

Abstract
The design, synthesis, X-ray crystal structure, molecular modeling, and biological evaluation of a series of new generation SARS-CoV PLpro inhibitors are described. A new lead compound 3 (6577871) was identified via high-throughput screening of a diverse chemical library. Subsequently, we carried out lead optimization and structure-activity studies to provide a series of improved inhibitors that show potent PLpro inhibition and antiviral activity against SARS-CoV infected Vero E6 cells. Interestingly, the (S)-Me inhibitor 15h (enzyme IC50 = 0.56 μM; antiviral EC50 = 9.1 μM) and the corresponding (R)-Me 15g (IC50 = 0.32 μM; antiviral EC50 = 9.1 μM) are the most potent compounds in this series, with nearly equivalent enzymatic inhibition and antiviral activity. A protein-ligand X-ray structure of 15g-bound SARS-CoV PLpro and a corresponding model of 15h docked to PLpro provide intriguing molecular insight into the ligand-binding site interactions.
Introduction
Severe Acute Respiratory Syndrome (SARS) was first reported in Guangdong province, China, in November 2002.1 SARS is a contagious respiratory illness with no effective treatment to date. SARS affected three continents, infecting more than 8,000 individuals and causing nearly 800 deaths. Fortunately, the spread of SARS-CoV was contained after the initial outbreaks through public health measures. As it turned out, the etiological agent of SARS is a novel coronavirus, SARS-CoV.2,3 There have been no known new cases of SARS since 2005. However, recent isolation of strains from zoonotic origins thought to be the reservoir for SARS-CoV raises the possibility of a reemergence of SARS and related ailments.4,5 Consequently, design and development of antivirals effective against SARS-CoV should be an important priority against future outbreaks.
Biochemical events critical to the viral replication revealed a number of important targets for therapeutic intervention of SARS.7 Most notably, two cysteine proteases, a papain-like protease (PLpro) and a 3C-like protease (3CLpro), play a critical role in the virus-mediated RNA replication. Not surprisingly, numerous studies related to the development of SARS-CoV 3CLpro inhibitors have already been reported.8,9 In contrast, very few inhibitor design efforts against SARS-CoV PLpro have been reported. We recently reported the discovery and design of a series of unprecedented noncovalent SARS-CoV PLpro inhibitors displaying antiviral activity against SARS-CoV with no associated cytotoxicity.10 Subsequently, a protein-ligand X-ray structure provided important molecular insights for further design and optimization of inhibitors.10 This initial work demonstrated that PLpro is a viable target for the development of anti-SARS therapeutics.
Besides viral peptide cleavage, recent structural and functional studies demonstrated that PLpro is involved in a number of other important biochemical events, such as deubiquitination, deISGylation, and involvement in the virus evasion from the innate immune response.11,12 The homologous enzyme, PLP2, from the human coronavirus 229E, has been shown to be critical to 229E viral replication.13 In addition, recent studies have shown that human deubiquitinating enzymes are potential anticancer drug-design targets. Thus, PLpro is a significant target for drug development for a variety of human diseases.
Recently, our primary screening of a library of 50,080 diverse, drug-like compounds, led to the identification of two compounds after lead validation. Both leads reproducibly inhibited PLpro in a dose dependent manner in the absence and presence of Triton-X. Subsequently, our optimization efforts of the most potent lead, 1 (7724772), containing a benzamide scaffold (IC50 = 20.1 ± 1.1 μM) led to the design of novel PLpro inhibitor 2 and related derivatives which displayed antiviral activity against SARS-CoV. We recently reported a detailed study describing synthesis, biological studies and X-ray structure of the protein-ligand complex of 2-bound PLpro.10 In our continuing studies toward the development of non-covalent/reversible PLpro inhibitors, we have now investigated the potential of the second and less potent lead that evolved from our high-throughput screening efforts. The second HTS lead, compound 3 (Figure 1), contains a piperidine carboxamide scaffold and exhibited an IC50 value of 59 μM. Our subsequent lead optimization efforts led to the design of potent inhibitor 15g (IC50 = 0.32 μM) which inhibited SARS-CoV viral replication in Vero cells with an EC50 value of 9.1 μM. The corresponding enantiomer 15h has shown slightly less potent enzyme inhibitory activity (IC50 = 0.56 μM) and similar antiviral potency. A protein-ligand X-ray structure of 15g-bound SARS-CoV PLpro was determined. Interestingly, this structure revealed a unique mode of binding with SARS-CoV PLpro and that key molecular interactions of inhibitor 15g are quite different from the active-site interactions with inhibitor 2. Herein we describe the design, synthesis, structure-activity studies, molecular modeling, protein-ligand X-ray structure, and biological evaluation of a series of novel and noncovalent inhibitors of SARS-CoV PLpro.
Figure 1.

Structures of PLpro inhibitors 1-3 and 15g
Chemistry
To ascertain the importance of the position of the methoxy substituent in lead inhibitor 3, we have synthesized the corresponding 2-methoxy and 3-methoxybenzyl derivatives. As shown in Scheme 1, Boc-piperidine-4-carboxylic acid 4 was coupled with 2- and 3-methoxybenzylamines 5a and 5b using N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDCI) and 1-hydroxybenzotriazole hydrate (HOBT) in the presence of N-methylmorpholine (NMM) in CH2Cl2 to provide coupling products 6a and 6b in 92% and 94% yield, respectively. Removal of Boc-group by exposure to trifluoroacetic acid (TFA) in CH2Cl2 at 0 °C to 23 °C for 6 h afforded the respective amine. Reductive amination of these amines with 1-naphthaldehyde using Na(OAc)3BH in the presence of acetic acid furnished inhibitors 7a and 7b in 70% and 71% yield, respectively.
Scheme 1.

Reagents and conditions: (a) 5a or 5b, EDCI, HOBT, NMM, CH2Cl2, 23 °C, 5 h; (b) TFA, 0 °C to 23 °C, 6 h; (c) 1-naphthaldehyde, Na(OAc)3BH, AcOH, CH2Cl2, 23 °C, 12 h.
For structure-activity studies and optimization of potency, we planned to synthesize derivatives of both 1- and 2-naphthylethyl-piperidin-4-carboxylic acids and coupled them with various substituted benzylamine derivatives. The synthesis of substituted piperidine-4-carboxylic acids is shown in Scheme 2. Alkylation of dimethylmalonate 8 with commercially available 2-bromomethyl-1,3-dioxolane 9 in the presence of KOtBu in DMSO at 23 °C afforded malonate derivative 10 as described previously.14 Deprotection of the ketal functionalities was carried out by treatment of 10 with 10% aqueous HCl in THF at 23 °C. The reaction was quenched with solid NaHCO3 and the resulting crude dialdehyde was used directly for the subsequent condensation reaction. Condensation of the dialdehyde with various optically active (S)- and (R)- 1-methyl-1-naphthylmethyl amines, 1-methyl-2-naphthylmethyl amines, 2-naphthylmethyl amine, 1-naphthylmethyl amine and dimethyl-1-naphthylmethyl amine 11g10 in aqueous THF for 16 h afforded dihydropyridines 12a-g in 39-62% yield. Catalytic hydrogenation of dihydropyridines 12a-f in ethylacetate at 23 °C provided various piperidine derivatives 13a-f in 60-94% yield.
Scheme 2.

Reagents and conditions: (a) KOt-Bu, DMSO, 23 °C, 48 h (b) 10% HCl, THF, 23 °C, 18 h; (c) NaHCO3, 23 °C, 16 h; (c) H2, PtO2, EtOAc, 23 °C, 2 h.
The synthesis of various test inhibitors is shown in Scheme 3. Treatment of diester 13a-f with NaCN in DMF at reflux for 16 h provided methyl ester 14a-f in 38-92% yield. Dihydropyridine derivative 12g was similarly converted to methyl ester 14g in a two-step sequence. Saponification of 14a-g with aqueous LiOH in a mixture (3:1:1) of THF, methanol, and water at 23 °C for 16 h afforded the corresponding carboxylic acids. Coupling of these resulting carboxylic acids with benzylamine derivatives 5a-d utilizing EDCI in the presence of diisopropylethylamine as described above furnished various inhibitors 15a-k in excellent yield (80-99%).
Scheme 3.

Reagents and conditions: (a) NaCN, DMF, reflux, 16 h; (b) LiOH·H2O, THF/MeOH/H2O (3:1:1), 23 °C, 16 h; (c) 5a-d, EDCI, HOBT, DIPEA, CH2Cl2/DMF (9:1), 23 °C, 15 h.
To evaluate the effect of the corresponding piperazine derivatives, we sought to synthesize racemic piperazine derivative 20 and the synthesis is outlined in Scheme 4. Reductive amination of Boc-piperazine 16 with 1-acetonaphthone 17 using sodium cyanoborohydride in a mixture (50:1) of methanol and acetic acid at 23 °C for 48 h afforded 18 in 24% yield. Removal of the Boc-group by treatment with trifluoroacetic acid in CH2Cl2 at 23 °C for 2 h provided amine 19. Treatment of 4-methoxybenzylamine 5c in the presence of N,N′-carbonyldiimidazole in CH2Cl2 followed by addition of 19 and stirring of the resulting mixture at 23 °C for 4 h afforded piperazine derivative 20 in 90% yield.
Scheme 4.

Reagents and conditions: (a) NaBH3CN, MeOH/AcOH (50:1), 23 °C, 48 h; (b) TFA, CH2Cl2, 23 °C, 2 h; (c) 5c, N,N′-carbonyldiimidazole, CH2Cl2, 23 °C, 4 h.
Results and discussion
The second HTS lead 3 is considerably weaker than the first lead inhibitor 1, a benzamide derivative of 2-naphthyl ethylamine. To enhance activity, we first investigated the effect of 2-methoxy and 3-methoxy derivatives 7a and 7b on PLpro inhibitory activity. As shown in Table 1, 2-methoxy derivative 7a showed a very poor inhibitory activity. The 3-methoxy derivative, 7b, however, displayed slightly better activity than the starting lead 3. Our previous structure-activity of lead 1 established that 1-naphthyl ethyl amides were significantly more potent than the corresponding 2-naphthyl derivative. The X-ray structure of 2 bound to PLpro demonstrated that a (R)-1-naphthylethylamide forms hydrophobic interactions with the Tyr-265 and Tyr-269 aromatic rings and with side chains of Pro-248 and Pro-249.10 The preference for (R)-methyl was also documented as it points into the interior of the enzyme between Tyr-265 and Thr-302. Based upon this ligand-binding site interaction, we elected to incorporate the (R)-methyl group. As shown in Table 1, the (R)-methyl derivative 15a displayed an IC50 value of 1.2 μM. To ascertain the importance of the position of the methoxy group, we synthesized o-methoxy and m-methoxy derivatives. Interestingly, m-methoxy derivative 15b exhibited improvement of enzyme inhibitory activity with an IC50 value of 0.34 μM. The corresponding p-methoxy derivative 15c have also shown similar potency enhancement (>170-fold over 3). However, the 2-methoxy derivative 15a showed a 3-fold reduction in potency over 15b and 15c. We then examined the effect of 2-(R)-naphthylethyl derivatives on potency. As shown, both m-methoxy and o-methoxy benzylamides 15d and 15e displayed significant reductions in potency compared to the 1-(R)-naphthylethyl derivatives 15b and 15a, respectively. Interestingly, the 2-(S)-naphthylethyl derivative 15f is 2-fold more potent than the 2(R)-derivative 15d.
