HPLC (Supplementary Fig. 4). The ACPI, ACPII and ACPIII have nearly identical amino acid sequences, and each was efficiently loaded with the HMG substrate. Thus, for convenience we chose excised CurA ACPII, as well as ACP3, for subsequent enzyme assays.
Fourier transform ion cyclotron resonance mass spectrometry (FTICR-MS) and infrared multiphoton dissociation (IRMPD) methods were applied to detect mass changes to the HMG substrate covalently linked to the ACP phosphopantetheine (PPant) arm33. Cur ER function was assessed by coupling it with the Cur ECH1 and ECH2 reactions. As reported, Cur ECH1 catalyzed the reversible dehydration of 1-ACPII to generate 2-ACPII, and Cur ECH2 catalyzed decarboxylation of 2-ACPII to generate 3-ACPII, corresponding to 18- and 62-Dalton mass losses from 1-ACPII, respectively (Fig. 2b and 2c). Cur ECH1 shows substrate preference for (S)-HMG-ACPII over (R)-HMG-ACPII (Supplementary Fig. 5a, 5b and 5c), which is consistent with our previous results using the CoA-linked substrates14. With Cur ER and NADPH, a 2-Dalton mass addition was observed for 3-ACPII (Fig. 2d and Supplementary Fig. 6a), corresponding to saturation of the α,β enoyl thioester to generate 4-ACPII.
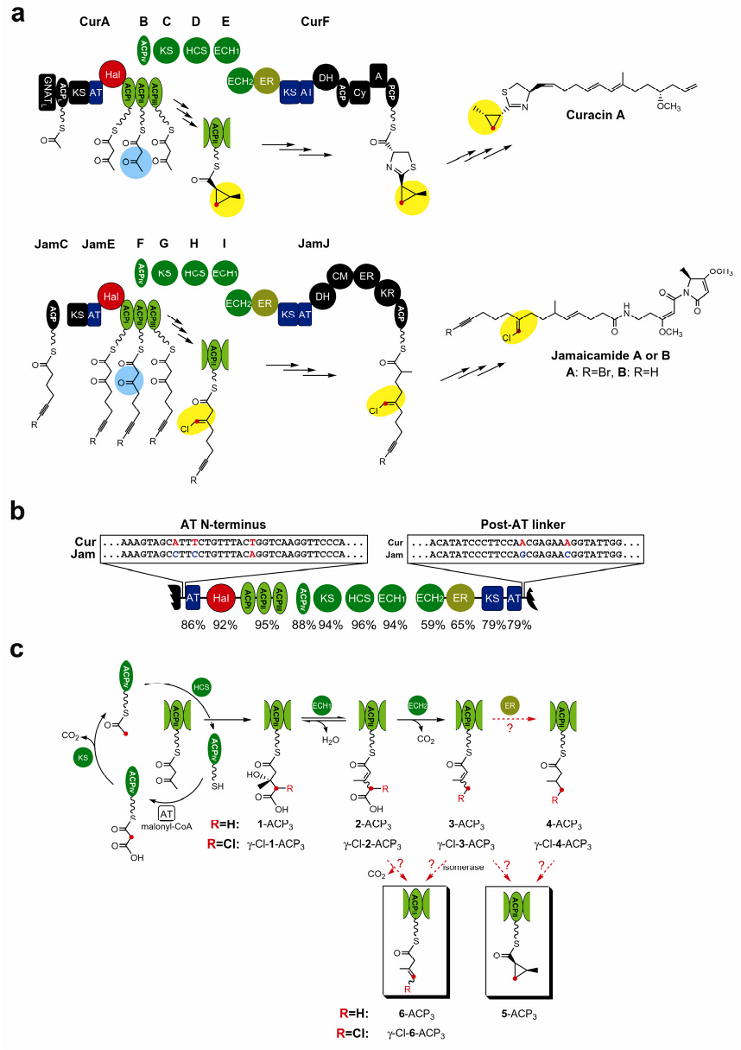
Figure 2. Halogenation and cyclopropanation in the Cur pathway.
a-h, Partial FTICR mass spectra (12+ charge state of ACPII) for Cur ECH1, ECH2 and ER reactions excluding (a-d) or including (e-h) the Cur Hal chlorination step. 1-ACPII was incubated with Cur Hal for 2 h to generate the γ-Cl-1-ACPII substrate. Reactions were incubated at 30°C for 2 h for the 1-ACPII substrate and 30 min for the γ-Cl-1-ACPII substrate. Asterisks denote unidentified species. i, GC-MS analysis of the enzyme products after butylamine cleavage, and comparison with authentic standards. For optimal sensitivity, the chromatograms were recorded at selective ion mode (SIM) by monitoring 55, 57, 83, 115, 155 and 157 atomic mass unit (amu). Retention times of the products were confirmed by coinjection with the authentic standards.
To confirm the structure of the Cur ER reaction product tethered to ACP3, we cleaved it from the PPant arm with butylamine to generate the corresponding butylamide derivative. Gas chromatography (GC)-MS analysis34 was performed and the readily separable isomers were compared and correlated with authentic standards by mass spectra and coinjection. We used 1-ACPII and 1-ACP3 as substrates for Cur ECH1, ECH2 and ER reactions, and their products were confirmed as 4 (Fig. 2i, upper trace). However, the relatively poor efficiency of Cur ER-catalyzed reduction of 3-ACPII (see below), suggested that it was unlikely to be the natural substrate. Moreover, the timing and function of Cur Hal remained to be established, and a key involvement in the β-branching scheme was hypothesized.
Halogenation and cyclopropane ring formation
Bioinformatic analysis and presence of the vinyl chloride in jamaicamide suggest that the Jam and Cur Hal domains might be α-KG-dependent non-haem halogenases19-21. In the Jam pathway, chlorination evidently occurs on the pendant carbon generated by β-branching. Thus for Cur Hal, we reasoned that cyclopropane ring formation likely involves transient halogenation as in coronatine biosynthesis, where the chloride serves as a leaving group20. However, the timing of the chlorination step remained to be established, and the identity of the Cur pathway cyclopropane ring-forming enzyme was not evident from examination of the Cur gene cluster.
