

Summary
Although autism is one of the most heritable neuropsychiatric disorders, its underlying genetic architecture has largely eluded description. To comprehensively examine the hypothesis that common variation is important in autism, we performed a genome-wide association study (GWAS) using a discovery dataset of 438 autistic Caucasian families and the Illumina Human 1M beadchip. 96 single nucleotide polymorphisms (SNPs) demonstrated strong association with autism risk (p-value < 0.0001). The validation of the top 96 SNPs was performed using an independent dataset of 487 Caucasian autism families genotyped on the 550K Illumina BeadChip. A novel region on chromosome 5p14.1 showed significance in both the discovery and validation datasets. Joint analysis of all SNPs in this region identified 8 SNPs having improved p-values (3.24E-04 to 3.40E-06) than in either dataset alone. Our findings demonstrate that in addition to multiple rare variations, part of the complex genetic architecture of autism involves common variation.
Introduction
Autism is a neurodevelopmental disorder characterized by three primary areas of impairment: social interaction, communication, and restricted and repetitive patterns of interest or behavior (Centers for Disease Control 2008). It is among a spectrum of disorders (ASDs) with symptoms that may range from quite severe (autistic disorder) to relatively mild (Asperger syndrome). With improved surveillance and a broadening of the diagnostic criteria, the most recent prevalence studies suggest that ASDs may affect as many as 1 in 150 children in the U.S. making it one of the most common neurodevelopmental disorders (NCBI 2008). ASDs are most often diagnosed before age four, and are at least three to four times more frequent in males than females (NCBI 2008).
Overwhelming evidence from twin and sibling studies demonstrates that autism is highly heritable (Steffenburg et al. 1989, Bailey et al. 1995, Bolton et al. 1994), but there is no consensus on the underlying genetic architecture. There are two alternative proposals, one involving numerous rare genetic mutations and the other involving fewer but more common genetic variations. Supporting the rare mutation hypothesis are mutations in several genes and rare structural DNA variations both of which have been identified, although the pervasiveness of these effects remains controversial (Weiss et al. 2008, Sebat et al. 2007). Data supporting the effect of common variation has been more difficult to find. Several genome-wide linkage screens and focused candidate gene association studies have been performed in autism (Shao et al. 2002, Szatmari et al. 2007, International Molecular Genetic Study of Autism Consortium (IMGSAC) 2001), but the results have been disappointing and no universally accepted susceptibility polymorphism has yet emerged. Collectively these data have suggested that the common variant hypothesis may not be relevant to autism genetics.
A recent study by Arking et al. (Arking et al. 2008) combining linkage and genome-wide association in 72 multiplex autism families identified a common variant in the CNTNAP2 gene that was associated with autism primarily in families where all affected individuals were male (male only families). This association was also seen by Alarcon et al. (Bakkaloglu et al. 2008) and similar to Arking et al. (Arking et al. 2008), the effect was primarily in male only autism families. However, this association has not been widely replicated.
Materials And Methods
Ascertainment and Sample description
We ascertained autism patients and their affected and unaffected family members as part of the Collaborative Autism Project (CAP) through four clinical groups at the Miami institute for Human Genomics (MIHG, Miami, Florida), University of South Carolina (Columbia, South Carolina), W.S. Hall Psychiatric Institute (Columbia, South Carolina) and Vanderbilt Center for Human Genetics Research (Vanderbilt University, Nashville, Tennessee) Participating families were enrolled through a multi-site study of autism genetics and recruited via support groups, advertisements, and clinical and educational settings. All participants and families were ascertained using a standard protocol. These protocols were approved by appropriate Institutional Review Boards. Written informed consent was obtained from parents as well as from minors who were able to give informed consent; in individuals unable to give assent due to age or developmental problems, assent was obtained whenever possible.
Core inclusion criteria were as follows: (1) Chronological age between 3 and 21 years of age; (2) Presumptive clinical diagnosis of autism; (3) Expert clinical determination of autism diagnosis using DSM-IV criteria supported by the Autism Diagnostic Interview (ADI-R) in the majority of cases and all available clinical information. The ADI-R is a semi-structured diagnostic interview which provides diagnostic algorithms for classification of autism (Autism Genetics Resource Exchange 2008). All ADI-R interviews were conducted by formally trained interviewers who have achieved reliability according to established methods. Thirty-eight individuals were missing an ADI-R. For those cases we implemented a best estimate procedure to determine a final diagnosis using all available information from the research record and data from other assessment procedures. This information was reviewed by a clinical panel led by an experienced clinical psychologist and included two other psychologists and a pediatric medical geneticist— all of whom were experienced in autism. Following review of case material the panel discussed the case until a consensus diagnosis was obtained. Only those cases in which a consensus diagnosis of autism was reached were included (4) Minimal developmental level of 18 months as determined by the Vineland Adaptive Behavior Scale (VABS) (Sparrow, Balla & Cicchetti 1984) or the VABS-II (Sparrow, Cicchetti & Balla 2005) or IQ equivalent > 35. These minimal developmental levels assure that ADI-R results are valid and reduce the likelihood of including individuals with severe mental retardation only. We excluded participants with severe sensory problems (e.g., visual impairment or hearing loss), significant motor impairments (e.g., failure to sit by 12 months or walk by 24 months), or identified metabolic, genetic, or progressive neurological disorders.
A total of 487 Caucasian families (1537 individuals) were genotyped. This dataset consisted of 80 multiplex families (more than one affected individual) and 407 singleton (parent-child trio) families. In addition, GWAS data were obtained from the Autism Genetic Resource Exchange (AGRE) (Autism Genetics Resource Exchange 2008) for use as a validation dataset. The full AGRE dataset is publicly available and contains families with the full spectrum of autism spectrum disorders. We selected only families with one or more individuals diagnosed with autism (using DSM-IV and ADI); affecteds with non-autism diagnosis within these families were excluded from the analysis. This resulted in a confirmation dataset of 680 multiplex families (3512 individuals) from the AGRE “SingleAllAgre” beadstudio file (Autism Genetics Resource Exchange 2008). Family and individual identifiers for all AGRE samples passed our quality control are listed in supplementary table 3.
Genotyping of the discovery dataset
Genomic DNA was purified from whole blood using Puregene chemistry on the Qiagen Autopure LS according to standard automated Qiagen protocols (Valencia, CA). DNA samples were quantitated via the ND-8000 spectrophotometer and DNA quality was evaluated via gel electrophoresis on a 0.8% agarose gel. The concentration for all qualified samples was normalized to 50 ng/ul and samples were arrayed in Matrix 0.5ml 2D barcoded tubes in racks of 96. Sample identity was confirmed by genotyping 8 SNPs using Taqman allelic discrimination assays (Applied Biosystems; CA) and assessing for concordance with historical data.
Samples that passed the above exclusion criteria were genotyped using Illumina's Human 1Mv1 Beadchip, containing 1,072,820 SNPs (of those 258,665 loci are in reported and new CNV regions). The samples have been processed according to Illumina Procedures for processing of the Infinium II ® assay.
The above protocol is automated using the Tecan EVO-1 to further enhance the efficiency and consistency of the assay. Samples are processed in batches of 48 at a time. The same Quality Control DNA sample is repeated during each run to ensure reproducibility of results between runs. Data is extracted by the Illumina ® Beadstudio software from data files created by the Illumina BeadArray reader. Samples and markers with call rates below 95% were excluded from analysis and a GenCall cutoff score of 0.15 was used for all Infinium II ® products.