Table 1.
Structure and activity of 1- and 2-naphthylmethyl derivatives
| Compound | Structure | IC50 (μM) |
|---|---|---|
| 3 | 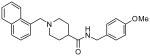 |
59.2 ± 7.8 |
| 7a | 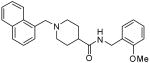 |
116 ± 30 |
| 7b | 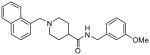 |
30 ± 3 |
| 15a | 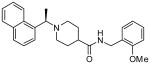 |
1.21 ± 0.04 |
| 15b | 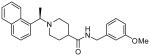 |
0.34 ± 0.01 |
| 15c | 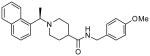 |
0.34 ± 0.01 |
| 15d | 13.2 ± 0.6 | |
| 15e | 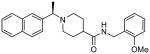 |
34.8 ± 4.0 |
| 15f | 5.8 ± 0.1 | |
| 20 | 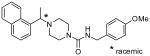 |
>100 |
NA= not active
Table 2.
Structure and activity of benzodioxolane derivartives
| Compound | Structure | IC50 (μM) |
|---|---|---|
| 15g | 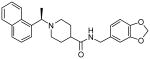 |
0.32 ± 0.01 |
| 15h | 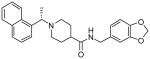 |
0.56 ± 0.03 |
| 15i | 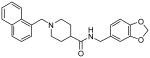 |
∼ 45 |
| 15j | ∼ 100 | |
| 15k | 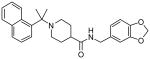 |
>200 |
We next examined the effect of a piperazine ring in place of piperidine in 15c by preparing compound 20. However, this piperazine derivative showed no activity against PLpro. Most likely, the piperazine derivative showed no activity against PLpro due to the structural constraints imposed by the carbon to nitrogen replacement on this ring. The new nitrogen is then attached to the amide group, forming a urea moiety. This urea moiety will tend to be planar, imposing a flexibility constraint. GOLD docking shows the amide to rotate ∼90 degrees away from the optimal hydrogen-bonding orientation (data not shown) of the other active compounds described here.
Our structure-activity studies established that both m-methoxy and p-methoxy derivatives (15b and 15c) are equally potent. Our preliminary modeling studies indicated that either methoxy oxygen (meta or para) is within proximity to form a hydrogen bond with the Gln-270 carboxamide side chain. Based upon these possible interactions, we incorporated a benzodioxolane ring and examined its effect on inhibitory potency. As shown in Table 2, dioxolane derivative 15g exhibits potency comparable to the corresponding m- and p-derivatives 15b and 15c. The corresponding (S)-derivative 15h also shows comparable enzyme inhibitory activity. To examine the preference for a methyl group over a hydrogen at the 1- and 2-naphthylmethyl positions, we have synthesized and evaluated the corresponding unsubstituted derivatives 15i and 15j. As shown, both compounds displayed significant reduction in potency, indicating the importance of the methyl group. We have also examined the corresponding gem-dimethyl derivative 15k. Interestingly, this compound is inactive, indicating that both methyl groups cannot be accommodated by the PLpro active site.
Antiviral activities of selected PLpro inhibitors were determined, and the results are shown in Table 3. The compounds were assayed for their ability to rescue a Vero cell culture from SARS-CoV infection. The viability of virus-infected Vero E6 cells as a function of inhibitor concentration was measured relative to mock-infected cells using a luminescence assay. This protocol allows for the evaluation of both inhibitor efficacy and cytotoxicity. As can be observed from the data presented in Table 3, the original HTS lead (3) does not show any antiviral activity. However, all 2-, 3- and 4-methoxy derivatives 15a-c show comparable antiviral activity. Inhibitor 15f with a 2-naphthyl substituent displayed no antiviral activity. While the (R)-methyl derivative 15g showed slightly better enzyme activity than the (S)-methyl derivative 15h, both inhibitors exhibited the same antiviral potency (EC50 = 9.1 μM). Interestingly, both dioxolane derivatives 15g and 15h showed antiviral activity approximately comparable to the corresponding methoxy or benzamide derivatives reported in our previous studies.10
Table 3.
Evaluation of compounds as inhibitors of SARS-CoV replication in a cell-based assay.
| Compound | IC50 (μM) | EC50 (μM) |
|---|---|---|
| 3 | 59.2 ± 7.8 | NI |
| 15a | 1.21 ± 0.04 | 11.6 ± 0.6 |
| 15b | 0.34 ± 0.01 | 9.7 ± 0.3 |
| 15c | 0.34 ± 0.01 | 10.2 ± 0.5 |
| 15f | 5.8 ± 0.1 | > 25 |
| 15g | 0.32 ± 0.01 | 9.1 ± 0.5 |
| 15h | 0.56 ± 0.03 | 9.1 ± 0.3 |
To obtain molecular insight into the ligand-binding site interactions, the X-ray crystal structure of 15g bound to PLpro was determined. Interestingly, the binding mode and key molecular interactions of inhibitor 15g are quite different than predicted and are different from the active-site interactions with the benzamide-derived inhibitors we previously reported. As shown in Figure 2, the inhibitor binds to the active via a series of interactions including a hydrogen-bond formed between the carboxamide NH of the inhibitor and the backbone carbonyl of Tyr-269, with 15g wrapped around the beta-turn. The 15g bound PLpro crystal structure also confirms the presence of a few structural water molecules conserved between the apo enzyme (PDB id:2FE8) and inhibitor 2 bound PLpro (PDB id: 3E9S). One of the conserved water molecules sits in the P5 pocket shown in Figure 2 as spheres between residues Asp-165, Asp-303 and Thr-302 preventing the inhibitor naphthyl rings from occupying this pocket. In the stereo image of 15g bound PLpro we also show two other water molecules near residue Leu-163 and Lys-158 that may prevent the benzodioxolane ring from flipping down toward Lys-158.
Figure 2.
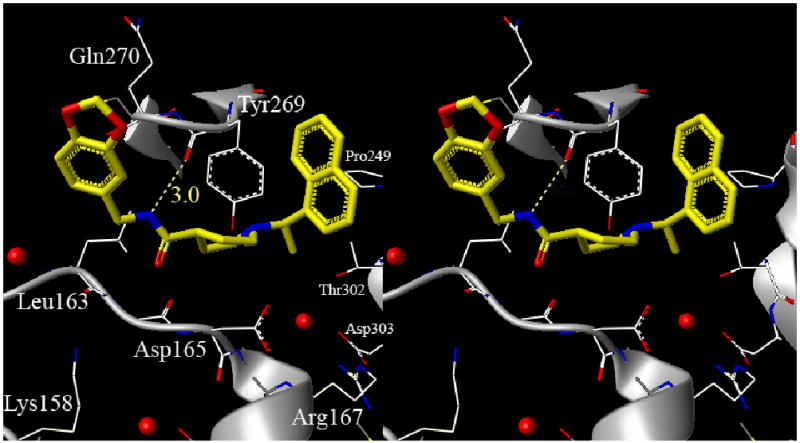
Stereo representation of 15g bound to PLpro, including the conserved waters adjacent to the binding site that may influence the binding configuration, as described in the text.
Figure 3 superimposes 15g and our previously developed inhibitor, 2,10 and demonstrates that the binding mode differs significantly between the two inhibitors. Interestingly, the turn region between Tyr-269 and Gln-270 also shows significant flexibility, particularly in the case of inhibitor 2 (PDB id: 3E9S), where the peptide bond between Tyr-269 and Gln-270 flips by 180 degrees to enable a hydrogen bond interaction between the backbone nitrogen of Tyr-269 and the carboxamide oxygen in inhibitor 2. The carboxyamide nitrogen makes a hydrogen bond with the side chain carboxylate of Asp-165. The carboxy amide nitrogen of inhibitor 15g (yellow) forms a hydrogen bond with the backbone carbonyl oxygen of Tyr-269 (protein shown in grey). The naphthyl rings of both inhibitors 2 and 15g align in a similar fashion in the hydrophobic pocket formed by residues Tyr-269, Tyr-265, Pro-248-249, and Thr-302. The overlapping position of one conserved water molecule observed for both the inhibitor 2-bound PLpro (oxygen atom shown as sphere in pink) crystal structure and inhibitor 15g-bound PLpro (oxygen atom shown as sphere in red) crystal structure is shown as overlapping spheres.
Figure 3.
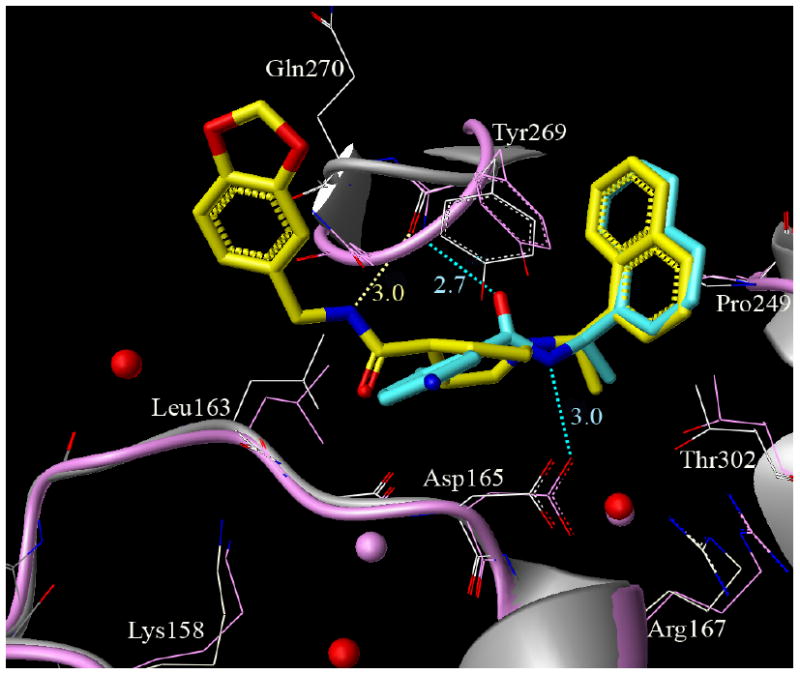
The X-ray structure of inhibitor 15g-bound (yellow) PLpro (grey) (PDB id: WXYZ) superimposed on the X-ray structure of inhibitor 2-bound (cyan) PLpro (pink) (PDB id: 3E9S).
Modeling Studies
To understand the SAR of the analogs of HTS hit compound 3, we used computer modeling to explore the interactions of this series of inhibitors with PLpro. The activity of this series of compounds is independent of stereoisomerism in contrast to the series of compounds synthesized from the first HTS hit compound 1.10 GOLD re-docking of inhibitor 15g into the PLpro crystal structure described above produces a heavy atom RMSD of 1.7Å with the crystal structure conformation of 15g, indicating that docking satisfactorily reproduces the experimental structure. When the inhibitors 15g, 15h and 15k are docked into the ligand removed 15g-bound PLpro crystal structure (with residues Tyr-269 and Gln-270 flagged as flexible), the internal strain scores of the compounds correlate very well with their enzymatic activities. The conserved overlapping water molecules observed in both chains A and B of the 15g bound PLpro crystal structure were included for all docking studies.
