

Abstract
There is increasing evidence that Prolactin (PRL), a hormone/cytokine, plays a role in breast, prostate and colorectal cancers via local production or accumulation. Elevated levels of serum PRL in ovarian and endometrial cancers have been reported indicating a potential role for prolactin in endometrial and ovarian carcinogenesis. In this study, we demonstrate that serum PRL levels are significantly elevated in women with a strong family history of ovarian cancer. We demonstrate dramatically increased expression of PRL receptor (PRLR) in ovarian and endometrial tumors as well as in endometrial hyperplasia signifying the importance of PRL signaling in malignant and premalignant conditions. PRL mRNA was expressed in ovarian and endometrial tumors indicating the presence of an autocrine loop. PRL potently induced proliferation in several ovarian and endometrial cancer cell lines. Binding of PRL to its receptor was followed by rapid phosphorylation of ERK1/2, MEK-1, STAT3, CREB, ATF-2, and p53, and activation of 37 transcription factors in ovarian and endometrial carcinoma cells. PRL also activated Ras oncogene in these cells. When human immortalized normal ovarian epithelial (NOE) cells were chronically exposed to PRL a malignant transformation occurred manifested by the acquired ability of transformed cells to form clones, grow in soft agar, and form tumors in SCID-beige mice. Transformation efficiency was diminished by a Ras inhibitor providing proof that PRL-induced transformation utilizes the Ras pathway. In summary, we present findings that indicate an important role for PRL in ovarian and endometrial tumorigenesis. PRL may represent a risk factor for ovarian and endometrial cancers.
Introduction
Prolactin (PRL) is a 23 kD protein that has a dual function – as a circulating hormone and as a cytokine. PRL is reportedly involved in more than 300 separate functions including development of the mammary gland, lactation, implantation and pregnancy, angiogenesis, and regulation of immune function (reviewed in (1)). PRL is secreted by the pituitary gland and by multiple non-pituitary sites including human ovarian follicular cells, decidualized stromal cells of the human endometrium, and normal peripheral blood lymphocytes (reviewed in (2)). The synthesis of extrapituitary PRL is driven by a different promoter than its pituitary counterpart (3) although the amino acid structure of pituitary and extrapituitary PRL appears to be identical (4).
PRL initiates signaling by binding to its cognate receptor, PRLR. The proximal transduction pathways activated during PRLR-associated signaling include the tyrosine kinases Jak2, Fyn and Tec, the phosphatase SHP-2, the guanine nucleotide exchange factor Vav, and the signaling suppressor SOCS (5). In addition, as demonstrated in rat GH4 neuroendocrine cells, the Ras/Raf/MAP kinase pathway is also activated by PRL and may be involved in the proliferative effects of the hormone (6).
There is increasing evidence that PRL plays a causal role in several types of cancer via local production or accumulation. In rodents, hyperprolactinemia correlates with increased mammary tumorigenesis (7, 8), and PRL administration induces mammary tumors and sustains carcinogen-induced tumor growth (9). Dopamine is the physiological inhibitor of PRL production. Recently, in a large retrospective cohort study, the use of dopamine antagonists (antipsychotics) resulted in a 16% increase in breast cancer, with a dose-response relationship between cumulative doses and greater risk (10). In a nested case-control study within the prospective Nurses' Health Study cohort a significant positive association was observed between plasma levels of PRL and postmenopausal breast cancer risk (11, 12). Association of elevated PRL serum levels with prostate cancer has also been documented (13).
Elevated PRL levels were recently reported in the sera of patients with ovarian (14) and endometrial (15) cancers. We hypothesize that elevated serum PRL could be a risk factor for ovarian and endometrial cancers. We propose a mechanism by which PRL promotes tumorigenesis by activating Ras oncogene thus inducing malignant transformation of cells carrying mutations in tumor suppressor genes. We test this hypothesis by analyzing the differential expression of and response to the PRL/PRLR axis in human ovarian and endometrial carcinoma cells.
Materials and Methods
Patient population
Sera from 167 patients with endometrial cancer were provided by Dr. Karen Lu (MD Anderson Cancer Center) and Gynecologic Oncology Group Blood Bank (Columbus, OH). Sera from 273 patients with endometrial cancer, 215 patients with stage I-II and 118 patients with stage III-IV ovarian cancer, 141 patients with benign pelvic disease, and 470 healthy controls were obtained from the Gynecologic Oncology Group (GOG) Blood Bank. Sera from patients with high risk of developing ovarian cancer were from Drs. Godwin (n=20) and Fishman (n=94). Sera of age-matched women with lung cancer (n=75) were provided by UPCI, Pittsburgh, PA (Dr. Jill Siegfried), sera from patients with breast cancer (n=219) were from Duke Medical Center (Dr. Jeffrey Marks), and sera from women with pancreatic cancer (n=101) were provided by Drs. Herbert Zeh and Randall Brand (UPCI). All sera were collected during day time prior to surgery and anesthesia according to the same protocol.
Cell lines
Human endometrial carcinoma cancer cell lines, HEC-1A, AN3 CA, and RL 95-2, and human ovarian carcinoma cell lines, OVCAR3 and SKOV3, were obtained from the American Type Culture Collection (ATCC, Rockville, MD, USA). Immortalized human normal ovarian epithelial cells, T29 and T80, were provided by Dr. Jinsong Liu, and immortalized non-cancerous endometrial fibroblasts were provided by Dr. Gil Mor.
Reagents
Cisplatin and Hoechst 33342 were purchased from Sigma-Aldrich (St. Louis, MO). Mouse monoclonal antibodies against human PRL (catalog no. 53797) and PRLR (catalog no. 53799) were from AnaSpec (San Jose, CA), and Ras inhibitor, α-hydroxy farnesyl phosphonic acid was from Cayman Chemicals (Ann Arbor, MI). Cancer and normal tissue microarrays (TMAs) were purchased from US Biomax, Inc. (Rockville, MD). The ovarian cancer TMA contained 5 sections of clear cell, 49 of endometrioid, 25 of mucinous, and 50 of serous adenocarcinoma (grades I-III), and 8 of healthy ovaries. The endometrial cancer TMA contained cores from 25 endometrial adenocarcinoma, 19 cores of endometrial hyperplasia, 3 cores from healthy secretory endometrium, and 3 cores of healthy proliferative endometrium.