Sample quality control
After genotyping, samples were subject of a battery of a quality control (QC) tests. We used the same protocol for both the discovery and validation datasets. Reported and genetic gender were examined using X-chromosome linked SNPs. Relatedness between samples, sample contaminations, miss identification and duplications were tested using genome-wide IBD estimation; inconsistent samples were dropped from the analysis. The numbers of remaining samples are listed in supplementary table 1.
As a next step we tested for Mendelian inconsistencies on all SNPs and samples. Mendelian errors (ME) can emerge from sample misidentification, DNA contamination, copy-number variation (CNV), genotype calling errors and other reasons. The median of ME per family in both investigated cohorts was below 0.005%. More than 99% of the discovery families and 98% of the validation families had ME below 0.02%. We excluded families with ME > 2% from the analysis. This threshold would still allow for small deletions and duplications that are common in human genome.
SNP quality control
SNPs were subject to QC before analysis. We removed SNPs with minor allele frequencies below 5% because of restricted power in the discovery sample.
As expected, we observed negative correlation between the proportion of ME per SNP and p-value for HWE. To minimize genotyping errors we excluded SNPs with p-value <10-6 for HWE and ME > 4%. Remaining erroneous genotypes were set as missing. PLINK software was used for quality control steps described above (Purcell et al. 2007).
Illumina provides information on which 1M BeadChip SNPs were located within known common CNV-regions. We compared the distribution of ME per family and per SNP. No significant differences between ME per SNP in the known CNV regions and the remaining markers were identified. The same quality criteria were used for both the discovery and the validation datasets. The summary of SNPs is presented in supplemental table 2.
Population Stratification
Although population substructure does not cause type I error in family-based association tests, multiple founder effects could result in reduced power to detect an association in a heterogeneous disease such as autism. Thus we conducted EIGENSTRAT (Patterson, Price & Reich 2006) analysis on all parents from analyzed families for evidence of population substructure using the 491,664 SNPs genotyped in both the discovery and validation datasets. To ensure the most homogeneous groups for association screening and replication, we excluded all families with outliers defined by EIGENSTRAT (Patterson, Price & Reich 2006) out of 4 standard deviations of principle components 1 and 2. After all QC steps, 1,390 samples from 438 autistic families were remained in the final discovery dataset and 2,390 samples from 457 autistic families (supplemental table 1 and 3) in the validation dataset. The average genotyping rate in the remaining individuals was 99.8 %.
Genotype Imputation
Since the validation dataset was genotyped on a different GWAS SNP panel with a smaller number of SNPs (558183), The genotypes from our data and the data from the AGRE were imputed independently by the program IMPUTE (Marchini et al. 2007) using a phased CEU HapMap dataset as a reference (International HapMap Consortium et al. 2007). Individual genotypes with probability less than 0.90 were not included. All individuals were treated independently while doing imputation. Mendelian inconsistencies were zeroed out in PLINK (Purcell et al. 2007). The results for the imputation are found in table 1. Results on imputed SNPs missing more than 10% of the genotypes were labeled in the table 1 and should be interpreted with caution because of possible bias.
Table 1.
Association results on top 96 SNPs.
| Chromosome | SNP | Position | MAF | P-Value HWE | Allele | P-value# discovery | P-value# validation | P-value joint# |
|---|---|---|---|---|---|---|---|---|
| 1 | rs201171 | 6573680 | 0.14 | 0.62 | T/C | 6.74E-05 | 0.72 | 1.49E-02 |
| 1 | rs4394668 | 12593816 | 0.21 | 1 | C/T | 4.89E-06 | 2.78E-02 | 1.31E-05 |
| 1 | rs4618985 | 12593911 | 0.27 | 0.94 | C/A | 2.04E-05 | 2.67E-02 | 4.71E-05 |
| 1 | rs1831870 | 57399791 | 0.45 | 1 | C/T | 3.27E-05 | 0.69 | 1.18E-02 |
| 1 | rs155288 | 57405075 | 0.45 | 0.95 | T/C | 2.84E-05 | 0.63 | 9.28E-03 |
| 1 | rs5008948 | 80011986 | 0.29 | 0.02 | C/T | 6.11E-05 | 0.69 | 0.05 |
| 1 | rs12024204 | 80015589 | 0.45 | 0.91 | A/G | 2.50E-05 | 0.83 | 0.04 |
| 1 | rs11162822 | 80018536 | 0.26 | 0.03 | C/T | 8.78E-05 | 0.72 | 0.14 |
| 1 | rs6424674 | 80047434 | 0.47 | 1 | G/A | 3.97E-05 | 0.81 | 0.07 |
| 1 | rs10493644 | 80056276 | 0.43 | 0.72 | A/C | 7.03E-05 | 0.91 | 0.06 |
| 1 | rs17425287 | 80058999 | 0.46 | 0.95 | T/C | 2.40E-05 | 0.8 | 0.06 |
| 1 | rs7523086 | 1.16E+08 | 0.38 | 0.19 | A/G | 7.27E-05 | 0.02 | 1 |
| 1 | rs16833075 | 2.34E+08 | 0.16 | 0.75 | G/T | 6.81E-05 | 0.44 | 7.15E-03 |
| 2 | rs492780 | 44997398 | 0.05 | 0.01 | C/T | 8.78E-05 | 0.34 | 7.81E-03 |
| 2 | rs1467068 | 60716378 | 0.32 | 0.74 | T/C | 3.35E-05 | 0.11 | 0.51 |
| 2 | rs6732653 | 60717337 | 0.32 | 0.79 | A/G | 3.50E-05 | 0.11 | 0.51 |
| 2 | rs1866206 | 60720330 | 0.32 | 0.69 | T/C | 4.92E-05 | 0.11 | 0.54 |
| 2 | rs4852531 | 79903226 | 0.45 | 0.2 | C/T | 5.93E-05 | 0.26 | 2.63E-03 |
| 2 | rs11679682 | 1.74E+08 | 0.23 | 0.25 | C/T | 7.80E-05 | 0.78 | 0.03 |
| 2 | rs11689493 | 1.74E+08 | 0.38 | 0.95 | C/T | 4.31E-05 | 0.39 | 5.59E-03 |
| 2 | rs4129081 | 1.91E+08 | 0.26 | 0.82 | A/C | 3.56E-05 | 0.71 | 0.07 |
| 2 | rs6707773 | 1.91E+08 | 0.26 | 0.71 | T/C | 7.44E-05 | 0.68 | 0.09 |
| 2 | rs13019278 | 1.91E+08 | 0.26 | 0.76 | A/G | 8.14E-05 | 0.9 | 4.85E-02 |
| 2 | rs12622496 | 1.91E+08 | 0.25 | 0.64 | C/T | 9.66E-06 | 0.97 | 2.18E-02 |