To investigate structural basis of the potency insensitivity to the (R)-Me (15g) versus (S)-Me (15h) configuration, we show the docked model of inhibitor 15h superimposed on the crystal structure of 15g-bound PLpro in Figure 4A. From this model, we observe an inversion of the piperidine ring between the (R)-Me and (S)-Me binding modes that allows the naphthyl rings of both isomers to be accommodated in the active site in very similar orientations. The flexible piperidine ring also acts as a spacer group that enables the carboxamide NH of both 15h and 15g to hydrogen bond with the backbone carbonyl oxygen of Tyr-269 in a similar fashion, thereby retaining the potency of both enantiomers. However, the gem-dimethyl substitution in 15k decreases the freedom around the carbon atom and locks the compound in a conformation where one of the methyl groups exhibits a bumping collision with the side chain of Asp-165. One of the methyl groups in 15k shifts almost 1.2 Å toward residue Asp-165 when compared to the single methyl substitution (R)-Me in 15g, as can be seen in Figure 4B. It is important to note that the side chain of this Asp-165 is locked in its position by a hydrogen bond with the backbone NH of Arg-167. Hence, the gem-dimethyl substitution is not favorably accommodated in the active site, because in order to fit the hydrophobic methyl group near the hydrophilic residue, the aspartic acid side chain would have to move out, thereby breaking structural hydrogen bonding with Arg-167.
Figure 4.
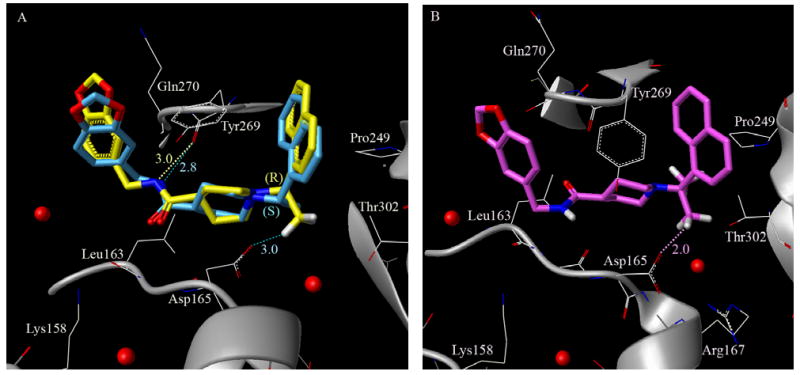
(A) Superposition of enantiomer 15h (blue) with the crystal structure of 15g-bound (yellow) PLpro. (B) Docked alignment of the gem-dimethyl substituted compound in the 15g-ligand removed PLpro crystal structure. The bumping collision of one of the methyl groups of the gem-dimethyl (magenta) 15k with the Asp-165 carboxylate is noted.
This hypothesis is further validated by the GoldScore scoring function of GOLDv4.1 during the docking study. Compound 15k is heavily penalized due to an unfavorable internal energy term (-12 compared to ∼ -6 for both 15g and 15h) which is a sum of the internal torsional strain and internal van der Waals energy terms of the ligand. Docking with flexible residues also suggests that the Gln-270 side chain may adopt conformations that might enable hydrogen bonding interactions with one of the 1,3 benzodioxolane oxygens in 15g and 15h (within 3Å). However, all docked conformations generated for 15k shows a loss of this hydrogen bonding interaction. The closest benzodioxolane oxygen of 15k is at least 4.8Å away from the side chain of Gln-270 (not shown). Figure 4B highlights the potential bumping collision of one of the methyl groups of 15k with Asp-165, demonstrating that two methyl groups cannot be accommodated favorably at this position.
In our previous study, we discussed the SAR of the analogs of our first HTS hit 1 and the evolution of inhibitor 2 in great detail.10 In distinct contrast to the present work, that series of compounds is extremely sensitive to the enantiomeric form of the compound. From docking studies we concluded that the (R)-Me form was active whereas the (S)-Me was inactive because the (S)-Me conformation pushed the carboxamide group of the inhibitor away from the backbone NH of Tyr-269, inhibiting hydrogen bond formation with the loop residue.
Conclusion
In conclusion, we have designed, synthesized and evaluated a novel series of SARS-CoV PLpro inhibitors. Initial lead structure 3 (IC50 = 59.2 μM) was discovered via high-throughput screening of a library of diverse compounds. Our preliminary structure-activity studies and systematic modification guided by X-ray crystal structure of 2-bound PLpro and subsequent molecular modeling resulted in a potent inhibitor 15g with enzyme inhibitory IC50 value of 320 nM and antiviral EC50 value of 9.1 μM in SARS-CoV- infected Vero E6 cells. Interestingly, the corresponding (S)-isomer 15h is only slightly less potent (IC50 = 560 nM) in PLpro inhibitory assays but equipotent in antiviral assay. The corresponding gem-dimethyl derivative 15k is significantly less potent. A protein-ligand X-ray structure of 15g-bound PLpro was determined to 2.6 Å resolution. This structure provided critical molecular insight into the ligand binding site interactions. It appears that the key active site interactions are quite different from the earlier series of inhibitors. Further design of improved reversible SARS-CoV PLpro inhibitors is currently underway in our laboratories.
Experimental Section
Chemistry
1H-NMR and 13C-NMR spectra were recorded on Varian Oxford 300 and Bruker Avance 400 spectrometers. Optical rotations were recorded on a Perkin-Elmer 341 polarimeter. Anhydrous solvent was obtained as follows: CH2Cl2 by distillation from CaH2, THF by distillation from Na and benzophenone. All other solvents were reagent grade. Column chromatography was performed with Whatman 240-400 mesh silica gel under low pressure of 3-5 psi. TLC was carried out with E. Merck silica gel 60-F-254 plates. Purity of all test compounds were determined by HRMS and HPLC analysis in the different solvent systems. All test compounds showed ≥95% purity.
1-(t-Butoxycarbonyl)-4-[(3-methoxybenzylamino)carbonyl]piperidine (6b)
To a solution of 1-(t-butoxycarbonyl)piperidine-4-carboxylic acid (344 mg, 1.5 mmol) in dry CH2Cl2 (5 mL), 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC.HCl) (287 mg, 1.5 mmol), 1-hydroxybenzotriazole hydrate (HOBt.H2O) (203 mg, 1.5 mmol), N-methylmorpholine (NMM) (0.16 mL, 1.5 mmol) and 3-methoxybenzyl amine (0.13 mL, 1 mmol) were added successively at 23 °C under argon atmosphere and the resulting reaction mixture was stirred for 5 h at the same temperature. The reaction mixture was quenched with aqueous NaOH solution and extracted with CH2Cl2. The organic layers were dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (40% EtOAc/Hexanes) to furnish 6b (327 mg, 94%) as a viscous liquid. 1H NMR (400 MHz, CDCl3): δ 7.24 (t, J = 7.6 Hz, 1H), 6.76-6.86 (m, 3H), 5.78 (br, 1H), 4.41 (d, J = 5.6 Hz, 2H), 4.12 (br, 2H), 3.79 (s, 3H), 2.73 (br t, J = 11.2 Hz, 2H), 2.25 (tt, J = 4.0 and 11.6 Hz, 1H), 1.82 (br d, J = 12.0 Hz, 2H), 1.65 (ddd, J = 4.1, 12.2 and 24.8 Hz, 2H), 1.45 (s, 9H); 13C NMR (100 MHz, CDCl3): δ 174.1, 159.9, 154.6, 139.7, 129.8, 119.9, 113.4, 112.9, 79.6, 55.2, 43.5, 43.4, 28.6, 28.4.
1-(t-Butoxycarbonyl)- 4-[(2-methoxybenzylamino)carbonyl]piperidine (6a)
The title compound 6a was obtained as described for compound 1-(t-butoxycarbonyl)-4-[(3-methoxybenzylamino)carbonyl]piperidine in 92% yield (viscous liquid). 1H NMR (400 MHz, CDCl3): δ 7.22 (br t, J = 7.2 Hz, 2H), 6.83-6.92 (m, 2H), 6.09 (br, 1H), 4.41 (d, J = 5.8 Hz, 2H), 4.09 (br, 2H), 3.83 (s, 3H), 2.70 (br t, J = 11.1 Hz, 2 H), 2.20 (tt, J = 3.7 and 11.6 Hz, 1H), 1.77 (br d, J = 12.0 Hz, 2H), 1.59 (ddd, J = 4.4, 12.0 and 24.8 Hz, 2H), 1.43 (s, 9H); 13C NMR (100 MHz, CDCl3): δ 173.9, 157.5, 154.6, 129.6, 128.8, 126.1, 120.6, 110.3, 79.5, 55.3, 43.2, 39.2, 28.5, 28.3.
1-[(1-Naphthyl)methyl]- 4-[(3-methoxybenzylamino)carbonyl]piperidine (7b)
To the solution of 1-(t-butoxycarbonyl)-4-[(3-methoxybenzylamino)carbonyl]piperidine (100 mg, 0.287 mmol) in CH2Cl2 (3 mL), trifluoroacetic acid (0.15 mL) was added at 0 °C and the resulting mixture was stirred for 6 h at 23 °C. The reaction mixture was diluted with CH2Cl2 and basified by slow addition of saturated NaHCO3 solution. The layers were separated and the aqueous layer was extracted several times with ethyl acetate. The combined organic layers were dried over anhydrous Na2SO4. The solvent was removed under reduced pressure to furnish the amine. To the crude amine in dry CH2Cl2 (5 mL), 1-naphthaldehyde (77μL, 0.57 mmol), Na(OAc)3BH (121 mg, 0.57 mmol), and AcOH (33 μL, 0.57 mmol) were added successively at 23 °C and the resulting mixture was stirred for 12 h at 23 °C. The reaction mixture was basified with 2N NaOH and diluted with CH2Cl2 and H2O. Organic layer was separated and the aqueous layer extracted with CH2Cl2. The combined organic layers were dried over anhydrous Na2SO4. Solvent was removed under reduced pressure and the resulting residue was purified by column chromatography over silica gel (2% MeOH/CH2Cl2) to provide 1-[(1-naphthyl)methyl]-4-[(3-methoxybenzylamino)carbonyl]piperidine as a viscous liquid (79 mg, 71%). 1H NMR (400 MHz, CDCl3): δ 8.28-8.33 (m, 1H), 7.82-7.88 (m, 1H), 7.77 (dd, J = 2.2 and 7.1 Hz, 1H), 7.44-7.53 (m, 2H), 7.36-7.43 (m, 2H), 7.23 (t, J = 7.8 Hz, 1H), 6.77-6.86 (m, 3H), 5.79 (br, 1H), 4.40 (d, J = 5.7 Hz, 2H), 3.88 (s, 2H), 3.78 (s, 3H), 2.94-3.04 (m, 2H), 2.15 (tt, J = 4.2 and 11.4 Hz, 1 H), 2.06 (dt, J = 2.7 and 11.3 Hz, 2H), 1.72-1.88 (m, 4H); 13C NMR (100 MHz, CDCl3): δ 174. 9, 159.8, 139.9, 134.3, 133.8, 132.5, 129.7, 128.3, 127.8, 127.2, 125.7, 125.6, 125.0, 124.8, 119.9, 113.3, 112.9, 61.3, 55.2, 53.3, 43.6, 43.3, 29.1. IR (neat): 3290, 2922, 1644, 1598,1263 cm-1; MS (ESI): m/z 389 [M+H]+.