Immunohistochemical (IHC) staining of formalin fixed paraffin embedded (FFPE) tissue microarray slides
FFPE sections were deparaffinized and rehydrated in xylene, ethanol, and H2O. Heat induced antigen retrieval was performed using EDTA buffer, pH8.0. After unmasking, slides were blocked with H2O2, and EnVision+ System (Dako, Carpinteria, CA) was used for staining as follows. Non-specific binding was blocked with a serum-free protein blocker, and sections were incubated with mouse anti-human prolactin receptor Ab for 30 min followed by incubation with labeled polymer-horseradish peroxidase (HRP) anti-mouse Ab and then with DAB-3,3′-diaminobenzidin. Sections were counterstained with Meyers hematoxylin and mounted in mounting medium. In negative control primary Ab was omitted.
RT-PCR analysis
Total RNA was isolated from normal pituitary tissue, normal endometrial and ovarian tissue, normal tissue adjacent to endometrial and ovarian tumor, and endometrial and ovarian tumor using RNAzol procedure (Ambion, Austin, TX). The normal ovarian total RNA represented a pool of 5 individuals, all other samples were from single donors. RT-PCR was performed according to the method developed by Shaw-Bruha et al. (16). The protocol utilized sense primers specific for pituitary specific proximal (P, 5′–ATCAAAGGATCGCCATGGAAAGG-3′) promoter and extrapituitary distal (D, 5′-CATTCCAGAAGTACCCTCAAAGAC-3′) along with a common antisense primer 5′-TGTGAATCCCTGCGTAGGCA-3′.
Staining of cultured cells
Cells grown in 96-well plates were incubated with antibodies against PRLR for 1 h and fixed in 2% PFA for 20 min. For PRL staining, cells were incubated with monensin for 24 h, and stained with anti-PRL monoclonal Ab for 1 h followed with the secondary antibody conjugated with Alexa 488 (Molecular Probes/Invitrogen) for 1 h. Cell nuclei were stained with Hoechst 33342 at 2 μg/ml for 20 min to identify individual cells and to optimize focusing.
Cellomics ArrayScan automated imaging
The Cellomics ArrayScan HCS Reader (Cellomics/ThermoFisher, Pittsburgh, PA) was utilized to collect information on distribution of fluorescently labeled components in stained cells. Data were captured, extracted, and analyzed with ArrayScan II Data Acquisition and Data Viewer version 3.0 (Cellomics) and Quattro Pro version 10.0.0 (Corel, Ottawa, Ontario, Canada).
Flow cytometry analysis
Cells were preincubated in PBS/0.1% BSA for 20 min followed by incubation with primary antibody (1:100 dilution in PBS/0.1% BSA) for 20 min. Flow cytometry was performed by FACScan using CellQuest software (Becton Dickinson, Mountain View, CA).
Proliferation assays
Cancer cells were plated onto 96-well plates at 2×103 cells per well. Next day media was changed to media with 2% FBS, human recombinant PRL (hrPRL) was added to the final concentration of 0 -100 ng/ml, and cells were grown for additional 72 h. Cells were fixed, stained with Hoechst 33342, and counted using the Cellomics ArrayScan HCS Reader (10× objective) as previously described (17).
Apoptosis assays
Apoptosis was analyzed by flow cytometry using FITC-conjugated Annexin V and propidium iodide (PI) and by analysis of caspase activation using Fluorescent Poly-Caspases FLICA Apoptosis Detection Kit FAM-VAD-FMK (Immunochemistry Technologies) as previously described (17).
Multiplex analysis of phosphoproteins
Analysis of phosphoproteins was performed using multiplexing xMAP technology (Luminex Corp., Austin, TX). Tumor cells were stimulated with 10 ng/ml of human recombinant PRL for 0-30 min; cell lysates were prepared using Bio-Rad Bio-Plex Cell Lyses Kit, and analyzed using Bio-Rad 17-Plex Phosphoprotein kit for testing phosphoproteins, Akt, ATF-2, ERK1/2, GSK-3a/B, JNK, p38 MAPK, STAT3, STAT6, CREB, HSP27, IRS-1, MEK1, NFkB p65, p53, p70 S6 kinase, p90RSK and TrkA according to manufacturer's protocol.
Multiplex analysis of transcription factors
Tumor cells growing in 6 well plates were treated with 10 ng/ml PRL for 0-30 min. Nuclear extracts were prepared and transcription factor (TF) analysis was performed according to the manufacturer's protocol using the Procarta Multiplex Transcription Factor Assay Kit (Panomics, Fremont, CA) designed for measuring activities of NF-kB, AP1, AP2, CREB, HIF-1, STAT1, STAT3, STAT4, STAT5, Oct, GATA, ELK-1, FAST-1, p53, PAX-3, NF-1, NF-E2, NF-E1/YY1, ATF-2, ISRE, PAX-5, AR, ETS/PEA, Nkx-2.5, E2F1, MyoD, PPAR, SMAD, RUNX/AML, BRN-3, CEBP, NF-Y, c-myb, ER, GR/PR, FKHR, HNF-1, MEF-2, NFAT and IRF TFs.
Clonogenic/colony formation assays
Cells were plated at a density of 0.5 cells/well onto 96-well plates and cultured for 7-10 days. For colony counting, cells were fixed and stained with Coomassie brilliant blue. The ability of cells to form colonies in soft agar was assessed in methyl cellulose-based (MC-based) medium as described (18). Briefly, cells were resuspended in 0.8% MC-based medium (Stem Cell Technologies, Vancouver, Canada) and plated at 102 cells per well in six-well plates coated with poly2-hydroxyethyl methacrylate (Sigma). EGF and bFGF were added every day for 2 weeks.
Ras activation
Cells grown in 100 mm2 Petri dishes (75% of culture confluence) were treated with 10 ng/ml PRL for 0-30 min. Cell lysates were prepared and analyzed according to RasGTPase Activation ELISA kit Upstate (Millipore).
Analysis of tumorigenic properties of tumor cells treated with PRL
Experiments were carried out in accordance with guidelines provided by the Pittsburgh University Institutional Animal Care and Use Committee (IACUC) and the National Institute of Health Guide for the Care and Use of Laboratory Animals. SCID-beige mice, 7–8 weeks old (Jackson Laboratories, Bar Harbor, ME), were maintained in the animal facility of the University of Pittsburgh Cancer Institute (UPCI). To compare tumorigenic properties, H29 cells untreated or treated with PRL were harvested and injected subcutaneously (s.c.) into SCID mice (5×106 cells per mouse in 0.2 ml of PBS) without Matrigel. Ten mice were inoculated into the left back with T29 untreated cells and into the right back with T29 cells treated in vitro with PRL. Tumor appearance and tumor growth was monitored twice a week. Mice were sacrificed when their tumors reached above 1.5 cm in diameter or became ulcerated.