| 2 | rs3731723 | 2.31E+08 | 0.41 | 0.04 | T/C | 3.20E-05 | 0.07 | 4.08E-04 |
| 2 | rs722555 | 2.31E+08 | 0.41 | 0.06 | G/A | 2.59E-05 | 0.09 | 4.10E-04 |
| 2 | rs6436915 | 2.31E+08 | 0.44 | 0.51 | T/G | 1.67E-05 | 0.14 | 5.51E-04 |
| 3 | rs2279977 | 3389586 | 0.45 | 0.52 | C/A | 7.52E-05 | 0.48 | 5.13E-03 |
| 3 | rs9822786 | 1.13E+08 | 0.08 | 1.27E-03 | T/G | 2.98E-07 | 0.46 | 0.45 |
| 3 | rs12491012 | 1.55E+08 | 0.1 | 0.76 | A/G | 5.44E-05 | 0.25 | 4.21E-03 |
| 3 | rs1811763 | 1.74E+08 | 0.14 | 0.33 | A/G | 1.65E-05 | 0.31 | 1.66E-03 |
| 5 | rs1896731 | 25934777 | 0.36 | 1 | C/T | 7.67E-05 | 8.00E-03 | 1.90E-05 |
| 5 | rs10038113 | 25938099 | 0.41 | 0.23 | C/T | 2.75E-05 | 3.28E-03 | 3.40E-06 |
| 5 | rs11739167 | 25945521 | 0.45 | 0.6 | T/C | 1.79E-05 | 1.98E-02 | 3.05E-05 |
| 5 | rs7447989 | 25950789 | 0.38 | 0.66 | G/A | 3.29E-05 | 0.16 | 1.07E-03 |
| 5 | rs6873221 | 25964323 | 0.45 | 0.45 | A/G | 5.07E-06 | 9.06E-03 | 5.90E-06 |
| 5 | rs12187724 | 25970827 | 0.38 | 0.66 | C/A | 4.78E-05 | 0.08 | 5.02E-04 |
| 5 | rs1423435 | 37940845 | 0.47 | 0.86 | T/C | 5.70E-05 | 0.68 | 1.47E-02 |
| 5 | rs12153325 | 51897032 | 0.45 | 0.27 | C/T | 3.89E-05 | 0.58 | 8.10E-03 |
| 5 | rs10461556 | 51907079 | 0.45 | 0.32 | T/C | 8.76E-06 | 0.45 | 2.63E-03 |
| 5 | rs350436 | 51932211 | 0.46 | 0.27 | C/T | 5.31E-05 | 0.6 | 9.84E-03 |
| 5 | rs830907 | 51940364 | 0.45 | 0.27 | T/C | 7.48E-05 | 0.6 | 0.01 |
| 5 | rs315717 | 1.7E+08 | 0.48 | 0.73 | T/C | 3.06E-05 | 0.94 | 0.05 |
| 5 | rs3804254 | 1.7E+08 | 0.48 | 0.64 | T/C | 4.24E-05 | 0.9 | 4.38E-02 |
| 6 | rs2317222 | 778579 | 0.19 | 0.51 | G/A | 9.97E-05 | 0.57 | 1.60E-02 |
| 6 | rs1936022 | 72723073 | 0.4 | 0.01 | T/C | 7.10E-05 | 0.19 | 0.38 |
| 6 | rs6907646 | 72736738 | 0.39 | 0.03 | A/G | 9.20E-05 | 0.15 | 0.47 |
| 6 | rs12529724 | 91590812 | 0.25 | 0.14 | A/G | 1.64E-05 | 0.9 | 2.36E-02 |
| 6 | rs1504279 | 91596967 | 0.3 | 0.13 | G/A | 1.19E-06 | 0.6 | 4.35E-03 |
| 6 | rs1504281 | 91597253 | 0.43 | 0.31 | T/C | 8.03E-05 | 0.51 | 1.13E-02 |
| 6 | rs2799644 | 96464883 | 0.34 | 0.52 | T/C | 5.90E-05 | 0.73 | 0.11 |
| 7 | rs320813 | 9751778 | 0.35 | 0.9 | T/C | 7.23E-05 | 0.59 | 1.27E-02 |
| 7 | rs1529001 | 52748818 | 0.06 | 0.02 | A/G | 4.30E-05 | - | - |
| 7 | rs11765584 | 63222219 | 0.32 | 0.46 | A/G | 8.07E-05 | 0.89 | 4.97E-02 |
| 7 | rs1529813 | 63255903 | 0.31 | 0.63 | A/G | 1.78E-05 | 0.95* | 2.31E-02 |
| 7 | rs2528795 | 73111430 | 0.23 | 0.87 | G/A | 7.78E-05 | 0.14* | 5.24E-05 |
| 7 | rs1000058 | 1.32E+08 | 0.08 | 0.11 | T/G | 7.11E-05 | 0.17* | 0.06 |
| 7 | rs1149558 | 1.33E+08 | 0.4 | 0.59 | C/T | 5.11E-05 | 0.48 | 0.15 |
| 7 | rs1130496 | 1.58E+08 | 0.43 | 0.18 | A/G | 4.70E-05 | 0.89* | 3.82E-03 |
| 8 | rs12155975 | 18823406 | 0.13 | 0.2 | T/G | 1.10E-05 | 0.85 | 3.95E-02 |
| 8 | rs16930253 | 64276820 | 0.07 | 0.67 | T/G | 7.96E-05 | 0.51 | 0.2 |
| 8 | rs2082804 | 64323440 | 0.08 | 0.44 | A/G | 6.68E-05 | 0.32 | 0.36 |
| 8 | rs4739071 | 64333676 | 0.08 | 0.56 | C/A | 8.89E-05 | 0.34 | 0.36 |
| 8 | rs6991229 | 64348114 | 0.08 | 0.69 | C/T | 5.84E-05 | 0.28 | 0.36 |
| 10 | rs1865641 | 77612829 | 0.17 | 0.47 | C/A | 3.50E-05 | 0.62 | 1.56E-02 |
| 10 | rs1865638 | 77625472 | 0.19 | 0.71 | T/C | 4.27E-05 | 0.75 | 2.37E-02 |
| 10 | rs11001685 | 77632360 | 0.19 | 1 | C/T | 2.34E-05 | 0.75 | 1.93E-02 |
| 10 | rs881631 | 77636503 | 0.17 | 0.53 | G/A | 3.39E-05 | 0.66 | 1.55E-02 |
| 10 | rs12245799 | 77637321 | 0.19 | 0.77 | C/T | 2.74E-05 | 0.77 | 2.26E-02 |
| 10 | rs12249859 | 77641198 | 0.19 | 0.78 | C/A | 4.79E-05 | 0.84 | 2.86E-02 |
| 10 | rs10748804 | 1.03E+08 | 0.45 | 0.86 | G/A | 8.07E-05 | 0.17 | 1.33E-03 |
| 10 | rs7910491 | 1.03E+08 | 0.45 | 0.68 | G/A | 5.51E-05 | 0.1 | 4.43E-04 |
| 11 | rs10840070 | 8524783 | 0.39 | 1 | T/C | 3.36E-05 | 0.67 | 0.1 |
| 11 | rs11021927 | 11533125 | 0.33 | 0.02 | A/C | 7.03E-05 | 0.67 | 1.50E-02 |
| 11 | rs9919560 | 11547547 | 0.28 | 0.17 | A/C | 1.13E-06 | 0.83 | 1.26E-02 |
| 12 | rs12366827 | 7834103 | 0.43 | 0.11 | C/T | 8.79E-05 | - | - |
| 12 | rs1894827 | 7834643 | 0.43 | 0.1 | A/G | 7.43E-05 | 0.89 | 0.08 |
| 12 | rs11055784 | 7836092 | 0.43 | 0.12 | G/T | 6.74E-05 | - | - |
| 12 | rs11055786 | 7836651 | 0.43 | 0.11 | T/C | 8.79E-05 | 0.81 | 0.1 |
| 12 | rs11117003 | 84639343 | 0.36 | 0.75 | T/G | 4.57E-05 | 0.25 | 6.91E-03 |
| 12 | rs10492086 | 1.01E+08 | 0.19 | 0.4 | G/A | 5.36E-05 | 0.08 | 1.85E-04 |
| 12 | rs10444509 | 1.01E+08 | 0.18 | 0.92 | C/T | 3.36E-05 | 0.09 | 2.16E-04 |
| 13 | rs11616562 | 83656026 | 0.06 | 0.05 | G/A | 4.51E-05 | 0.84 | 3.59E-02 |
| 13 | rs7325257 | 87203088 | 0.38 | 0.95 | T/C | 6.51E-05 | 0.96 | 4.85E-02 |
| 15 | rs11636552 | 23686048 | 0.15 | 0.2 | G/T | 3.27E-05 | 0.42 | 0.17 |
| 16 | rs1532926 | 8013919 | 0.38 | 0.81 | C/A | 4.71E-05 | 0.52 | 1.58E-02 |
| 16 | rs4456502 | 10257638 | 0.35 | 0.44 | G/A | 8.33E-05 | 0.51* | 2.77E-02 |
| 16 | rs4148358 | 16094676 | 0.23 | 0.04 | T/C | 8.50E-05 | 0.49 | 0.21 |
| 17 | rs891754 | 29834556 | 0.43 | 0.91 | C/A | 6.96E-05 | 0.59 | 0.12 |
| 18 | rs8088001 | 277705 | 0.07 | 0.36 | G/T | 6.23E-05 | 0.88 | 0.1 |
| 18 | rs547668 | 10466821 | 0.16 | 0.59 | T/G | 8.50E-05 | 0.09 | 0.8 |
| 18 | rs1144093 | 48371078 | 0.23 | 0.5 | A/G | 6.34E-05 | - | - |
| 18 | rs637644 | 61049646 | 0.44 | 0.48 | C/T | 1.57E-05 | 0.31 | 0.18 |
| 20 | rs6049129 | 23723819 | 0.39 | 3.90E-03 | C/T | 7.18E-05 | 0.31 | 0.35 |
| 20 | rs742759 | 47494552 | 0.22 | 0.8 | A/G | 1.52E-05 | 0.24 | 0.26 |
| 20 | rs171415 | 58229934 | 0.13 | 0.25 | T/G | 2.03E-06 | 0.27* | 1.24E-05 |
MAF: minor allele frequency in discovery dataset
P-value HWE: Hardy-Weinberg Equilibrium test p-value in discovery dataset
In italic and bold are the p-values for markers not genotyped on 550K Illumina panel. Genotypes for these markers were imputed.