1-[(1-Naphthyl)methyl]-4-[(2-methoxybenzylamino)carbonyl]piperidine (7a)
The title compound 7a was obtained as described for compound 7b in 70% yield (viscous liquid). 1H NMR (400 MHz, CDCl3): δ 8.30 (d, J = 7.9 Hz, 1H), 7.84 (d, J = 7.1 Hz, 1H), 7.77 (d, J = 7.1 Hz, 1H), 7.44-7.53 (m, 2H), 7.37-7.43 (m, 2H), 7.21-7.30 (m, 2H), 6.83-6.94 (m, 2H), 5.98 (br s, 1H), 4.43 (d, J = 5.6 Hz, 2H), 3.87 (s, 2H), 3.84 (s, 3H), 2.98 (d, J = 11.2 Hz, 2H), 2.01-2.20 (m, 3H), 1.68-1.84 (m, 4H); 13C NMR (100 MHz, CDCl3): δ 174.6, 157.5, 134.3, 133.8, 132.5, 129.8, 128.8, 128.3, 127.8, 127.2, 126.3, 125.7, 125.6, 125.1, 124.8, 120.7, 110.3, 61.3, 55.3, 53.4, 43.6, 39.3, 29.0. IR (neat): 3305, 1643, 1600, 1242 cm-1; MS (ESI): m/z 389 [M+H]+.
1-[(R)-1-(1-Naphthyl)ethyl]-4,4-bis(methoxycarbonyl)-1,4-dihydropyridine (12a)
A solution of malonate 10 (1.8 g, 5.92 mmol) in 10% hydrochloric acid solution (35 mL) and THF (35 mL) was stirred for 18 h at 23 °C. The solution was neutralized with powdered sodium hydrogen carbonate, and then 1-(R)-naphthylmethylamine 11a (1.0 g, 5.84 mmol) in THF (5 mL) was added. After being stirred for 16 h at 23 °C. The reaction was extracted with EtOAc, and dried over Na2SO4. Removal of the solvent afforded the residue, which was purified by silica gel column chromatography to furnish compound 12a (1.1 g, 54%) as a colorless oil, Rf = 0.74 (hexane : EtOAc = 1:1), [α]20D -58 (c = 1, CHCl3); 1H NMR (300 MHz, CDCl3): δ 7.90 (d, 1H, J = 7.8 Hz), 7.84 (d, 1H, J = 7.8 Hz), 7.80-7.75 (m, 1H), 7.54-7.40 (m, 4H), 6.21 (d, 2H, J = 8.3 Hz), 5.16 (q, 1H, J = 6.6 Hz), 4.77 (d, 2H, J = 8.3 Hz), 3.69 (s, 6H), 1.67 (d, 3H, J = 6.6 Hz); 13C NMR (75 MHz, CDCl3): δ 171.4, 136.2, 133.7, 130.8, 129.2, 128.7, 128.4, 126.3, 125.5, 124.9, 123.7, 122.8, 95.3, 56.8, 54.0, 52.4, 19.4. IR (neat): 2951, 1736, 1249, 1069 cm-1; MS (EI): m/z 352 [M+H]+; HRMS (EI), calcd for C21H22NO4 352.1549, found 352.1553.
1-[(R)-1-(2-Naphthyl)ethyl]-4,4-bis(methoxycarbonyl)-1,4-dihydropyridine (12b)
The title compound was obtained as described in compound 12a in 58% yield (colorless oil). Rf = 0.79 (hexane : EtOAc = 1:1), [α]20D +32 (c 1, CHCl3); 1H NMR (300 MHz, CDCl3): δ 7.84-7.78 (m, 3H), 7.66 (s, 1H), 7.49-7.43 (m, 2H), 7.33 (dd, 1H, J = 1.5 and 8.7 Hz), 6.21 (d, 2H, J = 8.3 Hz), 4.78 (d, 2H, J = 8.3 Hz), 4.59 (q, 1H, J = 6.9 Hz), 3.72 (s, 6H), 1.64 (d, 3H, J = 6.9 Hz); 13C NMR (75 MHz, CDCl3): δ 171.6, 139.2, 133.1, 132.6, 129.6, 128.4, 127.9, 127.7, 127.5, 126.2, 125.9, 124.8, 95.3, 60.4, 54.1, 52.6, 19.5. IR (neat): 2952, 1732, 1253, 1069 cm-1; MS (EI): m/z 292 [M-CO2Me]+; HRMS (EI), calcd for C19H18NO2 292.1337, found [M-CO2Me]+ 292.1345.
1-[(S)-1-(2-Naphthyl)ethyl]-4,4-bis(methoxycarbonyl)-1,4-dihydropyridine (12c)
The title compound was obtained as described in compound 12a in 54% yield (colorless oil). Rf = 0.73 (hexane : EtOAc = 1:1), [α]20D -32 (c 1, CHCl3); MS (EI): m/z 351 [M]+; HRMS (EI), calcd for C21H21NO4 351.1471, found [M]+ 351.1477.
1-[(S)-1-(1-Naphthyl)ethyl]-4,4-bis(methoxycarbonyl)-1,4-dihydropyridine (12d)
The title compound was obtained as described in compound 12a in 42% yield (colorless oil). Rf = 0.77 (hexane : EtOAc = 1:1), [α]20D +57 (c 1, CHCl3); MS (ESI): m/z 374 [M+Na]+; HRMS (ESI), calcd for C21H21NO4Na 374.1368, found 374.1371.
1-(1-Naphthylmethyl)-4,4-bis(methoxycarbonyl)-1,4-dihydropyridine (12e)
The title compound was obtained as described in compound 12a in 39% yield (colorless oil). Rf = 0.82 (hexane : EtOAc = 1:1); 1H NMR (300 MHz, CDCl3): δ 7.86-7.80 (m, 2H), 7.77 (d, 1H, J = 8.7 Hz), 7.54-7.48 (m, 2H), 7.42 (t, 1H, J = 8.3 Hz), 7.30 (d, 1H, J = 6.9 Hz), 6.15 (d, 2H, J = 8.3 Hz), 4.82 (d, 2H, J = 8.3 Hz), 4.74 (s, 2H), 3.73 (s, 6H); 13C NMR (75 MHz, CDCl3): δ 171.6, 133.5, 132.6, 131.1, 130.7, 128.7, 128.2, 126.4, 125.8, 125.4, 125.1, 122.5, 95.3, 54.5, 53.7, 52.7. IR (neat): 2951, 1735, 1253, 1067 cm-1; MS (EI): m/z 278 [M-CO2Me]+; HRMS (EI), calcd for C18H16NO2 278.1181, found 278.1185.
1-(2-Naphthylmethyl)-4,4-bis(methoxycarbonyl)-1,4-dihydropyridine (12f)
The title compound was obtained as described in compound 12a in 62% yield (colorless oil). Rf = 0.80 (hexane : EtOAc = 1:1); 1H NMR (300 MHz, CDCl3): δ 7.80-7.77 (m, 3H), 7.60 (s, 1H), 7.48-7.41 (m, 2H), 7.28 (d, 1H, J = 1.8 Hz), 6.16 (d, 2H, J = 8.0 Hz), 4.81 (d, 2H, J = 8.0 Hz), 4.41 (s, 2H), 3.73 (s, 6H); 13C NMR (75 MHz, CDCl3): δ 171.5, 134.9, 133.1, 132.6, 131.2, 128.5, 127.7, 127.5, 126.2, 125.9, 125.8, 124.8, 95.3, 56.9, 53.6, 52.6. IR (neat): 2950, 1731, 1253, 1066 cm-1; MS (EI): m/z 278 [M-CO2Me]+; HRMS (EI), calcd for C18H16NO2 278.1181, found 278.1184.
1-[1-Methyl-1-(1-naphthyl)ethyl]-4,4-bis(methoxycarbonyl)-1,4-dihydropyridine (12g)
The title compound was obtained as described in compound 12a in 41% yield (colorless oil). Rf = 0.77 (hexane : EtOAc = 1:1); 1H NMR (300 MHz, CDCl3): δ 8.20-8.16 (m, 1H), 7.89 (d, 1H, J = 7.8 Hz), 7.84-7.80 (m, 1H), 7.77 (d, 1H, J = 7.8 Hz), 7.52-7.36 (m, 4H), 6.27 (d, 2H, J = 8.1 Hz), 4.77 (d, 2H, J = 8.1 Hz), 3.69 (s, 6H), 1.77 (s, 6H); 13C NMR (75 MHz, CDCl3): δ 171.6, 140.1, 134.7, 130.5, 129.1, 129.0, 127.7, 126.1, 126.0, 125.3, 124.7, 124.0, 96.0, 61.9, 53.8, 52.5, 28.7. IR (neat): 2951, 1736, 1252, 1062 cm-1; MS (ESI): m/z 388 [M+Na]+; HRMS (ESI), calcd for C22H23NO4Na 388.1525, found 388.1529.
1-[(R)-1-(1-Naphthyl)ethyl]-4,4-bis(methoxycarbonyl)piperidine (13a)
To a stirred solution of dihydropyridine 12a (1.1 g, 3.1 mmol) in EtOAc (75 mL) was added platinum(IV) oxide (100 mg) and it was allowed to stir for 2 h at 23 °C under H2 atmosphere. The reaction was filtered through celite pad and the filtrate was concentrated under reduced pressure. The residue was purified by silica gel column chromatography to furnish compound 13a (942 mg, 88%) as a colorless oil, Rf = 0.7 (hexane : EtOAc = 1:1); [α]20D +9 (c 1, CHCl3); 1H NMR (300 MHz, CDCl3): δ 8.44-8.41 (m, 1H), 7.85-7.81 (m, 1H), 7.73 (d, 1H, J = 8.1 Hz), 7.56 (d, 1H, J = 6.9 Hz), 7.50-7.39 (m, 3H), 4.03 7.73 (q, 1H, J = 6.3 Hz), 3.72 (s, 6H), 2.58-2.56 (m, 2H), 2.47-2.40 (m, 2H), 2.20-2.04 (m, 4H), 1.44 (d, 3H, J = 6.3 Hz); 13C NMR (75 MHz, CDCl3): δ 171.8, 140.7, 134.0, 131.5, 128.6, 127.3, 125.4, 125.3, 125.2, 124.5, 124.1, 61.7, 53.3, 52.4, 47.9, 31.2, 18.7. IR (neat): 2952, 1734, 1251 cm-1; MS (ESI): m/z 356 [M+H]+; HRMS (ESI), calcd for C21H26NO4 356.1862, found 356.1866.
1-[(R)-1-(2-Naphthyl)ethyl]-4,4-bis(methoxycarbonyl)piperidine (13b)
The title compound was obtained as described in compound 13a in 74% yield (colorless oil). Rf = 0.48 (hexane: EtOAc = 1:1), [α]20D +23 (c 1, CHCl3); 1H NMR (400 MHz, CDCl3): δ 7.83-7.80 (m, 3H), 7.72 (s, 1H), 7.53 (dd, 1H, J = 1.3 and 8.7 Hz), 7.49-7.42 (m, 2H), 3.73 (s, 6H), 3.49 (q, 1H, J = 6.7 Hz), 2.57-2.55 (bm, 2H), 2.47-2.42 (m, 2H), 2.24-2.13 (m, 4H), 1.42 (d, 3H, J = 6.7 Hz); 13C NMR (100 MHz, CDCl3): δ 171.7, 142.1, 133.3, 132.7, 128.0, 127.7, 127.5, 125.8, 125.8, 125.4, 64.8, 53.2, 52.5, 47.9, 31.1, 19.6. IR (neat): 2951, 1731, 1250, 1073 cm-1; MS (EI): m/z 355 [M]+; HRMS (EI), calcd for C21H25NO4 355.1784, found 355.1781.