Statistical analysis of data
The Wilcoxon rank sum test was used to determine statistical significance of differences in biomarker concentrations between patient groups. All in vitro experiments were repeated at least three times. Comparisons between values were performed using a two-tailed Student's t-test. For the comparison of multiple groups, one- or two-way ANOVA test was applied.
Results
PRL in sera of patients with ovarian and endometrial cancer
Circulating PRL concentrations were determined in sera of 167 patients with stage I-IV endometrial cancer, 273 patients with ovarian cancer, 141 patients with benign pelvic disease, and 470 age-matched healthy controls. Serum concentrations of PRL were significantly elevated in patients with endometrial and ovarian cancers and in women with benign pelvic disease as compared to healthy controls (p<0.001; Figure 1). Mean serum PRL was significantly higher in women with ovarian and endometrial cancers as compared to women with benign pelvic disease (P<0.001). Serum PRL concentrations were equally high in patients with early and late stage cancers (p > 0.1, data not shown). PRL levels in sera of age-matched women with lung, breast, and pancreatic cancers were analyzed for comparison (Figure 1). Although mean circulating PRL concentrations in pancreatic and, to a lesser extent, lung and breast cancers, were significantly higher than in the healthy group (P<0.001), serum PRL levels were the highest in endometrial and ovarian cancers. This may indicate a specific role for PRL in peritoneal gynecological cancers.
Figure 1. Serum PRL levels in healthy women, women with endometrial, ovarian, breast, lung, and pancreatic cancers, and in women with strong family history of ovarian cancer.
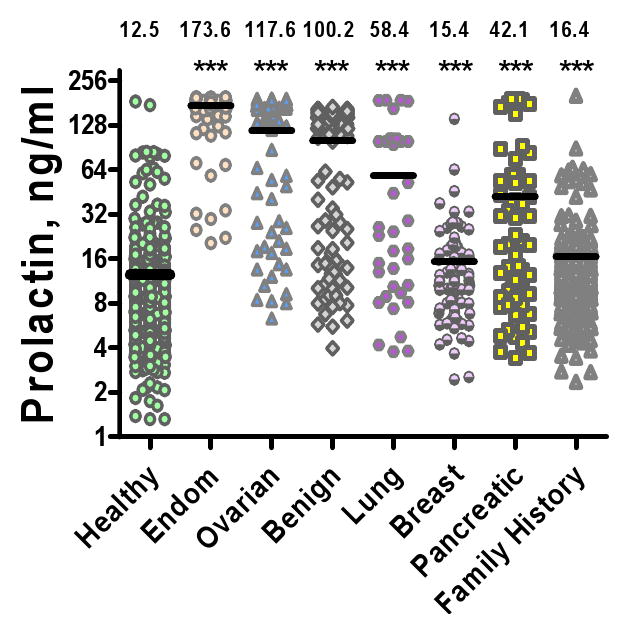
Horizontal lines and numbers over each distribution denote mean concentrations. *** denotes significance of differences between cases and controls at P < 0.001. PRL concentrations are significantly (p<0.001) higher in ovarian and endometrial cancers as compared to other cancers.
PRL levels are elevated in women with a family history of ovarian cancer
We have observed significantly elevated PRL levels in sera of 112 women with a strong (2 or more first relatives affected) family history of ovarian cancer (Figure 1). Sixty seven of 112 (60%) women demonstrated PRL levels above 14 ng/ml, whereas in the general population only 22% of women have serum PRL levels above 14 ng/ml. The mean PRL concentration in the high risk group was significantly (P<0.001) higher as compared to general population controls. This finding could be an important clue into the pathological mechanisms underlying predispositions to cancer and pre-neoplastic processes.
PRL mRNA is expressed in endometrial or ovarian tumors
Since positive PRL protein staining could reflect PRL locally produced by tumor and by other sources, we investigated PRL expression in endometrial and ovarian tumors by analysis of PRL mRNA. Total RNA was isolated from normal pituitary tissue, normal ovary and endometrium, normal tissue surrounding ovarian and endometrial tumors, as well as ovarian and endometrial tumors. RT-PCR was performed using sense primers specific for pituitary proximal promoter and extrapituitary distal according to previously published procedure (16) and the results are presented in Figure 2A. Pituitary gland expresses mostly proximal 617bp transcript along with low levels of the distal 780bp form. The normal endometrial tissue expressed both forms of PRL mRNA while the non-tumor adjacent tissue sample expressed only the distal transcript, at high levels. All tested endometrial tumor samples expressed the proximal transcript with 3 out of 5 also expressing the distal transcript, an example of which is shown. Normal ovary expressed low levels of proximal transcript and no expression of the distal transcript while the non-tumor adjacent ovarian tissue expressed both transcripts with the proximal being more prevalent. Tissue from five distinct ovarian tumors was analyzed with each expressing a unique mixture of expected and unexpected transcripts. The proximal transcript was observed in 4 out of 5 tumors, although in two of these, the expected band was present as a doublet (Figure 2A and data not shown). One of these tumors expressed high levels of the proximal transcript (Figure 2A). None of the tumors expressed appreciable levels of the distal transcript, although several products of unexpected sizes were observed from the RT-PCR reaction. The characterization of the unexpected transcripts and proximal transcript doublet observed here is the focus of ongoing investigation.
Figure 2. Expression of PRL and PRLR in endometrial and ovarian cancer.