“-” data could not been imputed because no genotypes are available for the reference dataset.
Allele: Minor Allele/Major Allele in discovery dataset
Shaded: Marker with improved P-value in validation dataset.
The Pedigree Disequilibrium Test (PDT) was performed on all SNPs for association testing.
the imputed markers missing more than 10% of genotypes
Distance: the distance (basepair) between gene and marker
Association Analysis
Association analysis was performed using the pedigree disequilibrium test (PDT) (Martin et al. 2000, Martin, Bass & Kaplan 2001). This method provides valid and robust tests for allelic association across both trio and extended families. Only autosomal markers were tested for association. The estimation of odds ratios and 95% confidence interval calculations were performed using UNPHASED (Dudbridge et al. 2008). Power calculations for association analysis were performed using the Genetic Power Calculator (Purcell S, Cherny SS, Sham PC. 2008).
Linkage disequilibrium
Linkage disequilibrium (LD) patterns and haplotype block delineation were determined by using Haploview 4.1 (Choi et al. 2001). Blocks were defined using the confidence interval method described by Gabriel et al. (Gabriel et al. 2002). Pair-wise LD measures (r2) were calculated in the 3,822 unrelated founders of the join sample
Results
To more comprehensively test the common variant hypothesis, we performed an unbiased genome-wide association study of common variation using as a discovery dataset the Caucasian autistic families from the Collaborative Autism Project (CAP). We validated our findings using an independent publicly available family-based Genome-Wide Association Study (GWAS) dataset from the Autism Genome Research Exchange (AGRE) (Autism Genetics Resource Exchange 2008). Quality-control (QC) procedures were applied to the more than 1,000,000 single nucleotide polymorphisms (SNPs) in the discovery dataset and 550,000 SNPs in the validation dataset.
After applying QC filters, 775,311 common autosomal SNPs remained in the discovery dataset with an average genotyping rate of 99.80% and 500,100 common autosomal SNPs remained in the validation dataset with an average genotyping rate of 99.82%. To account for possible population stratification, we excluded families if the values for the top two principal components for either of the probands' parents were > 4 standard deviations from the core Caucasian cluster generated in EIGENSTRAT (Patterson, Price & Reich 2006). The final datasets included 1,390 samples from 438 autistic families in the discovery dataset and 2,390 samples from 457 autistic families in the validation dataset. For any SNP of interest in the discovery dataset not directly genotyped in the validation dataset, imputation of genotypes was performed in the validation dataset using the program IMPUTE (Marchini et al. 2007). The Pedigree Disequilibrium Test (PDT) (Martin et al. 2000, Martin, Bass & Kaplan 2001) was used for all association analyses. The distribution of p-values examined in the discovery dataset demonstrated a close match to that expected for a null distribution except at extreme tail of low p-values (Figure 1). This is expected if there is little residual error in the data and common variants of modest effect sizes are acting in autism. In the discovery dataset, none of the p-values met the stringent and overly conservative Bonferroni correction for genome-wide significance (Figure 2).
Figure 1. Quantile-Quantile (Q-Q) plot of PDT p-values for the discovery dataset.
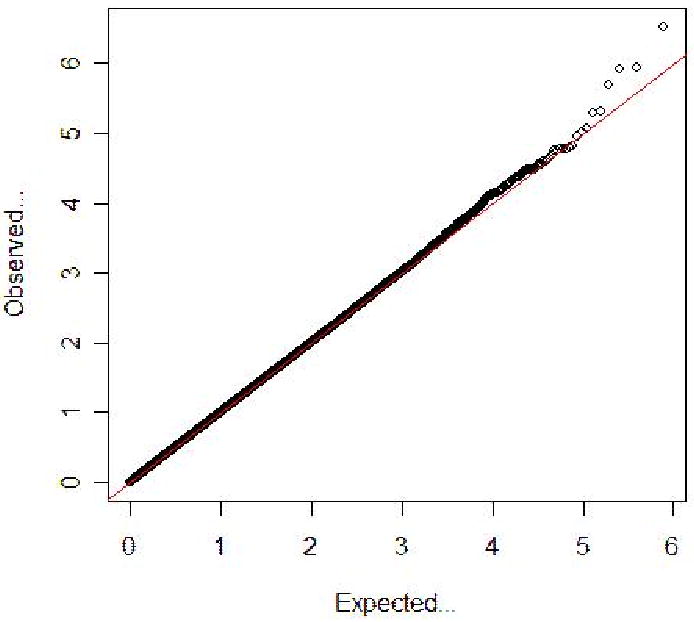
Note: The Q-Q plot measures deviation from the expected deviation of P-values. The horizontal (red) line represents the expected (null) distribution. The slight deviation of the observed values above expected values at the tail of the distribution is consistent with modest genetic effects.
Figure 2. Genome-wide plot of association p-values in the discovery dataset.