1-[(S)-1-(2-Naphthyl)ethyl]-4,4-bis(methoxycarbonyl)piperidine (13c)
The title compound was obtained as described in compound 13a in 67% yield (colorless oil). Rf = 0.48 (hexane : EtOAc = 1:1), [α]20D -24 (c 1, CHCl3); MS (EI): m/z 355 [M]+; HRMS (EI), calcd for C21H25NO4 355.1784, found 355.1786.
1-[(S)-1-(1-Naphthyl)ethyl]-4,4-bis(methoxycarbonyl)piperidine (13d)
The title compound was obtained as described in compound 13a in 87% yield (colorless oil). Rf = 0.7 (hexane : EtOAc = 1:1), [α]20D -9 (c 1, CHCl3); MS (ESI): m/z 356 [M+H]+; HRMS (ESI), calcd for C21H26NO4 356.1862, found 356.1865.
1-(1-Naphthylmethyl)-4,4-bis(methoxycarbonyl)piperidine (13e)
The title compound was obtained as described in compound 13a in 60% yield (colorless oil). Rf = 0.70 (hexane : EtOAc = 1:1); 1H NMR (300 MHz, CDCl3): δ 8.29-8.26 (m, 1H), 7.84-7.81 (m, 1H), 7.77-7.73 (m, 1H), 7.52-7.43 (m, 2H), 7.38-7.36 (m, 2H), 3.84 (s, 2H), 3.72 (s, 6H), 2.48 (t, 4H, J = 5.4 Hz), 2.13 (t, 4H, J = 5.4 Hz); 13C NMR (75 MHz, CDCl3): δ 171.7, 134.1, 133.8, 132.5, 128.3, 127.9, 127.2, 125.6, 125.5, 125.0, 124.8, 61.2, 53.3, 52.5, 50.6, 31.0. IR (neat): 2950, 1732, 1254, 1072 cm-1; MS (EI): m/z 341 [M]+; HRMS (EI), calcd for C20H23NO4 341.1627, found 341.1630.
1-(2-Naphthylmethyl)-4,4-bis(methoxycarbonyl)piperidine (13f)
The title compound was obtained as described in compound 13a in 94% yield (colorless oil). Rf = 0.48 (hexane : EtOAc = 1:1); 1H NMR (400 MHz, CDCl3): δ 7.83-7.78 (m, 3H), 7.72 (s, 1H), 7.50-7.42 (m, 3H), 3.74 (s, 6H), 3.61 (s, 2H), 2.48 (bm, 4H), 2.19 (t, 4H, J = 5.5 Hz); 13C NMR (100 MHz, CDCl3): δ 171.7, 135.9, 133.2, 132.7, 127.8, 127.6, 127.6, 127.5, 127.2, 125.9, 125.5, 63.2, 53.2, 52.5, 50.5, 30.9. IR (neat): 2950, 1732, 1254, 1073 cm-1; MS (EI): m/z 341 [M]+; HRMS (EI), calcd for C20H23NO4 341.1627, found [M]+ 341.1626.
1-[1-Methyl-1-(1-naphthyl)ethyl]-4,4-bis(methoxycarbonyl)piperidine (13g)
The title compound 13 g was obtained as described in compound 13a in 93% yield (colorless oil). Rf = 0.29 (hexane : EtOAc = 9:1), 1H NMR (400 MHz, CDCl3): δ 9.59 (d, 1H, J = 7.9 Hz), 7.84 (dd, 1H, J = 2.2 and 7.0 Hz), 7.74 (d, 1H, J = 8.0 Hz), 7.52-7.44 (m, 3H), 7.37 (t, 1H, J = 7.7 Hz), 3.74 (s, 6H), 2.63 (bm, 4H), 2.14 (bm, 4H), 1.59 (s, 6H); 13C NMR (100 MHz, CDCl3): δ 171.8, 143.7, 134.9, 132.0, 128.6, 128.2, 128.0, 125.1, 124.7, 124.4, 123.6, 62.2, 53.6, 52.4, 43.4, 31.8, 23.6. IR (neat): 2973, 1735, 1251, 1124, 780 cm-1; MS (ESI): m/z 370 [M+H]+; HRMS (ESI), calcd for C22H28NO4 370.2018, found 370.2013.
1-[(R)-1-(1-Naphthyl)ethyl]-4-methoxycarbonylpiperidine (14a)
To a stirred solution of dimethylmalonate 13a (917 mg, 2.58 mmol) in DMF (25 mL) was added sodium cyanide (190 mg, 3.88 mmol) at 23 °C and it was allowed to stir for 16 h at reflux temperature. The reaction was diluted with EtOAc and washed with water. The organic layer was dried over Na2SO4, filtered and concentrated. The residue was purified by silica gel column chromatography to furnish compound 14a (704 mg, 92%) as a colorless oil, Rf = 0.56 (CH2Cl2: MeOH = 9:1), [α]20D +2 (c 1, CHCl3); 1H NMR (400 MHz, CDCl3): δ 8.48 (dd, 1H, J = 1.2 and 7.6 Hz), 7.87 (d, 1H, J = 7.1 Hz), 7.76 (d, 1H, J = 8.1 Hz), 7.60 (d, 1H, J = 7.1 Hz), 7.53-7.43 (m, 3H), 4.12 (q, 1H, J = 6.7 Hz), 3.68 (s, 3H), 3.17-3.15 (m, 1H), 2.87-2.84 (m, 1H), 2.35-2.27 (m, 1H), 2.10 (ddd, 2H, J = 2.6, 11.2 and 19.8 Hz), 1.97-1.92 (m, 1H), 1.83-1.71 (m, 3H), 1.49 (d, 3H, J = 6.7 Hz); 13C NMR (100 MHz, CDCl3): δ 175.8, 140.8, 134.0, 131.6, 128.6, 127.2, 125.4, 125.3, 125.3, 124.5, 124.2, 61.6, 51.5, 49.1, 41.3, 28.7, 28.6, 18.6. IR (neat): 2950, 1732, 1169, 780 cm-1; MS (EI): m/z 297 [M]+; HRMS (EI), calcd for C19H23NO2 297.1729, found 297.1730.
1-[(R)-1-(2-Naphthyl)ethyl]-4-methoxycarbonylpiperidine (14b)
The title compound was obtained as described in compound 14a in 78% yield (colorless oil). Rf = 0.43 (CH2Cl2: MeOH = 9:1), [α]20D +16 (c 1, CHCl3); 1H NMR (400 MHz, CDCl3): δ 7.84-7.80 (m, 3H), 7.72 (s, 1H), 7.53 (dd, 1H, J = 1.1 and 8.4 Hz), 7.50-7.43 (m, 2H), 3.67 (s, 3H), 3.57 (q, 1H, J = 6.8 Hz), 3.08-3.06 (m, 1H), 2.86-2.83 (m, 1H), 2.29-2.22 (m, 1H), 2.09-1.91 (m, 3H), 1.87-1.71 (m, 3H), 1.45 (d, 3H, J = 6.8 Hz); 13C NMR (100 MHz, CDCl3): δ 175.7, 141.7, 133.3, 132.7, 127.8, 127.7, 127.5, 126.0, 125.9, 125.8, 125.4, 64.7, 51.5, 50.5, 49.6, 41.2, 28.5, 19.3. IR (neat): 2949, 1732, 1258, 1172 cm-1; MS (EI): m/z 297 [M]+; HRMS (EI), calcd for C19H23NO2 297.1729, found [M]+ 297.1732.
1-[(S)-1-(2-Naphthyl)ethyl]-4-methoxycarbonylpiperidine (14c)
The title compound was obtained as described in compound 14a in 90% yield (colorless oil). Rf = 0.47 (CH2Cl2 : MeOH = 9:1), [α]20D -15 (c 1, CHCl3); MS (EI): m/z 297 [M]+; HRMS (EI), calcd for C19H23NO2 297.1729, found 297.1731.
1-[(S)-1-(1-Naphthyl)ethyl]-4-methoxycarbonylpiperidine (14d)
The title compound was obtained as described in compound 14a in 76% yield (colorless oil). Rf = 0.57 (CH2Cl2 : MeOH = 9:1); [α]20D -2 (c 1, CHCl3); MS (EI): m/z 297 [M]+; HRMS (EI), calcd for C19H23NO2 297.1729, found 297.1729.
1-(1-Naphthylmethyl)-4-methoxycarbonylpiperidine (14e)
The title compound was obtained as described in compound 14a in 38% yield (colorless oil). Rf = 0.53 (CH2Cl2 : MeOH = 9:1); 1H NMR (400 MHz, CDCl3): δ 8.31 (d, 1H, J = 7.8 Hz), 7.86 (dd, 1H, J = 1.6 and 7.1 Hz), 7.78 (d, 1H, J = 7.1 Hz), 7.53-7.47 (m, 2H), 7.44-7.39 (m, 2H), 3.89 (s, 2H), 3.68 (s, 3H), 2.94-2.88 (m, 2H), 2.37-2.30 (m, 1H), 2.12 (dt, 2H, J = 1.6 and 11.2 Hz), 1.90-1.86 (m, 2H), 1.81-1.72 (m, 2H); 13C NMR (100 MHz, CDCl3): δ 175.7, 134.2, 133.7, 132.5, 128.3, 127.8, 127.2, 125.6, 125.5, 125.0, 124.7, 61.3, 53.1, 51.5, 41.1, 28.3. IR (neat): 2949, 1736, 1167, 788 cm-1; MS (EI): m/z 283 [M]+; HRMS (EI), calcd for C18H21NO2 283.1572, found 283.1569.
1-(2-Naphthylmethyl)-4-methoxycarbonylpiperidine (14f)
The title compound was obtained as described in compound 14a in 47% yield (colorless oil). Rf = 0.44 (CH2Cl2 : MeOH = 9:1); 1H NMR (400 MHz, CDCl3): δ 7.84-7.80 (m, 3H), 7.73 (s, 1H), 7.51-7.43 (m, 3H), 3.68 (s, 3H), 3.65 (s, 2H), 2.93-2.88 (m, 2H), 2.36-2.28 (m, 1H), 2.08 (dt, 2H, J = 2.2 and 11.4 Hz), 1.94-1.75 (m, 4H); 13C NMR (100 MHz, CDCl3): δ 175.6, 135.9, 133.2, 132.7, 127.8, 127.6, 127.6, 127.5, 127.3, 125.9, 125.5, 63.3, 52.9, 51.6, 41.0, 28.2. IR (neat): 2948, 1736, 1167 cm-1; MS (ESI): m/z 284 [M+H]+; HRMS (ESI), calcd for C18H22NO2 284.1651, found 284.1652.