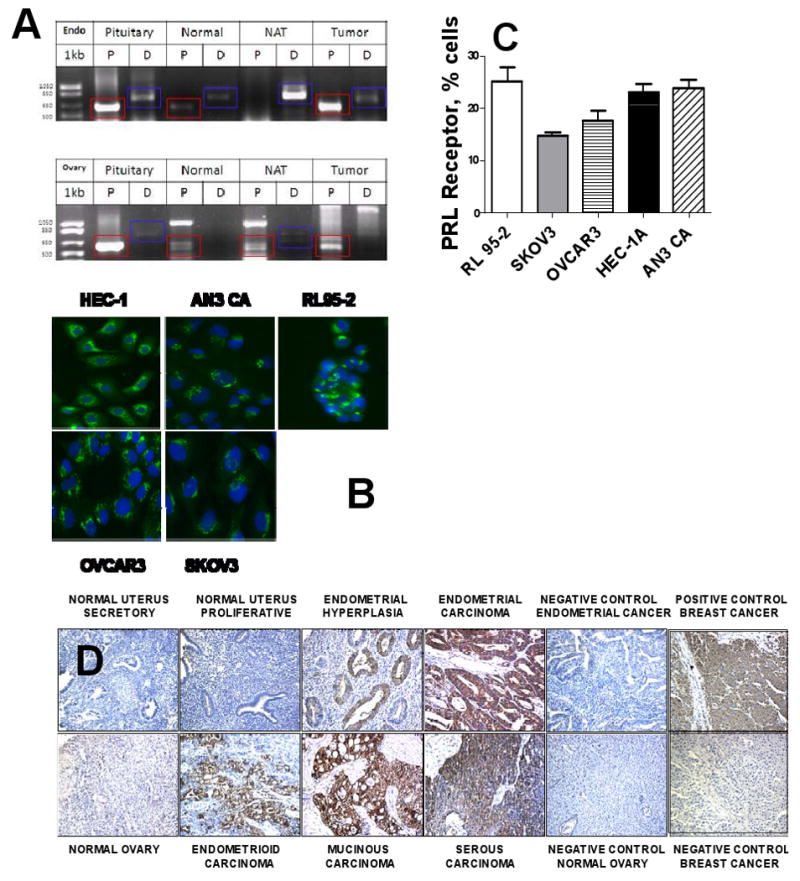
A. RT-PCR analysis. Total RNA was isolated from normal pituitary tissue (Pit), normal endometrium (Normal), non-tumor adjacent tissue surrounding endometrial (Endo) and ovarian tumors (NAT), and endometrial and ovarian tumors (Tumor). P-proximal transcript, expected size of 617bp (red rectangle); D-distal transcript, expected size of 780bp (blue rectangle). B. Prolactin expression by endometrial and ovarian carcinoma. Ovarian carcinoma cells, OVCAR3 and SKOV3, and endometrial carcinoma cells, HEC-1A, AN3 CA, and RL95-2 were stained with anti-PRL monoclonal Ab. Isotype-matched nonspecific Ab produced no staining (not shown); C. Expression of prolactin receptor in four human carcinoma cell lines. Cells were stained with rabbit polyclonal anti-PRLR Ab. Surface expression of prolactin receptor was measured by flow cytometry. Horizontal bars indicate SE. D. Expression of prolactin receptor in tumors and healthy tissues. Ovarian cancer, endometrial cancer, and endometrial progression (hyperplasia) TMAs were stained with anti-PRLR antibody. Representative cores are presented.
Expression of PRL and PRLR in cultured endometrial and ovarian carcinoma cells
Five human carcinoma cell lines were analyzed, endometrial carcinoma: HEC-1A, AN3 CA, and RL 95-2, and ovarian carcinoma: OVCAR3 and SKOV3. PRL expression was assessed using Cellomics imaging cytometry, and expression of PRLR was analyzed by flow cytometry. All five cell lines expressed PRL protein (Figure 2B) and PRLR (Figure 2C) although the expression levels varied.
Expression of PRLR in ovarian and endometrial tumors
Expression of PRLR in tissue sections was analyzed by IHC on TMA of ovarian tumors, normal ovarian epithelia, endometrial tumors, normal endometrium, non-cancerous cancer-adjacent endometrium, endometrial hyperplasia, and endometritis. Expression of PRLR increased dramatically in ovarian and endometrial tumor (Figure 2D). PRLR was highly expressed in all cancerous ovarian and endometrial tissues regardless of histology and grade (Table 1). Specifically, 80% of ovarian tumors were positive for PRLR with staining intensity varying from weak (W) to moderate (M) to strong (S). Serous, mucinous, and endometrioid tumors were mostly positive for PRLR (Table 1). None of 10 normal ovarian epithelia expressed PRLR. In endometrial cancer, 77% of tumors stained positively for PRLR with staining intensity varying from W to M to S, whereas only 13% of normal endometrial tissues (not cancer adjacent) were positive with staining intensity W-M. Non-cancerous tumor-adjacent endometrium displayed 27% expression of PRLR with staining intensity varying from W to M. Furthermore, increased expression of PRLR was observed in endometrial hyperplasia (Figure 2D, Table 1) with 75% staining positive with an intensity of W-M-S. We have additionally observed high expression of PRLR in 75% of endometritis with weak staining intensity. In both ovarian and endometrial cancers PRLR expression was decreased in tumors of higher grade (Table 1). No association with stage could be observed in either cancer (Table 1).
Table 1. Expression of PRLR in normal and cancerous ovarian and endometrial tissue.
| OVARIAN | Total N | Positive N | % Positive |
|---|---|---|---|
| Tumor | |||
| Total | 69 | 56 | 80% |
| CCC | 3 | 1 | 33% |
| Endo | 3 | 2 | 67% |
| M | 8 | 8 | 100% |
| S | 53 | 42 | 79% |
| Grade | |||
| 1 | 12 | 12 | 100% |
| 2 | 29 | 21 | 72% |
| 3 | 24 | 20 | 83% |
| Stage | |||
| 1 | 13 | 3 | 77% |
| 2 | 27 | 22 | 82% |
| 3-4 | 30 | 24 | 80% |
| Healthy | 10 | 0 | 0% |
| ENDOMETRIAL | |||
| Tumor | |||
| Total | 57 | 44 | 77% |
| Grade | |||
| 1 | 32 | 29 | 90.1% |
| 2 | 32 | 24 | 75% |
| 3 | 23 | 15 | 65% |
| Stage | |||
| 1 | 52 | 45 | 81% |
| 2-3 | 8 | 6 | 75% |
| Healthy | 15 | 2 | 13% |
|
Tumor-Adjacent Non-cancerous |
11 | 3 | 27% |
| Hyperplasia | 20 | 15 | 75% |
| Endometritis | 5 | 3 | 75% |
PRL induces proliferation in ovarian and endometrial carcinoma cell lines and protects cells from chemotherapy induced apoptosis
PRL at concentrations of 0.1-10 ng/ml was able to stimulate proliferation in all five studied cell lines (Figure 3A). Preincubation with 1 ng/ml PRL for 1 hr significantly protected cells in all five lines from cisplatin-induced apoptosis (Figure 3B for OVCAR3, similar results for other cell lines are not shown). Inhibition of PRL signaling by neutralizing antibody in unstimulated cells resulted in slower proliferation (Figure 3C), but did not potentiate cisplatin induced apoptosis or induce apoptosis/necrosis (data not shown).
Figure 3. Effects of PRL in ovarian and endometrial carcinoma cells.