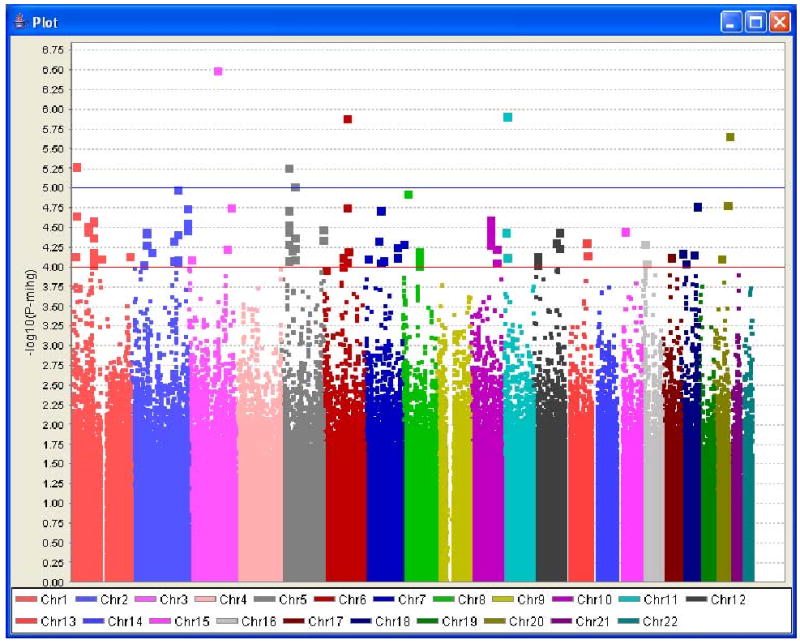
Note: -log10(p-value) for all 775, 311 tested SNPs in 438 families are plotted against their genomic location. 96 SNPs have p-values <1.0e10−4 (horizontal red line) and 6 SNPs have p-values <1.0e10−5 (blue horizontal line). Individual chromosomes are demarked by different colors. Details of the analysis are presented in the supplemental material.
Examination of the 651 SNPs in the CNTNAP2 gene (Arking et al. 2008, Bakkaloglu et al. 2008) in our discovery dataset revealed only eight genotyped SNPs that were nominally significant (p-values=0.002-0.04). The results did not significantly improve in male only families (data not shown). The tagging SNP, rs270102, reported by Alarcon et al. (Bakkaloglu et al. 2008), was not significant in either the overall or male only family dataset. SNP rs7794745 showing linkage in Arking et al. (Arking et al. 2008) study was not genotyped in our dataset. Association of imputed genotypes for this SNP was not significant (p=0.62). None of tested markers met gene-wide (CNTNAP2) significance after correction (data not shown).
Despite no genome-wide significant association, 96 SNPs showed strongly suggestive association with autism risk (Table 1, p<0.0001) and met our initial criteria for follow-up. Among the 96 top hits, 2 SNPs, residing in 5p14.1, had improved p-values in the joint analysis and also had nominally significant association signal in the validation dataset encouraging us to look at this region in more detail. Therefore, we examined every SNP (n=46) genotyped in this region (25830kb to 26100kb) in both datasets regardless of their initial p-value. Analyses of these data revealed a cluster of 19 SNPs including 8 imputed SNPs showing nominally significant association (p<0.05) in the validation dataset (data not shown). Eight SNPs on chromosome 5p14.1 (Table 2) showed improved association signals in the joint dataset. Risk was associated with the same allele for these eight SNPs in both datasets and the p-values became more significant (p-values: 3.24E-04 to 3.40E-06) in the joint analysis, with the most significant p-value coming from one of the top 96 hits rs10038113. The odds ratios for the major alleles ranged from 0.75 to 1.32 (Table 2).
Table 2.
Association statistics for validated SNPs on chromosome 5p 14.1:
| SNP number | SNP | Position | Allele | MAF | p-value HWE | p-value discovery | OR-discovery | p-value validation | p-value joint | OR-joint |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | rs10065041 | 25876207 | T/C | 0.39 | 0.76 | 5.85E-03 | 1.30[1.08-1.56] | 1.07E-02 | 3.24E-04 | 1.21[1.08-1.36] |
| 2 | rs7704909 | 25934678 | C/T | 0.36 | 0.32 | 9.06E-03 | 1.29[1.07-1.55] | 4.92E-04 | 1.53E-05 | 1.30[1.15-1.46] |
| 3 | rs1896731 | 25934777 | C/T | 0.36 | 1.00 | 7.67E-05 | 0.67[0.55-0.82] | 8.00E-03 | 1.90E-05 | 0.76[0.67-0.85] |
| 4 | rs10038113 | 25938099 | C/T | 0.41 | 0.23 | 2.75E-05 | 0.67[0.56-0.81] | 3.28E-03 | 3.40E-06 | 0.75[0.70-0.90] |
| 5 | rs6894838 | 25980703 | T/C | 0.38 | 0.14 | 0.02 | 1.25[1.05-1.50] | 1.58E-03 | 8.00E-05 | 1.26[1.12-1.42] |
| 6 | rs12518194 | 25987318 | G/A | 0.36 | 0.41 | 9.33E-03 | 1.29[1.07-1.55] | 2.67E-04 | 8.34E-06 | 1.31[1.16-1.49] |
| 7 | rs4307059 | 26003460 | C/T | 0.36 | 0.45 | 0.01 | 1.28[1.06-1.54] | 3.44E-04 | 1.29E-05 | 1.31[1.16-1.48] |
| 8 | rs4327572 | 26008578 | T/C | 0.36 | 0.18 | 7.34E-03 | 1.29[1.07-1.56] | 1.63E-04 | 4.05E-06 | 1.32[1.17-1.49] |
MAF: minor allele frequency in discovery dataset
P-value hwe: Hardy-Weinberg Equilibrium test P-value in the discovery dataset
Allele: minor allele/major allele based on discovery dataset.
OR: Odds Ratios for joint sample for major allele, minor allele used as a reference allele
Note: Nine SNPs in 5p14.1 had p-values <0.05 in both the discovery and the validation datasets and generated improved p-values in the joint analysis.
To determine if we might miss a strong signal by only using the CAP dataset as the discovery dataset, we also reversed the datasets for discovery and validation and used our same two stage approach. 21 SNPs had p-values < 0.0001 in the AGRE dataset but none of them could be replicated in CAP dataset even with a nominal significance of p<0.05. We computed the power of the TDT in 438 triad families that approximates a lower bound for power of the PDT in our discovery sample. Given a prevalence of autism of 0.0066 (Chakrabarti, Fombonne 2005), a SNP in LD (D'=1) with a risk allele frequency 0.6, we expect 84% power to detect an association at p =0.0001 under a recessive model (GRRAA=2, GRRAa=1) and 33% under additive model (GRRAA=2, GRRAa=1.5). These are consistent with the allelic GRR's estimated for the chromosome 5 region. The power to detect a Bonferroni-corrected genome-wide significance (p = 0.05 / 775311 SNPs = 6.4×10-8) drops to 30% and 2.5%, respectively, for recessive and additive models.
Discussion
We examined the linkage disequilibrium (LD) pattern among the eight replicated SNPs with improved P-values (Figure 3) to gain a better understanding of the association. Seven of these SNPs form two tightly linked LD blocks. Given that none of these SNPs reside within known genes or known regulatory sequences, the clustering of association signals suggests that one or more nearby functional variants is responsible for the signal. A survey of the genomic landscape surrounding this region of association reveals several interesting avenues for further molecular investigation. There are numerous sequence segments exhibiting a high degree of evolutionary conservation, suggesting potential regulatory, but currently undetermined, functions. In addition, there are three known copy number variants (CNVs) in proximity of the most significant SNPs (Table 2). Preliminary investigation of these CNVs in the discovery dataset is not suggestive of a causal relationship with autism (data not shown). Exhaustive molecular analysis of the candidate region is ongoing. In addition, although the immediate 1 Mb vicinity of the association region contains no known genes, flanking the region are CDH9 and CDH10, two genes belonging to the cadherin family, a group of proteins containing members that are involved in calcium-dependent cell-cell junctions in the nervous system (Liu et al. 2006, Pokutta, Weis 2007) and possible targets of regulatory action.
Figure 3. Linkage Disequilibrium pattern among validated SNPs on chromosome 5p14.1.