1-[1-Methyl-1-(1-naphthyl)ethyl]-4-methoxycarbonylpiperidine (14g)
The title compound was obtained as described in compound 14a in 87% yield (colorless oil). Rf = 0.43 (hexane : EtOAc = 9:1), 1H NMR (400 MHz, CDCl3): δ 9.63-9.60 (m, 1H), 7.87-7.84 (m, 1H), 7.76 (d, 1H, J = 8.0 Hz), 7.51-7.46 (m, 3H), 7.39 (t, 1H, J = 7.8 Hz), 3.69 (s, 3H), 2.96 (bs, 2H), 2.34-2.28 (m, 3H), 1.84-1.75 (bm, 4H), 1.61 (s, 6H); 13C NMR (100 MHz, CDCl3): δ 175.9, 144.1, 134.8, 132.0, 128.6, 128.4, 127.9, 125.1, 124.7, 124.4, 123.5, 62.3, 51.5, 45.9, 41.9, 29.2, 22.7. IR (neat): 2950, 1736, 1171, 780 cm-1; MS (EI): m/z 311 [M]+; HRMS (EI), calcd for C20H25NO2 311.1885, found 311.1891.
1-[(R)-1-(1-Naphthyl)ethyl]-4-(2-methoxybenzylamino)carbonylpiperidine (15a)
To a stirred solution of ester 14a (106 mg, 0.36 mmol) in THF/MeOH/H2O (3:1:1) (8 mL) was added LiOH·H2O (22.4 mg, 0.53 mmol) at 0 °C and it was allowed to stir for 16 h at 23 °C. The reaction was concentrated and added saturated NaHCO3 solution. The mixture was extracted with Et2O. The aqueous layer was adjusted to pH 2 with 10% HCl solution and extracted with EtOAc. The organic layers were dried over Na2SO4, filtered and concentrated to give the corresponding acid as a colorless oil. To a solution of acid (0.36 mmol), N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDCI) (138.0 mg, 0.72 mmol) and 1-hydroxybenzotriazole hydrate (HOBT) (97.3 mg, 0.72 mmol) in dry CH2Cl2/DMF (9:1) (8 mL) was added piperonylamine 6d (52.7 μL, 0.40 mmol) and diisopropylethylamine (0.38 mL, 2.2 mmol) at 0 °C under argon atmosphere and it was allowed to stir for 15 h at 23 °C. The reaction mixture was quenched with water and extracted with CH2Cl2. The organic layers were dried over Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by silica gel column chromatography to furnish compound 15a (143 mg, 99%) as a white amorphous solid, Rf = 0.42 (CH2Cl2 : MeOH = 9:1); [α]20D -2 (c 1, CHCl3); 1H NMR (400 MHz, CDCl3): δ 8.46 (d, 1H, J = 7.8 Hz), 7.86-7.84 (m, 1H), 7.74 (d, 1H, J = 8.1 Hz), 7.58 (d, 1H, J = 7.0 Hz), 7.51-7.41 (m, 3H), 7.23 (d, 1H, J = 7.3 Hz), 6.90 (t, 1H, J = 7.3 Hz), 6.85 (d, 1H, J = 8.4 Hz), 6.17 (bt, 1H, J = 5.8 Hz), 4.44 (d, 2H, J = 5.8 Hz), 4.10 (q, 1H, J = 6.6 Hz), 3.81 (s, 3H), 3.23-3.20 (m, 1H), 2.87-2.85 (m, 1H), 2.12-1.95 (m, 3H), 1.89-1.68 (m, 4H), 1.47 (d, 3H, J = 6.6 Hz); 13C NMR (100 MHz, CDCl3): δ 174.9, 157.5, 140.9, 134.1, 131.7, 129.6, 128.7, 128.7, 127.3, 126.5, 125.5, 125.4, 125.3, 124.5, 124.3, 120.7, 110.3, 61.7, 55.3, 52.0, 49.2, 43.7, 39.1, 29.3, 18.7. IR (neat): 3302, 2938, 1650, 1243, 753 cm-1; MS (ESI): m/z 403 [M+H]+; HRMS (ESI), calcd for C26H31N2O2 403.2386, found 403.2388.
1-[(R)-1-(1-Naphthyl)ethyl]-4-(3-methoxybenzylamino)carbonylpiperidine (15b)
The title compound was obtained as described in compound 15a in 95% yield (white amorphous solid). Rf = 0.49 (CH2Cl2 : MeOH = 9:1); [α]20D -2 (c 1, CHCl3); 1H NMR (400 MHz, CDCl3): δ 8.45 (d, 1H, J = 7.7 Hz), 7.86-7.84 (m, 1H), 7.74 (d, 1H, J = 8.1 Hz), 7.58 (d, 1H, J = 7.1 Hz), 7.51-7.41 (m, 3H), 6.83-6.78 (m, 3H), 6.12 (bt, 1H, J = 5.7 Hz), 4.37 (d, 2H, J = 5.7 Hz), 4.10 (q, 1H, J = 6.6 Hz), 3.76 (s, 3H), 3.23-3.20 (m, 1H), 2.90-2.85 (m, 1H), 2.14-1.95 (m, 3H), 1.89-1.69 (m, 4H), 1.47 (d, 3H, J = 6.6 Hz); 13C NMR (100 MHz, CDCl3): δ 175.2, 159.8, 140.7, 140.1, 134.0, 131.6, 129.6, 128.6, 127.2, 125.4, 125.3, 125.3, 124.5, 124.2, 119.8, 113.2, 112.7, 61.6, 55.1, 51.8, 49.1, 43.5, 43.2, 29.3, 18.6. IR (neat): 3293, 2937, 1644, 1263, 781 cm-1; MS (EI): m/z 402 [M]+; HRMS (EI), calcd for C26H30N2O2 402.2307, found 402.2303.
1-[(R)-1-(1-Naphthyl)ethyl]-4-(4-methoxybenzylamino)carbonylpiperidine (15c)
The title compound was obtained as described in compound 15a in 88% yield (white amorphous solid). Rf = 0.56 (CH2Cl2 : MeOH = 9:1); [α]20D -2 (c 1, CHCl3); 1H NMR (400 MHz, CDCl3): δ 8.45 (d, 1H, J = 7.7 Hz), 7.86-7.84 (m, 1H), 7.74 (d, 1H, J = 8.1 Hz), 7.57 (d, 1H, J = 7.1 Hz), 7.51-7.41 (m, 3H), 7.27 (d, 2H, J = 8.5 Hz), 6.84 (d, 2H, J = 8.5 Hz), 5.97 (bt, 1H, J = 5.6 Hz), 4.33 (d, 2H, J = 5.6 Hz), 4.10 (q, 1H, J = 6.7 Hz), 3.77 (s, 3H), 3.23-3.20 (m, 1H), 2.89-2.86 (m, 1H), 2.13-1.95 (m, 3H), 1.89-1.68 (m, 4H), 1.47 (d, 3H, J = 6.7 Hz); 13C NMR (100 MHz, CDCl3): δ 175.0, 158.9, 140.7, 134.0, 131.6, 130.5, 129.0, 128.6, 127.2, 125.4, 125.3, 125.3, 124.5, 124.2, 114.0, 61.6, 55.2, 51.8, 49.1, 43.5, 42.8, 29.3, 18.6. IR (neat): 3292, 2932, 1513, 1644, 1249, 781 cm-1; MS (EI): m/z 402 [M]+; HRMS (EI), calcd for C26H30N2O2 402.2307, found 402.2299.
1-[(R)-1-(2-Naphthyl)ethyl]-4-(3-methoxybenzylamino)carbonylpiperidine (15d)
The title compound was obtained as described in compound 15a in 94% yield (white amorphous solid). Rf = 0.43 (CH2Cl2 : MeOH = 9:1); [α]20D +10 (c 1, CHCl3); 1H NMR (400 MHz, CDCl3): δ 7.82-7.79 (m, 3H), 7.70 (s, 1H), 7.52 (dd, 1H, J = 1.2 and 8.4 Hz), 7.49-7.42 (m, 2H), 7.22 (t, 1H, J = 7.6 Hz), 6.83-6.79 (m, 3H), 5.95 (bt, 1H, J = 5.7 Hz), 4.38 (d, 2H, J = 5.7 Hz), 3.77 (s, 3H), 3.56 (q, 1H, J = 6.7 Hz), 3.16-3.14 (m, 1H), 2.90-2.88 (m, 1H), 2.11-1.91 (m, 3H), 1.89-1.70 (m, 4H), 1.44 (d, 3H, J = 6.7 Hz); 13C NMR (100 MHz, CDCl3): δ 175.0, 159.8, 141.7, 140.0, 133.2, 132.6, 129.6, 127.8, 127.7, 127.5, 125.9, 125.9, 125.8, 125.4, 119.8, 113.2, 112.8, 64.7, 55.1, 50.8, 49.7, 43.4, 43.2, 29.1, 19.3. IR (neat): 3296, 2932, 1645, 1264 cm-1; MS (ESI): m/z 403 [M+H]+; HRMS (ESI), calcd for C26H31N2O2 403.2386, found 403.2390.
1-[(R)-1-(2-Naphthyl)ethyl]-4-(2-methoxybenzylamino)carbonylpiperidine (15e)
The title compound was obtained as described in compound 15a in 98% yield (white amorphous solid). Rf = 0.47 (CH2Cl2 : MeOH = 9:1); [α]20D +12 (c 1, CHCl3); 1H NMR (400 MHz, CDCl3): δ 7.82-7.79 (m, 3H), 7.70 (s, 1H), 7.51 (d, 1H, J = 8.4 Hz), 7.49-7.43 (m, 2H), 7.28-7.24 (m, 2H), 6.93-6.85 (m, 2H), 6.02 (bt, 1H, J = 5.7 Hz), 4.43 (d, 2H, J = 5.7 Hz), 3.84 (s, 3H), 3.57 (q, 1H, J = 6.7 Hz), 3.16-3.14 (m, 1H), 2.90-2.88 (m, 1H), 2.08-1.86 (m, 4H), 1.84-1.64 (m, 3H), 1.45 (d, 3H, J = 6.7 Hz); 13C NMR (100 MHz, CDCl3): δ 174.6, 157.5, 141.6, 133.2, 132.6, 129.7, 129.0, 128.8, 128.2, 127.8, 127.7, 127.5, 126.3, 126.0, 125.8, 125.4, 120.6, 110.2, 64.7, 55.2, 50.8, 49.8, 43.5, 39.2, 29.1, 19.3. IR (neat): 3306, 2933, 1645, 1243, 751 cm-1; MS (ESI): m/z 403 [M+H]+; HRMS (ESI), calcd for C26H31N2O2 403.2386, found 403.2394.
1-[(S)-1-(2-Naphthyl)ethyl]-4-(3-methoxybenzylamino)carbonylpiperidine (15f)
The title compound was obtained as described in compound 15a in 83% yield (white amorphous solid). Rf = 0.37 (CH2Cl2 : MeOH = 9:1); [α]20D -10 (c 1, CHCl3); MS (ESI): m/z 403 [M+H]+; HRMS (ESI), calcd for C26H31N2O2 403.2386, found 403.2392.