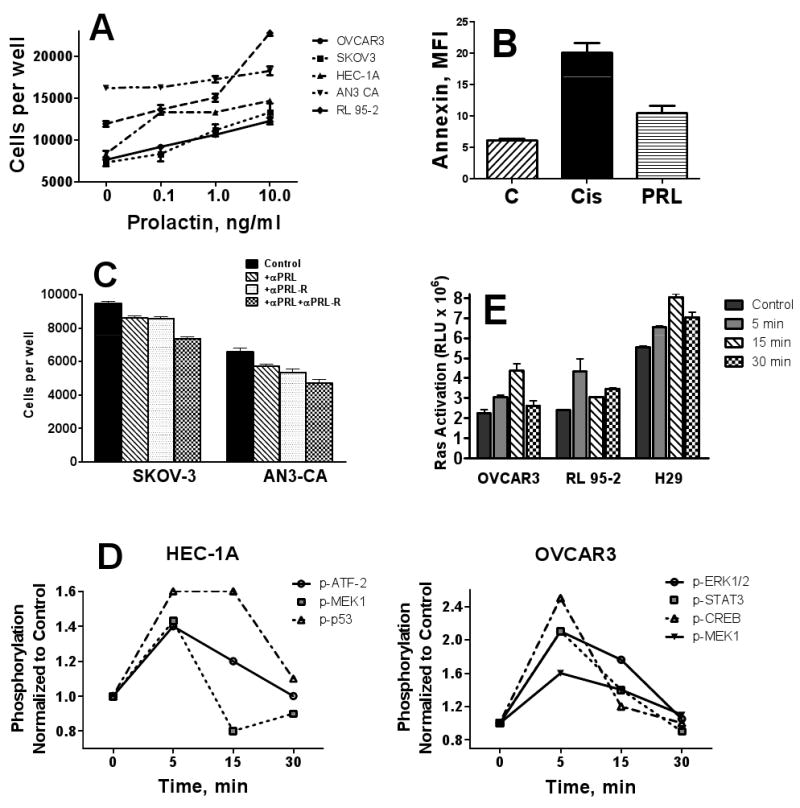
A, PRL induces proliferation in ovarian and endometrial cancer cell line. Ovarian carcinoma cells, OVCAR3 and SKOV3, and endometrial carcinoma cells, HEC-1A, AN3 CA, and RL95-2 were treated with 0-10 ng/ml hrPRL for 72 hr, and cell counts were determined as described in Methods; B, Prolactin protects ovarian and endometrial tumor cells from chemotherapy-induced apoptosis. HEC-1B cells were untreated (C), treated with 5 μM Cisplatin (Cis) for 8 hr, or pre-treated with 10 ng/ml PRL for 1 hr before treating with Cis, stained with Annexin V/PI, and analyzed with flow cytometry as described in Methods. Similar data for OVCAR3 cells are not shown. C, Effects of neutralizing anti-PRL and anti-PRLR antibodies on proliferation of ovarian carcinoma cells. OVCAR-3 cells were incubated with neutralizing antibodies to PRL, PRLR, or their combination or IgG isotype control for 72 h and cell proliferation was assessed by imaging cytometry. D, Prolactin activates phosphokinases in ovarian and endometrial carcinoma cells. E, Ras activation. Cells were treated with 10 ng/ml hrPRL for 0-30 min and Ras activity was determined as described in Methods. * P < 0.05; *** P < 0.001. Horizontal bars denote SE.
Analysis of PRL signal transduction pathways in ovarian and endometrial carcinoma cell lines
Phosphorylation of 17 phosphokinases (see Methods) was evaluated using multiplex bead-based immunoassay after 0-30 min incubation with 10 ng/ml of hrPRL. Of these 17 phosphokinases, ERK1/2, MEK-1, STAT3, and CREB were phosphorylated within 5 min in OVCAR3 in response to PRL treatment, whereas in HEC-1A cells, ATF-2, MEK1, and p53 were phosphorylated (Figure 3D).
Activation of 37 transcription factors (TFs) (see Methods) following 5-30 min incubation with 10 ng/ml of hrPRL was analyzed using a multiplexed bead-based assay. All 37 TFs were activated following 5-15 min incubation with hrPRL in both ovarian and endometrial carcinoma cells indicating direct effects of PRLR signaling on TF activity (Supplementary Table I).
Activation of Ras oncogene by PRL in ovarian and endometrial cells
The Ras gene family has been implicated in the development of endometrial and ovarian cancers (19-21). Ras genes often represent the last ‘hit’ for complete transformation of SV40-immortalized but not yet transformed human ovarian cells (22). We have analyzed Ras activity in ovarian and endometrial cancer cell lines, OVCAR3 and RL-95-2, and in an immortalized NOE cell line, T29, incubated with 10 ng/ml PRL for 0-30min. PRL significantly activated Ras by 5 min of incubation in all three cell lines (Figure 3E).
Chronic exposure to PRL induces malignant transformation in immortalized NOE and endometrial cell lines
To ascertain potential oncogenic function of PRL, we took advantage of the existing in vitro model developed by Liu et al. 2004 wherein normal human ovarian epithelial cells were immortalized (but not transformed) by transfection with SV40 and hTERT cDNA which discrupted the p53 and Rb pathways (22). We speculated that chronic exposure to PRL may activate Ras and result in malignant transformation of these cells. T29 cells were cultured in the presence of exogenous PRL at 10 and 100 ng/ml. After 2 weeks of incubation, we observed morphological changes indicative of malignant transformation, namely uncontrolled growth of treated T29 cells seeded onto a confluent T29 monolayer, and small rounded appearance of these cells (Figure 4A). No such changes were observed in untreated cells even after prolonged (> 8 weeks) culture. As an additional control, cells were also treated with CCL11, a chemokine that potently induces growth of ovarian cancer cells (23). No transformation was observed (data not shown). To confirm that PRL induced malignant transformation of immortalized NOE cells, their ability to form clones and to grow in soft agar was evaluated. PRL treated cells demonstrated a dramatically elevated capacity for both clonogenic growth and colony formation in semi-solid medium. Whereas parental immortalized cells produced 0.33±0.21 clones per plate, modified cells produced 20.2±0.87 clones per plate (Figure 4B). Similarly, while parental T29 cells did not form any colonies in semi-solid agar, PRL-modified cells formed on average 55.0±4.73 colonies per well of a 6-well plate (Figure 4C). Incubation of T29 cells with PRL in the presence of a specific Ras inhibitor, α-hydroxy farnesyl phosphonic acid, dramatically abrogated the transforming effects of PRL (Table 2). Similar results were obtained using endometrial fibroblasts immortalized with hTERT (24) (data not shown).