An important clue about the timing of chlorination at the β-branching carbon came from previous precursor-incorporation studies in curacin A biosynthesis. NMR data on curacin A labeling by [2H3,2-13C]acetate indicated that the β-branching carbon that forms cyclopropane was labeled by only one deuterium atom (α-isotope chemical shift at C20, 0.295)18, which was previously interpreted as an anomalous result. However, these data are consistent with chlorination occurring on the HMG β-branching intermediate before ECH2 catalyzed decarboxylation. Otherwise, the pendant carbon atom would be labeled by either one or two deuterium atoms in a 2:1 ratio (Supplementary Fig. 7).
To identify the function of Cur Hal, we constructed both Hal and the tetradomain Hal-ACP3 as N-terminal His-tagged proteins (Supplementary Fig. 3). Cur Hal eluted as a dimer from an analytical size-exclusion column. Following His-tag removal by thrombin cleavage, metal content of the protein was analyzed by inductively coupled plasma (ICP)-MS. After reconstitution with a mixture of metal ions and α-KG, more than 90% of Hal was bound to Fe2+ (Supplementary Methods), which indicated that it functions as an FeII-dependent enzyme. Thus, anaerobic purification coupled with α-KG/FeII-reconstitution was performed, as was previously reported to retain optimal activities of α-KG-, O2- and FeII-dependent halogenases20-22.
Seven acyl-ACP substrates bearing the target pendant β-branching carbon were tested to establish the substrate identity for Cur Hal, including malonyl-ACPIV, acetyl-ACPIV, 1-ACPII, 2-ACPII, 3-ACPII, 4-ACPII and 6-ACPII (Fig. 1c). Consistent with the [2H3,2-13C]acetate precursor incorporation experiment noted above, we observed formation of the mono-chlorinated species exclusively on 1-ACPII to generate γ-Cl-1-ACPII. The chlorinated product was confirmed by FTICR-MS and IRMPD analysis (Fig. 2e and Supplementary Fig. 6a), and corroborated by GC-MS results (see below, Fig. 3h). As expected, Cur Hal showed the same selectivity for (S)-HMG-ACPII (1-ACPII) as Cur ECH1 (Supplementary Fig. 5a, 5b and 5c). In the absence of α-KG or O2, no chlorinated product was detected with Cur Hal in the presence of HMG substrate (Supplementary Fig. 5d). Notably, the chlorination on the carboxylated γ-carbon of HMG is unusual for α-KG-dependent non-haem halogenases, which have been previously limited to catalyzing modification of unactivated carbons atoms20-24.
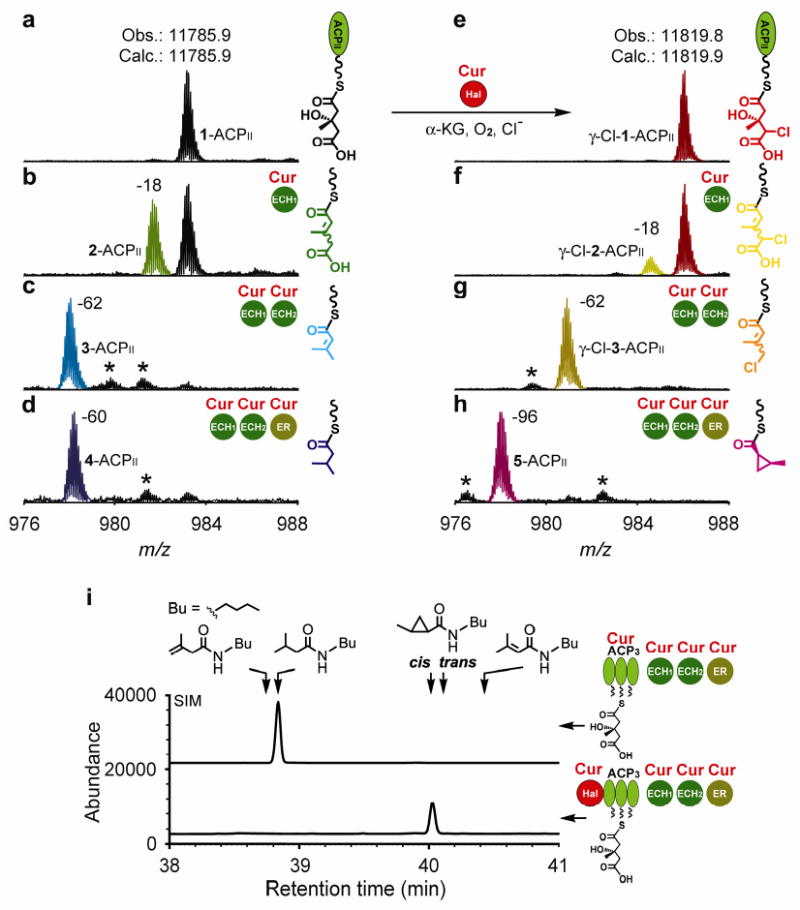
Figure 3. Comparison of ECH2s and ERs in Cur and Jam pathways.
a-f, Partial FTICR mass spectra (12+ charge state of ACPII) for Cur and Jam ECH1, ECH2 and ER reactions with the γ-Cl-1-ACPII substrate. The reactions were incubated at 30°C for 30 min. g, Comparison of catalytic efficiencies for cyclopropanation and saturation by Cur and Jam ERs. The product yields in the time-course studies were measured by IRMPD-based quantification. 3-ACPII was used as substrate for Cur ER saturation, and γ-Cl-3-ACPII was used as substrate for Cur ER cyclopropanation and Jam ER saturation. Assays were performed in triplicate, and standard deviation error bars are shown. h, GC-MS analysis to identify the structures of Cur and Jam ECH2 products. The chromatograms were recorded at SIM by monitoring 57, 117, 154 and 189 amu. The retention times of products were confirmed by coinjection with the authentic standards.