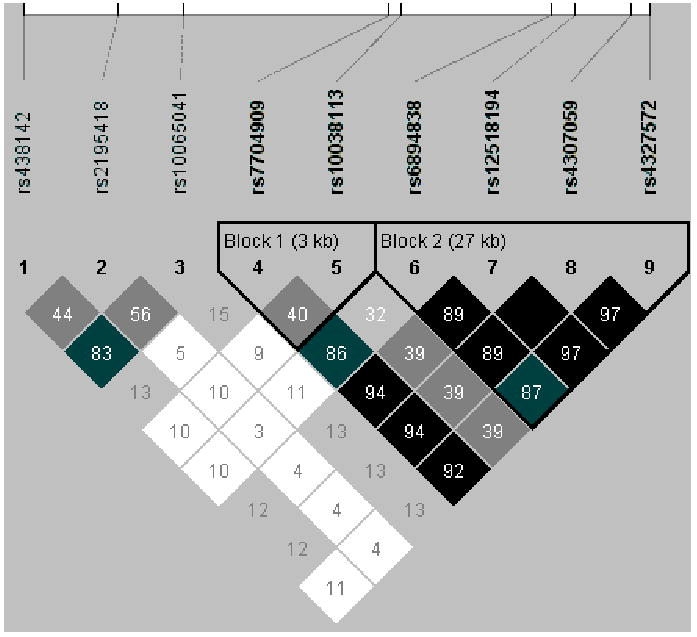
Note: Linkage disequilibrium (LD) was measured as r2 values, which range from 0 (no correlation) to 100 (complete correlation). LD was calculated between each pair of SNPs. Two blocks of strong LD are observed and span 3 Kb (SNPs 4-5) and 63 Kb (SNPs 6-9). Details of the analysis are given in the supplemental material.
Our power calculation shows that stringent adjustments for multiple testing provide power only to detect loci with large effects given our sample size. Lowering the threshold for significance allows detection of loci with relatively small effects (such as the chromosome 5 locus), while also relying on replication to limit the false positives. We note that this region of 5p14.1 did not generate exceptional p-values in our initial GWAS, suggesting that a strong single gene association, such as those seen with APOE gene in Alzheimer disease { and CFH gene in age related macular degeneration (International Multiple Sclerosis Genetics Consortium et al. 2007) is highly unlikely in autism. The absence of a large effect is consistent with the results of previously published linkage studies (Ma et al. 2007, Allen-Brady et al. 2008). Only through the analysis of the validation dataset were we able to identify this replicated signal, highlighting the value of both a validation dataset and of joint analyses. Two additional datasets have found association of autism at 5p14.1. These include a cohort of 1,241 ASD cases and 6,491 control subjects and a cohort of 108 ASD cases and 540 controls. The combined p-values for SNPs in the 5p14.1 region in these datasets combined with ours, which includes over 10,000 subjects, range from 7.4×10-8 to 2.1×10-10. These results survive stringent Bonferroni correction. (Wang et al. 2008)
Our approach, which uses a validation set as indication of a true association, has proven successful in other GWAS as exemplified by the identification of IL7RA and IL2RA susceptibility alleles in multiple sclerosis (MS) where no SNPs in either gene met genome-wide significance in the discovery dataset, but were confirmed through validation in an additional dataset (International Multiple Sclerosis Genetics Consortium et al. 2007). These MS findings recently have been confirmed across numerous datasets (International Multiple Sclerosis Genetics Consortium (IMSGC) 2008). We also note that other such common variants are likely to exist in autism and further GWAS studies are warranted.
Our identification and replication of common variation on chromosome 5p14.1 associated with autism is a promising development in the struggle to understand the genetics of autism. It also highlights the power of GWAS for detecting moderate genetic effects in neurobehavioral phenotypes. Our results suggest that in combination with the multiple rare variants already identified, that the genetic architecture of autism is as exquisitely complex as is its clinical phenotype.
Supplementary Material
Acknowledgments
We thank the patients with autism and their family members who participated in this study and personnel at the Miami Institute for Human Genomics (MIHG) including Sol Kissner from the MIHG Genetic Epidemiology and Statistical Genetics Core; Rachel Henson and Daniela Martinez from the MIHG Genotyping Core; staff at the MIHG Biorepository especially Sandra West; members of the MIHG and the Vanderbilt Center for Human Genetics Research autism ascertainment teams especially Laura Nations, Sandra Brinkley, Shannon Donnelly and Genea Crockett, Noelle Blackburn as well as Mary Margaret Welch for her expert editing and proofing of this paper. We would also like thank Dr. Michael Schmidt for his contributions to the data analysis and Drs. Jeffery M Vance, and Stephan Zuchner for their helpful comments and advice. This research was supported by grants from the National Institutes of Health (NIH) (NS26630, NS36768 and MH080647) and by a gift from the Hussman Foundation. Data management and analysis were performed in part using the Computational Genomics Core of the Vanderbilt Center for Human Genetics Research. We also acknowledge the partial support of the Autism Genome Project (AGP) which is supported by Autism Speaks. We also wish to gratefully acknowledge the resources provided by the AGRE consortium and the participating Autism Genetic Resource Exchange (AGRE) families. The AGRE resource is supported by Autism Speaks. A subset of the participants was ascertained while Dr Pericak-Vance was a faculty member at Duke University.
References
- Allen-Brady K, Miller J, Matsunami N, Stevens J, Block H, Farley M, Krasny L, Pingree C, Lainhart J, Leppert M, McMahon WM, Coon H. A high-density SNP genome-wide linkage scan in a large autism extended pedigree. Molecular psychiatry. 2008 doi: 10.1038/mp.2008.14. [DOI] [PubMed] [Google Scholar]
- Arking DE, Cutler DJ, Brune CW, Teslovich TM, West K, Ikeda M, Rea A, Guy M, Lin S, Cook EH, Chakravarti A. A common genetic variant in the neurexin superfamily member CNTNAP2 increases familial risk of autism. American Journal of Human Genetics. 2008;82(no. 1):160–164. doi: 10.1016/j.ajhg.2007.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Autism Genetics Resource Exchange. AGRE [Homepage of Autism Speaks] 2008 2008-last update. [Online]. Available: http://www.agre.org/ [2008, July]
- Bailey A, Le Couteur A, Gottesman I, Bolton P, Simonoff E, Yuzda E, Rutter M. Autism as a strongly genetic disorder: evidence from a British twin study. Psychological medicine. 1995;25:63–77. doi: 10.1017/s0033291700028099. [DOI] [PubMed] [Google Scholar]
- Bakkaloglu B, O'Roak BJ, Louvi A, Gupta AR, Abelson JF, Morgan TM, Chawarska K, Klin A, Ercan-Sencicek AG, Stillman AA, Tanriover G, Abrahams BS, Duvall JA, Robbins EM, Geschwind DH, Biederer T, Gunel M, Lifton RP, State MW. Molecular cytogenetic analysis and resequencing of contactin associated protein-like 2 in autism spectrum disorders. American Journal of Human Genetics. 2008;82(no. 1):165–173. doi: 10.1016/j.ajhg.2007.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolton P, Macdonald H, Pickles A, Rios P, Goode S, Crowson M, Bailey A, Rutter M. A case-control family history study of autism. Journal of Child Psychology & Psychiatry & Allied Disciplines. 1994;35:877–900. doi: 10.1111/j.1469-7610.1994.tb02300.x. [DOI] [PubMed] [Google Scholar]