Figure 4. Prolactin promotes malignant transformation in immortalized NOE H29 cells.
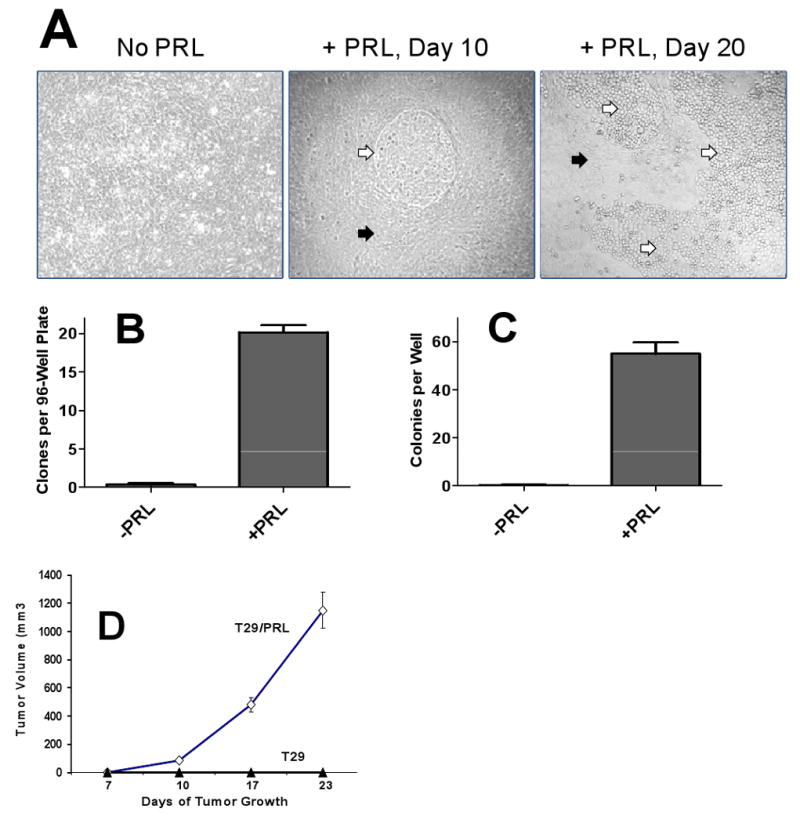
A, Effects of chronic exposure to PRL on morphology of immortalized normal ovarian epithelial cells. H29 cells were incubated with 100 ng/ml of PRL for 0-20 days. The experiment was repeated eighteen times. Black arrows – untransformed parental T29 cells; white arrows – PRL-transformed cells. B,C, PRL-modified H29 exhibit higher capacity for clonogenic growth (B) and semi-solid medium colony formation (C); D, Dynamics of tumor growth in SCID mice injected with PRL-transformed and parental T29 cells.
Table 2. Effect of Ras inhibition on the prolactin induced transformation of T29 cells.
| Experimental Conditions | Number of Transformed Clones/Plate | ||
|---|---|---|---|
| T-29, control | 0 | 0 | 0 |
| T-29 +PRL, | 8 | 12 | 15 |
| T-29 +lnhibitor | 0 | 0 | 0 |
| T-29 +PRL+inhibitor | 3 | 2 | 1 |
Growth of PRL transformed T29 in nude mice
We assessed the tumorigenic potential of parental and PRL transformed T29 cells in a SCID-beige mouse model. Ten mice received bilateral subcutaneous injections of 5×106 PRL-transformed or non-transformed H29 cells. Two weeks after injection, tumors appeared at the site of inoculation of PRL modified cells. These tumors grew progressively in all mice (Figure 4D). Mice were sacrificed when tumor exceeded 1.5 cm in diameter. In contrast, none of the 10 mice demonstrated tumor formation at the site of inoculation of PRL untreated parental T29 cells.
Discussion
In this study, we demonstrate the potential critical role of a hormone/cytokine, PRL, as a tumor promoter and risk factor in ovarian and endometrial cancers. Serum PRL levels are dramatically elevated in women suffering from ovarian and endometrial cancers making it a strong biomarker for these cancers (14, 15). The use in this study of samples drawn prior to the admission of anesthesia and surgery eliminates the effect of these confounders.shown to boost serum PRL (25). Although one may suggest that elevated blood PRL may reflect an increase in production of pituitary hormones as a result of stress related to cancer diagnosis, several lines of evidence argue against it. First, as we demonstrate here, circulating PRL is elevated to a different extent in different cancers. Secondly, several studies of stress-induced increase in blood PRL in healthy subjects concluded that PRL responses to purely psychological stress are rarely seen (26, 27).
We observed a dramatic increase in expression of PRLR in both ovarian and endometrial tumors as compared to healthy tissues indicating a critical role of PRL signaling for tumor growth and maintenance. Although PRL itself was not overexpressed in either cancerous endometrium or ovarian epithelium, high serum levels of PRL should be sufficient for activating PRL signaling in tumors. These increased levels of serum PRL must originate either from pituitary or from alternative extrapituitary sources such as lymphocytes.
Significant upregulation of PRL receptors could be responsible for increased PRL signaling in ovarian and endometrial tumors leading to increased tumor proliferation and cell survival. In addition, PRL may play several other active roles in tumor development by stimulating angiogenesis (28), activating pathways involved in cellular adhesion and motility (29, 30). We observed a reverse correlation between tumor grade and expression of PRLR in both ovarian and endometrial tumors, and no association of PRLR with cancer stage. A similar relationship with tumor grade was reported in prostate cancer where foci within high grade tumors demonstrated lower receptor expression in comparison to lower Gleason grade carcinomas (31). These results may suggest that prolactin is important in the development in early neoplastic transformation but not in the progression of higher grade tumors. In breast cancer, no association was found between expression of PRLR and either cancer stage or tumor grade (32). We have observed high expression of PRLR in endometrial hyperplasia, which is considered to be the pathologic precursor to endometrial cancer (33). These results may indicate that activation of PRL signaling could be a risk factor for the development of endometrial cancer. Although expression of PRLR was also elevated in endometritis, the staining intensity was weak. PRLR is induced by inflammatory cytokines, TNF-α, IL-1β, and IFNγ (34) and this association may indicate a possible mechanism by which inflammation can contribute to carcinogenesis.