Next, we sought to investigate how chlorination of 1-ACPII affects efficiency of the downstream reaction sequence with the HMG cassette enzymes. 1-ACPII was converted to γ-Cl-1-ACPII by Cur Hal (Fig. 2e), and reacted sequentially with Cur ECH1, ECH2 and ER. ECH1 dehydrated γ-Cl-1-ACPII and the γ-Cl-2-ACPII product was decarboxylated by ECH2 to generate γ-Cl-3-ACPII (Fig. 2f and 2g). The ECH1/ECH2-coupled dehydration and decarboxylation with γ-Cl-1-ACPII was shown to be ∼4-fold faster compared to 1-ACPII (Supplementary Fig. 8), which might be due either to the electron-withdrawing effect of the γ-chlorine atom to stabilize the negative charge on the intermediate of ECH2 decarboxylation or to a more effective binding position of the chlorinated versus non-chlorinated substrate in the enzyme active sites. Unexpectedly, when Cur ER was added with Cur ECH1 and ECH2 in the presence of γ-Cl-1-ACPII, no saturation product was obtained. Instead, we observed a 34-Dalton mass reduction from γ-Cl-3-ACPII (Fig. 2h and Supplementary Fig. 6), demonstrating the elimination of chlorine in the product. This result suggests that the product could be 3-ACPII, 5-ACPII or 6-ACPII (Fig. 1c).
The experimental design to determine the final product in the presence of both Hal and HMG-cassette enzymes was streamlined by a one-pot reaction using Cur Hal-ACP3, ECH1, ECH2 and ER. Cur (apo) Hal-ACP3 was loaded from 1-CoA, desalted, and mixed with Cur ECH1, ECH2 and ER in an anaerobic environment. The reaction was initiated by exposing the mixture to air. To confirm product structure, the acyl groups linked to Hal-ACP3 were cleaved with butylamine and the butylamide derivatives were compared with the authentic standards by GC-MS. Direct correlation was confirmed by the mass spectra and coinjection with authentic standards to a single species identified as the cis-2-methylcyclopropane-1-carboxyl compound (Fig. 2i, lower trace), demonstrating the formation of 5-ACP by an unprecedented ER-catalyzed cyclopropanation reaction, presumably via an intramolecular nucleophilic substitution. The internal nucleophile, presumably the resonance-stabilized α-carbanion, is believed to be generated by ER-catalyzed hydride transfer from NADPH to the β-carbon of enoyl thioester35,36.
Functional differentiation of ERs
Due to the similarity between Cur and Jam ERs (∼65% sequence identity; higher than those between Cur/Jam ER with other PKS ERs in the Cur and Jam pathways), we sought to test whether Jam ER can catalyze the same cyclopropanation when presented with γ-Cl-3-ACPII. Likewise, Jam ER was prepared (Supplementary Fig. 3) and assayed in the same way as the Cur ER. Unexpectedly, only the saturated product, γ-Cl-4-ACPII, was observed (Fig. 3c and Supplementary Fig. 6b), indicating that its activity is typical of a canonical PKS ER.
The distinct functions of Cur and Jam ERs motivated us to compare the catalytic efficiencies of Cur ER cyclopropanation vs. Jam ER saturation of the chlorinated substrate, and the efficiencies of cyclopropanation of the chlorinated substrate vs. saturation of the non-chlorinated substrate by Cur ER. This was accomplished using time-course studies by measuring product yields under uniform reaction conditions. It was not possible to measure enzyme kinetic parameters (kcat and KM) due to the tendency of ER to aggregate and the solubility limits of ACP-tethered substrates. Thus, γ-Cl-3-ACPII was employed to assess cyclopropanation by Cur ER, compared to reduction by Jam ER, and 3-ACPII was used to compare reductive efficiency of the Cur and Jam ERs. IRMPD-based MS analysis (e.g. peak abundance of PPant ejection products (PEPs)33) provided a convenient method to quantify the yields of ER saturation products that correspond to a 2-Dalton mass change (Supplementary Fig. 9 and 10; see Supplementary Methods). Similarly, Cur ER-catalyzed cyclopropanation was quantified by preparing 4-ACPII, as an internal standard for 5-ACPII.
We found that Jam ER saturation and Cur ER cyclopropanation of γ-Cl-3-ACPII are faster by ∼400-fold and ∼50-fold, respectively, than is Cur ER saturation of 3-ACPII under identical experimental conditions (Fig. 3g). For 3-ACPII, Jam ER saturation is ∼240-fold faster than is Cur ER saturation (Supplementary Fig. 11). This comparison confirmed that Jam ER has retained canonical function as an α,β enoyl reductase, in contrast to the Cur ER as a cyclopropanase. Given the proposed hydride-transfer step for both Cur and Jam ERs, their mechanisms are likely differentiated after formation of the α-carbanion intermediate, which functions as an intramolecular nucleophile (Cur ER) or is protonated (Jam ER) (Fig. 5b).
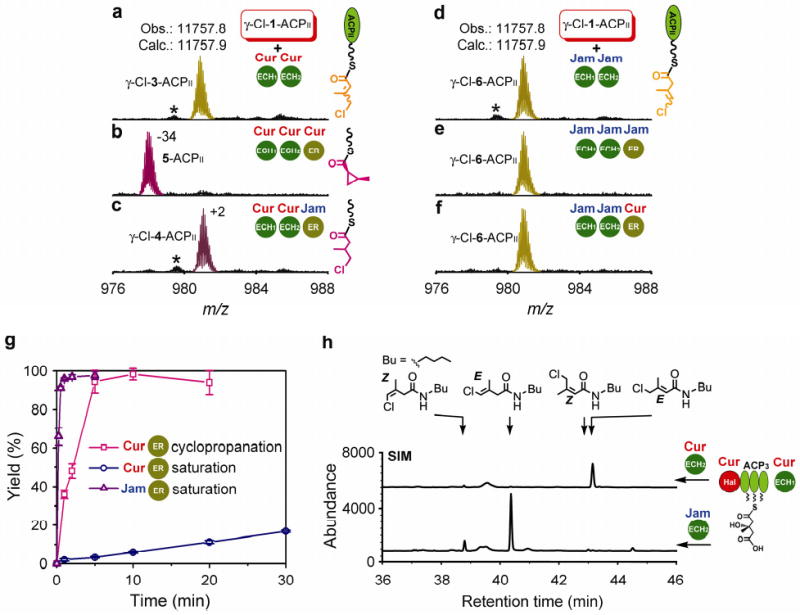
Figure 5. Impact of enzyme assembly evolution on β-branching chemical diversity.
a, Proposed ancestral forms of the enzyme assemblies in Cur and Jam pathways. b, The functional diversification of ERs. c, Differential regiochemical control by ECH2s.