- Centers for Disease Control. Autism [Homepage of Centers for Disease Control] 2008 2008-last update. [Online]. Available: http://www.cdc.gov/ncbddd/autism/ [2008, July]
- Chakrabarti S, Fombonne E. Pervasive developmental disorders in preschool children: confirmation of high prevalence. The American Journal of Psychiatry. 2005;162:1133–1141. doi: 10.1176/appi.ajp.162.6.1133. [DOI] [PubMed] [Google Scholar]
- Choi IJ, Jeong HJ, Han DS, Lee JS, Choi KH, Kang SW, Ha SK, Lee HY, Kim PK. An analysis of 4,514 cases of renal biopsy in Korea. Yonsei medical journal. 2001;42:247–254. doi: 10.3349/ymj.2001.42.2.247. [DOI] [PubMed] [Google Scholar]
- Dudbridge F. Likelihood-based association analysis for nuclear families and unrelated subjects with missing genotype data. Human Heredity. 2008;66(no. 2):87–98. doi: 10.1159/000119108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabriel SB, Schaffner SF, Nguyen H, Moore JM, Roy J, Blumenstiel B, Higgins J, DeFelice M, Lochner A, Faggart M, Liu-Cordero SN, Rotimi C, Adeyemo A, Cooper R, Ward R, Lander ES, Daly MJ, Altshuler D. The structure of haplotype blocks in the human genome. Science. 2002;296(no. 557):2225–2229. doi: 10.1126/science.1069424. [DOI] [PubMed] [Google Scholar]
- International HapMap Consortium. Frazer KA, Ballinger DG, Cox DR, Hinds DA, Stuve LL, Gibbs RA, Belmont JW, Boudreau A, Hardenbol P, Leal SM, Pasternak S, Wheeler DA, Willis TD, Yu F, Yang H, Zeng C, Gao Y, Hu H, Hu W, Li C, Lin W, Liu S, Pan H, Tang X, Wang J, Wang W, Yu J, Zhang B, Zhang Q, Zhao H, Zhao H, Zhou J, Gabriel SB, Barry R, Blumenstiel B, Camargo A, Defelice M, Faggart M, Goyette M, Gupta S, Moore J, Nguyen H, Onofrio RC, Parkin M, Roy J, Stahl E, Winchester E, Ziaugra L, Altshuler D, Shen Y, Yao Z, Huang W, Chu X, He Y, Jin L, Liu Y, Shen Y, Sun W, Wang H, Wang Y, Wang Y, Xiong X, Xu L, Waye MM, Tsui SK, Xue H, Wong JT, Galver LM, Fan JB, Gunderson K, Murray SS, Oliphant AR, Chee MS, Montpetit A, Chagnon F, Ferretti V, Leboeuf M, Olivier JF, Phillips MS, Roumy S, Sallee C, Verner A, Hudson TJ, Kwok PY, Cai D, Koboldt DC, Miller RD, Pawlikowska L, Taillon-Miller P, Xiao M, Tsui LC, Mak W, Song YQ, Tam PK, Nakamura Y, Kawaguchi T, Kitamoto T, Morizono T, Nagashima A, Ohnishi Y, Sekine A, Tanaka T, Tsunoda T, Deloukas P, Bird CP, Delgado M, Dermitzakis ET, Gwilliam R, Hunt S, Morrison J, Powell D, Stranger BE, Whittaker P, Bentley DR, Daly MJ, de Bakker PI, Barrett J, Chretien YR, Maller J, McCarroll S, Patterson N, Pe'er I, Price A, Purcell S, Richter DJ, Sabeti P, Saxena R, Schaffner SF, Sham PC, Varilly P, Altshuler D, Stein LD, Krishnan L, Smith AV, Tello-Ruiz MK, Thorisson GA, Chakravarti A, Chen PE, Cutler DJ, Kashuk CS, Lin S, Abecasis GR, Guan W, Li Y, Munro HM, Qin ZS, Thomas DJ, McVean G, Auton A, Bottolo L, Cardin N, Eyheramendy S, Freeman C, Marchini J, Myers S, Spencer C, Stephens M, Donnelly P, Cardon LR, Clarke G, Evans DM, Morris AP, Weir BS, Tsunoda T, Mullikin JC, Sherry ST, Feolo M, Skol A, Zhang H, Zeng C, Zhao H, Matsuda I, Fukushima Y, Macer DR, Suda E, Rotimi CN, Adebamowo CA, Ajayi I, Aniagwu T, Marshall PA, Nkwodimmah C, Royal CD, Leppert MF, Dixon M, Peiffer A, Qiu R, Kent A, Kato K, Niikawa N, Adewole IF, Knoppers BM, Foster MW, Clayton EW, Watkin J, Gibbs RA, Belmont JW, Muzny D, Nazareth L, Sodergren E, Weinstock GM, Wheeler DA, Yakub I, Gabriel SB, Onofrio RC, Richter DJ, Ziaugra L, Birren BW, Daly MJ, Altshuler D, Wilson RK, Fulton LL, Rogers J, Burton J, Carter NP, Clee CM, Griffiths M, Jones MC, McLay K, Plumb RW, Ross MT, Sims SK, Willey DL, Chen Z, Han H, Kang L, Godbout M, Wallenburg JC, L'Archeveque P, Bellemare G, Saeki K, Wang H, An D, Fu H, Li Q, Wang Z, Wang R, Holden AL, Brooks LD, McEwen JE, Guyer MS, Wang VO, Peterson JL, Shi M, Spiegel J, Sung LM, Zacharia LF, Collins FS, Kennedy K, Jamieson R, Stewart J. A second generation human haplotype map of over 3.1 million SNPs. Nature. 2007;449(no. 7164):851–861. doi: 10.1038/nature06258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- International Molecular Genetic Study of Autism Consortium (IMGSAC) A genomewide screen for autism: strong evidence for linkage to chromosomes 2q, 7q, and 16p. American Journal of Human Genetics. 2001;69(no. 3):570–581. doi: 10.1086/323264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- International Multiple Sclerosis Genetics Consortium (IMSGC) Refining genetic associations in multiple sclerosis. Lancet neurology. 2008;7(no. 7):567–569. doi: 10.1016/S1474-4422(08)70122-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- International Multiple Sclerosis Genetics Consortium. Hafler DA, Compston A, Sawcer S, Lander ES, Daly MJ, De Jager PL, de Bakker PI, Gabriel SB, Mirel DB, Ivinson AJ, Pericak-Vance MA, Gregory SG, Rioux JD, McCauley JL, Haines JL, Barcellos LF, Cree B, Oksenberg JR, Hauser SL. Risk alleles for multiple sclerosis identified by a genomewide study. The New England journal of medicine. 2007;357(no. 9):851–862. doi: 10.1056/NEJMoa073493. [DOI] [PubMed] [Google Scholar]
- Liu Q, Duff RJ, Liu B, Wilson AL, Babb-Clendenon SG, Francl J, Marrs JA. Expression of cadherin10, a type II classic cadherin gene, in the nervous system of the embryonic zebrafish. Gene expression patterns : GEP. 2006;6(no. 7):703–710. doi: 10.1016/j.modgep.2005.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma DQ, Cuccaro ML, Jaworski JM, Haynes CS, Stephan DA, Parod J, Abramson RK, Wright HH, Gilbert JR, Haines JL, Pericak-Vance MA. Dissecting the locus heterogeneity of autism: significant linkage to chromosome 12q14. Molecular psychiatry. 2007;12(no. 4):376–384. doi: 10.1038/sj.mp.4001927. [DOI] [PubMed] [Google Scholar]
- Marchini J, Howie B, Myers S, McVean G, Donnelly P. A new multipoint method for genome-wide association studies by imputation of genotypes. Nature genetics. 2007;39(no. 7):906–913. doi: 10.1038/ng2088. [DOI] [PubMed] [Google Scholar]