1-[(R)-1-(1-Naphthyl)ethyl]-4-[3,4-(methylenedioxy)benzylamino]carbonylpiperidine (15g)
The title compound was obtained as described in compound 15a in 93% yield (white amorphous solid). Rf = 0.47 (CH2Cl2 : MeOH = 9:1); [α]20D -2 (c 1, CHCl3); 1H NMR (400 MHz, CDCl3): δ 8.45 (d, 1H, J = 7.5 Hz), 7.86-7.83 (m, 1H), 7.73 (d, 1H, J = 8.1 Hz), 7.56 (d, 1H, J = 6.9 Hz), 7.51-7.40 (m, 3H), 6.73-6.66 (m, 3H), 6.20 (bt, 1H, J = 5.7 Hz), 5.89 (s, 2H), 4.29 (d, 2H, J = 5.7 Hz), 4.08 (q, 1H, J = 6.7 Hz), 3.22-3.19 (m, 1H), 2.88-2.85 (m, 1H), 2.13-1.94 (m, 3H), 1.87-1.67 (m, 4H), 1.46 (d, 3H, J = 6.7 Hz); 13C NMR (100 MHz, CDCl3): δ 175.2, 147.8, 146.8, 140.7, 134.0, 132.4, 131.6, 128.6, 127.2, 125.4, 125.3, 125.3, 124.5, 124.2, 120.8, 108.2, 100.9, 61.6, 51.8, 49.1, 43.5, 43.0, 29.3, 18.6. IR (neat): 3294, 2924, 1644, 1489, 1252, 1040, 781 cm-1; MS (ESI): m/z 417 [M+H]+; HRMS (ESI), calcd for C26H29N2O3 417.2178, found 417.2178.
1-[(S)-1-(1-Naphthyl)ethyl]-4-[3,4-(methylenedioxy)benzylamino]carbonylpiperidine (15h)
The title compound was obtained as described in compound 15a in 80% yield (white amorphous solid). Rf = 0.56 (CH2Cl2 : MeOH = 9:1); [α]20D +2 (c 1, CHCl3); MS (EI): m/z 417 [M+H]+; HRMS (EI), calcd for C26H29N2O3 417.2178, found 417.2173.
1-(1-Naphthylmethyl)-4-[3,4-(methylenedioxy)benzylamino]carbonylpiperidine (15i)
The title compound was obtained as described in compound 15a in >99% yield (white amorphous solid). Rf = 0.48 (CH2Cl2 : MeOH = 9:1); 1H NMR (400 MHz, CDCl3): δ 8.30-8.28 (m, 1H), 7.84 (d, 1H, J = 7.2 Hz), 7.77 (d, 1H, J = 7.8 Hz), 7.52-7.45 (m, 2H), 7.41-7.37 (m, 2H), 6.74-6.67 (m, 3H), 5.91 (bm, 1H), 5.91 (s, 2H), 4.30 (d, 2H, J = 5.7 Hz), 3.87 (s, 2H), 2.99-2.96 (m, 2H), 2.35-2.29 (m, 1H), 2.18-2.01 (m, 3H), 1.88-1.72 (m, 3H); 13C NMR (100 MHz, CDCl3): δ 174.9, 147.8, 146.8, 134.1, 133.7, 132.5, 132.2, 128.3, 127.8, 127.2, 126.0, 125.6, 125.5, 125.0, 124.7, 120.9, 108.2, 101.0, 61.2, 53.3, 43.4, 43.1, 28.9. IR (neat): 3307, 2924, 1645, 1490, 1252, 1040 cm-1; MS (ESI): m/z 403 [M+H]+; HRMS (ESI), calcd for C25H27N2O3 403.2022, found 403.2025.
1-(2-Naphthylmethyl)-4-[3,4-(methylenedioxy)benzylamino]carbonylpiperidine (15j)
The title compound was obtained as described in compound 15a in 88% yield (white amorphous solid). Rf = 0.42 (CH2Cl2 : MeOH = 9:1); 1H NMR (400 MHz, CDCl3): δ 7.82-7.78 (m, 3H), 7.72 (s, 1H), 7.49-7.42 (m, 3H), 6.74-6.68 (m, 3H), 6.01 (t, 1H, J = 5.6 Hz), 5.91 (s, 2H), 4.31 (d, 2H, J = 5.6 Hz), 3.63 (s, 2H), 2.97-2.94 (m, 2H), 2.15-2.07 (m, 1H), 2.04-1.98 (m, 2H), 1.83-1.77 (m, 4H); 13C NMR (100 MHz, CDCl3): δ 174.9, 147.8, 146.8, 135.9, 132.7, 132.3, 127.8, 127.6, 127.6, 127.4, 127.3, 125.9, 125.5, 120.9, 108.2, 101.0, 63.2, 53.1, 43.3, 43.1, 28.9. IR (neat): 3293, 2923, 1643, 1490, 1252, 1040 cm-1; MS (ESI): m/z 403 [M+H]+; HRMS (ESI), calcd for C25H27N2O3 403.2022, found 403.2025.
1-[1-Methyl-1-(1-naphthyl)ethyl]-4-[3,4-(methylenedioxy)benzylamino]carbonylpiperidine (15k)
The title compound was obtained as described in compound 15a in 90% yield (white amorphous solid). Rf = 0.51 (hexane : EtOAc = 1:1); 1H NMR (400 MHz, CDCl3): δ 9.56-9.53 (m, 1H), 7.82-7.79 (m, 1H), 7.72 (d, 1H, J = 7.9 Hz), 7.46-7.41 (m, 3H), 7.36 (t, 1H, J = 7.6 Hz), 6.73-6.71 (m, 2H), 6.67 (dd, 1H, J = 1.1 and 8.2 Hz), 6.00 (bt, 1H, J = 5.7 Hz), 5.90 (s, 2H), 4.29 (d, 2H, J = 5.7 Hz), 2.92 (bs, 2H), 2.21 (bm, 2H), 2.09-2.01 (m, 1H), 1.72 (bm, 4H), 1.56 (s, 6H); 13C NMR (100 MHz, CDCl3): δ 175.2, 147.8, 146.8, 144.0, 134.8, 132.4, 132.0, 128.5, 128.3, 127.9, 125.1, 124.7, 124.4, 123.5, 120.8, 108.2, 100.9, 62.3, 46.1, 44.0, 43.1, 29.8, 24.7. IR (neat): 3294, 2974, 1642, 1490, 1253, 1041, 780 cm-1; MS (ESI): m/z 311 [M+H]+; HRMS (ESI), calcd for C27H31N2O3 431.2335, found 431.2330.
1-[1-(2-Naphthyl)ethyl]-4-t-butoxycarbonylpiperazine (18)
To a solution of N-Boc-piperazine 16 (107 mg, 0.58 mmol) and 1-acetonaphtone 17 (0.10 mL, 0.69 mmol) in MeOH/AcOH (50:1) (4 mL) was added sodium cyanoborohydride (38 mg, 0.58 mmol) at 0 °C and it was allowed to stir for 48 h at 23 °C. The reaction was quenched with saturated NaHCO3 solution and it was extracted with CH2Cl2. The organic layers were dried over Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by silica gel column chromatography to furnish compound 18 (46 mg, 24%) as a colorless oil, Rf = 0.75 (hexane : EtOAc = 1:1); 1H NMR (300 MHz, CDCl3): δ 8.42 (d, 1H, J = 7.2 Hz), 7.86-7.83 (m, 1H), 7.74 (d, 1H, J = 10.8 Hz), 7.57 (d, 1H, J = 7.2 Hz), 7.50-7.39 (m, 3H), 4.09 (q, 1H, J = 6.6 Hz), 3.45-3.33 (m, 4H), 2.53 (bm, 2H), 2.42-2.35 (m, 2H), 1.47 (d, 3H, J = 6.6 Hz), 1.44 (s, 9H); 13C NMR (75 MHz, CDCl3): δ 154.7, 140.1, 134.0, 131.5, 128.7, 127.4, 125.5, 125.3, 125.3, 124.6, 124.0, 79.4, 61.5, 50.5, 43.8, 28.4, 18.7.
1-[1-(2-Naphthyl)ethyl]-4-(4-methoxybenzylamino)carbonylpiperazine (20)
To a solution of Boc 18 (32 mg, 0.094 mmol) in dry-CH2Cl2 (2 mL) was added trifluoroacetic acid (0.3 mL) at 0 °C and it was allowed to stir for 1 h at 23 °C. The reaction was concentrated under reduced pressure. The residue was added toluene and concentrated under reduced pressure to give crude compound 19. To a solution of 1,1′-carbonyldiimidazole (18.6 mg, 0.11 mmol) in dry-CH2Cl2 (2 mL) was added dropwise 4-methoxybenzylamine 6c (15.7 μL, 0.12 mmol) at 0 °C under argon atmosphere and it was allowed to stir for 4 h at 23 °C. The mixture was added dropwise a solution of 19 in dry-CH2Cl2 (1 mL) and it was allowed to stir for 24 h at 23 °C. The reaction was concentrated under reduced pressure. The residue was purified by silica gel column chromatography to furnish compound 20 (34 mg, 90%) as a white amorphous solid, Rf = 0.57 (CH2Cl2 : MeOH = 9:1); 1H NMR (400 MHz, CDCl3): δ 8.32 (d, 1H, J = 7.5 Hz), 7.81-7.78 (m, 1H), 7.69 (d, 1H, J = 8.1 Hz), 7.52 (d, 1H, J = 7.1 Hz), 7.44-7.36 (m, 3H), 7.13 (d, 2H, J = 8.6 Hz), 6.78 (d, 2H, J = 8.6 Hz), 4.22 (s, 2H), 4.07 (q, 1H, J = 6.6 Hz), 3.72 (s, 3H), 3.34-3.21 (m, 4H), 2.55-2.50 (m, 2H), 2.39-2.34 (m, 2H), 1.42 (d, 3H, J = 6.6 Hz); 13C NMR (100 MHz, CDCl3): δ 160.1, 160.0, 159.4, 141.1, 135.4, 132.9, 130.2, 130.1, 130.0, 128.9, 127.0, 126.8, 126.0, 125.2, 115.2, 62.6, 56.6, 51.7, 45.4, 45.2, 20.0. IR (neat): 3340, 2925, 1618, 1512, 1248 cm-1; MS (ESI): m/z 404 [M+H]+; HRMS (ESI), calcd for C25H30N3O2 404.2338, found 404.2336.
Molecular modeling
Computational analyses utilized the Sybyl 8.1 suite (Tripos, Inc.) and the GOLD docking program (CCDC) following previously described protocols.10
X-ray crystallography
The complex of inhibitor 15g with purified PLpro was formed prior to crystallization by incubating 10 mg/mL PLpro (in 20 mM Tris, pH 7.5, 10 mM DTT) with 2 mM 15g at 4°C for 16h. Diffraction-quality crystals grew from a sitting drop containing 5 mg/mL PLpro, 1 mM 15g, 1 M (NH4)2SO4, 50 mM MES, pH 6.5, and 2.5% PEG 400. Crystals were flash-frozen in liquid nitrogen and then transferred into a dry nitrogen stream at 100 K for X-ray data collection. The data set of the complex was collected at the Southeast Regional Collaborative Access Team (SER-CAT) beamline at the Advanced Photon Source, Argonne National Laboratory. Data were processed and scaled using the HKL2000 program suite. Crystals belonged to the spacegroup C2, with two monomers in the asymmetric unit. The inhibitor-complexed structure was solved to 2.63 Å by molecular replacement using the SARS-CoV PLpro apoenzyme structure (PDB entry: 2FE8) as a search model in the AMoRe program of the CCP4 suite. Manual model building was performed using Wincoot, and iterative rounds of positional & B-factor refinement and map building were performed using CNS. The structure was deposited under PDB Code: 3MJ5.