Regiochemical control by ECH2s
Ascertaining the role of chlorination in cyclopropane ring formation during curacin biosynthesis strongly suggested that a similar chlorination event occurs in the Jam pathway. Given the extraordinarily high similarity between Cur and Jam Hals (92% sequence identity), we surmised that the two pathways diverge after the halogenation step, resulting in differential catalytic processes leading to the vinyl chloride moiety in jamaicamides. In the two high similar enzyme assemblies, the Cur and Jam ECH2 domains have lowest sequence identities (59%) (Fig. 1b), and likely function as a key branch-point determinant. Accordingly, Jam ECH2 was prepared in the same way as the Cur ECH2 (Supplementary Fig. 3). We also expressed and purified Jam ECH1 to generate substrate for Jam ECH2. The function of these Jam enzymes were subsequently investigated with Cur substrates starting from 1-ACP or γ-Cl-1-ACPII to establish what controls introduction of the β,γ vinyl chloride group.
For both 1-ACPII and γ-Cl-1-ACPII substrates, Jam ECH1 and ECH2 catalyzed successive dehydration and decarboxylation steps as expected (Fig. 3d, Supplementary Fig. 12b, 12c, 12f, and 12g). However, when Jam ER was added, only ∼20% of the saturated product was detected for the non-chlorinated substrate (derived from 1-ACPII, Supplementary Fig. 12i). No mass change was observed for the corresponding chlorinated substrate (derived from γ-Cl-1-ACPII, Supplementary Fig. 12j), indicating that the Jam ECH2 product is not a substrate for Jam ER. In addition, the ECH1s were switched in the Cur and Jam ECH1/ECH2/ER coupled reactions with γ-Cl-1-ACPII substrate, and no change in the product profile was observed, suggesting that both Cur and Jam ECH1s generate the same product (γ-Cl-2-ACPII), consistent with their 94% sequence identity. Thus, the Jam ECH2-catalyzed decarboxylation product of γ-Cl-2-ACPII was predicted to be γ-Cl-6-ACPII (β,γ C=C; Fig. 3d) with a vinyl chloride group, instead of γ-Cl-3-ACPII (α,β C=C; Fig. 3a). UV spectral comparison of Cur and Jam ECH2 decarboxylation products revealed that their UV absorption patterns are slightly different between 250 and 280 nm (Supplementary Fig. 13b), which reflects isomeric α,β or β,γ C=C (enoyl thioester) functionality in the molecules.
To determine the structures of the decarboxylation products, one-pot reactions using Cur Hal-ACP3, Cur ECH1 and Cur or Jam ECH2s, and GC-MS analysis were performed as described above. For the reaction including Cur ECH2, the main product contained primarily an α,β C=C in the E configuration, with trace amounts of the β,γ C=C isomer (Fig. 3h, upper trace) quantified to be ∼3% (see below, Fig. 4b). In contrast, reactions using Jam ECH2 showed high regiochemical control to generate exclusively the β,γ C=C product, with ∼85% in the E configuration and ∼15% of the Z isomer (Fig. 3h, lower trace). The exclusive E configuration of the vinyl chloride C=C in jamaicamide natural products16 suggests that the small amount of Z double bond product is likely due to utilization of the curacin substrate, which is less sterically hindered than the jamaicamide substrate (Fig. 1a). Notably, Jam ECH2 decarboxylation had lower regiochemical control using the non-chlorinated substrate, and generated ∼80% β,γ C=C and ∼20% α,β C=C products, which further explains the partial enoyl reduced product observed following Jam ECH1, ECH2 and ER reactions with this substrate (Supplementary Fig. 12i). Given the normal function of ER to catalyze only α,β C=C (enoyl thioester) saturation, the selective formation of β,γ C=C product by Jam ECH2 renders Jam ER superfluous in the biosynthesis of jamaicamides. In general, α,β C=C ECH2 products are energetically preferred and frequently identified or predicted in other pathways15,27,37-40, except the pathways of pederin and its structural analogs41,42, which are predicted to generate β,γ C=C products (Supplementary Fig. 14a).
Figure 4. Loss of Cur ECH2-mediated regiochemical control by site-directed mutagenesis.
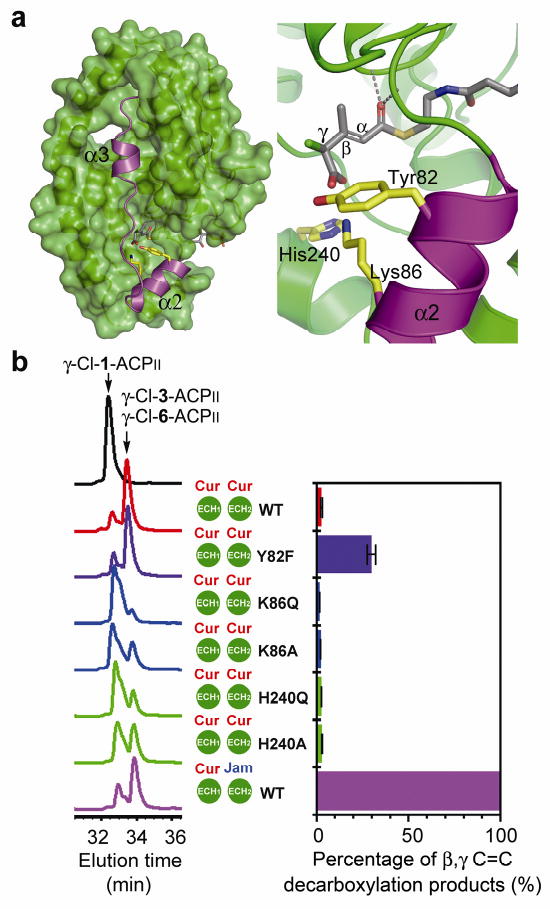
a, The hypervariable region (in magenta) of Cur ECH2 and the active site chamber modeled with the chlorinated substrate. The S-configuration of the HMG γ-carbon is preferred based on modeling results. b, Activity and regiochemical control of ECH2 WT and Cur ECH2 mutants. γ-Cl-1-ACPII was used as the substrate for all reactions. (Left) HPLC analysis for ECH1/ECH2 coupled dehydration and decarboxylation. All reactions were quenched after 10 min incubation at 30°C. (Right) IRMPD-based quantification to measure the percentage of β,γ C=C products. The coupled ECH1/ECH2 reactions were incubated for 45 min before treated with Jam ER for 45 min at 30°C. Assays were performed in triplicate, and standard deviation error bars are shown.