- Martin ER, Bass MP, Kaplan NL. Correcting for a potential bias in the pedigree disequilibrium test. Am J Hum Gen. 2001;68:1065–1067. doi: 10.1086/319525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin ER, Monks SA, Warren LL, Kaplan NL. A test for linkage and association in general pedigrees: the pedigree disequilibrium test. American Journal of Human Genetics. 2000;67:146–154. doi: 10.1086/302957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NCBI. Online Mendelian Inheritance of Man, Autism [Homepage of John's Hopkins University] 2008 2008-last update. [Online]. Available: http://www.ncbi.nlm.nih.gov/entrez/dispomim.cgi?id=209850 [2008, July]
- Patterson N, Price AL, Reich D. Population Structure and Eigenanalysis. PLoSGenet. 2006;2(no. 1):e190. doi: 10.1371/journal.pgen.0020190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pokutta S, Weis WI. Structure and mechanism of cadherins and catenins in cell-cell contacts. Annual Review of Cell and Developmental Biology. 2007;23:237–261. doi: 10.1146/annurev.cellbio.22.010305.104241. [DOI] [PubMed] [Google Scholar]
- Purcell S, Cherny SS, Sham PC. Genetic Power Calculator [Homepage of Harvard University] 2008 [Online]. Available: http://pngu.mgh.harvard.edu/∼purcell/gpc/ [2008,
- Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, Sham PC. PLINK: a tool set for whole-genome association and population-based linkage analyses. American Journal of Human Genetics. 2007;81(no. 3):559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebat J, Lakshmi B, Malhotra D, Troge J, Lese-Martin C, Walsh T, Yamrom B, Yoon S, Krasnitz A, Kendall J, Leotta A, Pai D, Zhang R, Lee YH, Hicks J, Spence SJ, Lee AT, Puura K, Lehtimaki T, Ledbetter D, Gregersen PK, Bregman J, Sutcliffe JS, Jobanputra V, Chung W, Warburton D, King MC, Skuse D, Geschwind DH, Gilliam TC, Ye K, Wigler M. Strong association of de novo copy number mutations with autism. Science (New York, N Y) 2007;316(no. 5823):445–449. doi: 10.1126/science.1138659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao Y, Wolpert CM, Raiford KL, Menold MM, Donnelly SL, Ravan SA, Bass MP, McClain C, von Wendt L, Vance JM, Abramson RH, Wright HH, Ashley-Koch A, Gilbert JR, DeLong RG, Cuccaro ML, Pericak-Vance MA. Genomic screen and follow-up analysis for autistic disorder. American Journal of Medical Genetics. 2002;114:99–105. doi: 10.1002/ajmg.10153. [DOI] [PubMed] [Google Scholar]
- Sparrow SS, Balla D, Cicchetti D. Vineland Adaptive Behavior Scales, Interview Edition. AGS Publishing; Circle Pines, MN: 1984. [Google Scholar]
- Sparrow SS, Cicchetti DV, Balla D. Vineland Adaptive Behavior Scales--Second Edition. AGS; Circle Pines, MN: 2005. [Google Scholar]
- Steffenburg S, Gillberg C, Hellgren L, Andersson L, Gillberg IC, Jakobsson G, Bohman M. A twin study of autism in Denmark, Finland, Iceland, Norway, and Sweden. Journal of child psychology and psychiatry, and allied disciplines. 1989;30:405–416. doi: 10.1111/j.1469-7610.1989.tb00254.x. [DOI] [PubMed] [Google Scholar]
- Szatmari P, Paterson AD, Zwaigenbaum L, Roberts W, Brian J, Liu XQ, Vincent JB, Skaug JL, Thompson AP, Senman L, Feuk L, Qian C, Bryson SE, Jones MB, Marshall CR, Scherer SW, Vieland VJ, Bartlett C, Mangin LV, Goedken R, Segre A, Pericak-Vance MA, Cuccaro ML, Gilbert JR, Wright HH, Abramson RK, Betancur C, Bourgeron T, Gillberg C, Leboyer M, Buxbaum JD, Davis KL, Hollander E, Silverman JM, Hallmayer J, Lotspeich L, Sutcliffe JS, Haines JL, Folstein SE, Piven J, Wassink TH, Sheffield V, Geschwind DH, Bucan M, Brown WT, Cantor RM, Constantino JN, Gilliam TC, Herbert M, Lajonchere C, Ledbetter DH, Lese-Martin C, Miller J, Nelson S, Samango-Sprouse CA, Spence S, State M, Tanzi RE, Coon H, Dawson G, Devlin B, Estes A, Flodman P, Klei L, McMahon WM, Minshew N, Munson J, Korvatska E, Rodier PM, Schellenberg GD, Smith M, Spence MA, Stodgell C, Tepper PG, Wijsman EM, Yu CE, Roge B, Mantoulan C, Wittemeyer K, Poustka A, Felder B, Klauck SM, Schuster C, Poustka F, Bolte S, Feineis-Matthews S, Herbrecht E, Schmotzer G, Tsiantis J, Papanikolaou K, Maestrini E, Bacchelli E, Blasi F, Carone S, Toma C, van Engeland H, de Jonge M, Kemner C, Koop F, Langemeijer M, Hijimans C, Staal WG, Baird G, Bolton PF, Rutter ML, Weisblatt E, Green J, Aldred C, Wilkinson JA, Pickles A, Le Couteur A, Berney T, McConachie H, Bailey AJ, Francis K, Honeyman G, Hutchinson A, Parr JR, Wallace S, Monaco AP, Barnby G, Kobayashi K, Lamb JA, Sousa I, Sykes N, Cook EH, Guter SJ, Leventhal BL, Salt J, Lord C, Corsello C, Hus V, Weeks DE, Volkmar F, Tauber M, Fombonne E, Shih A. Mapping autism risk loci using genetic linkage and chromosomal rearrangements. Nature genetics. 2007;39:319–328. doi: 10.1038/ng1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Haitao Z, Ma D, Bucan M, Glessner JT, Abrahams BS, Salyakina D, Imielinski M, Bradfield JP, Sleiman PMA, Kim CE, Chiavacci R, Lajonchere C, Munson J, Estes A, Korvatska O, Piven J, Sonnenblick LI, Alvarez Retuerto AI, Herman EI, Dong H, Hutman T, Sigman M, Ozonoff S, Klin A, Owley T, Sweeney JA, Brune CW, Cantor RM, Bernier R, Gilbert JR, Cuccaro ML, Wassink TH, McMahon WM, Coon H, Miller J, Nurnberger JI, State MW, Haines JL, Sutcliffe JS, Cook E, Minshew N, Buxbaum JD, Dawson G, Grant SFA, Geschwind DH, Pericak-Vance MA, Schellenberg GD, Hakonarson H. Common genetic variation in the intragenic region between CDH10 and CDH9 is associated with susceptibility to autism spectrum disorders. Nature 2008 In Press. [Google Scholar]
- Weiss LA, Shen Y, Korn JM, Arking DE, Miller DT, Fossdal R, Saemundsen E, Stefansson H, Ferreira MA, Green T, Platt OS, Ruderfer DM, Walsh CA, Altshuler D, Chakravarti A, Tanzi RE, Stefansson K, Santangelo SL, Gusella JF, Sklar P, Wu BL, Daly MJ the Autism Consortium. Association between Microdeletion and Microduplication at 16p11.2 and Autism. The New England journal of medicine. 2008 doi: 10.1056/NEJMoa075974. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.