We further demonstrated that exogenous PRL can, in fact, drive transformation of ovarian epithelial cells via Ras activation. The process of carcinogenesis could be divided into at least three stages: initiation, promotion, and progression (reviewed in (35)). Full transformation requires overexpression of at least two oncogenes in combination with inactivation of tumor suppressor genes and other growth regulatory elements. We hypothesized that by potently activating Ras oncogene PRL may play a role in promotion of ovarian or endometrial tumorigenesis. We utilized two human cell lines, ovarian epithelial cells and uterine fibroblasts immortalized by transfection with hTERT cDNA (22, 24). Additional introduction of H-ras or K-ras into these immortalized cells resulted in complete transformation (22). We therefore expected that chronic exposure to PRL may activate Ras and result in malignant transformation of these cells. When immortalized cells were subjected to prolonged incubation with PRL, we observed changes in cell morphology accompanied by a dramatic increase in clonogenic capacity and the ability to support colony formation in semi-solid agar. We conclude that these cells were driven to complete transformation by exogenous PRL. The ability of PRL-treated cells to form tumors in SCID mice provides further proof of this transforming activity. The Ras-mediated transforming activity of PRL could be further amplified via several mechanisms. Ras activation can lead to PRLR stabilization (36). PRL can induce IGF-I and -2 signaling (37) that in turn stimulates PRL gene expression by a Ras-dependent mechanism (38).
In summary, our findings indicate an important role for PRL in ovarian and endometrial tumorigenesis. Ongoing efforts should seek to characterize the role of PRL in specific and non-specific tumorigenic pathways. Such work could lead to the use of PRL as a risk factor for ovarian, endometrial and potentially other cancers.
Acknowledgments
We are grateful to Gynecologic Oncology Group Blood Bank, Drs. Karen Lu, Jeffrey Marks, Jill Siegfried, Herbert Zeh, III, and Randall Brand for providing serum samples. This work was supported by NIH grants: R03 CA13601, RO1 CA098642, R01 CA108990, P50, and UPCI Hillman Fellows Award (AEL) and DOD, IDEA Award BC051720, Harry J. Lloyd Charitable Trust and Hillman Foundation (EG)
References
- 1.Goffin V, Binart N, Touraine P, Kelly PA. Prolactin: the new biology of an old hormone. Ann Rev Physiology. 2002;64:47–67. doi: 10.1146/annurev.physiol.64.081501.131049. [DOI] [PubMed] [Google Scholar]
- 2.Ben-Jonathan N, Liby K, McFarland M, Zinger M. Prolactin as an autocrine/paracrine growth factor in human cancer. Trends Endocrinol Metabolism. 2002;13:245–50. doi: 10.1016/s1043-2760(02)00603-3. [DOI] [PubMed] [Google Scholar]
- 3.Gerlo S, Davis JR, Mager DL, Kooijman R. Prolactin in man: a tale of two promoters. Bioessays. 2006;28:1051–5. doi: 10.1002/bies.20468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Takahashi H, Nabeshima Y, Nabeshima Y, Ogata K, Takeuchi S. Molecular cloning and nucleotide sequence of DNA complementary to human decidual prolactin mRNA. J Biochem. 1984;95:1491–9. doi: 10.1093/oxfordjournals.jbchem.a134757. [DOI] [PubMed] [Google Scholar]
- 5.Clevenger CV, Kline JB. Prolactin receptor signal transduction. Lupus. 2001;10:706–18. doi: 10.1191/096120301717164949. [DOI] [PubMed] [Google Scholar]
- 6.Pickett CA, Gutierrez-Hartmann A. Epidermal growth factor and Ras regulate gene expression in GH4 pituitary cells by separate antagonistic signal transduction pathways. Mol Cell Biol. 1995;15:6777–84. doi: 10.1128/mcb.15.12.6777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Buckley AR, Putnam CW, Russell DH. Prolactin as a mammalian mitogen and tumor promoter. Adv Enzyme Regulation. 1988;27:371–91. doi: 10.1016/0065-2571(88)90027-1. [DOI] [PubMed] [Google Scholar]
- 8.Wennbo H, Gebre-Medhin M, Gritli-Linde A, Ohlsson C, Isaksson OG, Tornell J. Activation of the prolactin receptor but not the growth hormone receptor is important for induction of mammary tumors in transgenic mice. J Clin Investigation. 1997;100:2744–51. doi: 10.1172/JCI119820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nandi S, Guzman RC, Yang J. Hormones and mammary carcinogenesis in mice, rats, and humans: a unifying hypothesis. Proc Natl Acad Sci USA. 1995;92:3650–7. doi: 10.1073/pnas.92.9.3650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang DY, De Stavola BL, Bulbrook RD, et al. Relationship of blood prolactin levels and the risk of subsequent breast cancer. Intl J Epidemiol. 1992;21:214–21. doi: 10.1093/ije/21.2.214. [DOI] [PubMed] [Google Scholar]
- 11.Hankinson SE, Willett WC, Michaud DS, et al. Plasma prolactin levels and subsequent risk of breast cancer in postmenopausal women. J Natl Cancer Inst. 1999;91:629–34. doi: 10.1093/jnci/91.7.629. [DOI] [PubMed] [Google Scholar]
- 12.Tworoger SS, Hankinson SE. Prolactin and breast cancer risk. Cancer Letters. 2006;243(2):160–9. doi: 10.1016/j.canlet.2006.01.032. [DOI] [PubMed] [Google Scholar]
- 13.Stattin P, Rinaldi S, Stenman UH, et al. Plasma prolactin and prostate cancer risk: A prospective study. Intl J Cancer. 2001;92:463–5. doi: 10.1002/ijc.1191. [DOI] [PubMed] [Google Scholar]
- 14.Mor G, Visintin I, Lai Y, et al. Serum protein markers for early detection of ovarian cancer. Proc Natl Acad Sci USA. 2005;102:7677–82. doi: 10.1073/pnas.0502178102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yurkovetsky Z, Ta'asan S, Skates S, et al. Development of multimarker panel for early detection of endometrial cancer. High diagnostic power of prolactin. Gynecol Oncol. 2007;107:58–65. doi: 10.1016/j.ygyno.2007.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shaw-Bruha CM, Pirrucello SJ, Shull JD. Expression of the prolactin gene in normal and neoplastic human breast tissues and human mammary cell lines: promoter usage and alternative mRNA splicing. Breast Cancer Res Treat. 1997;44:243–53. doi: 10.1023/a:1005879103367. [DOI] [PubMed] [Google Scholar]