Loss of regiochemical control by mutation
To understand the regiochemical control of ECH2-catalyzed decarboxylation, the previously solved Cur ECH2 structure32 was modeled with the chlorinated substrate (Fig. 4a). The catalytic efficiencies of the wild type (WT) and mutants of Cur ECH2 were compared by performing the ECH1/ECH2 coupled assay with γ-Cl-1-ACPII substrate. Moreover, we measured the ratios of the two possible decarboxylation products (α,β and β,γ C=C), γ-Cl-3-ACPII and γ-Cl-6-ACPII.
Based on our results, the catalytic activities of WT and mutant Cur ECH2s were significantly increased with the chlorinated substrate, possibly due to γ-Cl stabilization of the carbanion intermediate (Fig. 4b, left panel). However, their relative catalytic activities are similar to our previous results for the non-chlorinated substrate32. Cur ECH2 Y82F and Jam ECH2 WT had activities close to Cur ECH2 WT, indicating that Tyr82 is not essential for decarboxylation.
Next, we measured the ratio of α,β and β,γ C=C decarboxylation products to investigate whether the site-directed mutations can elucidate a basis for double bond regiochemical control by Cur ECH2. Changes in the ratio of α,β and β,γ C=C products were assessed by measuring UV absorbance ratios (A280nm/A250nm, Supplementary Fig. 13) for HPLC peaks corresponding to ECH2 decarboxylation products (Fig. 4b). Measured peak ratios for Cur ECH2 WT, K86Q, K86A, H240Q and H240A are ∼1.75, for Jam ECH2 WT the ratio is 2.23, but for Cur ECH2 Y82F it is 1.85. The intermediate value for Cur ECH2 Y82F suggests a mixture of α,β and β,γ C=C products. These products can be distinguished directly by using Jam ER as a reagent to selectively reduce α,β C=C (Fig. 3c) followed by IRMPD to quantify product ratios (Supplementary Fig. 9c). The level of β,γ C=C product (γ-Cl-6-ACPII) for Cur ECH2 WT, K86Q, K86A, H240Q and H240A was ∼3% of the total product formed, but was ∼30% of total product generated by Cur ECH2 Y82F (Fig. 4b). Based on the site-directed mutagenesis results, positioning of the Tyr82 hydroxyl group seems crucial for regioselectivity (α or γ position, Fig. 4a) of the protonation step following collapse of the presumed enolate intermediate (Fig. 5c). The Tyr82 resides in a hypervariable region (Fig. 4a, α2–loop–α3, in magenta) and is a non-conserved residue for ECH2 enzymes32. Our results suggest that ECH2 regiochemical control might be easily affected by mutations that occur in this hypervariable region, thus serving as a facile strategy to introduce functional group diversification.
Discussion
The Cur and Jam pathways enable us to witness the remarkable process of evolutionary diversification in secondary metabolism based on comparative biochemical analysis of two parallel β-branching enzyme assemblies (Fig. 5a). DNA duplications and insertions in natural product pathway evolution are readily identified in these systems, but the mutations in enzyme assemblies leading to generation of chemical diversity require direct analysis to elucidate function. Here we show at the biochemical level that subtle changes in amino acid sequences of only two members (e.g. ECH2 and ER) of the 10 component β-branching enzyme system are ultimately responsible for distinct chemical outcomes.
Both Cur and Jam enzyme assemblies contain Hal domains that were evidently recruited and embedded in a modular PKS to impart new chemical diversity. Recent studies on this class of α-KG dependent non-haem halogenases have been reported as discrete enzymes in secondary metabolite pathways6,20-23, but this integrated domain represents an unprecedented example of pathway diversification. Cur and Jam are further diversified by the amino acid sequence variation in downstream enzymes to yield different catalytic activities. Specifically, the Cur ER domain was shown to be a cyclopropanase catalyzing nucleophilic displacement of the chlorine atom leading to a highly strained and unusual functional group (Fig. 5b). In contrast, the Jam ER domain was found to retain reductase function for the curacin α,β enoylthioester substrate, but is inactive against the corresponding β,γ enoylthioester isomer. Thus, in addition to the cyclopropanation strategies of Zn2+-dependent CmaC20,43 and the recently reported FAD-dependent dehydrogenase KtzA44, where chloride also serves as a leaving group, the NADPH dependent Cur ER-catalyzed cyclopropanation represents a new strategy for generating a thioester enolate and subsequent ring formation. Structural insights to reveal the sequence variations of Cur ER that stabilize the α-carbanion while supporting closure of the highly strained cyclopropane is key to understanding its functional evolution. Moreover, regiochemical control is presumed to be the result of a protonation step accompanying enolate collapse after ECH2-mediated decarboxylation (Fig. 5c). Thus, further pathway diversification is reflected in select amino acid sequence changes that direct alternative double bond regiochemistry in the jamaicamide products. These parallel yet distinct systems demonstrate the mutability of enzymes within complex metabolic pathways, and reveal their metamorphic properties for creating chemical diversity in biologically active natural products.
Methods
Chemicals
1-CoA and 2-CoA were enzymatically generated using HMG reductase14,45 and Cur ECH214. 6-CoA and the butylamide derivatives were synthesized as described in Supplementary Methods. (1R,2S)-2-methylcyclopropanecarboxylic acid was a gift from Timothy M. Ramsey (Novartis Institutes for Biomedical Research, Inc.). All other chemicals were from Sigma-Aldrich.