SARS-CoV Antiviral and PLpro inhibition assays
SARS-CoV antiviral assays and PLpro inhibition assays were performed as previously described.10
Supplementary Material
Acknowledgments
This work was supported by a National Institutes of Health Research Grant P01AI060915 (“Development of Novel Protease Inhibitors as SARS Therapeutics”). The authors gratefully acknowledge the synchrotron beamline personnel at Advanced Photon Source (SER-CAT) 22-ID and 22-BM beamlines. Use of the Advanced Photon Source was supported by the U. S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. W-31-109-Eng-38.
Abbreviations
- SARS
severe acute respiratory syndrome
- SARS-CoV
SARS-coronavirus
- 3CLpro
chymotrypsin-like protease
- PLpro
papain-like protease
- WHO
world health organization
Footnotes
The PDB accession code for 15g-bound PLpro X-ray structure is 3MJ5.
Supporting Information Available.
References
- 1.World Health Organization. Communicable Disease Surveillance & Response. website: http://www.who.int/csr/sars/archive/2003_05_07a/en and http://www.who.int/csr/sars/country/en/coutry2003_08_15.pdf.
- 2.Drosten C, Gunther S, Preiser W, van der Werf S, Brodt HR, Becker S, Rabenau H, Panning M, Kolesnikova L, Fouchier RA, Berger A, Burguiere AM, Cinatl J, Eickmann M, Escriou N, Grywna K, Kramme S, Manuguerra JC, Muller S, Rickerts V, Sturmer M, Vieth S, Klenk HD, Osterhaus AD, Schmitz H, Doerr HW. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N Engl J Med. 2003;348:1967–1976. doi: 10.1056/NEJMoa030747. [DOI] [PubMed] [Google Scholar]
- 3.(a) Ksiazek TG, Erdman D, Goldsmith CS, Zaki SR, Peret T, Emery S, Tong S, Urbani C, Comer JA, Lim W, Rollin PE, Dowell SF, Ling AE, Humphrey CD, Shieh WJ, Guarner J, Paddock CD, Rota P, Fields B, DeRisi J, Yang JY, Cox N, Hughes JM, LeDuc JW, Bellini WJ, Anderson LJ. A novel coronavirus associated with severe acute respiratory syndrome. N Engl J Med. 2003;348:1953–1966. doi: 10.1056/NEJMoa030781. [DOI] [PubMed] [Google Scholar]; (b) Peiris JS, Lai ST, Poon LL, Guan Y, Yam LYC, Lim W, Nicholls J, Yee WKS, Yan WW, Cheung MT, Cheng VC, Chan KH, Tsang DN, Yung RWH, Ng TK, Yuen KY. Coronavirus as a possible cause of severe acute respiratory syndrome. Lancet. 2003;361:1319–1325. doi: 10.1016/S0140-6736(03)13077-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li W, Shi Z, Yu M, Ren W, Smith C, Epstein JH, Wang H, Crameri G, Hu Z, Zhang H, Zhang J, McEachern J, Field H, Daszak P, Eaton BT, Zhang S, Wang LF. Bats are natural reservoirs of SARS-like coronaviruses. Science. 2005;310:676–679. doi: 10.1126/science.1118391. [DOI] [PubMed] [Google Scholar]
- 5.Lau SKP, Woo PCY, Li KSM, Huang Y, Tsoi HW, Wong BHL, Wong SSY, Leung SY, Chan KH, Yuen KY. Severe acute respiratory syndrome coronavirus-like virus in Chinese horseshoe bats. Proc Natl Acad Sci USA. 2005;102:14040–14045. doi: 10.1073/pnas.0506735102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.He JF, Peng GW, Min J, Yu DW, Liang WJ, Zhang SY, Xu RH, Zheng HY, Wu XW, Xu J, Wang ZH, Fang L, Zhang X, Li H, Yan XG, Lu JH, Hu ZH, Huang JC, Wan ZY, Hou JL, Lin JY, Song HD, Wang SY, Zhou XJ, Zhang GW, Gu BW, Zheng HJ, Zhang XL, He M, Zheng K, Wang BF, Fu G, Wang XN, Chen SJ, Chen Z, Hao P, Tang H, Ren SX, Zhong Y, Guo ZM, Liu Q, Miao YG, Kong XY, He WZ, Li YX, Wu CI, Zhao GP, Chiu RWK, Chim SSC, Tong YK, Chan PKS, Tam JS, Lo YMD. Molecular evolution of the SARS-coronavirus during the course of the SARS epidemic in China. Science. 2004;303:1666–1669. doi: 10.1126/science.1092002. [DOI] [PubMed] [Google Scholar]
- 7.Baker SC. Coronaviruses: Molecular Biology. Encyclopedia of Virology. (Third) 2008;1:554–562. [Google Scholar]
- 8.Ghosh AK, Xi K, Johnson ME, Baker SC, Mesecar AD. Progress in Anti-SARS Coronavirus Chemistry, Biology and Chemotherapy. Ann Rep Med Chem. 2006;41:183–196. doi: 10.1016/S0065-7743(06)41011-3. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Yang H, Bartlam M, Rao Z. Drug Design Targeting the Main Protease, the Achilles' Heel of Coronaviruses. Curr Pharm Des. 2006;12:4573–4590. doi: 10.2174/138161206779010369. [DOI] [PubMed] [Google Scholar]
- 9.(a) Ghosh AK, Xi K, Grum-Tokars V, Xu X, Ratia K, Fu W, Houser KV, Baker SC, Johnson ME, Mesecar AD. Structure-based design, synthesis, and biological evaluation of peptidomimetic SARS-CoV 3CLpro inhibitors. Bioorg Med Chem Lett. 2007;17:5876–5880. doi: 10.1016/j.bmcl.2007.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Jain RP, Pettersson HI, Zhang J, Aull KD, Fortin PD, Huitema C, Eltis LD, Parrish JC, James MNG, Wishart DS, Vederas JC. Synthesis and Evaluation of Keto-Glutamine Analogues as Potent Inhibitors of Severe Acute Respiratory Syndrome 3CLpro. J Med Chem. 2004;47:6113–6434. doi: 10.1021/jm0494873. [DOI] [PubMed] [Google Scholar]; (c) Vederas JC, Jain RP. Structural variations in keto-glutamines for improved inhibition against hepatitis A virus 3C proteinase. Bioorg Med Chem Lett. 2004;14:3655. doi: 10.1016/j.bmcl.2004.05.021. [DOI] [PubMed] [Google Scholar]
- 10.(a) Ratia K, Pegan S, Takayama J, Sleeman K, Coughlin M, Chaudhuri R, Fu W, Prabhakar BS, Johnson ME, Baker SC, Ghosh AK, Mesecar AD. A noncovalent class of papain-like protease/deubiquitinase inhibitors blocks SARS virus replication. Proc Natl Acad Sci USA. 2008;105:16119–16124. doi: 10.1073/pnas.0805240105. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ghosh AK, Takayama J, Aubin Y, Ratia K, Chaudhuri R, Baez Y, Sleeman K, Coughlin M, Nichols DB, Mulhearn DC, Prabhakar BS, Baker SC, Johnson ME, Mesecar AD. Structure-Based Design, Synthesis, and Biological Evaluation of a Series of Novel and Reversible Inhibitors for the Severe Acute Respiratory Syndrome-Coronavirus Papain-Like Protease. J Med Chem. 2009;52:5228–5240. doi: 10.1021/jm900611t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.(a) Devaraj SG, Wang N, Chen Z, Chen Z, Tseng M, Barretto N, Lin R, Peters CJ, Tseng CTK, Baker SC, Li K. Regulation of IRF-3-dependent Innate Immunity by the Papain-like Protease Domain of the Severe Acute Respiratory Syndrome Coronavirus. J Biol Chem. 2007;282:32208–32221. doi: 10.1074/jbc.M704870200. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ratia K, Saikatendu KS, Santarsiero BD, Barretto N, Baker SC, Stevens RC, Mesecar AD. Severe acute respiratory syndrome coronavirus papain-like protease: Structure of a viral deubiquitinating enzyme. Proc Natl Acad Sci USA. 2006;103:5717–5722. doi: 10.1073/pnas.0510851103. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Barretto N, Jukneliene D, Ratia K, Chen Z, Mesecar AD, Baker SC. The Papain-Like Protease of Severe Acute Respiratory Syndrome Coronavirus Has Deubiquitinating Activity. J Virol. 2005;79:15189–15198. doi: 10.1128/JVI.79.24.15189-15198.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.(a) Lindner HA, Fotouhi-Ardakani N, Lytvyn V, Lachance P, Sulea T, Ménard R. The Papain-Like Protease from the Severe Acute Respiratory Syndrome Coronavirus Is a Deubiquitinating Enzyme. J Virol. 2005;79:15199–15208. doi: 10.1128/JVI.79.24.15199-15208.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Sulea T, Lindner HA, Purisima EO, Ménard R. Deubiquitination, a New Function of the Severe Acute Respiratory Syndrome Coronavirus Papain-Like Protease? J Virol. 2005;79:4550–4551. doi: 10.1128/JVI.79.7.4550-4551.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ziebuhr J, Schelle B, Karl N, Minskaia E, Bayer S, Siddell SG, Gorbalenya AE, Thiel V. Human Coronavirus 229E Papain-Like Proteases Have Overlapping Specificities but Distinct Functions in Viral Replication. J Virol. 2007;81:3922–3932. doi: 10.1128/JVI.02091-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Oba M, Tanaka M, Takano Y, Suemune H. Concise synthetic strategy toward cyclic αα-disubstituted α-amino acids bearing a δ-nitrogen atom: chiral 1-substituted 4-aminopiperidine-4-carboxylic acids. Tetrahedron. 2005;61:593–598. [Google Scholar]
- 15.Gaspari P, Banerjee T, Malachowski WP, Muller AJ, Prendergast GC, DuHadaway J, Bennett S, Donovan AM. Structure-Activity Study of Brassinin Derivatives as Indoleamine 2,3- Dioxygenase Inhibitors. J Med Chem. 2006;49:684–692. doi: 10.1021/jm0508888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dolby LD, Biere H. The Total Synthesis of (±)-Epidasycarpidone. and (±)-Epiuleine. J Org Chem. 1970;35:3843–3845. [Google Scholar]
- 17.Yang Q, Ney JE, Wolfe JP. Palladium-Catalyzed Tandem N-Arylation/Carboamination Reactions for the Stereoselective Synthesis of N-Aryl-2-benzyl Pyrrolidines. Org Lett. 2005;7:2575–2578. doi: 10.1021/ol050647u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tahtaoui C, Parrot I, Klotz P, Guillier F, Galzi JL, Hibert M, Ilien B. Fluorescent Pirenzepine Derivatives as Potential Bitopic Ligands of the Human M1 Muscarinic Receptor. J Med Chem. 2004;47:4300–4315. doi: 10.1021/jm040800a. [DOI] [PubMed] [Google Scholar]
- 19.Matsuno K, Ichimura M, Nakajima T, Tahara K, Fujiwara S, Kase H, Ushiki J, Giese NA, Pandey A, Scarborough RM, Lokker NA, Yu JC, Irie J, Tsukuda E, Ide S, Oda S, Nomoto Y. Potent and Selective Inhibitors of Platelet-Derived Growth Factor Receptor Phosphorylation. 1. Synthesis, Structure-Activity Relationship, and Biological Effects of a New Class of Quinazoline Derivatives. J Med Chem. 2002;45:3057–3066. doi: 10.1021/jm010428o. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.