- 17.Levina V, Marrangoni AM, DeMarco R, Gorelik E, Lokshin AE. Multiple effects of TRAIL in human carcinoma cells: induction of apoptosis, senescence, proliferation, and cytokine production. Exp Cell Res. 2008;314:1605–1616. doi: 10.1016/j.yexcr.2007.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ignatova TN, Kukekov VG, Laywell ED, Suslov ON, Vrionis FD, Steindler DA. Human cortical glial tumors contain neural stem-like cells expressing astroglial and neuronal markers in vitro. Glia. 2002;39:193–206. doi: 10.1002/glia.10094. [DOI] [PubMed] [Google Scholar]
- 19.Al-Jehani RM, Jeyarajah AR, Hagen B, Hogdall EV, Oram DH, Jacobs IJ. Model for the molecular genetic diagnosis of endometrial cancer using K-ras mutation analysis. J Natl Cancer Inst. 1998;90:540–2. doi: 10.1093/jnci/90.7.540. [DOI] [PubMed] [Google Scholar]
- 20.Esteller M, Garcia A, Martinez-Palones JM, Xercavins J, Reventos J. The clinicopathological significance of K-RAS point mutation and gene amplification in endometrial cancer. Eur J Cancer. 1997;33:1572–7. doi: 10.1016/s0959-8049(97)00154-8. [DOI] [PubMed] [Google Scholar]
- 21.Pijnenborg JM, Dam-de Veen GC, Kisters N, et al. RASSF1A methylation and K-ras and B-raf mutations and recurrent endometrial cancer. Ann Oncol. 2007;18:491–7. doi: 10.1093/annonc/mdl455. [DOI] [PubMed] [Google Scholar]
- 22.Liu J, Yang G, Thompson-Lanza JA, et al. A genetically defined model for human ovarian cancer. Cancer Res. 2004;64(5):1655–63. doi: 10.1158/0008-5472.can-03-3380. [DOI] [PubMed] [Google Scholar]
- 23.Levina VN, Nolen BM, Marrangoni AM, et al. Role of CCL11/eotaxin-1 signaling in ovarian cancer. Clin Cancer Res. 2009 doi: 10.1158/1078-0432.CCR-08-2024. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Krikun G, Mor G, Lockwood C. The immortalization of human endometrial cells. Methods Mol Medicine. 2006;121:79–83. doi: 10.1385/1-59259-983-4:077. [DOI] [PubMed] [Google Scholar]
- 25.Yuen BH, McMorland G, Pudek M, Cannon W. Effect of general and peridural anesthesia on the concentrations of prolactin and cortisol in maternal plasma. Am J Obst Gynecology. 1981;141:483–6. doi: 10.1016/s0002-9378(15)33264-6. [DOI] [PubMed] [Google Scholar]
- 26.Delitala G, Tomasi P, Virdis R. Prolactin, growth hormone and thyrotropin-thyroid hormone secretion during stress states in man. Bailliere's Clin Endocrinol Metabolism. 1987;1:391–414. doi: 10.1016/s0950-351x(87)80069-1. [DOI] [PubMed] [Google Scholar]
- 27.Brooks JE, Herbert M, Walder CP, Selby C, Jeffcoate WJ. Prolactin and stress: some endocrine correlates of pre-operative anxiety. Clin Endocrinol. 1986;24:653–6. doi: 10.1111/j.1365-2265.1986.tb01661.x. [DOI] [PubMed] [Google Scholar]
- 28.Struman I, Bentzien F, Lee H, et al. Opposing actions of intact and N-terminal fragments of the human prolactin/growth hormone family members on angiogenesis: an efficient mechanism for the regulation of angiogenesis. Proc Natl Acad Sci USA. 1999;96:1246–51. doi: 10.1073/pnas.96.4.1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Canbay E, Norman M, Kilic E, Goffin V, Zachary I. Prolactin stimulates the JAK2 and focal adhesion kinase pathways in human breast carcinoma T47-D cells. Biochem J. 1997;324:231–6. doi: 10.1042/bj3240231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Maus MV, Reilly SC, Clevenger CV. Prolactin as a chemoattractant for human breast carcinoma. Endocrinology. 1999;140:5447–50. doi: 10.1210/endo.140.11.7245. [DOI] [PubMed] [Google Scholar]
- 31.Leav I, Merk FB, Lee KF, et al. Prolactin receptor expression in the developing human prostate and in hyperplastic, dysplastic, and neoplastic lesions. Am J Pathol. 1999;154:863–70. doi: 10.1016/S0002-9440(10)65333-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gill S, Peston D, Vonderhaar BK, Shousha S. Expression of prolactin receptors in normal, benign, and malignant breast tissue: an immunohistological study. J Clin Pathol. 2001;54:956–60. doi: 10.1136/jcp.54.12.956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kurman RJ, Kaminski PF, Norris HJ. The behavior of endometrial hyperplasia. A long-term study of “untreated” hyperplasia in 170 patients. Cancer. 1985;56:403–12. doi: 10.1002/1097-0142(19850715)56:2<403::aid-cncr2820560233>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 34.Corbacho AM, Macotela Y, Nava G, et al. Cytokine induction of prolactin receptors mediates prolactin inhibition of nitric oxide synthesis in pulmonary fibroblasts. FEBS Letters. 2003;544:171–5. doi: 10.1016/s0014-5793(03)00499-x. [DOI] [PubMed] [Google Scholar]
- 35.Boyd JA, Barrett JC. Genetic and cellular basis of multistep carcinogenesis. Pharmacol Therapeutics. 1990;46:469–86. doi: 10.1016/0163-7258(90)90028-z. [DOI] [PubMed] [Google Scholar]
- 36.Plotnikov A, Li Y, Tran TH, et al. Oncogene-mediated inhibition of glycogen synthase kinase 3 beta impairs degradation of prolactin receptor. Cancer Res. 2008;68:1354–61. doi: 10.1158/0008-5472.CAN-07-6094. [DOI] [PubMed] [Google Scholar]
- 37.Brisken C, Ayyannan A, Nguyen C, et al. IGF-2 is a mediator of prolactin-induced morphogenesis in the breast. Developmental Cell. 2002;3:877–87. doi: 10.1016/s1534-5807(02)00365-9. [DOI] [PubMed] [Google Scholar]
- 38.Castillo AI, Tolon RM, Aranda A. Insulin-like growth factor-1 stimulates rat prolactin gene expression by a Ras, ETS and phosphatidylinositol 3-kinase dependent mechanism. Oncogene. 1998;16:1981–91. doi: 10.1038/sj.onc.1200204. [DOI] [PubMed] [Google Scholar]