Construction of plasmids and overexpression and purification of proteins
The expression plasmids for Cur ACPIV, Cur ECH1 and Cur ECH2 were constructed in our previous work14. The expression plasmids for Cur ACP3, ACPI, ACPII and ACPIII were gifts from Christopher T. Walsh (Harvard Medical School). Cur Hal, Hal-ACP3 and ER genes were amplified from the cosmid pLM5418. Jam ECH1, ECH2 and ER genes were amplified from cosmid pJam316. The primers for the plasmid construction are listed in Supplementary Table 1. His-tagged proteins were expressed in E. coli BL21 (DE3) transformed with the corresponding plasmids.
His-tagged Cur and Jam ECH1s, ECH2s and ERs, as well as Cur ACP constructs were purified using Ni-nitrilotriacetate (Ni-NTA) HisTrap column followed by desalting or gel-filtration. The purification of Cur Hal and Hal-ACP3 was performed under inert atmosphere by using an ÄKTA FPLC (GE Healthcare) with its tubing linked to a glove box (Coy Laboratory Products), which is similar to the system previously described20,21,46. The Hal N-terminal His-tag was removed by thrombin for subsequent metal content analysis. See Supplementary Methods for detailed protocols.
Metal content analysis of Cur Hal
His-tag cleaved Cur Hal was anaerobically reconstituted with 1 mM α-KG and Fe2+ (or a metal mixture). The metal content of Cur Hal was measured by ICP-MS (Finnigan).
Cur Hal functional assays
1-ACPII was served as the substrate for Hal catalytic activity assays. Typically, ∼200 μl of reaction mixture containing 50 μM 1-ACPII, 5 μM Cur Hal, 50 μM fresh Fe(NH4)2(SO4)2, and 0.5 mM α-KG in 50 mM Tris-HCl buffer (pH 7.5) was prepared in a glove box. The reaction was initiated by exposing the mixture to air, and incubated at 30°C for 2 h to achieve full conversion to γ-Cl-1-ACPII. The product was detected by FTICR-MS and IRMPD as described in Supplementary Methods.
Cur and Jam ECH1/ECH2 functional assays
1-ACPII or γ-Cl-1-ACPII was served as the substrate for ECH1 and ECH2 assays. Typically, ∼50 μM 1-ACPII or γ-Cl-1-ACPII was incubated with 1 μM ECH1 or ECH1/ECH2 in 50 mM Tris-HCl buffer (pH 7.5) at 30°C. The reactions were examined by reverse-phase HPLC, and the products were detected by FTICR-MS and IRMPD.
Cur and Jam ER functional assays
Typically, ER reactions were performed by incubating ∼50 μM γ-Cl-3-ACPII or 3-ACPII with 1 μM ER and 0.5 mM NADPH in 50 mM Tris-HCl buffer (pH 7.5) at 30°C. Alternatively, the ER reaction was coupled with ECH1/ECH2 dehydration and decarboxylation. The products were analyzed by FTICR-MS and IRMPD.
One-pot reactions and GC-MS analysis
Each one-pot reaction was performed by incubating ∼50 μM ACP3 or Hal-ACP3 loaded with 1-CoA, and ∼10 μM Cur and/or Jam enzymes with the corresponding cofactors in 50 mM Tris-HCl buffer (pH 7.5) at 30°C for 5 min. The reactions were initiated by exposing the reaction mixture to air. The products were cleaved from Cur ACP3 and Hal-ACP3 PPant arms by butylamine aminolysis to generate the butylamide derivatives that were subsequently analyzed by GC/MS34 and compared with the authentic standards for structure determination. See Supplementary Methods for detailed protocols.
Analysis of regiochemical control by ECH2 WT and mutants
The ratios of α,β and β,γ C=C products of the ECH2 decarboxylation were measured for Cur ECH2 WT and mutants, and Jam ECH2 WT by a coupled ECH1/ECH2 dehydration and decarboxylation assay. 50 μM γ-Cl-1-ACPII was incubated with 2 μM Cur ECH1 and 2 μM Cur or Jam ECH2 in 50 mM Tris-HCl buffer (pH 7.5) at 30°C for 45 min. The reaction mixtures were treated with 2 μM Jam ER and subjected to IRMPD-based quantification as described Supplementary Methods.
Supplementary Material
Acknowledgments
We thank C. T. Walsh and C. T. Calderone for ACP constructs; S. M. Chernyak, H. Liu and J. Byun for mass spectrometry assistance; P. C. López for NMR assistance; T.M. Ramsey for chiral cyclopronanecarboxylic acid; D. L. Akey for discussion; This work was supported by grants from the National Institutes of Health (to D.H.S. and J.L.S.), a graduate fellowship from Eli Lilly & Co. and a Rackham Predoctoral Fellowship (to L.G.).
Footnotes
Author Contribution L.G., W.H.G and D.H.S. designed the experiments, analyzed data and wrote the paper; L.G. performed the experiments; B.W. and K.H. recorded FTICR mass spectra and analyzed the data; T.W.G. and J.L.S. modeled Cur ECH2 structure with the chlorinated substrate and designed site mutagenesis; A.K. and P.W. synthesized the chlorinated butylamide derivatives; R.V.G. and L.G. made Jam ECH1 and ECH2 constructs; W.H.G. provided DNA of Jam enzymes and analyzed NMR data for isotope-labeled curacin A.
Author Information The authors declare no competing financial interests.
Supplementary Information is linked to the online version of the paper at www.nature.com/nature.
References
- 1.Hartmann T. Diversity and variability of plant secondary metabolism: A mechanistic view. Entomol Exp Appl. 1996;80:177–188. [Google Scholar]
- 2.Firn RD, Jones CG. Natural products - a simple model to explain chemical diversity. Nat Prod Rep. 2003;20:382–391. doi: 10.1039/b208815k. [DOI] [PubMed] [Google Scholar]
- 3.Jenke-Kodama H, Mueller R, Dittmann E. Evolutionary mechanisms underlying secondary metabolite diversity. Prog Drug Res. 2008;119:121–140. doi: 10.1007/978-3-7643-8117-2_3. [DOI] [PubMed] [Google Scholar]
- 4.Fischbach MA, Walsh CT, Clardy J. The evolution of gene collectives: How natural selection drives chemical innovation. Proc Natl Acad Sci USA. 2008;105:4601–4608. doi: 10.1073/pnas.0709132105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Haslam E. Secondary metabolism - evolution and function: products or processes? Chemoecology. 1994;5:89–95. [Google Scholar]
- 6.Vaillancourt FH, Yeh E, Vosburg DA, Garneau-Tsodikova S, Walsh CT. Nature's inventory of halogenation catalysts: Oxidative strategies predominate. Chem Rev. 2006;106:3364–3378. doi: 10.1021/cr050313i. [DOI] [PubMed] [Google Scholar]
- 7.Fischbach MA, Clardy J. One pathway, many products. Nat Chem Biol. 2007;3:353–355. doi: 10.1038/nchembio0707-353. [DOI] [PubMed] [Google Scholar]
- 8.Haslam E. Secondary Metabolism - Fact and Fiction. Nat Prod Rep. 1986;3:217–249. [Google Scholar]
- 9.Austin MB, O'Maille PE, Noel JP. Evolving biosynthetic tangos negotiate mechanistic landscapes. Nat Chem Biol. 2008;4:217–222. doi: 10.1038/nchembio0408-217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Smith JL, Sherman DH. Biochemistry - An enzyme assembly line. Science. 2008;321:1304–1305. doi: 10.1126/science.1163785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Khosla C, Gokhale RS, Jacobsen JR, Cane DE. Tolerance and specificity of polyketide synthases. Annu Rev Biochem. 1999;68:219–253. doi: 10.1146/annurev.biochem.68.1.219. [DOI] [PubMed] [Google Scholar]
- 12.Moore BS, Hertweck C. Biosynthesis and attachment of novel bacterial polyketide synthase starter units. Nat Prod Rep. 2002;19:70–99. doi: 10.1039/b003939j. [DOI] [PubMed] [Google Scholar]
- 13.Fischbach MA, Walsh CT. Assembly-line enzymology for polyketide and nonribosomal peptide antibiotics: Logic, machinery, and mechanisms. Chem Rev. 2006;106:3468–3496. doi: 10.1021/cr0503097. [DOI] [PubMed] [Google Scholar]
- 14.Gu LC, et al. Metabolic coupling of dehydration and decarboxylation in the curacin A pathway: Functional identification of a mechanistically diverse enzyme pair. J Am Chem Soc. 2006;128:9014–9015. doi: 10.1021/ja0626382. [DOI] [PubMed] [Google Scholar]
- 15.Calderone CT, Kowtoniuk WE, Kelleher NL, Walsh CT, Dorrestein PC. Convergence of isoprene and polyketide biosynthetic machinery: Isoprenyl-S-carrier proteins in the pksX pathway of Bacillus subtilis. Proc Natl Acad Sci USA. 2006;103:8977–8982. doi: 10.1073/pnas.0603148103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Edwards DJ, et al. Structure and biosynthesis of the jamaicamides, new mixed polyketide-peptide neurotoxins from the marine cyanobacterium Lyngbya majuscula. Chem Biol. 2004;11:817–833. doi: 10.1016/j.chembiol.2004.03.030. [DOI] [PubMed] [Google Scholar]
- 17.Verdier-Pinard P, et al. Structure-activity analysis of the interaction of curacin A, the potent colchicine site antimitotic agent, with tubulin and effects of analogs on the growth of MCF-7 breast cancer cells. Mol Pharmacol. 1998;53:62–76. doi: 10.1124/mol.53.1.62. [DOI] [PubMed] [Google Scholar]
- 18.Chang Z, et al. Biosynthetic pathway and gene cluster analysis of curacin A, an antitubulin natural product from the tropical marine cyanobacterium Lyngbya majuscula. J Nat Prod. 2004;67:1356–1367. doi: 10.1021/np0499261. [DOI] [PubMed] [Google Scholar]
- 19.Blasiak LC, Vaillancourt FH, Walsh CT, Drennan CL. Crystal structure of the non-haem iron halogenase SyrB2 in syringomycin biosynthesis. Nature. 2006;440:368–371. doi: 10.1038/nature04544. [DOI] [PubMed] [Google Scholar]
- 20.Vaillancourt FH, Yeh E, Vosburg DA, O'Connor SE, Walsh CT. Cryptic chlorination by a non-haem iron enzyme during cyclopropyl amino acid biosynthesis. Nature. 2005;436:1191–1194. doi: 10.1038/nature03797. [DOI] [PubMed] [Google Scholar]
- 21.Vaillancourt FH, Yin J, Walsh CT. SyrB2 in syringomycin E biosynthesis is a nonheme FeII alpha-ketoglutarate- and O2-dependent halogenase. Proc Natl Acad Sci USA. 2005;102:10111–10116. doi: 10.1073/pnas.0504412102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Galonic DP, Vaillancourt FH, Walsh CT. Halogenation of unactivated carbon centers in natural product biosynthesis: Trichlorination of leucine during barbamide biosynthesis. J Am Chem Soc. 2006;128:3900–3901. doi: 10.1021/ja060151n. [DOI] [PubMed] [Google Scholar]
- 23.Chang Z, et al. The barbamide biosynthetic gene cluster: a novel marine cyanobacterial system of mixed polyketide synthase (PKS)-non-ribosomal peptide synthetase (NRPS) origin involving an unusual trichloroleucyl starter unit. Gene. 2002;296:235–247. doi: 10.1016/s0378-1119(02)00860-0. [DOI] [PubMed] [Google Scholar]
- 24.Flatt PM, et al. Characterization of the initial enzymatic steps of barbamide biosynthesis. J Nat Prod. 2006;69:938–944. doi: 10.1021/np050523q. [DOI] [PubMed] [Google Scholar]
- 25.Galonic DP, Barr EW, Walsh CT, Bollinger JM, Krebs C. Two interconverting Fe(IV) intermediates in aliphatic chlorination by the halogenase CytC3. Nat Chem Biol. 2007;3:113–116. doi: 10.1038/nchembio856. [DOI] [PubMed] [Google Scholar]
- 26.Krebs C, Fujimori DG, Walsh CT, Bollinger JM. Non-heme Fe(IV)-oxo intermediates. Acc Chem Res. 2007;40:484–492. doi: 10.1021/ar700066p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Calderone CT, Iwig DF, Dorrestein PC, Kelleher NL, Walsh CT. Incorporation of nonmethyl branches by lsoprenoid-like logic: Multiple beta-alkylation events in the biosynthesis of myxovirescin A1. Chem Biol. 2007;14:835–846. doi: 10.1016/j.chembiol.2007.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nordling E, Jornvall H, Persson B. Medium-chain dehydrogenases/reductases (MDR) - Family characterizations including genome comparisons and active site modelling. Eur J Biochem. 2002;269:4267–4276. doi: 10.1046/j.1432-1033.2002.03114.x. [DOI] [PubMed] [Google Scholar]
- 29.Tang YY, Kim CY, Mathews II, Cane DE, Khosla C. The 2.7-angstrom crystal structure of a 194-kDa homodimeric fragment of the 6-deoxyerythronolide B synthase. Proc Natl Acad Sci USA. 2006;103:11124–11129. doi: 10.1073/pnas.0601924103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jenke-Kodama H, Borner T, Dittmann E. Natural biocombinatorics in the polyketide synthase genes of the actinobacterium Streptomyces avermitilis. PloS Comput Biol. 2006;2:1210–1218. doi: 10.1371/journal.pcbi.0020132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ridley CP, Lee HY, Khosla C. Evolution of polyketide synthases in bacteria. Proc Natl Acad Sci USA. 2008;105:4595–4600. doi: 10.1073/pnas.0710107105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Geders TW, et al. Crystal structure of the ECH2 catalytic domain of CurF from Lyngbya majuscula - Insights into a decarboxylase involved in polyketide chain beta-branching. J Biol Chem. 2007;282:35954–35963. doi: 10.1074/jbc.M703921200. [DOI] [PubMed] [Google Scholar]
- 33.Dorrestein PC, et al. Facile detection of acyl and peptidyl intermediates on thiotemplate carrier domains via phosphopantetheinyl elimination reactions during tandem mass spectrometry. Biochemistry. 2006;45:12756–12766. doi: 10.1021/bi061169d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kopka J, Ohlrogge JB, Jaworski JG. Analysis of in-Vivo Levels of Acyl-Thioesters with Gas-Chromatography Mass-Spectrometry of the Butylamide Derivative. Anal Biochem. 1995;224:51–60. doi: 10.1006/abio.1995.1007. [DOI] [PubMed] [Google Scholar]
- 35.Quemard A, et al. Enzymatic characterization of the target for isoniazid in Mycobacterium tuberculosis. Biochemistry. 1995;34:8235–8241. doi: 10.1021/bi00026a004. [DOI] [PubMed] [Google Scholar]
- 36.Khosla C, Tang YY, Chen AY, Schnarr NA, Cane DE. Structure and mechanism of the 6-deoxyerythronolide B synthase. Annu Rev Biochem. 2007;76:195–221. doi: 10.1146/annurev.biochem.76.053105.093515. [DOI] [PubMed] [Google Scholar]
- 37.Butcher RA, et al. The identification of bacillaene, the product of the PksX megacomplex in Bacillus subtilis. Proc Natl Acad Sci USA. 2007;104:1506–1509. doi: 10.1073/pnas.0610503104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.El-Sayed AK, et al. Characterization of the mupirocin biosynthesis gene cluster from Pseudomonas fluorescens NCIMB 10586. Chemistry & Biology. 2003;10:419–430. doi: 10.1016/s1074-5521(03)00091-7. [DOI] [PubMed] [Google Scholar]
- 39.Simunovic V, et al. Myxovirescin A biosynthesis is directed by hybrid polyketide synthases/nonribosomal peptide synthetase, 3-hydroxy-3-methylglutaryl-CoA synthases, and trans-acting acyltransferases. Chembiochem. 2006;7:1206–1220. doi: 10.1002/cbic.200600075. [DOI] [PubMed] [Google Scholar]
- 40.Pulsawat N, Kitani S, Nihira T. Characterization of biosynthetic gene cluster for the production of virginiamycin M, a streptogramin type A antibiotic, in Streptomyces virginiae. Gene. 2007;393:31–42. doi: 10.1016/j.gene.2006.12.035. [DOI] [PubMed] [Google Scholar]
- 41.Piel J. A polyketide synthase-peptide synthetase gene cluster from an uncultured bacterial symbiont of Paederus beetles. Proc Natl Acad Sci USA. 2002;99:14002–14007. doi: 10.1073/pnas.222481399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Piel J, et al. Antitumor polyketide biosynthesis by an uncultivated bacterial symbiont of the marine sponge Theonella swinhoei. Proc Natl Acad Sci USA. 2004;101:16222–16227. doi: 10.1073/pnas.0405976101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kelly WL, et al. Characterization of the aminocarboxycyclopropane-forming enzyme CmaC. Biochemistry. 2007;46:359–368. doi: 10.1021/bi061930j. [DOI] [PubMed] [Google Scholar]
- 44.Neumann CS. & Walsh C.T. Biosynthesis of (-)-(1S,2R)-allocoronamic acyl thioester by an Fe(II)-dependent halogenase and a cyclopropane-forming flavoprotein. J Am Chem Soc. 2008 doi: 10.1021/ja8064667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bischoff KM, Rodwell VW. Biosynthesis and Characterization of (S) and (R)-3-Hydroxy-3-Methylglutaryl Coenzyme-A. Biochem Med Metab Biol. 1992;48:149–158. doi: 10.1016/0885-4505(92)90060-c. [DOI] [PubMed] [Google Scholar]
- 46.Vaillancourt FH, Han S, Fortin PD, Bolin JT, Eltis LD. Molecular basis for the stabilization and inhibition of 2,3-dihydroxybiphenyl 1,2-dioxygenase by t-butanol. J Biol Chem. 1998;273:34887–34895. doi: 10.1074/jbc.273.52.34887. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.